

Introduction
Cardiac fibrosis is the hallmark feature of heart failure [1, 2], which currently affects nearly 6 million people in the US alone [3]. The key cellular event underlying cardiac fibrosis is pathological activation and conversion of quiescent cardiac fibroblasts (CFs) into myofibroblasts, which contribute to dysregulation of ECM turnover and progressive deterioration of cardiac function [4–7]. Traditionally, cell sources including hematopoietic [8] and epicardial [9–11] cells and endothelial-to-mesenchymal transition (EndoMT) [12] were thought to significantly contribute to the accumulation of myofibroblasts in heart failure. However, recent studies with advanced transgenic mouse models have revealed that tissue-resident CFs are the predominant source of all activated myofibroblasts contributing to disease progression [13–15]. Still, whether all fibroblasts and myofibroblasts in the heart are equally pathogenic and responsive to specific types of cardiac stress remains unknown [16]. Therefore, better understanding of the diverse population of tissue-resident CFs and their potentially heterogenous responses to a cardiac insult is expected to facilitate the development of novel antifibrotic therapies to delay or reverse the progression of heart failure [17–19].
The frontline hurdle of cardiac fibroblast biology is the lack of specific and comprehensive markers to identify and isolate resident CFs [20–22]. While transgenic mouse lines harboring fluorescence-tagged Collagen type I (Col1a1) and platelet-derived growth factor alpha receptor (PDGFRα) enable reliable identification of tissue-resident CFs in vivo [13, 23–25], these markers cannot be used in antibody-based flow cytometry sorting applications due to the lack of robust antibodies (PDGFRα) or the intracellular nature of the antigen (Col1a1). Traditionally, the commonly used cell surface marker to sort resident fibroblasts in the heart is Thymocyte Differentiation Antigen 1 (Thy1, also known as CD90), a glycophosphatidylinositol (GPI)-anchored cell surface glycoprotein known to only recognize a subpopulation of CFs in ventricles [21, 26–28]. Only recently, the MEFSK4 has been discovered as a more inclusive surface marker than Thy1 to sort CFs, as evidenced by its highly overlapping expression with Col1α1 and PDGFRα [28]. This leaves a possibility that within the more inclusive MEFSK4+ population of CFs, Thy1 expression (or lack of it) may identify phenotypically distinct fibroblast subpopulations with diverse roles in cardiac function and disease.
Previously, Thy1 has been shown to have diverse biological functions in cell-cell and cell-matrix interactions, mechanotransduction, cell migration, differentiation, apoptosis, and regulation of outside-in signaling [29]. In particular, the Thy1 expression in tissue-resident fibroblasts appears to define response of these cells to inflammation and injury, with Thy1+ and Thy1− fibroblast subsets playing different roles in different organs [30–36]. In the context of lung pathophysiology, Thy1−, but not Thy1+, pulmonary fibroblasts represent the pathologically activated profibrotic subpopulation responsive to cytokines and fibrogenic stimuli [37–41]. We thus hypothesized that Thy1 expression (or lack thereof) in heart-resident fibroblasts may define molecularly and functionally distinct cell subpopulations in the setting of heart failure. To test this hypothesis, we performed a set of complementary studies using mouse transverse aortic constriction (TAC) model of pressure overload induced heart failure, freshly sorted ventricular fibroblasts, a physiologically relevant 3D tissue-engineered co-culture model system, and mice with global Thy1 knockout. Our studies for the first time indicate that in heart failure the lack of Thy1 expression can define a pathogenic subpopulation of ventricular fibroblasts causative of cardiomyocyte contractile dysfunction and tissue fibrosis.
Results
Thy1 expression of cardiac fibroblasts in response to pressure overload-induced heart failure.
To study the potential differences in CF makeup in pressure overload induced heart failure, we employed the surgical TAC mouse model [35, 42–47]. Consistent with previous studies, TAC animals exhibited progressive structural and function remodeling which by 8 weeks resulted in severe LV dilation and myocardial dysfunction compared to sham-operated (Sham) control animals (Supplemental Figure 1, A and B). This functional decline was associated with occurrence of cardiac fibrosis characterized by significantly increased expression of Collagen I and Vimentin (Supplemental Figure 1, C–E). Mice with LV fractional shortening of less than 30% at 7–9 weeks post-TAC were selected for the subsequent cell isolation by enzyme digestion followed by fluorescence-activated cell sorting (FACS) (Figure 1A). To isolate and characterize the CFs in TAC and Sham ventricles, freshly isolated cells were sorted for MEFSK4+ [28] and Thy1 (CD90)+ [48, 49] cells with exclusion of endothelial (CD31+) and immune system (CD45+) cells (Figure 1, A and B). The flow cytometry analysis revealed that the percentage of CD45+ immune cells was significantly higher while the percentage of CD31+ endothelial cells was significantly lower in TAC vs. Sham ventricles (Figure 1, C and D), as previously reported [50, 51]; however, the two groups did not differ in the percentage of CD45−/CD31− non-myocytes (Figure 1, C–E). Notably, we identified 2 distinct subsets of MEFSK4+/CD45−/CD31− CFs by analyzing their Thy1 expression, namely Thy1pos (Thy1+/MEFSK4+/CD45−/CD31−) and Thy1neg (Thy1−/MEFSK4+/CD45−/CD31−) CFs (Figure 1, A and B). Surprisingly, while the percentage of Thy1pos cells remained unchanged, Thy1neg percentage was significantly higher in TAC vs. Sham ventricles (Figure 1, B and F). Moreover, within the MEFSK4+/CD45−/CD31− CFs, Thy1pos fraction was more abundant in Sham ventricles, while the Thy1neg fraction become dominant in TAC ventricles (Figure 1G). Consistent with previous reports of TAC-induced CF proliferation [52–55], the total numbers of both Thy1pos and Thy1neg CFs were significantly higher in TAC compared to Sham ventricles; however, the Thy1neg CFs showed significantly greater increase than Thy1pos CFs (Figure 1H). Collectively, these results demonstrate that pressure overload-induced heart failure in mice leads to a disproportionate increase in the subpopulation of ventricular CFs negative for the expression of surface marker Thy1.
Figure 1. Two distinct subsets of cardiac fibroblasts in TAC and Sham ventricles distinguished by Thy1 expression.
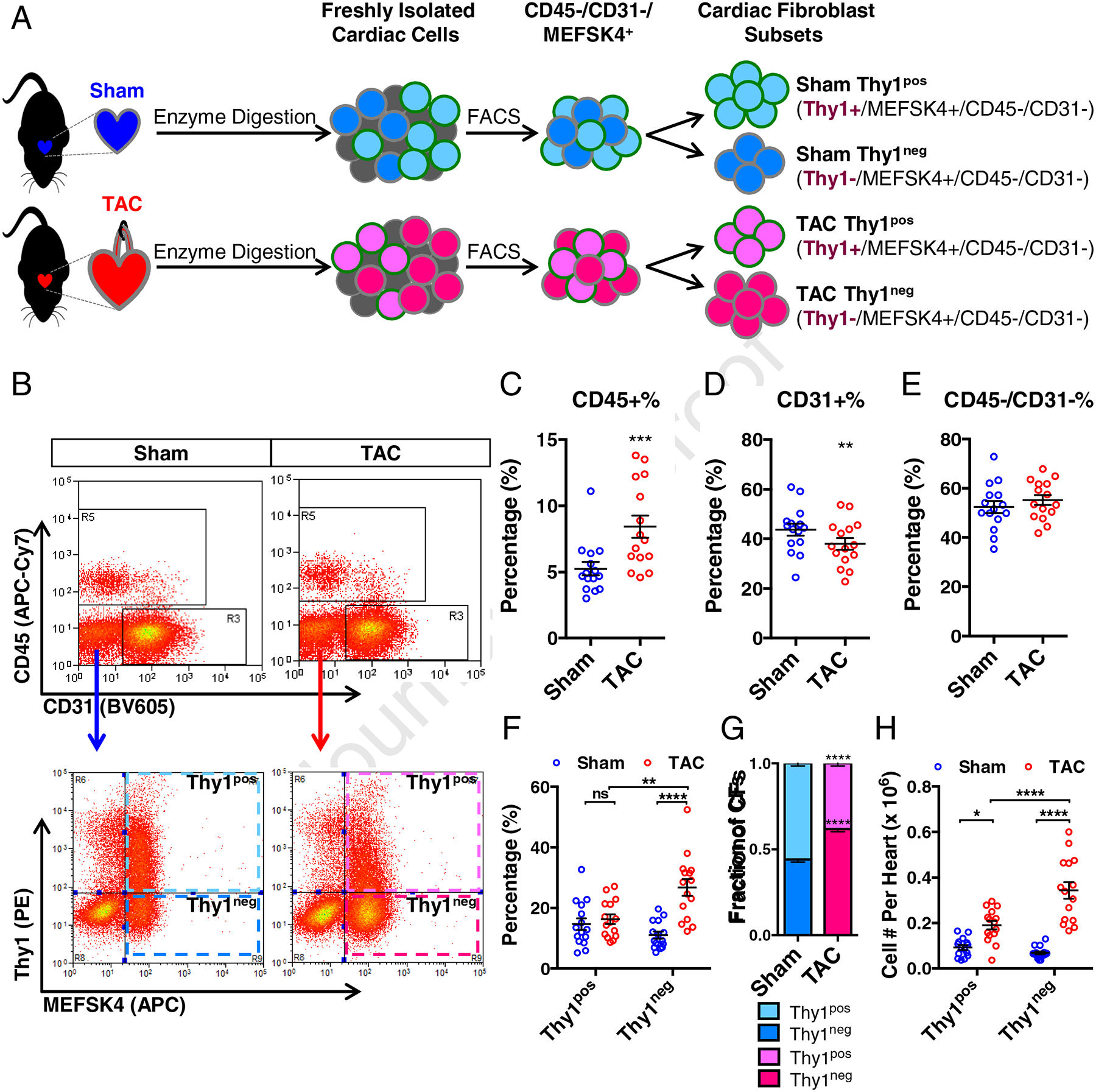
(A) Schematic depicting the isolation of cardiac fibroblasts (CFs) from murine ventricles undergoing heart failure induced by transverse aortic constriction (TAC) or age-matched sham-operated ventricles (Sham), followed by purification of different CF subsets by fluorescence-activated cell sorting (FACS). (B) Representative flow cytometry analyses of freshly isolated cells from Sham and TAC ventricles stained with APC-Cy7-conjugated CD45, BV605-conjugated CD31, PE-conjugated Thy1 and APC-conjugated MEFSK4. Quantification of the percentage of CD45+ (C), CD31+ (D), and CD45−/CD31− (E) populations from Sham and TAC ventricles. (F) Quantification of the percentages of cells from Sham and TAC ventricles labelled for Thy1+/MEFSK4+/CD45−/CD31− (namely Thy1pos CFs) and Thy1−/MEFSK4+/CD45−/CD31− (namely Thy1neg CFs). (G) The fractions of Thy1pos and Thy1neg CFs within the MEFSK4+/CD45−/CD31− CF populations from Sham and TAC ventricles. (H) The yield of Thy1pos and Thy1neg CFs per Sham and TAC heart (ventricles only). N=15 independent experiments. ns: not significant, **P < 0.01, ***P < 0.001, ****P < 0.0001 by paired two-tailed Student’s t test (C-E) or 2-way ANOVA with Bonferroni’s multiple comparisons test (F–H).
Expression of pro-fibrotic genes in Thy1pos and Thy1neg CFs.
To investigate whether the Thy1pos and Thy1neg CFs exhibit molecularly distinct phenotypes in response to pressure overload, we profiled the expression of a panel of key genes associated with pathological fibrosis in the freshly isolated CFs from TAC and Sham hearts. We found that the Thy1neg CFs from either TAC or Sham ventricles express negligible levels of Thy1 mRNA (Figure 2A), thus demonstrating the lack of Thy1 at the transcriptional level. Furthermore, the Thy1 expression in Thy1pos CFs was downregulated for TAC vs. Sham hearts (Figure 2A), suggesting that pressure overload is the negative regulator of Thy1. We further examined 2 markers of activated myofibroblasts: Periostin (Postn) and alpha smooth muscle actin (Acta2); the master regulator of cardiac fibrosis: Transforming Growth Factor Beta 1 (TGFβ1); and 3 extracellular matrix (ECM) genes: Collagen I (Col1a1), Collagen III (Col3a1), and Fibronectin (Fn1). In Sham ventricles, no expression difference was observed between Thy1pos and Thy1neg CFs for all of the genes examined (Figure 2, B–H). However, in TAC ventricles, the Thy1neg CFs showed significantly elevated expression of Postn, TGFb1, Col1a1 and Col3a1 compared to the Thy1pos CFs (Figure 2, B–E and H), while no difference was observed for Fn1 expression (Figure 2, F–H). Additionally, although not statistically significant, we observed a trend towards higher expression of Acta2 in Thy1neg vs. Thy1pos CFs in both Sham and TAC hearts (Figure 2G). Overall, this analysis suggested that in response to pressure overload, the previously unstudied Thy1neg CF population attains a more pro-fibrotic myofibroblast phenotype compared to Thy1pos population.
Figure 2. Increased expression of pro-fibrotic genes in Thy1neg CFs from TAC hearts.
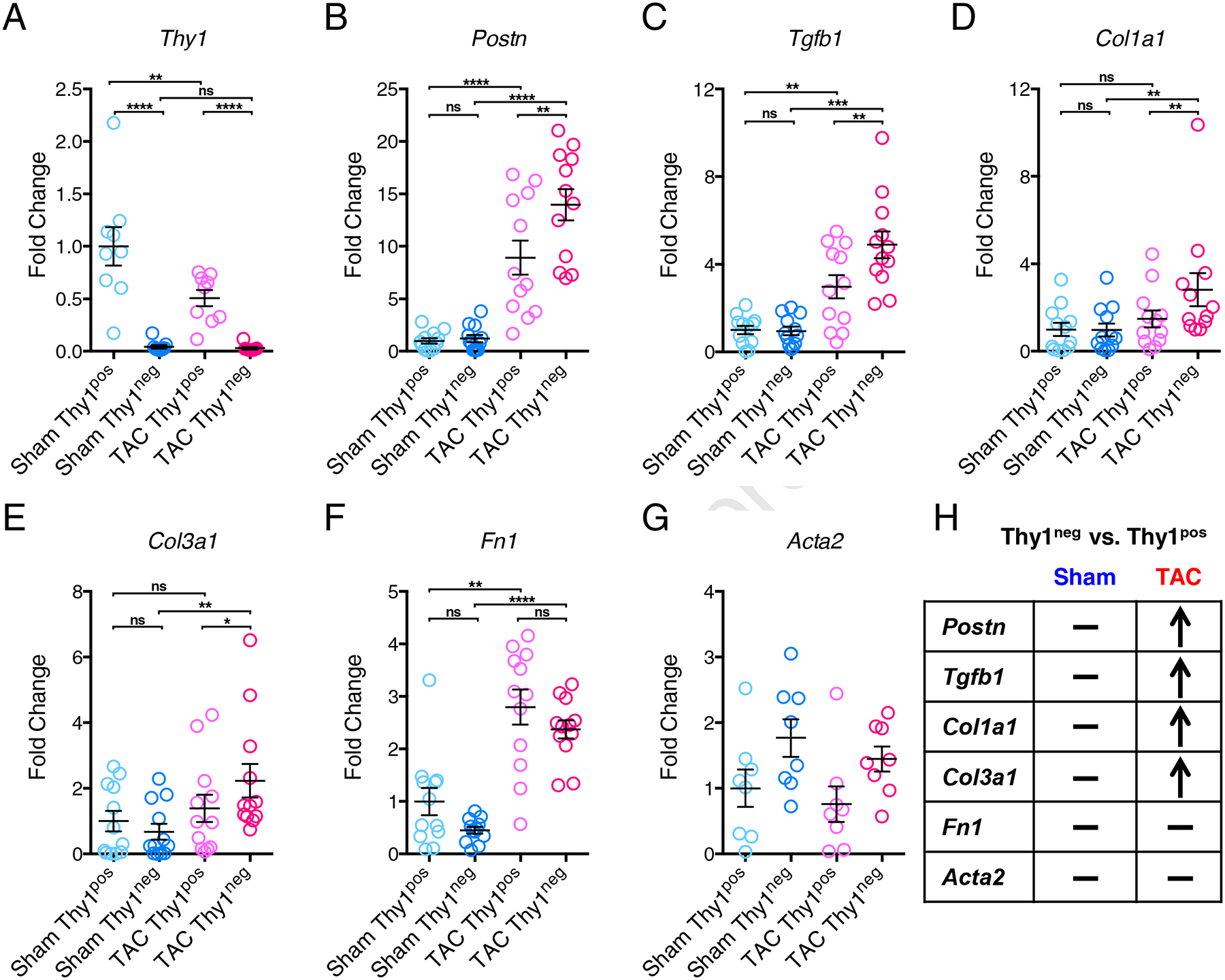
Expression of Thy1 (A), Postn (B), Tgfb1 (C), Col1a1 (D), Col3a1 (E), Fn1 (F) and Acta2 (G) genes in freshly isolated Thy1pos and Thy1neg CFs from Sham and TAC ventricles. (H) Summarized gene expression comparison between Thy1pos and Thy1neg CFs from Sham and TAC ventricles. “−” denotes no significant difference in Thy1pos vs. Thy1neg, “↑” denotes significant upregulation in Thy1pos vs. Thy1neg. The mRNA expression was normalized to house-keeping gene B2M and shown as fold change relative to Sham Thy1pos group. N=9 independent experiments for Thy1, N=8 independent experiments for Acta2, and N=12 independent experiments for all other genes. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA with Tukey’s multiple comparisons test.
Effects of Thy1pos and Thy1neg CFs on engineered cardiac muscle function.
To evaluate whether the Thy1pos and Thy1neg CFs from Sham and TAC hearts have distinct functional roles, we studied them in vitro using a physiologically relevant 3D co-culture system established in our previous studies [56, 57]. Based on the prior reports that the 2D culture alters CFs and could mask the phenotypic differences between fibroblasts from diseased and healthy hearts [58–60], we compared gene expression between 2D subcultured and freshly isolated CFs. Consistent with previous studies, we found that the initial gene expression differences among the freshly isolated CF populations were neutralized in 2D culture as early as after the first passage (Supplementary Figure 2). This finding prompted us to exclusively use freshly isolated CFs from TAC or Sham hearts to form 3D co-cultured engineered cardiac tissue bundles (Figure 3A) made with 0.3:1 ratio of CFs to neonatal rat ventricular myocytes (NRVMs). To evaluate the effects that different CF subsets exert on the contractile function of surrounding cardiomyocytes, we measured the contractile force (Figure 3B) and the active force-length relationships (Figure 3C) in the co-cultured bundles. The bundles containing NRVMs alone (namely NRVM bundles) served as the benchmark control to ensure relative consistency within each experiment, and expectedly generated significantly higher contractile forces than all the other 4 groups with added CFs (Figure 3D). Interestingly, bundles containing NRVMs and freshly isolated Thy1neg CFs from TAC heart (namely +TAC Thy1neg bundles) generated significantly lower contractile forces than all the other groups, whereas no significant difference was seen in the force generation among +Sham Thy1pos, +Sham Thy1neg and +TAC Thy1pos bundles (Figure 3D). Moreover, the +TAC Thy1neg bundles exhibited significantly prolonged contraction rise time compared to all other bundle groups (Figure 3E), while no significant differences among groups were found for the decay time (Figure 3F). Given that dysregulated calcium handling is the central cause of contractile dysfunction in heart failure [61, 62], we assessed Ca2+ transients in the bundles and found that the +TAC Thy1neg bundles exhibited significantly reduced Ca2+ transient amplitude compared to all other groups (Figure 3G). Together, these findings suggest that Thy1pos and Thy1neg CFs from failing TAC ventricle represent 2 functionally distinct CF subsets, and that the Thy1neg subset may be more detrimental to cardiomyocyte function in pressure overload-induced heart failure than the Thy1pos subset.
Figure 3. The detrimental effect of Thy1neg CFs on engineered cardiac bundle function.
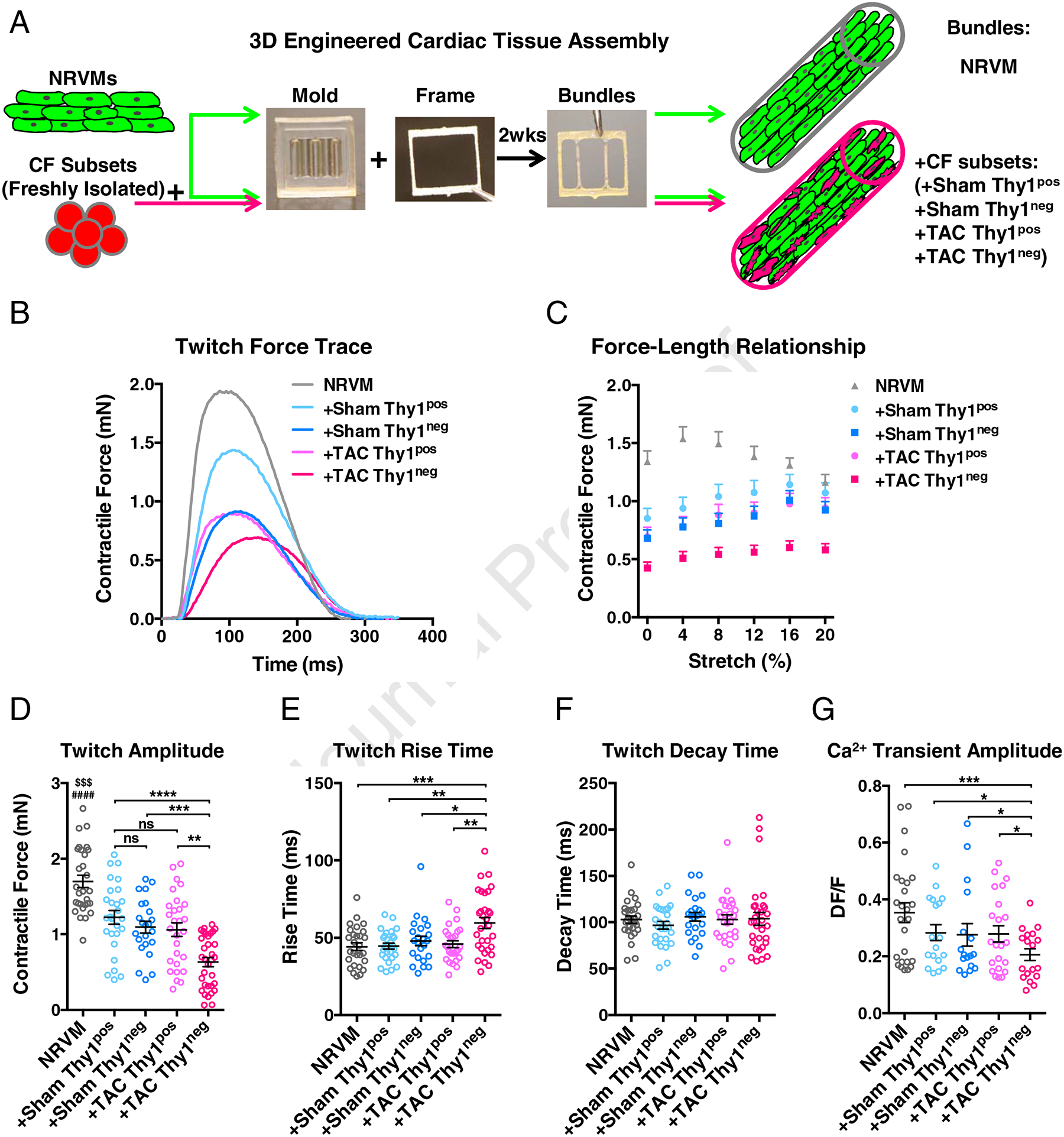
(A) Schematic describing the assembly of 3D engineered cardiac tissue bundles consisting of neonatal rat ventricular myocytes (NRVMs) alone or co-cultured with Thy1pos and Thy1neg CF subsets from Sham or failing TAC ventricles. Cell-hydrogel mixture was polymerized within PDMS mold with laser-cut Cerex frame, and after 2 weeks of free-floating dynamic culture, the bundles were subjected to functional and structural analyses. (B) Representative contractile force traces generated by the engineered cardiac bundles paced at 1 Hz. (C) Amplitude of contractile force as a function of applied bundle elongation in 4% strain increments. (D) Maximum contractile force amplitude at optimal tissue length shown for different bundle groups. $$$P < 0.001 NRVM vs. +Sham Thy1pos, ####P < 0.0001 NRVM vs. +Sham Thy1neg, or +TAC Thy1pos, or +TAC Thy1neg. (E) Twitch rise time measured between 10% and 90% of peak amplitude. (F) Twitch decay time measured between 90% and 10% of peak amplitude. N=6 independent experiments, n=23–33 bundles per group. (G) Calcium transient amplitude. N=4 independent experiments, n=17–26 bundles per group. Statistical analysis was performed on the data normalized for each experiment to the respective control. ns: not significant, **P < 0.01, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA with Tukey’s multiple comparisons test.
Effects of Thy1pos and Thy1neg CFs on engineered cardiac muscle structure.
To assess the effects of different CF subsets on 3D engineered cardiac bundle structure, we performed immunohistological analysis of the bundle transverse cross-sections (Figure 4A). Notably, we found significantly increased Collagen I expression in +TAC Thy1neg bundles compared to all other groups (Figure 4B). No significant difference was found in the number of Vimentin+ fibroblasts (Figure 4C) indicating that CFs from the 4 groups had similar rates of cell turnover (proliferation and death) within cardiac bundles. In addition, the total cross-sectional area of cardiomyocytes within the bundle (labelled by F-actin, Figure 4D) as well as the average cross-sectional area of individual cardiomyocyte (obtained by dividing total F-actin+ area with encompassed nuclei number, Figure 4E) [56, 57, 63] were similar for all groups. Moreover, no differences were observed in the numbers of apoptotic (TUNEL+) nuclei among the 4 CF groups (Supplementary Figure 3). Overall, these analyses showed that compared to other CF subsets added to the cardiac bundles, the Thy1neg CFs from failing TAC ventricles led to increased collagen 1 deposition without differentially affecting cardiac muscle mass, cardiomyocyte size, or apoptosis.
Figure 4. Effects of different CF subsets on the engineered cardiac bundle structure.
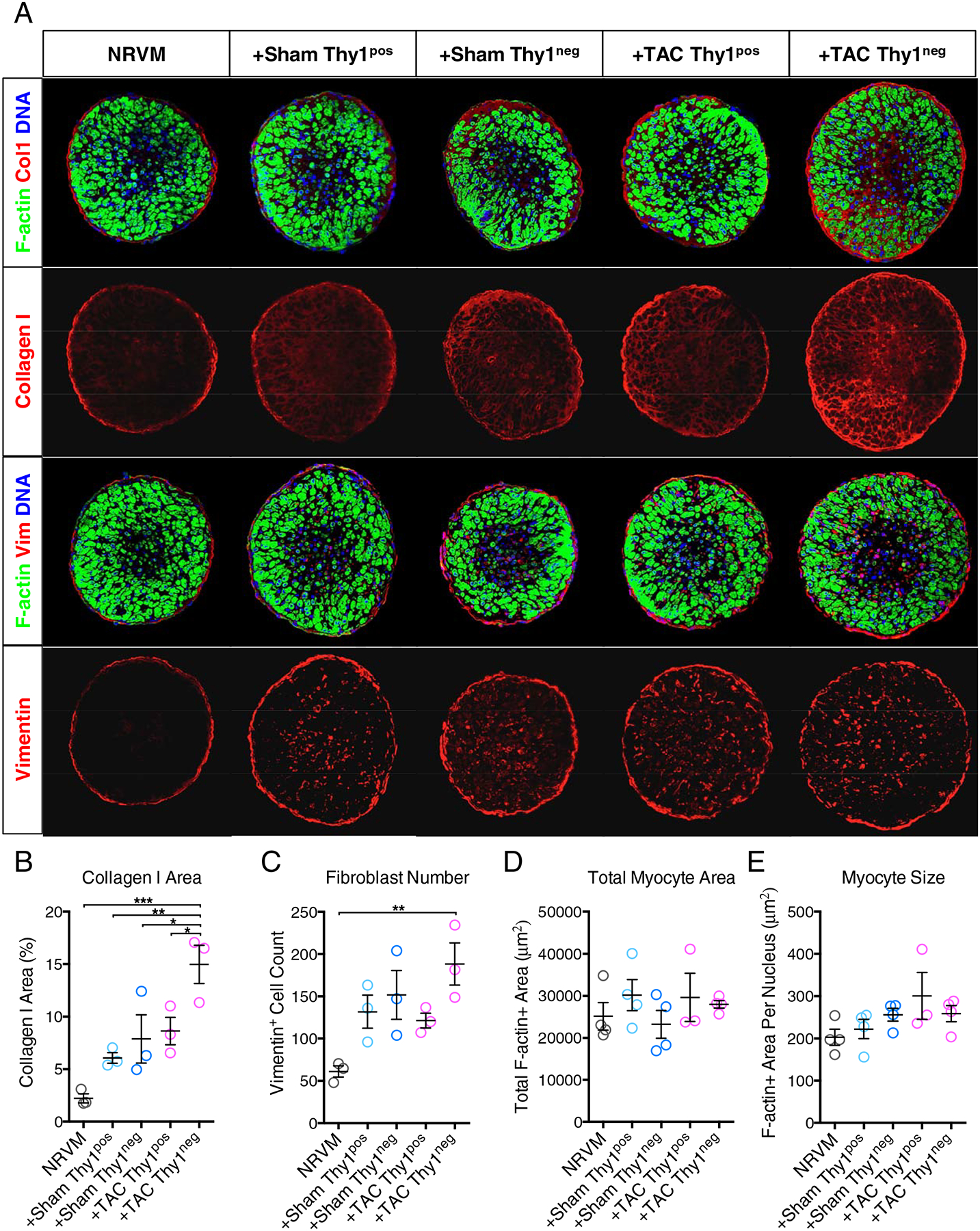
(A) Representative cross-sections of engineered cardiac bundles stained for filamentous actin (F-actin, green), Vimentin (Vim, red) or Collagen I (Col1, red), and DNA (nuclei, blue). Scar bar: 75 μm. (B–E) Quantification of bundle cross-section images for (B) percentage of Col1+ area, (C) total CF (Vim+) cell number, (D) total cardiomyocyte (F-actin+) area, and (E) average area of a cardiomyocyte (F-actin+ area per cardiomyocyte number). n=3–4 bundles per group. *P < 0.05, **P < 0.01, ***P < 0.001 by 1-way ANOVA with Bonferroni’s multiple comparisons test.
Global Thy1 knockout mice develop more severe cardiac dysfunction and fibrosis in response to TAC.
Given that the CFs lacking Thy1 isolated from mice with pressure-overload heart failure had an enhanced pro-fibrotic phenotype and exerted adverse effects on engineered cardiac tissue structure and function, we hypothesized that loss of Thy1 in all CFs may augment pressure-overload induced cardiac dysfunction and pathological fibrosis. To test this hypothesis, we utilized the already available global Thy1 knockout (Thy1KO) mouse model [64–66] to investigate its response to TAC in comparison to wild-type (WT) mice. The Thy1KO mice appear grossly normal at the basal conditions, but develop more severe histopathological lung fibrosis upon intratracheal bleomycin insult [64–66]. So far, neither the basal cardiac function nor the response to any cardiac insult has been explored in Thy1KO mice. The serial echocardiographic measurements revealed that Thy1KO mice displayed mild but non-significant reduction in fractional shortening (FS) compared to WT mice in basal Sham conditions (Figure 5A). In response to TAC, Thy1KO mice exhibited progressively more diminished FS compared to WT mice, which by 4 weeks post-TAC reached statistical significance (Figure 5, A–C; Supplementary Table 3). Moreover, other readouts of systolic function at 4 weeks post-TAC such as left ventricular end systolic dimension (LVESD) and the velocity of circumferential shortening corrected for heart rate (VcFc) were significantly increased and reduced, respectively in Thy1KO vs. WT mice, while no significant difference was seen in Sham conditions (Figure 5, D and E; Supplementary Table 3). The more severe cardiac dysfunction in response to TAC in Thy1KO mice was associated with significantly increased interstitial ventricular fibrosis compared to WT mice (Figure 5, F–G). Taken together, these functional and histopathological results in global Thy1KO mice further suggest a critical role of Thy1 expression in pathogenic response of CFs in pressure-overload induced heart failure.
Figure 5. Increased sensitivity of Thy1 KO mice to development of heart failure in response to TAC.
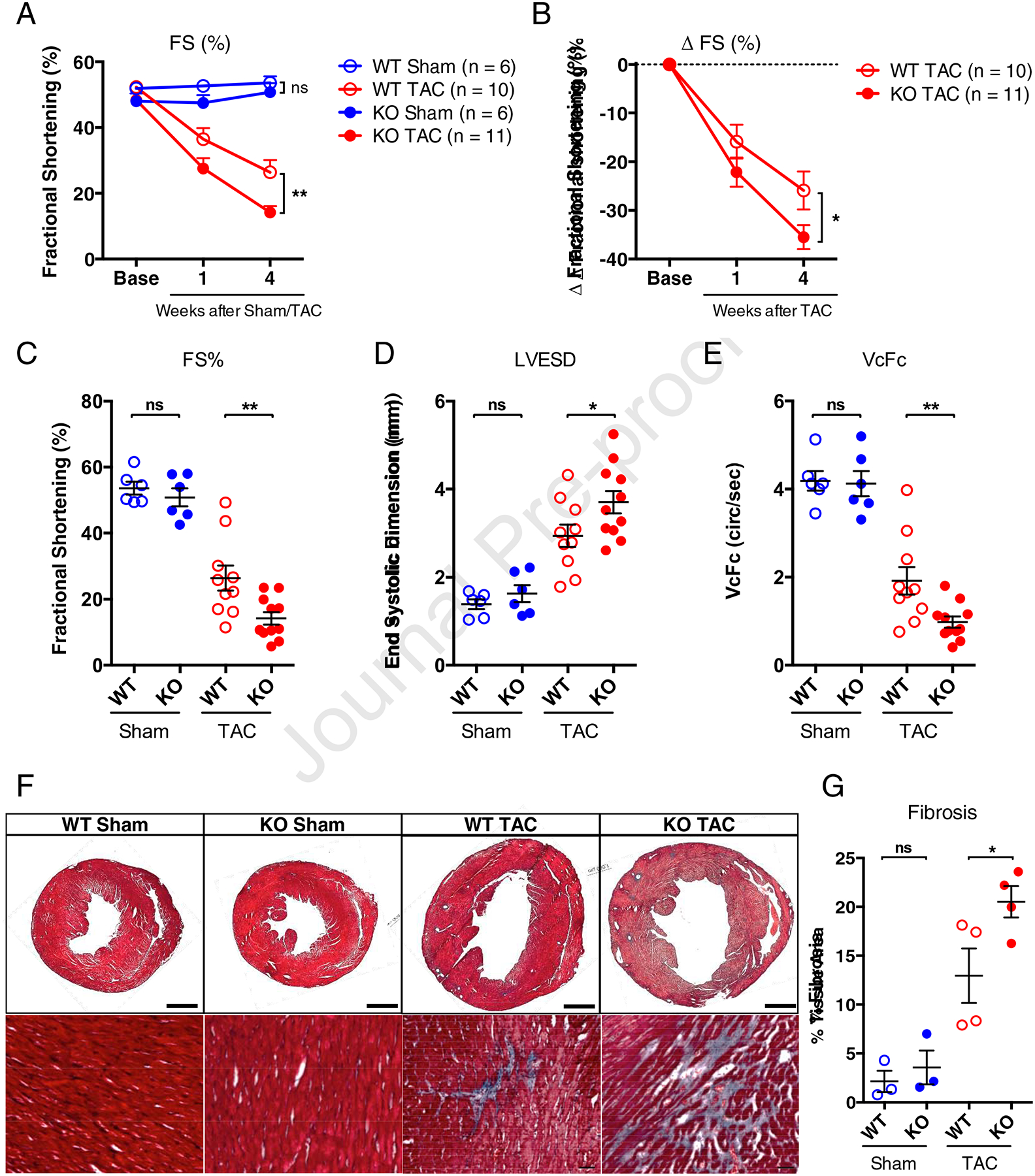
(A) Echocardiographic measurements of fractional shortening (FS) at pre-surgery (Base), and 1 week and 4 weeks post-surgery in wild-type (WT) and Thy1 knockout (KO) mice. (B) Change in FS from pre-surgery baseline calculated for each individual mouse and averaged. Fractional shortening (C), Left Ventricular End Systolic Dimension (LVESD) (D), and the velocity of circumferential shortening corrected for heart rate calculated as VcFc = (Fractional Shortening/Aortic Ejection Time)*(60/Heart Rate)0.5. (E) at 4 weeks post-surgery. n=6 mice per sham group, n=10 mice for WT TAC group, and n=11 mice for KO TAC group. ns: not significant, *P < 0.05, **P < 0.01 by 2-way ANOVA (A and B) or 1-way ANOVA (C, D, E) with Bonferroni’s multiple comparisons test. (F) Representative Masson’s Trichrome stains (transverse whole-ventricle cross-section shown at the top row with scale bar of 1 mm, and the magnified images shown at the bottom row with scale bar of 100 μm) at 4 weeks post-TAC surgery and in age-matched sham control mice. (G) Corresponding image quantification of the percentage of fibrosis in transverse ventricular cross-sections. n=3 hearts per sham group and n=4 hearts per TAC group. ns: not significant, *P < 0.05, by 1-way ANOVA with Bonferroni’s multiple comparisons test.
Cardiac fibroblasts from Thy1 KO mice express increased level of profibrotic genes in response to TAC.
To further determine whether the functional deficit in Thy1KO mice in response to TAC involves CF-specific mechanisms, we performed flow cytometry to sort freshly isolated CFs from Thy1KO and WT ventricles in TAC and Sham conditions (Figure 6A). The deletion of Thy1 expression in Thy1KO mice at the protein and mRNA level was validated by flow cytometry and qPCR (Figure 6, A and D). The flow cytometry analysis demonstrated no significant difference between Thy1KO and WT mice in the percentage of MEFSK4+/CD45−/CD31− CFs (Figure 6B). However, the number of MEFSK4+/CD45−/CD31− CFs per ventricle by flow cytometry sorting trended (P=0.08) to a higher value in Thy1KO compared to WT mice in response to TAC (Figure 6C). The gene expression analysis in the freshly isolated MEFSK4+/CD45−/CD31− CFs revealed that Postn, Tgfβ1, WNT1 Inducible Signaling Pathway Protein 1 (Wisp1), and Acta2 were significantly upregulated in Thy1KO compared to WT mice in response to TAC-induced heart failure, whereas no significant difference was detected in Sham condition (Figure 6, E–H). The same trend was also seen for the expression of Col1a1 (P=0.12).
Figure 6. Cardiac fibroblasts from Thy1 KO mice express increased level of profibrotic genes in response to TAC.
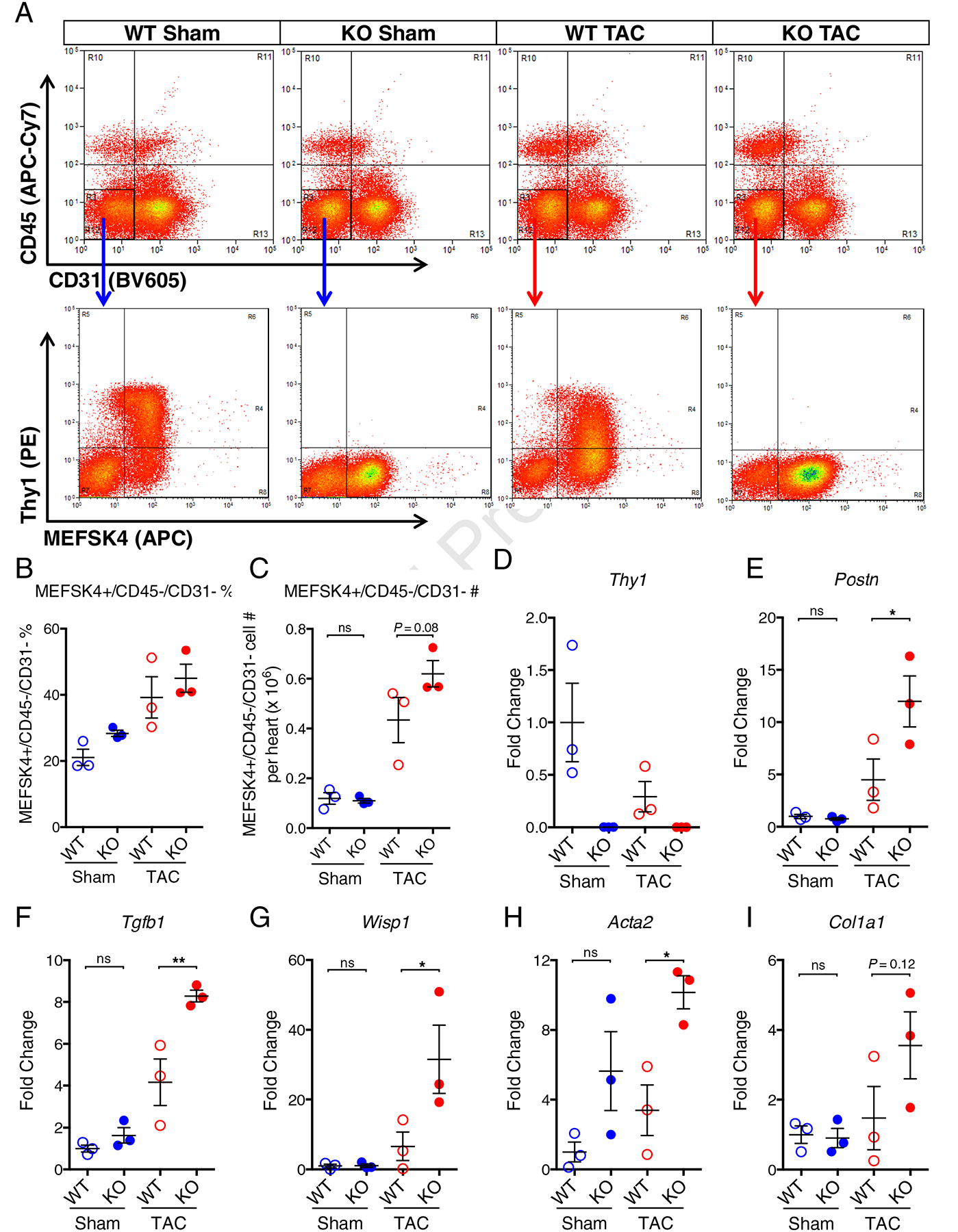
(A) Representative flow cytometry analysis of freshly isolated cells from WT and Thy1 KO mice in Sham or TAC conditions stained with APC-Cy7-conjugated CD45, BV605-conjugated CD31, PE-conjugated Thy1 and APC-conjugated MEFSK4. (B) Quantification of percentage fraction of MEFSK4+/CD45−/CD31− CFs in freshly isolated ventricular cells. (C) Quantification of MEFSK4+/CD45−/CD31− CFs cell number per heart (ventricles only) after FACS. Expression of Thy1 (D), Postn (E), Tgfb1 (F), Wisp1(G), Acta2 (H), and Col1a1 (I) genes in freshly isolated MEFSK4+/CD45−/CD31− CFs from WT and KO mice in Sham or TAC conditions. The mRNA expression was normalized to house-keeping gene B2M and shown as fold change relative to WT Sham group. N=3 independent experiments per group. ns: not significant, *P < 0.05, **P < 0.01 by 1-way ANOVA with Bonferroni’s multiple comparisons test.
Discussion
Pathologically activated myofibroblasts represent an attractive therapeutic target to delay or reverse the progression of heart failure and cardiac fibrosis [17–19]. In this study, we sought to determine whether Thy1 expression defines functionally distinct subsets of ventricular fibroblasts in response to pressure overload. We identified the previously uncharacterized Thy1neg (Thy1−/MEFSK4+/CD45−/CD31−) cell population in failing ventricles that displays a pro-fibrotic gene expression program and adversely affects contractile function of healthy cardiomyocytes in a 3D engineered co-culture system. Additionally, in response to pressure overload, mice with global knockout of Thy1 developed more severe cardiac dysfunction and fibrosis, further indicating that CFs lacking Thy1 expression may be important players in cardiac pathogenesis and worthy additional investigation as a potential therapeutic target.
The traditionally used cardiac fibroblast markers Thy1, Discoidin domain receptor 2 (DDR2), Fibroblast-specific protein 1 (Fsp1), Sca1, and Transcription factor 21 (Tcf21) label only a subpopulation of fibroblasts residing in the heart [13, 28, 67–69]. The more inclusive markers such as the intracellular marker Vimentin, extracellular marker Col1a1, and cell membrane marker PDGFRα are not suitable for sorting cardiac fibroblasts. By leveraging transgenic mouse lines harboring fluorescence-tagged Col1a1, PDGFRα, and Tcf21 [13, 23–25, 70, 71], Pinto and colleagues have demonstrated that use of MEFSK4 antibody currently represents the most inclusive strategy for labeling and sorting mouse heart-resident CFs [28]. Using MEFSK4 (and negative selection for CD45 and CD31) to sort CFs from ventricles allowed us to identify subpopulation of MEFSK4+ CFs that is negative for Thy1 (Thy1neg, Figure 1, A, B and F–H) and that exhibits an activated myofibroblast phenotype in response to pressure overload (Figure 1–4). Cardiac myofibroblasts were traditionally defined as αSMA-expressing cells; however recent studies in advanced transgenic mouse models revealed that αSMA labeling identifies only 15% of myofibroblasts after TAC [13] and 35% after AngII stimulation [27]. We only observed trends towards increased expression of Acta2 (αSMA) in Thy1neg vs. Thy1pos CFs in both Sham and TAC ventricles (Figure 2, A and G), which was further confirmed when comparing WT and Thy1KO groups (Figure 6, D and H). On the other hand, periostin has recently emerged as a pan-myofibroblast marker that identifies essentially all the activated fibroblasts in the mouse heart [15, 68]. In our study, we found that Postn is significantly upregulated in Thy1neg compared to Thy1pos CFs in TAC heart (Figure 2B), along with other signature pro-fibrotic factors such as TGFβ1, Col1a1 and Col3a1 (Figure 2, C–E and H).
Previous studies have established that the in vitro culture of CFs on non-physiologically rigid 2D substrates rapidly and massively upregulates the expression of αSMA, profoundly alters the cell phenotype compared to in vivo conditions [56, 58–60], and can mask the potential phenotypic differences between CFs from diseased and healthy hearts [58]. Consistent with these reports, we observed marked elevation of αSMA expression in subcultured CFs at passage 1 and 2 compared to freshly isolated CFs (Supplementary Figure 2G), along with upregulation of other key ECM genes Col1a1, Col3a1 and Fn1 (Supplementary Figure 2, D–F). Interestingly, the Thy1 expression also became upregulated in subcultured compared to freshly isolated CFs suggesting its plasticity and epigenetic sensitivity to environmental conditions. Importantly, the distinct differences in the expression of Thy1, Acta2, Postn, Tgfβ1, Col1a1, Col3a1 and Fn1 among the 4 freshly isolated CF groups were neutralized with cell subculture (Supplementary Figure 2). Furthermore, healthy adult rat fibroblasts subcultured in 2D have been shown to alter the morphology and negatively affect calcium transients of co-cultured cardiomyocytes similar to the freshly isolated myofibroblasts from TAC hearts [72]. In light of these findings, we ensured that only freshly isolated CFs rather than 2D-expanded CFs were directly utilized in our 3D co-culture experiments.
We have previously established a versatile 3D tissue-engineered co-culture system (cardiac bundles) that enabled us to systematically study the direct fibroblast-specific effects on the surrounding healthy neonatal rat ventricular myocytes (NRVMs) in a physiologically relevant microenvironment that better mimics the native myocardium than 2D cultures [56, 57, 73]. In the current study, we demonstrated that the co-cultured NRVM + TAC Thy1neg bundles exhibit significantly weaker contractions, prolonged contraction rise time, reduced calcium transient amplitude, and increased Collagen I deposition (Figure 3; Figure 4, A and B), all hallmarks of a failing heart phenotype (Fig. 1). Previous studies have shown that the proliferation of mouse ventricular CFs peaks in vivo within the first week after TAC, and resolves back to the basal level by 4 weeks post-TAC [13, 74, 75]. The facts that the CFs used in this study were isolated from 7–9-week post-TAC ventricles and that we found no difference in fibroblast numbers in bundles with different CF subsets, indicate that the deteriorating effects of Thy1neg vs. Thy1pos CFs were proliferation-independent and likely a result of their distinct fibrogenic phenotype and specific adverse paracrine effects on NRVMs. Interestingly, no cardiomyocyte hypertrophy was observed in bundles with different subsets of CFs (Figure 4, A and D), suggesting that either hypertrophic CM phenotype may require longer than 2 weeks to occur in vitro or hypertrophic remodeling in native heart in response to pressure overload requires complex environmental signals beyond CF-mediated effects, including mechanical stretch, specific metabolic substrates, and/or a multi-cellular rather than two-cellular crosstalk [76–81].
Although no previous reports linked the CF subpopulations distinguished by Thy1 expression to cardiac pathology, it has been well documented that Thy1+ and Thy1− lung fibroblasts have distinct morphology and inflammatory responses [30, 37–40, 65, 82]. Specifically, Thy1− but not Thy1+ mouse lung fibroblasts increase class II MHC (Ia) expression in response to interferon-γ stimulation [37] and Thy1− rat lung fibroblasts react similarly in response to bleomycin injury [30]. Moreover, Thy1− rat lung fibroblasts demonstrate a more pronounced myofibroblast phenotype and greater collagen gel contraction than Thy1+ fibroblasts when exposed to pro-fibrotic cytokines TGFβ, endothelin-1 (ET-1), or connective tissue growth factor (CTGF) [82]. Interestingly, rat strains with a naturally higher percentage of Thy1− lung fibroblasts develop more severe lung fibrosis [30, 83], further indicating that Thy1− rather than Thy1+ fibroblasts are the main responders to fibrogenic stimulation of lung. In the same mouse model of global Thy1 knockout as used in our study, Hagood and colleagues showed that these mice develop severe lung fibrosis with extensive collagen deposition and increased TGFβ activity in response to bleomycin injury [64, 65]. Similar to these conclusions, our ex vivo and in vitro studies with freshly isolated CFs as well as in vivo studies with global Thy1 knockout mice consistently point to the finding that Thy1neg cells represent a functionally distinct, pathogenic subpopulation of cardiac fibroblasts and that the absence of Thy1 expression in CFs leads to more severe cardiac dysfunction and fibrosis in response to pressure overload.
Thy1 is not exclusively expressed in fibroblasts, but also in neural cells, hematopoietic progenitor cells, T cells, and endothelial cells [84–88]. Since we used a global Thy1 knockout mouse model, it is possible that the observed cardiac functional deficit and fibrosis might result from more complex multi-cellular interactions. We showed that the pro-fibrotic factors including Postn, Tgfβ1, Wisp1, Acta2 and Col1a1 were upregulated in the freshly isolated MEFSK4+/CD45−/CD31− CFs from Thy1KO compared to WT mice in response to TAC (Figure 6, E–I), suggesting the involvement of a CF-mediated mechanism. Generation of CF-specific Thy1 deletion by crossing PDGFRα-Cre or Tcf21-Cre with Thy1fl/fl (currently unavailable) mice would be required to further dissect the fibroblast-specific roles of Thy1 in heart failure. Importantly, the findings that Thy1 KO hearts and engineered heart tissues with Thy1neg CFs exhibit aggravated cardiac dysfunction and fibrosis, along with proven causative roles of Thy1 in lung disease [89], hold promise that targeting Thy1-related pathogenic signals might lead to discovery of new therapeutics for heart failure. A necessary first step in this process will be to fully elucidate Thy1’s cis and trans regulatory functions [90, 91] and expression dynamics in cardiac fibroblasts at different stages of the disease.
In summary, our in vitro and in vivo studies of a previously unexplored population of Thy1neg ventricular cardiac fibroblasts have revealed that these cells exhibit a pronounced pathogenic phenotype in the setting of pressure overload-induced heart failure. Future investigations are warranted to better understand the functional roles of Thy1 in CFs and to elucidate if a similar cell population may play analogous roles in other cardiac fibrotic diseases (e.g. myocardial infarction, atrial fibrosis, and various cardiomyopathies).
Methods
Generation of pressure overload induced heart failure and echocardiography.
Eight to twelve-week-old C57BL/6 wild-type and global Thy1 knockout (KO) mice were subjected to TAC or sham-operated control surgery, which were carried out at the Duke Cardiovascular Physiology Core Facility as previously described [42, 92]. Echocardiography was performed with a Vevo 2100 high-resolution imaging system (VisualSonics) [92]. Mice that did not survive beyond 2 weeks after the surgery or were identified to have undergone unsuccessful TAC (<10% LV mass increase) were excluded from the study. Due to increased mortality at later time points, Thy1 KO mice were compared to wild-type controls only between 0 and 4 weeks post-TAC.
Cell isolation and culture.
CFs were isolated from murine hearts 4 to 9 weeks after TAC or sham procedure. Briefly, ventricles were dissected out and minced into pieces smaller than 1mm in diameter. The minced tissues were digested with 1 mg/ml Collagenase/Dispase (Sigma 11097113001) and 100 U/ml DNase I (Sigma 4536282001) in PBS, incubated at 37 °C for 20 min on a rocking platform, followed by gentleMACS Dissociator program m_neoheart_01_01. The supernatant was collected and diluted in fibroblast culture medium (DMEM (Sigma D6429) with 10% FBS and Penicillin/streptomycin) on ice. The 20-min digestion step was repeated for 5–7 times followed by a final dissociation step by gentleMACS Dissociator program m_heart_01.01. After the serial digestions, the cells were filtered through a 100 μm nylon cell strainer and centrifuged at 500×g for 5 min, and then resuspended in either PBS for fluorescence-activated cell sorting (FACS) or fibroblast culture medium for further culture. For the culture experiments, fibroblasts were seeded into 6-well plates and cultured until confluence with media changed every other day. Fibroblasts were trypsinized and sub-cultured at 1:3 ratio and used at passage 1 and 2 for qPCR analysis.
Flow cytometry.
Freshly isolated ventricular cells from TAC and Sham hearts were washed once in PBS and then resuspended in red blood cell lysing buffer (BD Biosciences 555899), then incubated for 5 min at room temperature with a gentle vortexing followed by a PBS wash. Cells were subsequently stained with antibodies for APC-Cy7-conjugated CD45, BV605-conjugated CD31, PE-conjugated Thy1 and APC-conjugated MEFSK4 for 30 min on ice in the dark. The specific antibody dilutions were listed in Supplementary Table 1. Upon antibody staining, cells were washed in the sorting buffer (PBS with 3% FBS) twice, filtered through a 30 μm CellTrics filter (Sysmex 04-004-2326), and resuspended in the sorting buffer. FACS was performed using either BD DiVA or B-C Astrios cell sorter at the Flow Cytometry Shared Resource Core Facility at Duke University. Cells were sorted into 15ml falcon tubes filled with fibroblast culture medium.
Gene expression analysis.
The total RNA from freshly isolated or cultured CFs was extracted using RNeasy Plus Mini Kit (Qiagen 74134) according to the manufacturer’s instructions. The RNA concentration was measured in NanoDrop at the Duke Center for Genomic and Computational Biology Core Facility. Total RNA was converted to cDNA using iScript cDNA synthesis kit (Bio-Rad 170–8891). Standard qPCR reactions were performed with iTaq Universal SYBR Green Supermix (Bio-Rad 172–5121) in Bio-Rad CFX Real-Time System. The primers are listed in Supplementary Table 2. The mRNA expression was normalized to the house-keeping gene Beta-2-Microglobulin (B2M) and shown as 2^-ΔCt.
Fabrication of 3D engineered cardiac tissue bundles.
The 3D engineered cardiac tissue bundles were generated as previously described [56, 57, 63]. Briefly, NRVMs were dissociated from the ventricles of 2-day-old Sprague Dawley rats. Each bundle was constructed by encapsulating 2.25 × 105 NRVMs with or without 0.675 × 105 freshly isolated CFs in a fibrin-based hydrogel consisting of 20 μl of 3D culture media (see below), 8 μl of 10 mg/ml Fibrinogen (Akron), 4 μl of Matrigel, 8 μl of 2× cardiac media (see below), and 0.4 μl of 50 unit/ml thrombin in 0.1% BSA in PBS [56]. The cell-hydrogel mixture was poured into polydimethylsiloxane (PDMS) molds pre-treated with 0.2% (w/v) pluronic F-127 (Invitrogen) with a laser-cut Cerex frame in individual wells of a 12-well plate and polymerized for 45 min at 37°C followed by addition of 2 ml culture media per well. The following day, frames with the attached bundles were removed from the molds and cultured in suspension dynamically at a rocking platform for 2 weeks. Culture media were changed every other day. 3D culture media: DMEM (ThermoFisher 11885 or Sigma D6046), 10% horse serum, 1% chick embryo extract, 5 U/ml penicillin, 2 μg/ml Vitamin B12, 1 mg/ml Aminocaproic Acid (ACA) and 50 μg/ml Ascorbic Acid 2-phosphate sesquimagnesium salt hydrate). 2x cardiac media: DMEM (ThermoFisher 31600–034), 10% horse serum, 2 mg/ml ACA, 10 U/ml penicillin and 4 μg/ml Vitamin B12.
Contractile force measurements.
The isometric contractile force generated by 2-week old engineered cardiac bundles was measured as previously described [56, 57, 63, 93]. Briefly, individual bundles were transferred to a chamber with Tyrode’s solution maintained at 37°C and mounted onto a force transducer connected to a computer-controlled linear actuator that applied 20% stretch above the resting length of the bundle in 4% increments. The contractile force traces were acquired during 2Hz electrical stimulation and analyzed for maximum peak force amplitude, contraction rise time (between 10% and 90% of peak amplitude) and decay time (between 90% and 10% of peak amplitude) using a custom MATLAB program [56, 57, 63, 93].
Calcium imaging.
Two-week cultured cardiac bundles were stained with intracellular calcium indicator Rhod-2 AM (ThermoFisher R1245MP) and calcium transients were recorded as previously described [56, 57, 94]. Briefly, bundles were placed in a 37°C live-imaging chamber in Tyrode’s solution with 10 μM blebbistatin, electrically stimulated at 2Hz, and calcium transient videos were acquired using an EMCCD camera (iXonEM., Andor) attached to a Nikon microscope, and analyzed using Andor Solis software. The changes in fluorescence signal were displayed as ΔF/F [56, 57, 94].
Histological analysis.
Cardiac bundles were washed with PBS and fixed with 2%v/v paraformaldehyde (Electron Microscopy Sciences) at 4°C on a rocker overnight. Fixed bundles were washed in PBS and embedded in optimal cutting temperature (OCT) medium and frozen in liquid Nitrogen. Five μm sections were obtained using a cryostat at −21°C and mounted onto slides, followed by permeabilization in 0.5% Triton X-100 for 30 min and blocking in 5% chick serum in PBS for 1 hr at room temperature. Sections were incubated with primary antibodies in blocking buffer on a rocker overnight at 4°C in indicated dilutions: Collagen I (Abcam ab34710, 1:200), Vimentin (Abcam ab92547, 1:400), α-smooth muscle actin (Abcam ab5694, 1:200). Alexa-Fluor conjugated secondary antibodies (Invitrogen), DAPI, and F-actin (Alexa Fluor 488 Phalloidin, Life Technologies A12379) were applied at a 1:400 dilution in blocking buffer for 2 hrs at room temperature. The TUNEL (Sigma 12156792910) assay was performed according to the manufacturer’s instructions. The immunostained cross-sections were imaged using a Leica inverted SP5 confocal microscope at the Duke Light Microscopy Core Facility. Masson’s Trichrome staining was performed by BioRepository and Precision Pathology Center at Duke University, and images were acquired using an Axio Imager upright microscope. Image analysis was performed using custom ImageJ software.
Statistics.
Statistical analysis was performed with the GraphPad Prism Software Package, version 6.0 (GraphPad Inc). A two-tailed Student’s t test was used for comparisons between 2 groups, and 1-way ANOVA with Tukey’s or Bonferroni’s multiple comparisons test was used for comparisons among more than 2 groups. A 2-way ANOVA with Bonferroni’s multiple comparisons test was used for time-course echocardiographic measurements. P < 0.05 was considered statistically significant. Results are expressed as mean ± SEM.
Study approval.
All animal procedures were performed according to approved protocols and in compliance with the Institutional Animal Care and Use Committee at Duke University and the NIH Guide for the Care and Use of Laboratory Animals. Global Thy1 knockout mice on a C57BL/6 background were described previously and provided Dr. James S. Hagood from the University of North Carolina, Chapel Hill [64, 65]. The wildtype C57BL/6 mice (strain code: 027) were obtained from Charles River Laboratories.
Supplementary Material
Acknowledgements
This study was supported by the National Institutes of Health grants HL126524, HL132389, HL134764 and grant from Foundation Leducq to NB and Morton Friedman Fellowship from Duke Biomedical Engineering and American Heart Association Pre-doctoral Fellowship to YL. We sincerely thank Dr. James S. Hagood (University of North Carolina, Chapel Hill) for providing the global Thy1 knockout mice. We would like to thank Dr. Howard A. Rockman for advice on the in vivo experiments, as well as Christopher P. Jackman, Hengtao Zhang, Sanjana Vasudevan, Azalia Maul, and Sumie Okuwa for technical support. We acknowledge the support of the Duke Cardiovascular Physiology Core for animal surgeries and echocardiography, as well as other core facilities including Light Microscopy Core Facility (LMCF), the Flow Cytometry Shared Resource, BioRepository and Precision Pathology Center, and Genomic and Computational Biology (GCB) at Duke University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors have declared no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations
References
- [1].Bernardo BC, Weeks KL, Pretorius L, McMullen JR, Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies, Pharmacology & therapeutics 128(1) (2010) 191–227. [DOI] [PubMed] [Google Scholar]
- [2].Piek A, de Boer RA, Sillje HH, The fibrosis-cell death axis in heart failure, Heart failure reviews 21(2) (2016) 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Roger VL, Epidemiology of heart failure, Circ Res 113(6) (2013) 646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baudino TA, Carver W, Giles W, Borg TK, Cardiac fibroblasts: friend or foe?, Am J Physiol Heart Circ Physiol 291(3) (2006) H1015–26. [DOI] [PubMed] [Google Scholar]
- [5].Zeisberg EM, Kalluri R, Origins of cardiac fibroblasts, Circ Res 107(11) (2010) 1304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Souders CA, Bowers SL, Baudino TA, Cardiac fibroblast: the renaissance cell, Circ Res 105(12) (2009) 1164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Leslie KO, Taatjes DJ, Schwarz J, vonTurkovich M, Low RB, Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit, Am J Pathol 139(1) (1991) 207–16. [PMC free article] [PubMed] [Google Scholar]
- [8].Abe R, Donnelly SC, Peng T, Bucala R, Metz CN, Peripheral blood fibrocytes: differentiation pathway and migration to wound sites, J Immunol 166(12) (2001) 7556–62. [DOI] [PubMed] [Google Scholar]
- [9].Zhou B, Pu WT, Epicardial epithelial-to-mesenchymal transition in injured heart, J Cell Mol Med 15(12) (2011) 2781–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].van Wijk B, Gunst QD, Moorman AF, van den Hoff MJ, Cardiac regeneration from activated epicardium, PloS one 7(9) (2012) e44692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhou B, Honor LB, He H, Ma Q, Oh JH, Butterfield C, Lin RZ, Melero-Martin JM, Dolmatova E, Duffy HS, Gise A, Zhou P, Hu YW, Wang G, Zhang B, Wang L, Hall JL, Moses MA, McGowan FX, Pu WT, Adult mouse epicardium modulates myocardial injury by secreting paracrine factors, J Clin Invest 121(5) (2011) 1894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R, Endothelial-to mesenchymal transition contributes to cardiac fibrosis, Nat Med 13(8) (2007) 952–61. [DOI] [PubMed] [Google Scholar]
- [13].Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM, Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis, J Clin Invest 124(7) (2014) 2921–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Muller AM, Volz KS, Tang Z, Red-Horse K, Ardehali R, Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation, Circ Res 115(7) (2014) 625–35. [DOI] [PubMed] [Google Scholar]
- [15].Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD, Genetic lineage tracing defines myofibroblast origin and function in the injured heart, Nat Commun 7 (2016) 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, Pinto AR, Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart, Cell reports 22(3) (2018) 600–610. [DOI] [PubMed] [Google Scholar]
- [17].Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, Tan J, Chothani SP, Ye L, Rackham OJL, Ko NSJ, Sahib NE, Pua CJ, Zhen NTG, Xie C, Wang M, Maatz H, Lim S, Saar K, Blachut S, Petretto E, Schmidt S, Putoczki T, Guimaraes-Camboa N, Wakimoto H, van Heesch S, Sigmundsson K, Lim SL, Soon JL, Chao VTT, Chua YL, Tan TE, Evans SM, Loh YJ, Jamal MH, Ong KK, Chua KC, Ong BH, Chakaramakkil MJ, Seidman JG, Seidman CE, Hubner N, Sin KYK, Cook SA, IL-11 is a crucial determinant of cardiovascular fibrosis, Nature 552(7683) (2017) 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gourdie RG, Dimmeler S, Kohl P, Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease, Nat Rev Drug Discov (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kong P, Christia P, Frangogiannis NG, The pathogenesis of cardiac fibrosis, Cell Mol Life Sci 71(4) (2014) 549–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bursac N, Cardiac fibroblasts in pressure overload hypertrophy: the enemy within?, J Clin Invest 124(7) (2014) 2850–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ivey MJ, Tallquist MD, Defining the Cardiac Fibroblast, Circ J 80(11) (2016) 2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lajiness JD, Conway SJ, Origin, development, and differentiation of cardiac fibroblasts, J Mol Cell Cardiol 70 (2014) 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P, Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms, Mol Cell Biol 23(11) (2003) 4013–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yata Y, Scanga A, Gillan A, Yang L, Reif S, Breindl M, Brenner DA, Rippe RA, DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells, Hepatology 37(2) (2003) 267–76. [DOI] [PubMed] [Google Scholar]
- [25].Smith CL, Baek ST, Sung CY, Tallquist MD, Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling, Circ Res 108(12) (2011) e15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD, Revisiting Cardiac Cellular Composition, Circ Res 118(3) (2016) 400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S, Wettschureck N, Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice, Circ Res 118(12) (2016) 1906–17. [DOI] [PubMed] [Google Scholar]
- [28].Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni M, Debuque RJ, Chandran A, Wang L, Arora K, Rosenthal N, Tallquist MD, Revisiting Cardiac Cellular Composition, Circ Res (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hagood JS, Thy-1 as an Integrator of Diverse Extracellular Signals, Front Cell Dev Biol 7 (2019) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McIntosh JC, Hagood JS, Richardson TL, Simecka JW, Thy1 (+) and (−) lung fibrosis subpopulations in LEW and F344 rats, Eur Respir J 7(12) (1994) 2131–8. [DOI] [PubMed] [Google Scholar]
- [31].Koumas L, Smith TJ, Phipps RP, Fibroblast subsets in the human orbit: Thy-1+ and Thy-1− subpopulations exhibit distinct phenotypes, Eur J Immunol 32(2) (2002) 477–85. [DOI] [PubMed] [Google Scholar]
- [32].Koumas L, King AE, Critchley HO, Kelly RW, Phipps RP, Fibroblast heterogeneity: existence of functionally distinct Thy 1(+) and Thy 1(−) human female reproductive tract fibroblasts, Am J Pathol 159(3) (2001) 925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kawka E, Witowski J, Bartosova M, Catar R, Rudolf A, Philippe A, Rutkowski R, Schafer B, Schmitt CP, Dragun D, Jorres A, Thy-1(+/−) fibroblast subsets in the human peritoneum, American journal of physiology. Renal physiology 313(5) (2017) F1116–F1123. [DOI] [PubMed] [Google Scholar]
- [34].Borrello MA, Phipps RP, Differential Thy-1 expression by splenic fibroblasts defines functionally distinct subsets, Cell Immunol 173(2) (1996) 198–206. [DOI] [PubMed] [Google Scholar]
- [35].Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, Nguyen HN, Noss EH, Turner JD, Earp BE, Blazar PE, Wright J, Simmons BP, Donlin LT, Kalliolias GD, Goodman SM, Bykerk VP, Ivashkiv LB, Lederer JA, Hacohen N, Nigrovic PA, Filer A, Buckley CD, Raychaudhuri S, Brenner MB, Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis, Nat Commun 9(1) (2018) 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Croft A, Campos J, Jansen K, Turner J, Marshall J, Attar M, Savary L, Wehmeyer C, Naylor A, Kemble S, Begum J, Dürholz K, Perlman H, Barone F, McGettrick H, Fearon D, Wei K, Raychaudhuri S, Korsunsky I, Brenner M, Coles M, Sansom S, Filer A, Buckley C, Distinct fibroblast subsets drive inflammation and damage in arthritis, Nature (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Phipps RP, Penney DP, Keng P, Quill H, Paxhia A, Derdak S, Felch ME, Characterization of two major populations of lung fibroblasts: distinguishing morphology and discordant display of Thy 1 and class II MHC, Am J Respir Cell Mol Biol 1(1) (1989) 65–74. [DOI] [PubMed] [Google Scholar]
- [38].Zhou Y, Hagood JS, Murphy-Ullrich JE, Thy-1 expression regulates the ability of rat lung fibroblasts to activate transforming growth factor-beta in response to fibrogenic stimuli, Am J Pathol 165(2) (2004) 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hagood JS, Miller PJ, Lasky JA, Tousson A, Guo B, Fuller GM, McIntosh JC, Differential expression of platelet-derived growth factor-alpha receptor by Thy-1(−) and Thy-1(+) lung fibroblasts, Am J Physiol 277(1) (1999) L218–24. [DOI] [PubMed] [Google Scholar]
- [40].Hagood JS, Mangalwadi A, Guo B, MacEwen MW, Salazar L, Fuller GM, Concordant and discordant interleukin-1-mediated signaling in lung fibroblast thy-1 subpopulations, Am J Respir Cell Mol Biol 26(6) (2002) 702–8. [DOI] [PubMed] [Google Scholar]
- [41].Phipps RP, Baecher C, Frelinger JG, Penney DP, Keng P, Brown D, Differential expression of interleukin 1 alpha by Thy-1+ and Thy-1− lung fibroblast subpopulations: enhancement of interleukin 1 alpha production by tumor necrosis factor-alpha, Eur J Immunol 20(8) (1990) 1723–7. [DOI] [PubMed] [Google Scholar]
- [42].Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J Jr., Chien KR, Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy, Proc Natl Acad Sci U S A 88(18) (1991) 8277–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ, Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy, Science 280(5363) (1998) 574–7. [DOI] [PubMed] [Google Scholar]
- [44].Choi DJ, Koch WJ, Hunter JJ, Rockman HA, Mechanism of beta-adrenergic receptor desensitization in cardiac hypertrophy is increased beta-adrenergic receptor kinase, J Biol Chem 272(27) (1997) 17223–9. [DOI] [PubMed] [Google Scholar]
- [45].Naga Prasad SV, Esposito G, Mao L, Koch WJ, Rockman HA, Gbetagamma-dependent phosphoinositide 3-kinase activation in hearts with in vivo pressure overload hypertrophy, J Biol Chem 275(7) (2000) 4693–8. [DOI] [PubMed] [Google Scholar]
- [46].Rapacciuolo A, Esposito G, Caron K, Mao L, Thomas SA, Rockman HA, Important role of endogenous norepinephrine and epinephrine in the development of in vivo pressure-overload cardiac hypertrophy, J Am Coll Cardiol 38(3) (2001) 876–82. [DOI] [PubMed] [Google Scholar]
- [47].Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA, Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress, Circulation 105(1) (2002) 85–92. [DOI] [PubMed] [Google Scholar]
- [48].Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, Srivastava D, Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling, Dev Cell 16(2) (2009) 233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D, In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes, Nature 485(7400) (2012) 593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang Z, Xu Y, Wang M, Ye J, Liu J, Jiang H, Ye D, Wan J, TRPA1 inhibition ameliorates pressure overload-induced cardiac hypertrophy and fibrosis in mice, EBioMedicine 36 (2018) 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang B, Yang Q, Bai WW, Xing YF, Lu XT, Sun YY, Zhao YX, Tongxinluo protects against pressure overload-induced heart failure in mice involving VEGF/Akt/eNOS pathway activation, PloS one 9(6) (2014) e98047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ, Nagai R, Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload, J Clin Invest 120(1) (2010) 254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Izumo S, Nadal-Ginard B, Mahdavi V, Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload, Proceedings of the National Academy of Sciences of the United States of America 85(2) (1988) 339–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cohn JN, Ferrari R, Sharpe N, Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling, J Am Coll Cardiol 35(3) (2000) 569–82. [DOI] [PubMed] [Google Scholar]
- [55].Grimm D, Huber M, Jabusch HC, Shakibaei M, Fredersdorf S, Paul M, Riegger GA, Kromer EP, Extracellular matrix proteins in cardiac fibroblasts derived from rat hearts with chronic pressure overload: effects of beta-receptor blockade, Journal of molecular and cellular cardiology 33(3) (2001) 487–501. [DOI] [PubMed] [Google Scholar]
- [56].Li Y, Asfour H, Bursac N, Age-dependent functional crosstalk between cardiac fibroblasts and cardiomyocytes in a 3D engineered cardiac tissue, Acta Biomater 55 (2017) 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jackman CP, Carlson AL, Bursac N, Dynamic culture yields engineered myocardium with near-adult functional output, Biomaterials 111 (2016) 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dawson K, Wu CT, Qi XY, Nattel S, Congestive Heart Failure Effects on Atrial Fibroblast Phenotype: Differences between Freshly-Isolated and Cultured Cells, PloS one 7(12) (2012) e52032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rohr S, Cardiac fibroblasts in cell culture systems: myofibroblasts all along?, J Cardiovasc Pharmacol 57(4) (2011) 389–99. [DOI] [PubMed] [Google Scholar]
- [60].Baum J, Duffy HS, Fibroblasts and myofibroblasts: what are we talking about?, Journal of cardiovascular pharmacology 57(4) (2011) 376–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yano M, Yamamoto T, Kobayashi S, Ikeda Y, Matsuzaki M, Defective Ca2+ cycling as a key pathogenic mechanism of heart failure, Circ J 72 Suppl A (2008) A22–30. [DOI] [PubMed] [Google Scholar]
- [62].Mora MT, Ferrero JM, Gomez JF, Sobie EA, Trenor B, Ca(2+) Cycling Impairment in Heart Failure Is Exacerbated by Fibrosis: Insights Gained From Mechanistic Simulations, Frontiers in physiology 9 (2018) 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, Bursac N, Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues, Nat Commun 8(1) (2017) 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hagood JS, Prabhakaran P, Kumbla P, Salazar L, MacEwen MW, Barker TH, Ortiz LA, Schoeb T, Siegal GP, Alexander CB, Pardo A, Selman M, Loss of fibroblast Thy-1 expression correlates with lung fibrogenesis, Am J Pathol 167(2) (2005) 365–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Liu X, Wong SS, Taype CA, Kim J, Shentu TP, Espinoza CR, Finley JC, Bradley JE, Head BP, Patel HH, Mah EJ, Hagood JS, Thy-1 interaction with Fas in lipid rafts regulates fibroblast apoptosis and lung injury resolution, Lab Invest 97(3) (2017) 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Rege TA, Hagood JS, Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses, Biochim Biophys Acta 1763(10) (2006) 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chen G, Bracamonte-Baran W, Diny NL, Hou X, Talor MV, Fu K, Liu Y, Davogustto G, Vasquez H, Taegtmeyer H, Frazier OH, Waisman A, Conway SJ, Wan F, Cihakova D, Sca-1(+) cardiac fibroblasts promote development of heart failure, Eur J Immunol 48(9) (2018) 1522–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kaur H, Takefuji M, Ngai C, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Holper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S, Wettschureck N, Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice, Circ Res (2016). [DOI] [PubMed] [Google Scholar]
- [69].Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC, Cardiac Fibrosis: The Fibroblast Awakens, Circ Res 118(6) (2016) 1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN, Tallquist MD, The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors, Development 139(12) (2012) 2139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN, Heart repair by reprogramming non-myocytes with cardiac transcription factors, Nature 485(7400) (2012) 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cartledge JE, Kane C, Dias P, Tesfom M, Clarke L, McKee B, Al Ayoubi S, Chester A, Yacoub MH, Camelliti P, Terracciano CM, Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators, Cardiovasc Res 105(3) (2015) 260–70. [DOI] [PubMed] [Google Scholar]
- [73].Li Y, Dal-Pra S, Mirotsou M, Jayawardena TM, Hodgkinson CP, Bursac N, Dzau VJ, Tissue-engineered 3-dimensional (3D) microenvironment enhances the direct reprogramming of fibroblasts into cardiomyocytes by microRNAs, Scientific reports 6 (2016) 38815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD, Resident fibroblast expansion during cardiac growth and remodeling, J Mol Cell Cardiol 114 (2018) 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Souders CA, Borg TK, Banerjee I, Baudino TA, Pressure overload induces early morphological changes in the heart, Am J Pathol 181(4) (2012) 1226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Komuro I, Molecular mechanism of mechanical stress-induced cardiac hypertrophy, Jpn Heart J 41(2) (2000) 117–29. [DOI] [PubMed] [Google Scholar]
- [77].Ruwhof C, van der Laarse A, Mechanical stress-induced cardiac hypertrophy: mechanisms and signal transduction pathways, Cardiovasc Res 47(1) (2000) 23–37. [DOI] [PubMed] [Google Scholar]
- [78].Frohlich ED, Overview of hemodynamic and non-hemodynamic factors associated with left ventricular hypertrophy, J Mol Cell Cardiol 21 Suppl 5 (1989) 3–10. [DOI] [PubMed] [Google Scholar]
- [79].Talman V, Kivela R, Cardiomyocyte-Endothelial Cell Interactions in Cardiac Remodeling and Regeneration, Front Cardiovasc Med 5 (2018) 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kamo T, Akazawa H, Komuro I, Cardiac nonmyocytes in the hub of cardiac hypertrophy, Circ Res 117(1) (2015) 89–98. [DOI] [PubMed] [Google Scholar]
- [81].Selvetella G, Hirsch E, Notte A, Tarone G, Lembo G, Adaptive and maladaptive hypertrophic pathways: points of convergence and divergence, Cardiovasc Res 63(3) (2004) 373–80. [DOI] [PubMed] [Google Scholar]
- [82].Sanders YY, Kumbla P, Hagood JS, Enhanced myofibroblastic differentiation and survival in Thy-1(−) lung fibroblasts, Am J Respir Cell Mol Biol 36(2) (2007) 226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].McIntosh JC, Simecka JW, Ross SE, Davis JK, Miller EJ, Cassell GH, Infection-induced airway fibrosis in two rat strains with differential susceptibility, Infect Immun 60(7) (1992) 2936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Rege TA, Hagood JS, Thy-1 as a regulator of cell-cell and cell-matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis, FASEB J 20(8) (2006) 1045–54. [DOI] [PubMed] [Google Scholar]
- [85].Haeryfar SM, Hoskin DW, Thy-1: more than a mouse pan-T cell marker, J Immunol 173(6) (2004) 3581–8. [DOI] [PubMed] [Google Scholar]
- [86].Saalbach A, Kraft R, Herrmann K, Haustein UF, Anderegg U, The monoclonal antibody AS02 recognizes a protein on human fibroblasts being highly homologous to Thy-1, Arch Dermatol Res 290(7) (1998) 360–6. [DOI] [PubMed] [Google Scholar]
- [87].Saalbach A, Wetzig T, Haustein UF, Anderegg U, Detection of human soluble Thy-1 in serum by ELISA. Fibroblasts and activated endothelial cells are a possible source of soluble Thy-1 in serum, Cell Tissue Res 298(2) (1999) 307–15. [DOI] [PubMed] [Google Scholar]
- [88].Craig W, Kay R, Cutler RL, Lansdorp PM, Expression of Thy-1 on human hematopoietic progenitor cells, J Exp Med 177(5) (1993) 1331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tan C, Jiang M, Wong SS, Espinoza CR, Kim C, Li X, Connors E, Hagood JS, Soluble Thy-1 reverses lung fibrosis via its integrin-binding motif, JCI Insight 4(21) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hu P, Barker TH, Thy-1 in Integrin Mediated Mechanotransduction, Front Cell Dev Biol 7 (2019) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Leyton L, Diaz J, Martinez S, Palacios E, Perez LA, Perez RD, Thy-1/CD90 a Bidirectional and Lateral Signaling Scaffold, Front Cell Dev Biol 7 (2019) 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Abraham DM, Lee TE, Watson LJ, Mao L, Chandok GS, Wang HG, Frangakis S, Pitt GS, Shah SH, Wolf MJ, Rockman HA, The two-pore-domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction, J Clin Invest (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Liau B, Zhang D, Bursac N, Functional cardiac tissue engineering, Regenerative medicine 7(2) (2012) 187–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Juhas M, Engelmayr GC Jr., Fontanella AN, Palmer GM, Bursac N, Biomimetic engineered muscle with capacity for vascular integration and functional maturation in vivo, Proc Natl Acad Sci U S A 111(15) (2014) 5508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.