

Abstract
Membrane transporters are key gatekeeper proteins at cellular membranes that closely control the traffic of materials. Their function relies on structural rearrangements of varying degrees that facilitate substrate translocation across the membrane. Characterizing these functionally important molecular events at a microscopic level is key to our understanding of membrane transport, yet challenging to achieve experimentally. Recent advances in simulation technology and computing power have rendered molecular dynamics (MD) simulation a powerful biophysical tool to investigate a wide range of dynamical events spanning multiple spatial and temporal scales. Here, we review recent studies of diverse membrane transporters using computational methods, with an emphasis on highlighting the technical challenges, key lessons learnt, and new opportunities to illuminate transporter structure and function.
Keywords: Membrane transporter, molecular dynamics simulation, conformational change, free energy calculation, lipid-protein interaction, drug target
1. Membrane Transporter: The Major Regulator of Solute Exchange
Living cells rely on the exchange of diverse materials across the cellular and organellar membranes for proper function. Transmembrane material exchange is regulated by membrane transport proteins, which serve as specialized gatekeepers to selectively mediate the translocation of a plethora of substrates ranging from single-atomic ions to large biological macromolecules. Among these structurally and mechanistically diverse gatekeepers, membrane transporters provide the molecular machinery to drive active transmembrane movement of substrates, often against their electrochemical gradient. This special feature is enabled through coupling with various forms of cellular energy (e.g., light, chemical reactions, or electrochemical gradients) that can be utilized to fuel the transport processes (Box 1). The fundamental role of transporters in governing the traffic of molecular species across the membrane has made them therapeutic drug targets for a wide range of diseases, which continues to stimulate mechanistic investigation of these important molecular devices.
Box 1. Knowledge Corner: Transporters and Alternating Access Mechanisms.
The cellular membranes are effective diffusion barriers that separate the interior of a cell from its surroundings. The entry and exit of solutes across the membranes are mediated by transport proteins embedded in membranes. Transporters are major transport proteins that can utilize energy to actively drive the transmembrane movement of solutes in a selective manner. The driving energy of the translocation reaction could be light or chemical energy (e.g., ATP) for primary transporters, or electrochemical gradients of other solutes (e.g., H+ and Na+ ions) for secondary transporters [3, 4, 72–74]. Depending on the direction of transport and whether a coupling solute is used, transporters can be classified into uniporters, symporters, and antiporters. Uniporters transport a single type of substrate down the concentration gradient; while symporters and anitporters transport a substrate against its concentration gradient by cotransport or countertransport of other solutes [3, 4, 72].
A model capturing the essence of the transport mechanism in transporters is known as the “alternating-access model” [1, 2], proposed by Jardetzky more than 50 years ago, which shaped the theoretical understanding of substrate transport across cell membranes. The essence of this model is that transporters can only accept or release the substrate from/at one side of the membrane at any time, and that the active transport is achieved by alternating between these conformations of different accessibility. In the outward-facing (OF) state, the substrate binding site is exclusively exposed to the extracellular side of the cell or the interior of an organelle. In the inward-facing (IF) state, the binding site is accessible only from the cytosol. Transporters transition through intermediate occluded state(s) between the OF and IF states, where the binding site is obstructed from both sides of the membrane.
Figure I.

A schematic illustration of the conformational changes in alternating-access models. The transport can occur through rocker-switch (left), rocking-bundle (middle), or elevator-type (right) mechanisms, during which the transporter alternates between the OF (top) and IF (bottom) states. Each transporter is schematically represented as two domains (pink and blue) with stationary domains in shaded colors. Conformational transitions are shown as the changes in the relative angles between the domains and the water accessibility of the transporters.
Depending on the nature of the structural rearrangement of the protein domains, alternating access transport can occur through “rocker-switch”, “rocking-bundle”, or “elevator-type” mechanisms (Figure I) [3, 4, 72]. The rocker-switch or rocking-bundle mechanisms involve domain rearrangements around a central substrate binding site, during which the bound substrate remains relatively static with respect to the membrane. The rocker-switch mechanism is used widely in the major facilitator superfamily (MFS), where the two domains move to switch the accessibility of the substrate binding site. The rocking-bundle mechanism is adopted mostly by LeuT-fold transporters, in which the bundle domain rearranges with respect to a rather static scaffold domain to expose the substrate binding site. The elevator-type mechanism describes a process in which the transport domain undergoes a more “vertical” translation (along the membrane normal) relative to a static scaffold domain to carry the cargo across the membrane. This mechanism is exemplified in glutamate transporter Gltph, dicarboxylate transporter VcINDY, and Na+/H+ antiporter NapA.
The function of transporters is tightly and particularly linked to their structural dynamics. The widely accepted “alternating-access model [1, 2]” proposes that transporters need to undergo conformational transitions among the outward-facing (OF), occluded, and inward-facing (IF) states to alternatively expose the substrate binding site to either side of the membrane (Box 1) (Figure 1, Key Figure). Generally speaking, this conformation-coupled mechanism effectively prevents the leakage of cellular components and is vital for uni-directional conveyance of substrates against their concentration gradient in active transport [3, 4]. While X-ray crystallography (see Glossary) and Cryo-electron microscopy (cryo-EM) have resulted in a growing collection of high-resolution structures of transporters in different conformational states (for example, for ABC transporters [5] and neurotransmitter-sodium symporters [6]), microscopic details of the transitions between these states, as well as the structural coupling of these transitions to the driving energy source—which is key to the transport mechanism, remain largely unknown.
Figure 1, Key figure. Major functional aspects of membrane transporters.

(A) Schematic representation of major conformational states visited by a membrane transporter during the alternating access mechanism (see Box 1) as it facilitates the substrate and/or ion transport. Protein is represented by a white cartoon while the membrane by a brown disc. Ions and substrate are represented by yellow circle and blue hexagon, respectively. (B) Local conformational changes. A typical monomer of fully-bound glutamate transporter (Glt) with scaffold domain shown in pink and transport domain in blue. HP1 and HP2 loops which control the accessibility of the substrate binding site and therefore involved in gating, are shown in green. In the fully bound state (with 3 bound Na+ and the substrate), the transporter remains closed throughout the 500 ns of MD simulations (starting snapshots of HP1 and HP2 are shown in grey). MD simulations of the partially bound state (with two bound Na+ ions) captures the opening of HP2 gate, thus highlighting the importance of substrate and third Na+ in locking the extracellular gate of the transporter. (C) Global structural transition. Crystal and cryo-EM structures of ABC-transporter Pgp in IF (left, PDB: 4M1M) and OF (right, PDB: 6C0V) states, respectively. Pgp is a heterodimer comprising of two pseudosymetric halves (shown in blue and pink surface representations, respectively) each containing a transmembrane domain (TMD) connected to a nucleotide-binding domain (NBD). Binding of ATP (shown in van der Waals) to the NBDs leads to their dimerization and transition of the transporter from the IF to the OF state. The arrows depict the binding of the substrate molecule to the lumen in the IF state and its release to the extracellular side in the OF state, respectively. (D) Lipid-protein interactions. Crystal structure of ABC transporter MsbA (left, PDB: 5TV4), an inner membrane lipid flippase, reveals a lipopolysaccharide (LPS) molecule bound deeply in the cavity, shedding light on the mechanism of MsbA-mediated LPS transport. The two monomers of the transporter are shown as blue and pink cartoon representations, respectively. The bound LPS is shown as sticks. Crystal structure of dopamine transporter (DAT) (right, PDB: 4M48), a neurotransmitter transporter, reveals a cholesterol molecule bound at the junction of TM5 (blue) and TM7 (pink). The protein is drawn in cartoon representation, whereas the cholesterol and the coordinating residues are shown as sticks. (E) Drug-transporter interactions. Interaction of serotonin transporter (SERT) with paroxetine (left, PDB: 5I6X) and ibogaine (right, PDB: 6DZZ) in its different functional states, respectively. The drugs wedge between scaffold (pink) and core helices (blue), potentially interfering with their structural transition during the transporter function. The bound drugs and the extracellular gating residues are highlighted as sticks. The blue curves indicate the exposure of the central binding pocket.
Computational techniques such as molecular dynamics (MD) simulations offer high spatial and temporal resolutions necessary to scrutinize mechanistically important aspects of membrane transporters, including translocation of the substrate, the structural interplay between the energy source and the protein, and the overall conformational transitions during the transport cycle. The increased availability of faster and more powerful computing resources, and the development of more efficient algorithms during the past decade, have made it possible now to perform long timescale simulations, investigate complex reaction processes, and accurately calculate the free energies associated with them. In this review, we present an overview of exemplary studies in recent years that demonstrate the power of MD-based methods in deciphering important elements contributing to the mechanism of transporters. The research covered here highlights how MD simulations can complement experimental techniques by providing a dynamic, microscopic view of the underlying molecular mechanisms, guide future studies, and aide the development of novel molecular therapeutics. In the following sections, we will first present studies in which the mechanisms of energy coupling were investigated. Then, we turn our attention to computational studies aiming at describing large-scale transitions of membrane transporters. The following section will focus on lipid-protein interactions and their functional ramifications, and in the last section we will describe simulation studies where the role of membrane transporters as major drug targets has been investigated.
2. Unveiling the Role of the Driving Forces
The transport cycles of membrane transporters involve highly diverse structural changes, ranging from local arrangements at substrate binding sites to global conformational transitions that switch the accessibility of the site to either side of the membrane. These structural changes are fueled by specific energy-providing sources, including electrochemical gradient of ions (most prominently Na+ and H+) for secondary active transporters, and chemical reactions (e.g., ATP hydrolysis) for primary active transporters (Box 1). The coupling between energy sources and protein dynamics, and how it affects the binding and translocation of the driven substrate, are the central questions underlying the active transport mechanism.
Na+-coupled transporters
Recent computational studies on various Na+-coupled transporters have illustrated the dynamic behaviors of Na + ions and their pivotal role in the transport cycle. For example, extended equilibrium simulations have not only captured the binding and unbinding of Na + ions, but also revealed the ensuing structural responses in the transporters (Figure 1B) [7–13].
Equilibrium simulations of leucine transporter LeuT in its OF state have successfully described a sequence of Na+ binding events and the coupled uptake of substrate [7], which cooperatively bridge an inter-domain interaction network essential for the cytoplasmic closure [8, 9]. Na+-triggered closing of the extracellular gate was also captured in the substrate-bound glutamate transporter GltTk, highlighting the coupling between the driving force and the driven substrate [10, 11]. A recent GltTk structure has provided an important clue on the role of a conserved methionine in the Na+ binding site. A more rigorous re-parametrization of this conserved methionine was shown to successfully reproduce the binding free energy of Na+ in close agreement with the experimental data [14], highlighting the importance of polarization effects, which are largely absent from standard classical simulations.
Attributed to the advances in the long-timescale simulations, stochastic release of Na+ and sugar into the cytosol was captured in sugar transporter vSGLT [13]. As yet another example, analysis of the Na + release kinetics using Markov state models (MSMs) (Table 1) enabled the identification of distinct Na + unbinding/release pathways from the human dopamine transporter hDAT [15, 16]. Besides conventional MD, the application of free energy perturbation (FEP) (Table 1) allows one to calculate the free energy differences between the binding of Na+ versus other cationic ions, highlighting, e.g., the functional specificity of Na+ in the multidrug transporter PfMATE [17] or characterizing cation binding sites in K+-dependent Na+/Ca2+ exchanger NCKX2 [18, 19].
Table 1:
Computational methods used by studies reported in this review.
| Acronym | Method | Synopsis | Application | Refs |
|---|---|---|---|---|
| QM/MM | Hybrid Quantum Mechanics / Molecular Mechanics | A subset of system simulated quantum mechanically (embedded in a larger MM environment) in order to allow bond breaking/formation, or to include molecules that cannot be adequately described by classical force fields | H+ transport | [21, 23, 24, 27] |
| ATP hydrolysis | [31] | |||
| MS-RMD | Multiscale Reactive Molecular Dynamics | Using purposely derived reactive force field to treat bond breaking/formation | H+ transport | [21, 24–27] |
| CpHMD | Constant pH Molecular Dynamics | MD simulation allowing H+ exchange at specified sites (e.g., titratable protein side chain) | Determine pKa of residue and pH-induced conformational transition | [35] |
| MDFF | Molecular Dynamics Flexible Fitting | Structural refinement method that dynamically fits an atomic structure into experimentally determined electron densities | Refine low resolution molecular structure | [20] |
| FEP/TI | Free Energy Perturbation / Thermodynamic Integration | Calculate the energy difference between two related states by alchemically morphing one into the other | Calculate binding energy | [10, 14, 17–19, 45, 64, 68] |
| determine residue pKa | [20] | |||
| SMD | Steered Molecular Dynamics | Applying a force or a moving restraint to bias the movement of a (group of) atom(s), either in real space or in collective variable space | Probe conformational transitions | [11, 20, 42–45] |
| Drug unbinding | [62, 64] | |||
| US | Umbrella Sampling | Restraining the system at multiple points along a specified reaction coordinate to calculate the potential of mean force | Free energy calculations along specified reaction coordinate | [24–26, 30, 45, 54] |
| MetaD | Metadynamics | Calculate potential of mean force along a collective variable by inversely summing Gaussian potentials added during simulation to overcome energy barriers | ||
| aMD | Accelerated Molecular Dynamics | Enhanced sampling method where a biasing potential is applied to the dihedral and/or total potential of the system | Substrate binding | [40] |
| Conformational change | [40, 41] | |||
| MSM | Markov State Model | Describe the kinetic of the process using the transition rates between different substates, typically calculated from very long or many discrete MD simulations | Ion release pathway | [16] |
| Conformation change | [41, 49] | |||
| String Method | Path search/optimization method to obtain a pathway connecting two states with a least barrier | Conformational change | [42, 45] | |
| Binding pathway | [64] | |||
| CG | Coarse Grained Simulation | Represent simulation systems with coarser particles, each representing a group of atoms | Lipid binding | [50, 53, 54] |
| Molecular Docking | Predict ligand binding pose and energy to a target protein | Predict substrate binding | [40] | |
| Predict lipid/detergent binding | [48] | |||
| Predict drug binding | [62, 67, 68] | |||
| Ensemble Docking | An ensemble of protein structures (often derived from MD simulations) are used for ligand docking, to account for the dynamics of the docking receptor | Predict drug binding | [63, 66] |
H+ -coupled transporters
For active transporters that are fueled by H+ , the effect of H+ binding (protonation) can be computationally probed by comparing the protein dynamics under different protonation states. This can be achieved by conventional classical MD simulations in which the protonation states of specific H+-binding residues/sites are altered in different simulations and the results compared. Using this method, recent studies on several H+-coupled transporters have demonstrated conformational changes induced by H + binding and highlighted the potential connection with substrate binding [20–22]. For example, a thorough sampling of all the possible protonation states of the two conformation-governing residues in multidrug resistance transporter EmrE revealed the coupling between dehydration of the lumen and protonation of these sites [20]. For uracil-H+ symporter UraA, protonating potential H+-binding residues within the substrate-binding region was shown to stabilize or disrupt substrate binding, implying the functional relevance of H+-mediated local structural rearrangements [22]. MD simulations were also utilized to determine the most probable protonation states of the key amino acids in PfMATE by comparing the local conformational stability in different protonation states [23].
To explicitly describe the process of H+ transfer (PT), advanced simulation techniques such as quantum-mechanics/molecular-mechanics (QM/MM) and multiscale reactive molecular dynamics (MS-RMD) (Table 1) are required [21, 23–26]. Combined with free energy or enhanced sampling methods such as umbrella sampling (US) and metadynamics (Table 1), QM/MM has been successfully utilized to quantify the free energy of localized PT in PfMATE, oligopeptide transporter PepTXc, and Cl−/H+ antiporter ClC-ec1 [21, 23, 24]. These studies demonstrated the coupling of PT to various conditions, including hydration level [21, 23], interaction with specific lipids [23], and substrate loading states [24]. In most QM/MM treatments, the QM subsystem is confined to a fixed selection of atoms, for example, the hopping H+ and its immediate surroundings, preventing material exchange between the QM region and the rest of the simulation system. Adaptive QM/MM (Table 1) addresses this limitation by allowing on-the-fly redefinition of the QM subsystem. Application of this technique to ClC-ec1 illustrated a stepwise PT [27] though a connected water wire previously discovered in conventional MD simulations [28, 29].
Due to the high computational demand, the free energy calculation in QM/MM simulations is usually limited to one reaction coordinate. To efficiently quantify the PT process and its coupled dynamics at a higher dimension, the MS-RMD method has been developed to accurately reproduce results from QM/MM calculations at a much lower cost. Recent studies employing MS-RMD in conjunction with enhanced sampling methods suggested that PT in ClC-ec1 might be coupled to the bound Cl− and revealed the underlying molecular basis for the outward and inward flux of H+ [24, 25]. A follow-up MD simulation study further confirmed that the Cl−-facilitated PT through the central region is partly or fully uncoupled when Cl− is substituted with other anions [26].
ATP-driven transporters
As a major driving force in primary transporters, the impact of ATP binding and hydrolysis has also been investigated computationally, revealing, e.g., the nucleotide-dependent dimerization and dissociation of the nucleotidebinding domains (NBDs) in ATP-binding cassette (ABC) transporters [30–32] (Figure 1C). Conventional MD combined with US showed that ATP binding stabilizes the NBD dimer by fostering an inter-NBD interaction network, which is disrupted or largely absent in ADP-bound or apo forms [30].
Consistently, the free energy of ATP hydrolysis calculated using QM/MM simulations suggested that the chemical energy released from the phosphodiester bond cleavage is not the main driving force for the NBD separation [31]. Rather, the phosphate unbinding [33] and water entropy change [34] may be the main elements contributing to this process. In addition, through analyses of equilibrium MD trajectories, the intrinsic dynamics of individual NBD monomers are found to be nucleotide-dependent, and shown to leads to an inter-NBD motion that supports an asymmetric ATP hydrolysis scheme in a symmetric NBD dimer [32].
The examples provided above well demonstrate that MD simulation offers a powerful tool to characterize the interplay between energy sources and diverse aspects of transporter function.
3. Characterizing Global Structural Dynamics
Capturing Spontaneous Dynamics with MD Simulations
Transporters operate by undergoing a series of transitions between major functional (i.e., IF, OF, and intermediate) states (Box 1, Fig. 1), and MD simulations are often used to investigate the global (and frequently large-scale) structural dynamics involved in these transitions. The most basic form such an investigation can take is an examination of the effect substrates, ions, inhibitors, lipids, etc. have on the global structural dynamics of a transporter. As an example, MD was recently used to study the effects different combinations of bound ligands have on the global dynamics of the two NBDs of the ABC transporter MJ0796 [32].
Accurate investigations into the global dynamics of transporters sometimes require more detail than standard MD provides, and a recent trend has been to use more advanced flavors of MD in these investigations. One example is constant pH MD (CpHMD; Table 1), a technique in which the protonation states of titratable groups (e.g., amino acids) are updated automatically throughout the simulation to reflect changes in their local environments. CpHMD simulations were recently used to determine the structural details of the transition between two functional states in the multidrug efflux transporter AcrB, which occurred spontaneously after a proton was released from the transporter [35].
When used to investigate the global structural dynamics of a transporter, MD simulations are most commonly performed with the transporter in a single functional state at a time. However, MD simulations do occasionally capture the spontaneous transition of a transporter between a pair of major functional states, and, with longer MD simulations being made possible, these spontaneous transitions are being observed with increasing frequency. Recently, such spontaneous transitions were observed in MD simulations of the dopamine transporter hDAT [36], the ABC transporter TM287/288 [37], and the sugar transporter SemiSWEET [38, 39].
Driving Conformational Changes with Enhanced Sampling MD Simulations
While it is possible to occasionally observe spontaneous transitions between major functional states of a transport cycle in standard MD simulations, the timescales of these transitions for most transporters (milliseconds to seconds or greater) are beyond the reach of standard MD simulations. To make it possible for MD simulations to capture full structural transitions and additionally obtain detailed thermodynamic information associated with the transitions, a variety of advanced MD-based techniques designed to enhance conformational sampling have been developed and applied to transporters. A small selection of these techniques include accelerated MD (aMD), steered MD (SMD), the string method, and US. In an exciting trend, the application of advanced MD-based techniques like these to transporters has become increasingly common as advances in computer hardware have made their application more computationally feasible.
In aMD, enhanced sampling is achieved by applying biases to energy terms in the MD simulation, most commonly the dihedral energy and/or total energy of the system. This technique has recently been used to study the transition between states of the dopamine transporter dDAT [40] and the oligopeptide transporter PepTSo [41]. In SMD, moving harmonic restraints along a set of one or more system-specific reaction coordinates are used to induce a transition between two states. SMD can be followed up by more advanced MD-based techniques to relax the transition pathway between the end states (e.g., using the string method) and obtain thermodynamic information associated with the transition pathway (e.g., using US). SMD and successive enhanced sampling techniques have been used on transitions in the glycerol-3-phosphate transporter GlpT (Fig. 2) [42], the Na+/aspartate symporter Gltph [43], the drug/metabolite transporter YddG [44], and AcrB [45].
Figure 2. MD simulations applied to study the energetics of large scale structural transition in an explicit membrane/solvent environment.
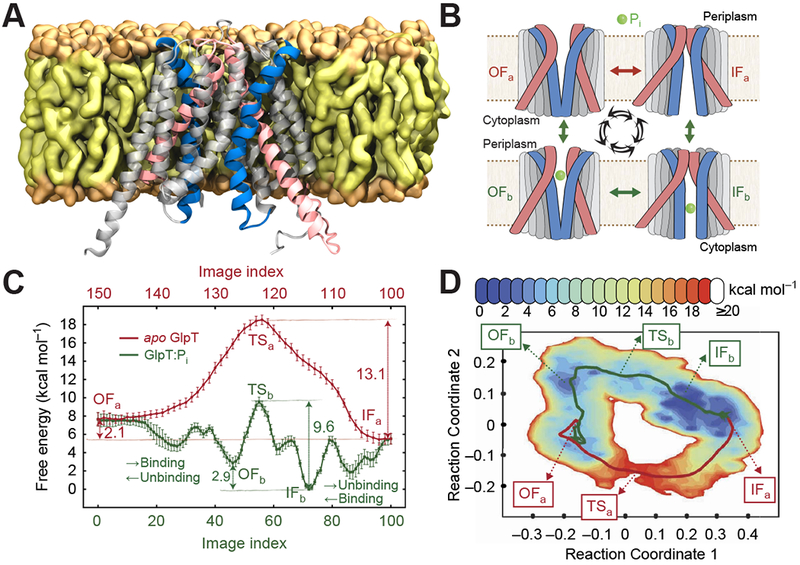
(A) GlpT in an inward facing (IF) open conformation is represented as cartoon, embedded in an explicit lipid bilayer which is shown in surface representation. The helices involved in biasing the transition are shown as pink and blue cartoon. (B) Schematic representation of the states involved in the thermodynamic cycle studied (colored as in A). (C) Free energy profile along the thermodynamic cycle shown in (A), involving both apo (red) and bound (green) GlpT. The free energy along the transition pathway (discretized into 150 images/windows) was computed employing an umbrella sampling method. The free energy calculations identify all the intermediate states involved in the transport cycle and show that the apo transition is unfavorable while the transition of the phosphate-bound (Pi-bound) state is feasible. (D) Potential of mean force (PMF) projected along the two reaction coordinates, showing two distinct pathways in red and blue. The blue pathway is the lower free energy pathway explaining the energetics of allowed GlpT transition in the presence of the substrate Pi. The red pathway, on the other hand, is a higher free energy pathway that explains forbidden transition of GlpT in the absence of substrate. Adapted with permission from ref [42]. Copyright 2015 Moradi et al. Licensed under a Creative Commons Attribution 4.0 International License.
As with all computational and experimental techniques, enhanced sampling MD techniques each come with their own unique set of strengths and limitations. For example, the effectiveness of SMD, the string method, and US is highly dependant on defining reaction coordinates that accurately, specifically, and comprehensively describe the transitions between transporters’ major functional states. Conversely, the biases applied in aMD are general, obviating the need to define reaction coordinates, but this general nature also means that there is no guarantee that an aMD simulation will sample the transitions of interest. To give another example of the strengths and limitations of enhanced sampling MD techniques, because such techniques necessarily attempt to speed up transitions, great care must be taken to ensure the transporters’ environments, especially the lipids in the membrane, are not pulled out of equilibrium during the simulation. Otherwise, the simulation will not be able to provide quantitatively meaningful results.
Global structural transitions underpin the function of all transporters, and MD is an excellent tool to obtain detailed structural and thermodynamic information about these functional underpinnings. As computer hardware continues to improve, the frequency of MD-based investigations into the global structural dynamics of transporters will only continue to increase.
4. Active Participation of Lipids in Transporter Function
Membrane lipids are now recognized to be active contributors to various transport processes by regulating the structure and function of transporters [46]. Recent MD simulations have successfully demonstrated that transporter dynamics may strongly depend on the physicochemical properties and lipid compositions of the membranes. Recent studies using various computational methods as well as the incorporation of experimental information have highlighted the regulatory role of lipids in modulating conformational transitions [23, 36, 47] and assisting substrate binding and translocation inside the transporters [16, 20, 48, 49].
Lipids as Functional Regulators
Latest studies revealed that lipids can regulate transport activity through diverse mechanisms, some of which involve direct interactions between specific lipid head groups and the functional residues of the transporter [23, 36, 47]. One example using MD simulations combined with hydrogen-deuterium exchange mass spectrometry (HDX-MS) showed howphos-phatidylethanolamine (PE) head groups can modulate the conformational equilibrium of major facilitator superfamily (MFS) transporters between the OF and IF states through interactions with specific salt bridge networks (Figure 3) [47]. Another QM/MM study found that phosphatidylcholine (PC) head groups can participate in the H+-transfer reaction in the H+-coupled multidrug antiporter PfMATE, thereby triggering the conformational change of the protein [23]. For the signaling lipid phosphatidylinositol-4,5-bisphosphate (PIP2), the highly-charged head group was found to electrostatically associate with specific structural motifs in hDAT, resulting in an inward opening of the transporter [36]. In contrast to PIP2, cholesterol binding to hDAT (Figure 1D) was shown to inhibit the OF-to-IF transition [50]. Following the PIP2-induced destabilization of the intracellular gates, multiple instances of Na+ release were captured in statistically independent simulation ensembles, demonstrating the allosteric coupling of the protein’s dynamics to substrate translocation [16]. Furthermore, the MSM calculations of the kinetics suggested that Na+ release pathways are strongly affected by the presence of PIP2 lipids [49].
Figure 3. Lipid-protein interactions modulate the conformational dynamics of membrane transporters.
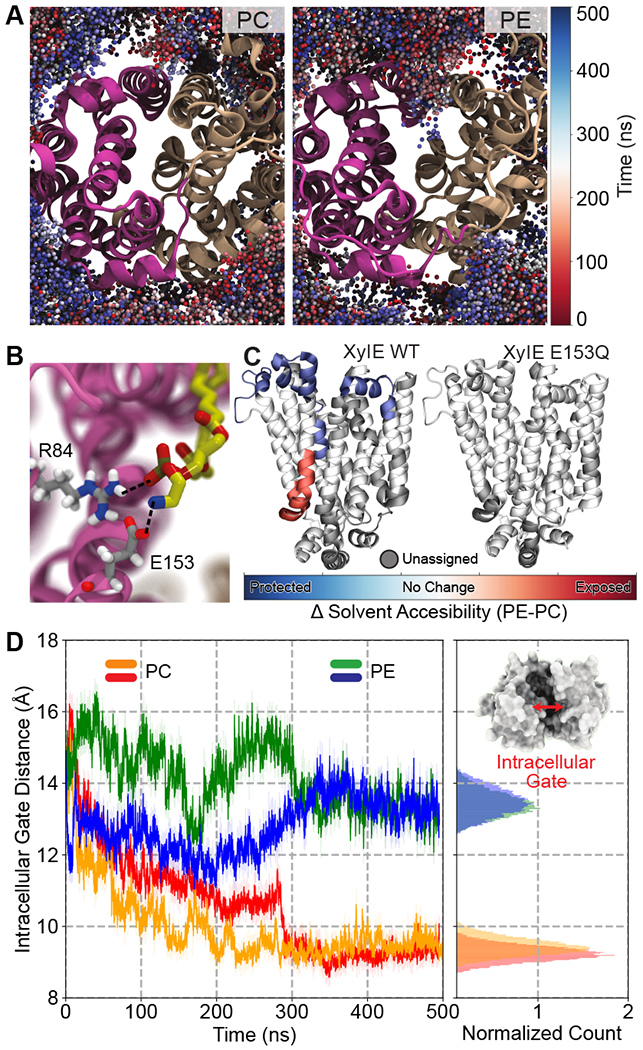
(A) Preferential interaction of PE lipids with IF XylE. Shown is the lipid headgroup sampling in PC (left) and PE (right). Spheres represent phosphorus atoms of PE/PC lipids and are color coded: red at t = 0 and blue at t = 500 ns. A significantly deeper penetration of PE lipids is discernable. (B) Zoomed in snapshot depicting specific interactions between a PE lipid headgroup and a salt bridge within the lumen of the protein, viewed from the intracellular side. No such interaction was observed for PC lipids, as they don’t visit this space. (C) Differential solvent accessibility in PE vs. PC lipid environment, for XylE wildtype and the E153Q mutant. Solvent accessibility, as measured by HDX-MS experiments, is mapped onto the structure depicted by the following color code: red (solvent exposed), blue (protected), white (no change) and grey (unassigned). (D) Time trace showing the intracellular gate distance over simulation time. PC simulation sets are colored orange and red, while the PE simulation sets are colored green and blue. Inset shows a representative snapshot of the protein viewed from the intracellular side highlighting the intracellular gate. Adapted with permission from ref [47]. Copyright 2018 Martens et al. Licensed under a Creative Commons Attribution 4.0 International License.
Lipids can also modulate or stabilize the transporter conformation by wedging into the interface between monomers or domains of the protein [20, 48]. Simulations of a refined EmrE model using molecular dynamics flexible fitting (MDFF) and interactive biased MD techniques (Table 1) showed that lipid intercalation into the dimer interface can wedge the monomers apart and consequently influence the substrate transport pathway [20]. For Gltph, which undergoes elevator-like motions (Box 1) during the transport cycle, the crevice opened at the domain interface in the IF state was compatible with the insertion of a lipid [48]. The inserted lipid was shown to stabilize the protein configuration and increase the solvent accessibility to the domain interface, which may in turn facilitate substrate release [48].
Lipids as Transporter Substrates
The interplay between the lipids and the protein is especially important for members of the ABC transporter family, due to their role in transport of lipids and lipid-like substrates. Simulations of the multidrug exporter P-glycoprotein (Pgp) in its IF state captured the entrance of inner leaflet lipids into the central lumen of the protein formed by the transmembrane domain [51]. Since transported drugs need to partition into membrane to gain access to the central lumen, the observed lipid translocation pathway suggests a putative pathway for direct drug recruitment from the membrane [51]. Simulation of Pgp OF state showed that lipid occupancy at the extracellular opening of the transmembrane domain may facilitate substrate exit into the outer leaflet by modulating the dynamics of the protein [51]. Moreover, an extensive set of microsecond-long atomistic simulations on ABC exporter Sav1866 revealed differential lipid binding for different protein conformations, in line with experimental studies of ABC transporters suggesting lipid-specific effects on the protein’s conformational dynamics and activity [52].
Although the increase in computing power now allows all-atom simulations to be routinely performed up to many microseconds in length, the application of coarse-grained (CG) methods has proven its efficiency in accelerating the system dynamics and probing biological processes that happen at much longer time scales. The enhanced sampling of lipid diffusion in CG simulations has enabled, e.g., the observation of simultaneous lipid uptake for PC and PE lipids [53], and the identification of potential cholesterol binding sites on Pgp [54].
Beside active transporters, intimate interactions between lipids and other membrane transport proteins have been also captured in MD simulations and emerged as an important topic for understanding physiologically relevant processes [46, 55]. These interactions include the transmembrane movement of phospholipid mediated by lipid scramblases [56–59] and the direct involvement of lipids in ion translocation through ion channels [57, 60].
5. Outlook for Drug Discovery
Transporters as Drug Targets
As membrane transporters are directly involved in a number of key physiological processes, they present promising therapeutic targets for a variety of diseases ranging from neurological disorders to cancers. Complementing experimental techniques, MD simulations and molecular docking are being increasingly utilized in identifying potential drug binding sites and developing potent drugs for these proteins (Figure 1E) [61–64]. For example, pharmacophore-based docking (Table 1) combined with SMD simulations suggested the existence of a secondary drug binding site in human serotonin transporter hSERT, the primary target for antidepressants [62]. The results from docking and simulations were then utilized to guide the design of molecular linked drug-analogs to investigate the positional relationship between this site and the well-documented primary drug binding site [62]. Another study on GABA transporter GAT1 showed that experimental data-guided docking and MD simulations can be used to derive a common binding mode shared by a specific class of inhibitors, which was utilized to identify a novel inhibitor drug from DrugBank [63].
Since transporters usually undergo large-scale conformational changes during the transport cycle, it is important to consider the protein dynamics when performing molecular docking. Using a collection of protein structures generated by different MD techniques, ensemble docking (Table 1) efficiently enhances the sampling of the protein conformational space by allowing the inclusion of both local and global flexibility of the protein [65]. This approach was exemplified in a recent study combining MD simulations and high-throughput virtual screening in targeting putative hidden binding pockets in amino acid transporter ASCT2, responsible for nutrient uptake in proliferating cancer cells [66]. The study successfully identified more efficient activators than any other known ASCT2 substrate, as well as molecules that inhibit the proliferation of a melanoma cancer cell line [66]. Another study on the multidrug resistance protein EmrE utilized the simulation-refined protein structure to guide the design of inhibitor models to interfere with protein dimerization and drug resistance [64]. Based on the binding free energy evaluated from FEP calculations, potential inhibitors were synthesized and verified experimentally for efflux inhibition in live Escherichia coli (E. coli) cells [64].
Transporters in Pharmacokinetics
In addition to being important drug targets, transporters also play an important role in the absorption, distribution, and elimination of drugs used for treating different diseases, as well as mediating drug-drug interactions. MD based computational methods are also widely utilized to complement the experimental binding and permeability studies of chemicals. For example, a combination of in silico modeling (molecular docking and free energy calculations) and experimental techniques (competition transport and tissue-based assays) was used in binding and permeability predictions of a triple formulation of antiretroviral drugs (tenofovir, darunavir, and dapivirine) with multidrug transporter Pgp [67]. A similar hybrid approach was utilized for calculating binding free energies of different peptides with PepT1 [68], a proton-dependent oligopeptide transporter responsible for the uptake of β-lactams and peptide-based drugs.
Computational models utilizing supervised learning methods like machine learning can also be employed in the prediction of pharmacokinetic properties of prospective drug candidates, providing scientists with new tools in tackling the same problem in the early stages of lead discovery. For example, the web-based tool SwissADME [69] allows the prediction of a compound being substrate or non-substrate of Pgp, key to assessing its potential efflux across the cellular membrane by this multidrug transporter.
6. Concluding Remarks and Future Perspectives
With the rapid technological advancement in computing and availability of an ever increasing number of high-resolution structures for biomolecular and cellular systems, MD simulations and related computational techniques that allow for surveying atomic-level motion of the system of interest have evolved into an essential biophysical tool for achieving complete and detailed characterization of biochemical systems and processes. The power of the methodology lies in the high spatial and temporal resolutions offered simultaneously, which enable it to achieve a dynamic description of the underlying phenomena at a microscopic level. Given the heavy involvement of conformational changes of varying forms and degrees in transporter function, mechanistic investigation of membrane transporters has particularly benefited from the application of MD simulations. The long timescale associated with the transport cycle in this important class of proteins, which stems from the involvement of complex, large-scale structural transitions in their mechanism, continues to pose major challenges to MD studies. Nevertheless, as exemplified in this review, we have already witnessed how MD simulations can bring an unparalleled perspective into the mechanistic studies of membrane transporters, particularly when applied in close concert to experimental approaches.
MD simulations have also proven highly valuable in integrating experimental information from various sources, including cryo-EM, single-molecule FRET and spectroscopic techniques like NMR, into structural models, in an approach collectively referred to as integrative modeling. While this area of modern structural biology is still in its infancy, there are already many examples in the literature of its successful applications, for example, when identifying the functionally important states of the transporter from its conformational ensemble using spin-label distance distributions calculated from double electron-electron resonance (DEER) spectroscopy [70]. Biasing the simulations using available experimental data provides the added value of further treating the systematic force-field and statistical sampling errors inherent to most of the computational techniques.
While the progress has been impressive, like any other methodology and scientific approach, there remains extensive room for improvement and for extending the scope of MD simulations, e.g., in terms of increasing the accuracy of the results, expanding the scope of the methodology towards more complex and larger systems and processes, and making the technology accessible to a wider range of researchers (See “Outstanding Questions”). Many of these aspects will continue to benefit from the continuous growth of our computing power and increased access to a richer pool of experimental data on structural and other biophysical properties on biomolecular systems and processes. Some others, however, would have to wait for substantially different and/or novel approaches, for example, more chemically informed treatments of the molecular representation included in the simulation.
Outstanding Questions.
Insufficient sampling continues to be one of the most important limitations of MD. Description of complete structural transitions in membrane transporters and their coupling to key chemical events in the natural environment of a membrane remains a challenging task due to the timescale and complexity of these processes. Moreover, the observation of a single spontaneous transition does not preclude the existence of other structural transition pathways that may be more commonly taken by a transporter in nature.
Accurate characterization of the energetics associated with the events involved in the complete mechanism of membrane transporters will require much longer simulations and can only be achieved once substantially more powerful computers are available.
Increasingly realistic representations for the biological context in which membrane transporters function would be highly desirable, both at the membrane and at the whole cell level. Among these, the effect of lipid composition of the membrane and specific interactions of special lipids with membrane transporters are particularly challenging to describe, both experimentally due to their semi-fluid and highly dynamic nature, and computationally given the long timescales associated with their diffusion and mixing.
The classical descriptors used in the majority of MD simulations applied to membrane transporters do not allow important details to be adequately accounted for. There remains a need for more complete treatments of forces governing the structure, dynamics, and interactions in a molecular system. Examples include force fields with more “chemical knowledge” (e.g., polarizable force fields) that more accurately describe polarization effects and the ability to account for the dynamic change of the protonation states of titratable groups in response to changes in the environment, for example as a result of a change in a transporter’s conformation.
Construction and validation of the structural models used for MD simulation, particularly for large assemblies and cellular scale systems, will largely benefit from, and in fact require, a more extensive set of experimental information.
The prospect for the application of computational techniques such as MD to biochemical questions is rather bright. New technological developments now allow easy access and sharing of large amounts of computational data on the web for interactive visualizations of the system and deeper understanding of complex mechanisms [71]. The field is witnessing an increasing number of researchers forming interdisciplinary approaches successfully combining experiments and computation; they have learned how to “talk” to each other, how to communicate and exchange data and inform one approach by the other, and how to define problems that can be tackled jointly by a concerted effort. We can only expect this trend to continue in the future, resulting in exciting discoveries in our understanding of the mechanism of membrane transporters, prime examples of highly specialized molecular machines at work in biology.
Highlights.
Membrane transporters regulate a wide range of biochemical and physiological processes, many of them with direct implications in human health and disease.
The high spatial and temporal resolutions offered by MD simulations allow the investigation of a wide range of processes critical to the function of membrane transporters.
MD simulations and related computational methods have become a powerful approach for in situ characterization of the structural and dynamical aspects of transporters within their natural membrane environment.
Recent advances in computer algorithms, force fields, and computing power have resulted in a quantum leap in computational characterization of membrane transporters.
Applications of integrative modeling techniques which incorporate experimental data from various sources into molecular models used by MD simulations open up new vistas to studying the functional cycles of transporters within their natural environment of a cell.
Acknowledgements
The authors acknowledge support by the National Institutes of Health under award numbers P41-GM104601 (to ET) and R01-GM123455 (to ET), the National Science Foundation Graduate Research Fellowship Program under Grant No. 1746047 (to NT), and the Beckman Institute Graduate Fellowship (to SP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and the National Science Foundation. We also acknowledge computing resources provided by Blue Waters at National Center for Supercomputing Applications, and Extreme Science and Engineering Discovery Environment (grant MCA06N060 to ET).
Glossary
- All-atom molecular dynamics (MD)
is a physics-based method to simulate the thermal fluctuations and movements in molecular systems. The simulated molecules are presented in structural models that compose of individual atoms, and their interactions defined in the Force field. Usually MD simulations are carried out under equilibrium (unbiased) conditions, however additional forces (biases) can be introduced for thermodynamic and/or kinetic characterizations.
- Cholesterol
is a lipid component that appears in eukaryotic membranes exclusively. It is believed to cluster with itself and sphingolipids, thereby locally rigidifying the membrane into a gel phase in contrast to the fluid, cholesterol-free phase of the membrane. The formation of cholesterol-rich microdomains influences the location and functional regulation of many membrane proteins.
- Coarse-grained molecular dynamics (CGMD)
simplifies simulation systems using reduced representations that typically represent a group of atoms with one interaction site, instead of individual atoms as in all-atom MD, thereby allowing the construction and simulation of very large systems and the study of long term dynamics efficiently. Since the reduced representations in CG models lack chemical detail, their structural and energetic accuracy is sacrificed to trade for the computational efficiency.
- Cryo-electron microscopy (Cryo-EM)
observes molecular structures using a transmission electron microscope under cryogenic conditions. Recent development in single particle cryo-EM allows structural determination at near-atomic resolutions for large biomolecular and cellular systems.
- Enhanced sampling methods
are often used to address the insufficient timescale of conventional (unbiased) MD at describing the functional dynamics of biomolecules. The approaches of enhanced sampling typically involve a structural and thermodynamic characterizations of a process along a specific pathway (defined by Reaction coordinate).
- Force field
in MD refers to the parameters describing the interactions between atoms or simulation particles. Typically, parameters of an all-atom force field consist of bonded terms (including bond stretching, bond angles and torsional angles) as well as non-bonded terms (van der Waals and electrostatic interactions).
- Hydrogen-deuterium exchange mass spectrometry (HDX-MS)
probes solvent accessibility of protein residues by measuring the exchange rate between polar protons of the protein and the deuterium of heavy water using protein mass spectrometry.
- Reaction coordinate
is the abstraction of the changes occurred during a process, usually mapped onto geometrical features of the molecular systems, which is used to represent the transition pathway associated with the process. For example, the distance between a substrate and its binding site residues can be used as a reaction coordinate for substrate unbinding process. Conversely, the relative orientation of two major structural domains in a transporter can be used to map its global conformational change during the transport cycle.
- X-ray crystallography
determines structures of biological molecules by resolving the X-ray defraction patterns from crystals of purified samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mitchell P (1957) A general theory of membrane transport from studies of bacteria. Nature 180, 134–136 [DOI] [PubMed] [Google Scholar]
- [2].Jardetzky O (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- [3].Drew D and Boudker O (2016) Shared molecular mechanisms of membrane transporters 35, 543–572 [DOI] [PubMed] [Google Scholar]
- [4].Henderson RK et al. (2019) Coupling efficiency of secondary active transporters 58, 62–71 [DOI] [PubMed] [Google Scholar]
- [5].Locher KP (2016) Mechanistic diversity in ATP-binding cassette (ABC) transporters 23, 487–493 [DOI] [PubMed] [Google Scholar]
- [6].Navratna V and Gouaux E (2019) Insights into the mechanism and pharmacology of neurotransmitter sodium symporters 54, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zomot E et al. (2015) Microseconds simulations reveal a new sodiumbinding site and the mechanism of sodium-coupled substrate uptake by LeuT. 290, 544–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tavoulari S et al. (2016) Two Na+ sites control conformational change in a neurotransmitter transporter homolog. 291, 1456–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang YW et al. (2018) Structural elements required for coupling ion and substrate transport in the neurotransmitter transporter homolog LeuT. Proc. Nat. Acad. Sci. USA , 283176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guskov A et al. (2016) Coupled binding mechanism of three sodium ions and aspartate in the glutamate transporter homologue GltTk 7, 13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Venkatesan S et al. (2015) Refinement of the central steps of substrate transport by the aspartate transporter GltPh: Elucidating the role of the Na2 sodium binding site 11, e1004551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wahlgren WY et al. (2018) Substrate-bound outward-open structure of a Na+-coupled sialic acid symporter reveals a new Na+ site 9, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Adelman JL et al. (2016) Stochastic steps in secondary active sugar transport. Proc. Nat. Acad. Sci. USA 113, E3960–E3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Setiadi J and Kuyucak S (2017) Elucidation of the role of a conserved methionine in glutamate transporters and its implication for force fields 121, 9526–9531 [DOI] [PubMed] [Google Scholar]
- [15].LeVine MV et al. (2018) Thermodynamic coupling function analysis of allosteric mechanisms in the human dopamine transporter 114, 10–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Razavi AM et al. (2017) A Markov state-based quantitative kinetic model of sodium release from the dopamine transporter 7, 40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ficici E et al. (2018) Broadly conserved Na+-binding site in the N-lobe of prokaryotic multidrug mate transporters. Proc. Nat. Acad. Sci. USA 115, E6172–E6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Marinelli F et al. (2014) Sodium recognition by the Na+/Ca2+ exchanger in the outward-facing conformation. Proc. Nat. Acad. Sci. USA 111, E5354–E5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhekova H et al. (2016) Characterization of the cation binding sites in the NCKX2 Na+ /Ca2+-K+ exchanger 55, 6445–6455 [DOI] [PubMed] [Google Scholar]
- [20].Vermaas JV et al. (2018) Electrostatic lock in the transport cycle of the multi-drug resistance transporter EmrE. Proc. Nat. Acad. Sci. USA 115, E7502–E7511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Parker JL et al. (2017) Proton movement and coupling in the POT family of peptide transporters. Proc. Nat. Acad. Sci. USA 114, 13182–13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yu X et al. (2017) Dimeric structure of the uracil: proton symporter UraA provides mechanistic insights into the SLC4/23/26 transporters 27, 1020–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nishima W et al. (2016) Mechanisms for two-step proton transfer reactions in the outward-facing form of MATE transporter 110, 1346–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee S et al. (2016) Multiscale simulations reveal key aspects of the proton transport mechanism in the ClC-ec1 antiporter 110, 1334–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee S et al. (2016) The origin of coupled chloride and proton transport in a Cl−/H+ antiporter 138, 14923–14930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang Z et al. (2018) Modulating the chemical transport properties of a transmembrane antiporter via alternative anion flux 140, 16535–16543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Duster AW et al. (2019) Adaptive partitioning QM/MM for molecular dynamics simulations: 6. proton transport through a biological channel 15, 892–905 [DOI] [PubMed] [Google Scholar]
- [28].Han W et al. (2014) Water access points and hydration pathways in CLC H+/Cl− transporters. Proc. Nat. Acad. Sci. USA 111, 1819–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang T et al. (2016) Molecular basis for differential anion binding and proton coupling in the Cl−/H+ exchanger ClC-ec1 138, 3066–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Szöllősi D et al. (2018) Dissecting the forces that dominate dimerization of the nucleotide binding domains of ABCB1 114, 331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Prieß M et al. (2018) Molecular mechanism of ATP hydrolysis in an ABC transporter 4, 1334–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jones PM and George AM (2017) How intrinsic dynamics mediates the allosteric mechanism in the ABC transporter nucleotide binding domain dimer 13, 1712–1722 [DOI] [PubMed] [Google Scholar]
- [33].Hofmann S et al. (2019) Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571, 580–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hayashi T et al. (2014) ATP-induced conformational changes of nucleotide-binding domains in an ABC transporter. importance of the water-mediated entropic force 118, 12612–12620 [DOI] [PubMed] [Google Scholar]
- [35].Yue Z et al. (2017) Constant pH molecular dynamics reveals how proton release drives the conformational transition of a transmembrane efflux pump 13, 6405–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Khelashvili G et al. (2015) Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus 6, 1825–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Göddeke H et al. (2018) Atomistic mechanism of large-scale conformational transition in a heterodimeric ABC exporter 140, 4543–4551 [DOI] [PubMed] [Google Scholar]
- [38].Latorraca NR et al. (2017) Mechanism of substrate translocation in an alternating access transporter 169, 96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bera I and Klauda JB (2018) Structural events in a bacterial uniporter leading to translocation of glucose to the cytosol 430, 3337–3352 [DOI] [PubMed] [Google Scholar]
- [40].Cheng MH and Bahar I (2015) Molecular mechanism of dopamine transport by human dopamine transporter 23, 2171–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Selvam B et al. (2018) Free energy landscape of the complete transport cycle in a key bacterial transporter 4, 1146–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moradi M et al. (2015) Atomic-level characterization of transport cycle thermodynamics in the glycerol-3-phosphate:phosphate transporter 6, 8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Machtens JP et al. (2015) Mechanisms of anion conduction by coupled glutamate transporters 160, 542–553 [DOI] [PubMed] [Google Scholar]
- [44].Tsuchiya H et al. (2016) Structural basis for amino acid export by DMT superfamily transporter YddG. Nature 534, 417–420 [DOI] [PubMed] [Google Scholar]
- [45].Matsunaga Y et al. (2018) Energetics and conformational pathways of functional rotation in the multidrug transporter AcrB 7, e31715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Muller MP et al. (2019) Characterization of lipid-protein interactions and lipid-mediated modulation of membrane protein function through molecular simulation 119, 6086–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Martens C et al. (2018) Direct protein-lipid interactions shape the conformational landscape of secondary transporters 9, 4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Akyuz N et al. (2015) Transport domain unlocking sets the uptake rate of an aspartate transporter. Nature 518, 68–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Razavi AM et al. (2018) How structural elements evolving from bacterial to human SLC6 transporters enabled new functional properties 16, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zeppelin T et al. (2018) A direct interaction of cholesterol with the dopamine transporter prevents its out-to-inward transition 14, e1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mayne CG et al. (2016) The cellular membrane as a mediator for small molecule interaction with membrane proteins 1858, 2290–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Immadisetty K et al. (2019) Lipid-dependent alternating access mechanism of a bacterial multidrug ABC exporter 5, 43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Barreto-Ojeda E et al. (2018) Coarse-grained molecular dynamics simulations reveal lipid access pathways in P-glycoprotein 150, 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Domicevica L et al. (2018) Multiscale molecular dynamics simulations of lipid interactions with p-glycoprotein in a complex membrane 80, 147–156 [DOI] [PubMed] [Google Scholar]
- [55].Wen PC et al. (2018) Microscopic view of lipids and their diverse biological functions 51, 177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bethel NP and Grabe M (2016) Atomistic insight into lipid translocation by a TMEM16 scramblase. Proc. Nat. Acad. Sci. USA 113, 14049–14054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jiang T et al. (2017) Lipids and ions traverse the membrane by the same physical pathway in the nhTMEM16 scramblase 6, e28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lee BC et al. (2018) Gating mechanism of the extracellular entry to the lipid pathway in a TMEM16 scramblase 9, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Morra G et al. (2018) Mechanisms of lipid scrambling by the G protein-coupled receptor opsin 26, 356–367. e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mansoor SE et al. (2016) X-ray structures of human P2X3 receptor define complete gating cycle and antagonist action. Nature 538, 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Leelananda SP and Lindert S (2016) Computational methods in drug discovery 12, 2694–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Brinkø A et al. (2016) Synthesis and inhibitory evaluation of 3-linked imipramines for the exploration of the S2 site of the human serotonin transporter 24, 2725–2738 [DOI] [PubMed] [Google Scholar]
- [63].Jurik A et al. (2015) A binding mode hypothesis of tiagabine confirms liothyronine effect on γ-aminobutyric acid transporter 1 (GAT1) 58, 2149–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ovchinnikov V et al. (2018) Structure of the EmrE multidrug transporter and its use for inhibitor peptide design. Proc. Nat. Acad. Sci. USA 115, 7932–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Amaro RE et al. (2018) Ensemble docking in drug discovery 114, 2271–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Colas C et al. (2015) Ligand discovery for the alanine-serine-cysteine transporter (ASCT2, SLC1A5) from homology modeling and virtual screening 11, e1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Swedrowska M et al. (2017) In silico and in vitro screening for p-glycoprotein interaction with tenofovir, darunavir, and dapivirine: An antiretroviral drug combination for topical prevention of colorectal HIV transmission 14, 2660–2669 [DOI] [PubMed] [Google Scholar]
- [68].Samsudin F et al. (2016) Accurate prediction of ligand affinities for a proton-dependent oligopeptide transporter 23, 299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Daina A et al. (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules 7, 42717–42720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Leone V et al. (2019) Interpretation of spectroscopic data using molecular simulations for the secondary active transporter betp 151, 381–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hildebrand PW et al. (2019) Bringing molecular dynamics simulation data into view In press [DOI] [PubMed]
- [72].Forrest LR et al. (2011) The structural basis of secondary active transport mechanisms 1807, 167–188 [DOI] [PubMed] [Google Scholar]
- [73].Enkavi G et al. (2014) A microscopic view of the mechanisms of active transport across the cellular membrane, in Annual Reports in Computational Chemistry, vol. 10 (Wheeler RA, ed.), chap. 4, Elsevier, pp. 77–125 [Google Scholar]
- [74].Vermaas JV et al. (2016) Microscopic characterization of membrane transporter function by in silico modeling and simulation, in Computational Approaches for Studying Enzyme Mechanism Part B, vol. 578 (Voth GA ed.), chap. 16, Academic Press, pp. 373–428 [DOI] [PMC free article] [PubMed] [Google Scholar]