

Abstract
Epidermal growth factor receptor variant III (EGFRvIII) seems to constitute the perfect therapeutic target for glioblastoma (GB), as it is specifically present on up to 28–30% of GB cells. In case of other tumor types, expression and possible role of this oncogene still remain controversial. In spite of EGFRvIII mechanism of action being crucial for the design of small active anticancer molecules and immunotherapies, i.e., CAR-T technology, it is yet to be precisely defined. EGFRvIII is known to be resistant to degradation, but it is still unclear whether it heterodimerizes with EGF-activated wild-type EGFR (EGFRWT) or homodimerizes (including covalent homodimerization). Constitutive kinase activity of this mutated receptor is relatively low, and some researchers even claim that a nuclear, but not a membrane function, is crucial for its activity. Based on the analyses of recurrent tumors that are often lacking EGFRvIII expression despite its initial presence in corresponding primary foci, this oncogene is suggested to play a marginal role during later stages of carcinogenesis, while even in primary tumors EGFRvIII expression is detected only in a small percentage of tumor cells, undermining the rationality of EGFRvIII-targeting therapies. On the other hand, EGFRvIII-positive cells are resistant to apoptosis, more invasive, and characterized with enhanced proliferation rate. Moreover, expression of this oncogenic receptor was also postulated to be a marker of cancer stem cells. Opinions regarding the role that EGFRvIII plays in tumorigenesis and for tumor aggressiveness are clearly contradictory and, therefore, it is crucial not only to determine its mechanism of action, but also to unambiguously define its role at early and advanced cancer stages.
1. EGFR: Parental Gene of EGFRvIII
Epidermal growth factor receptor (EGFR/ErbB1/HER1) is a member of a tyrosine kinase receptor family, also including ErbB2/HER2/Neu, ErbB3/HER3, and ErbB4/HER4 [1]. All these receptors are transmembrane glycoproteins with a molecular mass ranging from 170 to 185 kDa [2]. Activation of ErbB receptor may be triggered by one of 13 ligands, such as epidermal growth factor (EGF), transforming growth factor-α (TGF-α), amphiregulin, betacellulin, epiregulin, neuregulin 1–6, heparin-binding EGF-like growth factor (HB-EGF), or epigen, with the first five being EGFR-specific [3]. It is not clear how EGFR is activated and triggers a cascade of downstream signaling in cells. Generally, its activation involves ligand binding and subsequent receptor dimerization; however, it was also indicated that receptor may dimerize regardless of ligand presence [4, 5]. Intriguingly, dimers formed in such a ligand-independent manner remain inactive till the ligand is finally bound [4]. Activation of EGF receptor may induce signal in Ras/Raf/MAPK, PI3K/AKT, JAK/STAT, or PLC/PKC pathways [6, 7], having an impact on a variety of cellular processes, including proliferation, metabolism, apoptosis, cell survival, or differentiation [8, 9]. Termination of signaling cascade occurs after receptor internalization, mostly in clathrin-dependent endocytosis, leading to its trafficking into early endosomes. Further, receptor may be either transported back to the cell membrane or degraded in late endosomes and lysosomes [10].
Gene encoding EGFR is located on a short arm of chromosome 7 (p11.2) and consists of 28 exons [11]. Mature EGFR protein (1186 amino acids) is formed from a precursor one (1210 amino acids) following the removal of the N-terminal part [12]. From the N to the C-terminal end, EGFR is composed of extracellular domain involved in ligand binding and receptor dimerization (exons 1–16), hydrophobic transmembrane domain (exon 17), and intracellular domain with tyrosine kinase activity that is flanked by the linker region and the C-terminal part of the receptor (exons 18–28) (Figure 1(a)) [13]. Twelve out of 20 tyrosine residues of intracellular domain were demonstrated to undergo phosphorylation and these bind membrane-bound or cytoplasmic effector proteins that are recruited following receptor activation [14, 15].
Figure 1.
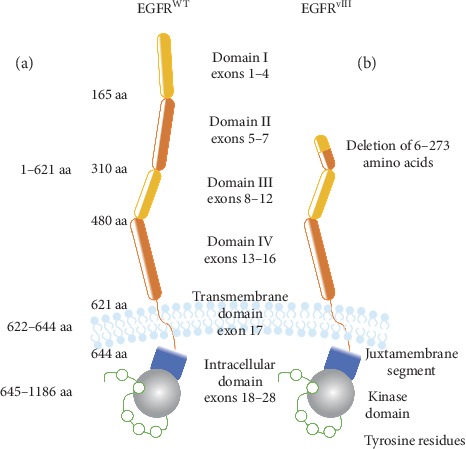
Schematic structure of EGFRWT (a) and EGFRvIII (b). Both receptors are composed of extracellular (I–IV), transmembrane, and intracellular domains, and the major difference is deletion of exons 2–7 encoding extracellular domains I and II in mutated receptor. As a result of deletion, EGFRvIII is unable to bind known ligands and shows enhanced stability in cell membrane.
2. EGFRvIII Alteration in Cancer
Overexpression of EGF receptor was detected in many tumor types and demonstrated to be associated with cancer cell resistance to chemo-, radio-, and/or hormone therapy. This receptor is often mutated in certain tumors, especially in extracellular and tyrosine kinase domains [16], resulting in elevated or prolonged EGFR signaling [17, 18]. Such abnormal signaling is associated not only with enhanced proliferation and apoptosis inhibition in tumor cells, but also with metastasis and angiogenesis [19, 20]. In case of glioblastoma (GB), EGFR amplification is in the majority of cases accompanied by gene rearrangements. Such alterations involve deletion of particular exons or exon parts and are designated as EGFRvI (deletion of N-terminal part), EGFRvII (deletion of exons 14 and 15), EGFRvIII (deletion of exons 2–7), EGFRvIV (deletion of exons 25–27), and EGFRvV (deletion of exons 25–28) [21–24]. One of the most commonly detected variants in GB cells is EGFRvIII [25–29] (Figure 1(b)).
Despite being mentioned in several articles [30], alternative splicing does not constitute key mechanisms for EGFRvIII expression in glioblastoma and other tumor types. There are only single reports indicating that this phenomenon may be involved in EGFRvIII generation in head and neck squamous cell carcinoma (HNSCC), but it is still not considered the major mechanism. Gene encoding EGFR is amplified in approximately 50% of GB patients, and in 50–60% of cases, amplification is accompanied by EGFRvIII expression that is tumor cell-specific, making this oncogenic protein a perfect therapeutic target [22, 31, 32]. Expression of mutated receptor is also detected in few percent of prostate, breast, or colon cancer cases, but only in trace cell populations [33–36]. Nevertheless, EGFRvIII expression in tumor types other than glioblastoma remains controversial and needs to be unequivocally assessed, as many contradictory data have been published so far [27, 37–41]. Such inconsistencies are particularly associated with technical limitations of applied methodological approaches. Data collected from several research centers indicate that results of EGFRvIII-related analyses tend to be even completely inconsistent. As an example, a research conducted by Moscatello et al. (1995) demonstrated EGFRvIII expression in 73% of ovarian cancer samples (Western blot analysis) and was completely contradictory to independent analysis, utilizing other methods, that indicated lack of this oncogene expression at both mRNA and protein levels in analyzed tumor samples, as well as cell lines [34, 42–44]. Similar inconsistencies were detected in case of colon or bladder cancer [43, 45–47] and most interestingly, in breast cancer, in which case EGFRvIII expression is in some reports estimated to be 20–78%, while in others not to exceed 0–4% [34, 36, 43, 48–50]. Nevertheless, various agents acting on EGFRWT or EGFRvIII are extensively studied in different types of cancers (summarized in Table 1).
Table 1.
Agents acting specifically on EGFRvIII or on both EGFRvIII and EGFRWT, based on the analysis of different cancer types.
| Specificity | Examined cancers | Activity | Stage of research | References | ||
|---|---|---|---|---|---|---|
| Agents acting only on EGFR vIII | ||||||
| Immunotherapy | ||||||
| ADC | AMG-595 | EGFRvIII | Glioblastoma | Potentially active | Phase I | [51] |
| CARs | CAR-T | e.g., EGFRvIII | Glioblastoma | Potentially active | Phase I | [52, 53] |
| Lung cancer | Potentially active | Preclinical | [54] | |||
| BiTE | bscEGFRvIII × CD3 | e.g., EGFRvIII | Glioma | Potentially active | Phase I | [55, 56, 57] |
| Vaccine | Rindopepimut | EGFRvIII | Glioblastoma | Inactive | Phase III | [58] |
|
| ||||||
| RNA interference | ||||||
| Ribozymes | e.g., EGFRvIII | Breast cancer | Potentially active | Preclinical | [59] | |
| Glioblastoma | Potentially active | [60] | ||||
| Antisense oligonucleotides | e.g., EGFRvIII | Glioblastoma | Potentially active | Preclinical | [61, 62] | |
| siRNA | e.g., EGFRvIII | Glioblastoma | Potentially active | Preclinical | [61, 63] | |
|
| ||||||
| Agents acting on EGFR vIII and EGFR WT | ||||||
| Tyrosine kinase inhibitors | ||||||
| First generation | Gefitinib | EGFR/HER1 | High-grade gliomas | Limited activity | Phase II | [64, 65] |
| Non-small-cell lung cancer | Active | Clinical use | [66, 67] | |||
| Salivary gland cancer | Potentially active | Phase II | [68] | |||
| Breast cancer | Potentially active | Phase II | [69] | |||
| Ovarian, peritoneal, or fallopian tube cancer | Potentially active | Phase I/II | [70] | |||
| Liver cancer | Potentially active | Phase II | [71] | |||
| Lapatinib | EGFR/HER1/HER2 | Glioblastoma | Inactive | Phase I/II | [72, 73] | |
| Breast cancer | Active | Clinical use | [74] | |||
| Gastric cancer | Limited activity | Phase II | [75] | |||
| Colorectal cancer | Potentially active | Phase II | [76] | |||
| Erlotinib | EGFR/HER1 | Gliomas | Limited activity | Phase II | [77, 78] | |
| Vulvar cancer | Potentially active | Phase II | [79] | |||
| Non–small-cell lung cancer | Active | Clinical use | [80, 81] | |||
| Pancreatic cancer | Active | Clinical use | [82] | |||
| Head and neck cancer | Limited activity | Phase II | [83, 84] | |||
| Second generation | Afatinib | EGFR/HER1/HER2/HER4 | Non-small-cell lung cancer | Active | Clinical use | [85] |
| Squamous cell carcinoma of the lung | Active | Clinical use | [85] | |||
| Head and neck cancer | Potentially active | Phase III | [86] | |||
| Glioblastoma | Limited activity | Phase I/II | [87, 88] | |||
| Breast cancer | Potentially active | Phase II | [89] | |||
| Colorectal cancer | Potentially active | Phase II | [90] | |||
|
| ||||||
| Immunotherapy | ||||||
| Antibodies | Cetuximab | EGFR/HER1/HER2 | Head and neck cancer | Active | Clinical use | [91] |
| Glioblastoma | Potentially active | Phase II | [92, 93, 94] | |||
| Colorectal cancer | Active | Clinical use | [95] | |||
| Esophageal and gastric cancer | Limited activity | Phase II | [96] | |||
| Non-small-cell lung cancer | Potentially active | Phase II | [97] | |||
| Breast cancer | Limited activity | Phase II | [98] | |||
| Prostate cancer | Inactive | Phase II | [99] | |||
| Cervical cancer | Inactive | Phase II | [100] | |||
| Panitumumab | EGFR/HER1 | Colorectal cancer | Active | Clinical use | [101, 102] | |
| Biliary tract cancer | Potentially active | Phase II | [103] | |||
| Head and neck cancer | Inactive | Phase II | [104, 105] | |||
| Glioblastoma | Potentially active | Phase II | [106, 107] | |||
| Breast cancer | Potentially active | Phase II | [108] | |||
| Nimotuzumab | EGFR/HER1 | Glioblastoma | Orphan status in Europe and USA | Clinical use | [109, 110] | |
| Head and neck cancer | Active | Phase II | [111, 112] | |||
| Pancreatic cancer | Orphan status in Europe | Clinical use | [110, 113] | |||
|
| ||||||
| ADC | ABT-414 | EGFR/EGFRvIII | Glioblastoma | Limited activity | Phase I | [114, 115] |
| Breast cancer | Limited activity | Phase I/II | [116] | |||
From a therapeutic point of view, glioblastoma seems to be the most important tumor type in terms of EGFRvIII because of the relatively high expression and frequency of occurrence of this oncogene and, most importantly, continuous lack of effective therapy for GB patients. Due to the deletion of 801 bp encoding N-terminal, domains I and II are lost and the mutated receptor becomes unable to bind ligands [8, 25]. Mechanisms leading to the formation of nucleotide sequence encoding EGFRvIII have not been completely elucidated yet; however, it seems plausible that deletion of receptor part is the result of recombination between Alu sequences flanking junctions in introns 1 and 7 of EGFR-encoding gene [117]. As EGFRvIII usually acts as an amplified gene, it may be suggested that increase in the number of gene copies will translate into increased mRNA levels of this oncogenic variant, but no such obvious dependence has been found, even in relation to EGFRWT levels [118, 119]. It may be associated with the fact that the main role of gene amplification in this case is not to provide additional gene copies that will increase mRNA levels of amplified gene. With the current focus on the field of extracellular vesicles (EVs), the fact that extrachromosomal amplicons may be transported between cells is gaining importance [120]. Therefore, in such a context, the role of amplicons is not to increase oncogene mRNA levels, but rather to enable more flexible regulation of gene expression as well as transfer of mutated gene to cells initially lacking such alteration. Moreover, detection of EGFRvIII in extrachromosomal amplicons derived from cerebrospinal fluid may constitute a highly specific and less invasive approach, to molecularly diagnose GB patients and make them candidates for currently developed anti-EGFRvIII-targeted therapies [118].
3. EGFRvIII Mechanism of Action
Compared to EGFRWT (normal EGFR protein), EGFRvIII signaling is considered to be elevated, due to its ability to dimerize in a ligand-independent manner. However, as it is not clear whether EGF is indeed crucial for EGFRWT dimerization or required only for a dimer to switch from inactive to active state, the role of EGFRvIII dimerization is becoming less evident [3, 4]. Loss of large extracellular receptor fragment makes it difficult to determine whether EGFRvIII dimerizes in tethered or untethered conformation or if such conformation resembles active or rather inactive EGFRWT [16]. Importantly, it has to be emphasized that EGFRvIII exhibits constitutive activity [121, 122]. Our team demonstrated that mutant phosphorylation is elevated when compared to nonstimulated EGFRWT, while data obtained by other research teams indicate that constitutive EGFRvIII signaling corresponds to low level of signal intensity induced by ligand-activated EGFRWT [17, 123–125]. Such data indicate that EGFRvIII dimer conformation resembles inactive dimers of EGFRWT, thus suggesting that impact of this oncogenic receptor on cell biology is not a result of some specific, dimerization-related kinase activity, but rather a consequence of unique membrane stability [121, 122]. Therefore, constitutive activity of EGFRvIII is not particularly high, but when combined with high membrane stability, it may enable triggering of some significant biological effects by this oncogenic receptor [17, 123–125].
Despite the fact that EGFRvIII stability seems to be more important than its kinase activity, the latter feature is still required to fully exhibit the oncogenic potential of this receptor. Such potential is dependent on signal transduction induced by phosphorylated tyrosines (at least in a model explaining EGFRvIII oncogenicity as a membrane receptor), especially as EGFRvIII was demonstrated to undergo constant phosphorylation and dephosphorylation cycles [123]. Additionally, our data indicate that EGFRvIII signaling may not be associated with slightly elevated kinase activity, but rather its minimally lower sensitivity to phosphatase activity, when compared to wild-type receptor [123]. Moreover, we indicated that enhanced phosphorylation of tyrosine 1045 did not result in EGFRvIII degradation [123], suggesting that the previous model of impaired EGFRvIII degradation requires an update [121]. Nevertheless, without membrane stability, EGFRvIII signaling will not be strong enough to induce a biological effect. So far, the reasons behind the unique EGFRvIII stability have not been fully elucidated. Initially, it was suggested that it is a result of different phosphorylation of tyrosine responsible for interaction with Cbl protein in mutated and wild-type receptors [121]. Additionally, the involvement of FHL2 in EGFRWT and EGFRvIII stabilization was suggested [126]. Stabilization and activity of EGFRvIII are mostly determined by the quaternary structure of the receptor. Long noncoding RNA (lncRNA) EGFR-AS1, an antisense transcript of EGFR, was suggested to be involved in EGFR folding [127], but it is still unknown whether this structure may have a different impact on mutated than wild-type receptor. Nevertheless, gene encoding this lncRNA is located on the same amplicon as EGFR and thus also undergoes amplification [127]. Actually, it is still unknown why EGFRvIII is much more stable than EGFRWT; however, such feature of this oncogenic receptor may be considered crucial, as the ability to trigger EGFRvIII degradation, considering its low kinase activity, may deprive this variant of oncogenic properties. Intriguingly, the impact of the dimerization process alone was suggested to be associated with increased EGFRvIII stability, especially since the involvement of the so-called “crypto” domains was described to have an impact on EGFRWT stabilization [128–132].
According to the majority of analyses, EGFRvIII is able to form both hetero- and homodimers [123, 133–140], and in the latter case, covalent and noncovalent dimers can be observed [17, 123]. Our analyses indicate that the most of EGFRvIII monomers are a part of covalent homodimers with covalent bonds formed with the involvement of free cysteine in position 16 of amino acid chain (Figure 2(a)) [123]. Nevertheless, some authors undermine the constitutive activity of the mutant, suggesting that EGFRvIII activity is mostly due to EGF-activated EGFRWT forming a heterodimer with EGFRvIII (Figure 2(b)) [138]. Additionally, it was suggested that only EGF-bound EGFRWT is able to phosphorylate EGFRvIII in a heterodimer, but not vice versa [138]. The way these receptors tend to dimerize is substantial, as molecules inhibiting dimerization may be plausibly used in anticancer therapy. Intriguingly, the possibility of EGFRvIIIcis-autophosphorylation is very rarely discussed in the literature [135, 141]. Cross-activation of EGFRWT kinases is well recognized as the mechanism crucial for their activation, clearly explaining why receptor dimerization is actually required. Nevertheless, a priori rejection of the hypothesis stating that some part of EGFRvIIIcis-autophosphorylates may be too hasty.
Figure 2.
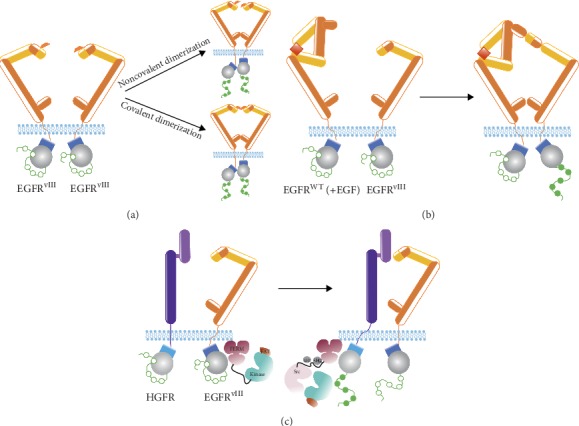
Currently proposed models of EGFRvIII dimerization. (a) Covalently or noncovalently linked EGFRvIII homodimers. In both cases, phosphorylation of tyrosine residues of both monomers can be observed. (b) Heterodimerization of EGFRvIII with ligand-activated (e.g., EGF-activated) EGFRWT. Only EGFRvIII phosphorylation is observed in such a case. (c) EGFRvIII dimers with monomers of other inactive receptors. Example of FAK-mediated EGFRvIII dimerization with HGFR, resulting in phosphorylation of HGFR tyrosine residues.
EGFRvIII is also suggested to dimerize with monomers of other inactive receptors (hepatocyte growth factor receptor, HGFR; platelet-derived growth factor receptor, PDGFR) or to have an indirect impact on their function (Figure 2(c)) [142, 143]. Regulation of other receptors may potentially constitute an additional mechanism for EGFRvIII-mediated activation of signal transduction pathways, especially in the absence of ligands. This hypothesis was more profoundly tested by Greenall et al., who demonstrated that EGFRvIII is able to activate HGFR via focal adhesion kinase (FAK); however, it was not clearly defined how such activation takes place on a FAK protein platform [139].
All doubts concerning the mechanism of EGFRvIII encourage researchers to search for mechanisms of oncogenic action of this protein that are independent of its membrane receptor activities. One of the most interesting analyses is focused on the nuclear role of EGFRvIII, as this function is suggested to be very relevant [144, 145]. Therefore, EGFRvIII interaction with oncostatin M receptor (OSMR) can be considered interesting, as it may be possible to design molecules inhibiting such interaction for therapeutic purposes [146]. In general, results of research conducted so far indicate that the majority of EGFRvIII activity is exhibited outside the nucleus, while its low kinase activity may be compensated by uniquely high stability [17, 121, 123–125, 147–149].
4. Biological Role of EGFRvIII
Despite the fact that cells with high expression of mutated receptor are unable to bind EGFR ligands, these cells are still characterized by increased invasiveness and enhanced proliferation rate when compared to cells with low EGFRvIII expression or EGFRvIII-negative ones [147]. Therefore, it is not only the mechanism of EGFRvIII action, but also biological changes triggered within EGFRvIII-positive cells, as well as the role such cells play in tumor as a whole, that are important when considering the impact of this mutated receptor. This aspect can be especially important, since EGFRvIII expression is not observed in all cells comprising the tumor [33]. Our data suggested that EGFRvIII acts as a classical oncogene, stimulating proliferation and inhibiting apoptosis of glioblastoma cells [147], while other studies indicated much more complicated influence of this oncogene. Considering the impact of EGFRvIII on cells, both autocrine and paracrine effects were investigated. As an example, EGFRvIII-positive cells may secrete leukemia inhibitory factor (LIF) and IL-6 that activate IL-6R/gp130 receptors present on the surface of EGFRWT-positive cells, promoting their proliferation. Moreover, by activation of NF-κB pathway and stimulation of survivin expression, IL-6 may make cells more resistant to apoptosis [150, 151].
EGFRvIII amplicons are present only in part of glioblastoma cells derived from patients and in stable DK-MG cell line (intratumoral heterogeneity). Moreover, only part of these amplicons is active (not epigenetically silenced) and enables expression of the mutated gene [31]. EGFRvIII expression alone is epigenetically controlled, as it was demonstrated that inhibition of histone deacetylation leads to decrease in expression of this oncogenic receptor. It may be explained by the fact that there is a relatively low EGFRvIII expression in tumor parts where high amplification level of this mutated gene is detected [31, 152].
Some researchers suggest that EGFRvIII expression may be present on the surface of brain cancer stem-like cells (bCSCs) that share some similarity with normal neural stem cells (NSCs) [153, 154]. The former cells are characterized by self-renewal potential as well as expression of markers characteristic for stem cells [155–157]. EGFRvIII is coexpressed with marker characteristic for nondifferentiated cells (CD133 and SOX2) [158, 159], and it is even indicated that this oncogenic receptor may be used to define CSC populations [158]. One can speculate that low kinase activity together with high stability of EGFRvIII is enough to inhibit cell differentiation. Interestingly, it can be also assumed that EGFRvIII epigenetically reprograms cells, depriving them of differentiation potential and, hence, following such process, this mutated receptor may be no longer needed. Brain CSCs are involved in initiation and progression stages of GB, mostly due to their impact on angiogenesis and treatment response [160, 161]. Moreover, presence of bCSCs may hinder long-term maintenance of therapeutic effect, as currently used compounds do not affect these cells, mostly due to very efficient DNA damage repair mechanisms [160, 161]. However, it was demonstrated that usage of bispecific antibodies directed against EGFRvIII and CD133 (CSCs marker) has a cytotoxic effect on bCSCs and impairs their self-renewal abilities [158]. Some researchers suggest that in vivo CSCs, but not other cancer cells, are mostly responsible for the process of tumor formation in SCID mice as well as for the propagation of intratumoral heterogeneity [162]. Our results clearly demonstrated SOX2 expression in high percentage of GB cells that, in our opinion, undermines the presence of only a minor stem cell population in glioblastoma tumors [163].
5. Intratumoral Heterogeneity of Glioblastoma in terms of EGFRvIII Expression
The fact that EGFRvIII is not present in all GB cells in tumor mass may complicate the perception of this receptor as a perfect therapeutic target. However, if cells expressing EGFRvIII are cancer stem cells [164] or EGFRvIII-negative cells are somehow dependent on EGFRvIII-positive ones, then discussed targeted therapy may turn out to be effective (Figure 3(a)). Our research indicates that EGFRvIII-negative cells may be indeed dependent on EGFRvIII-positive population. It is supported by the fact that we were unable to establish a subline of DK-MG cell line completely deprived of cell expressing this mutated oncogene, as at least small percentage of EGFRvIII-positive cells was necessary in order to maintain survival and proliferation [33, 147]. On the other hand, at least in 30% of cases, EGFRvIII expression is spontaneously lost in recurrent GB tumors, even when the treatment was not directed against the mutated receptor (Figure 3(b)) [119, 165]. Remarkably, there were also some cases in which EGFRvIII expression was detected only in recurrent GB tumors (Figure 3(c)) [119, 165]. Such observations are of utmost importance, as these enable to evaluate the relevance of EGFRvIII and indirectly cells expressing this mutated receptor, as therapeutic targets. If EGFRvIII is lost (not detected) in recurrent tumors due to the fact that it is present only in a small part of cells and EGFRvIII-negative cells are independent of the activity of this oncogenic variant, it undermines the validity of EGFRvIII-targeting therapies, for example, those based on CAR-T technology [166]. It may be associated with the fact that the expression of some oncogenes, including EGFRvIII, is crucial at earlier stages of neoplastic transformation, but not further during advanced cancer progression. Opinions on the role of EGFRvIII as well as EGFRvIII-positive cells are extremely different, as this oncogene is suggested either to play an insignificant role at the later stages of carcinogenesis, or, on the contrary, to be a marker of GB stem cells (Figures 3(a)–3(c)). Our analyses do not confirm the hypothesis stating that EGFRvIII is irrelevant in fully differentiated GB cells, as DK-MG cells deprived of this oncogene expression lose their proliferation abilities are more prone to apoptosis and unable to give rise to tumors in SCID mice models [147].
Figure 3.
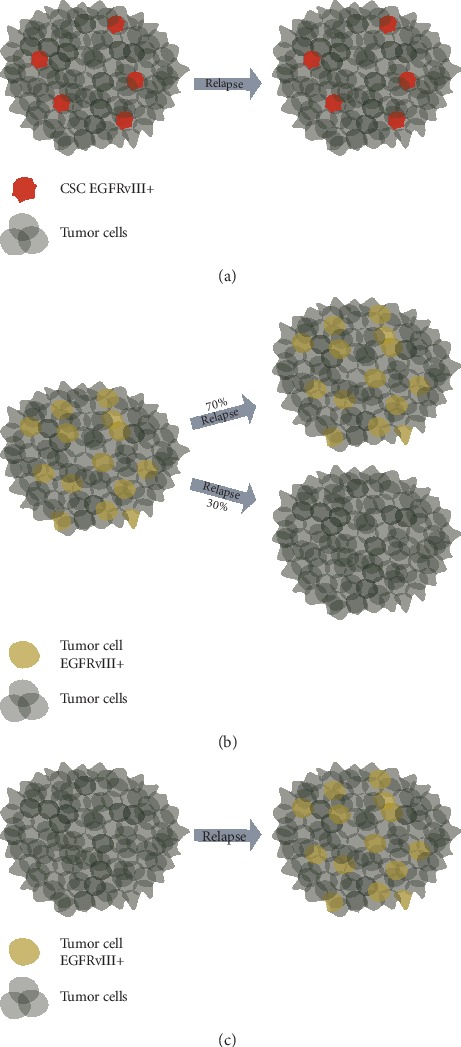
Hypotheses concerning the presence and role of EGFRvIII-positive cells in tumors, on the example of glioblastoma. (a) One of the hypotheses states that EGFRvIII is expressed on the surface of cancer stem cells (CSCs). In such a case, EGFRvIII-positive CSCs should be also detected in recurrent GB tumors [164]. Nevertheless, failure to detect such cells may be due to the exposure of primary tumor to therapeutic compounds. (b) Another hypothesis states that EGFRvIII-positive cells are only crucial during the early stages of carcinogenesis. It is supported by reports demonstrating loss of expression of this mutated oncogene in approx. 30% of patients with EGFRvIII-positive primary tumors [119, 165]. (c) Cells expressing EGFRvIII are also reported in recurrent tumors when primary GB was EGFRvIII-negative [119, 165].
Recently, a lot of attention is focused on the ability to transfer extrachromosomal vesicles containing various structures (including DNA amplicons) between cells. Obviously, extrusion of amplicons or decrease in their number during mitoses may lead to generation of cells without amplicons [120]. Simultaneously, amplicons may be transferred to cells initially lacking such structures. Derivation of amplicon-deprived cells from cells with amplicons, as well as “infection” of cells lacking amplicons with these elements of extrachromosomal DNA, is in favor of hypothesis stating that EGFRvIII-positive cells may, in a certain sense, play a role of precursor cells. It clearly emphasizes the biological role of EGFRvIII not at the protein, but DNA level, and it may partially explain why the expression of this mutated protein is in particular cases very low, almost at the detection level of protein analysis methods such as western blot. However, it should not be confused with the role played by so-called cancer stem cells.
The fact that intratumoral heterogeneity may constitute one of the mechanisms responsible for resistance of cancer cells to targeted therapies (including TKIs) was first demonstrated by Nathanson et al. and further confirmed by other research teams. Such a specific adaptation via changing cell phenotypes is mainly focused on achievement of an optimal balance for the unaltered proliferation of the overall population and is mostly due to dynamic regulation of extrachromosomal DNA encoding mutated EGFR [167–169].
6. Targeted Therapies Based on Tyrosine Kinase Inhibitors (TKI), Directed against EGFRWT and EGFRvIII
A wide variety of factors contribute to the fact that glioblastoma is one of the most difficult tumors from a clinical perspective and that effective therapies for patients diagnosed with this tumor type are still lacking. So far, many therapeutic approaches were developed to treat patients with EGFRvIII-positive glioblastoma (Table 1). Generally, average survival rate of GB patients does not exceed 12–14 months from the moment of diagnosis and there has been actually no improvement for many years [170]. Ideal drug directed against GB cells should be well tolerated by the patients, able to cross the blood-brain barrier, and specifically induce tumor cell death. Classical therapeutic regimen in case of GB consists of surgical resection with adjuvant radio- and chemotherapy with alkylating agent temozolomide [171]. Current clinical and preclinical trials concerning anti-EGFR/EGFRvIII therapies include small molecule tyrosine kinase inhibitors, antibodies, vaccines, as well as therapies based on RNA interference. As silencing of a single gene in a particular signaling pathway may not be sufficient to provide a therapeutic effect in GB patients, there is a need for a complex approach, focusing on several signal transduction pathways [172].
There have been several attempts to experimentally apply EGFR tyrosine kinase inhibitors, also inhibiting EGFRvIII, in glioblastoma therapy, as significant differences between kinase domains of mutated and wild-type receptor have not been described so far. A broad spectrum of anti-EGFR TKIs was developed, with the first-generation inhibitors (gefitinib, lapatinib, and erlotinib) binding reversibly and the second-generation inhibitors (afatinib and dacomitinib) binding covalently to the receptor [173–175]. Inhibitors of the third generation (rociletinib and osimertinib) covalently bind to ATP-binding site in cells with T790M EGFR mutation, conferring resistance to inhibitors of previous generations [176]. Second-phase clinical trial studies demonstrated that gefitinib, lapatinib, and erlotinib administered to patients with primary or recurrent GB tumors resulted in only marginal therapeutic response, when administered either in monotherapy or in combination [72, 77, 177]. Although osimertinib may be recognized as especially important in terms of EGFRvIII, as it was suggested to be efficiently delivered to cancer cells in brain [176], the activity of this compound against EGFRvIII-positive cells was lower when compared to afatinib. Since the kinase domain of this splice variant is structurally close to EGFR wild type, this was not unexpected [176].
In case of TKI-based therapy, alterations downstream to EGFRvIII, including PTEN mutations, should be taken into account. Despite the fact that inactivating mutations of PTEN have an impact on only one of EGFR-regulated pathways (AKT), it was demonstrated that such mutation is able to hinder the impact of erlotinib on GB cells [178]. Considering this aspect, immunotherapies may possibly outperform small molecule-based approaches. Despite the wide availability of TKIs clinically approved in oncological treatment, none of these inhibitors is used as a standard approach in GB treatment [64, 72, 77, 179]. As EGFRvIII is a key oncogene with kinase activity-dependent function, it seems reasonable to consider whether the efficacy of TKI-based therapies should not be greater, especially since it has been postulated that the blood-brain barrier in advanced GB is disrupted and thus should not enable for crossing of small molecules [180]. It is well established that EGFR-targeting TKIs improve the progression-free survival of patients with EGFR-mutated non-small-cell lung cancers (NSCLCs) [181, 182]. Therefore, the verification whether glioblastoma patients with high frequency of EGFR mutations respond to TKIs is completely justified, even despite different EGFR mutational spectrum. This becomes even more important since Orellana et al. showed that ectodomain EGFR mutations including those leading to EGFRvIII may sensitize tumor cells to tyrosine kinase inhibitors [183]. Reports from in vitro studies conducted on EGFRvIII-expressing cell lines tend to be contradictory. Some results indicate EGFRvIII sensitivity, while the others demonstrate that EGFRvIII, in contrast to EGFRWT, appears to be relatively resistant to EGFR-TKIs [87]. By now, several TKI-involving clinical trials on glioblastoma were completed or terminated, however still without any significant patients' benefits [65, 72, 77, 88, 178, 184–188]. Moreover, it was demonstrated that although cetuximab binds to EGFRvIII and decreases expression and leads to overall downregulation of this mutated receptor, it does not inhibit the proliferation of EGFRvIII-expressing GB cells and is not effective in GB clinical trials [189–191]. Therefore, it seems that so far neither EGFR-TKIs nor monoclonal antibodies such as cetuximab are effective therapeutic options in glioblastoma patients, irrespective of EGFRvIII occurrence in tumor [177, 188]. Thus, it is difficult to speculate whether EGFRvIII affects EGFR-targeted treatment, as no treatment approach was truly effective in patients, in spite of quite encouraging results from in vitro studies. As EGFRvIII presence in other tumor types is highly debatable, there were no clinical trials to investigate the issue of EGFRvIII-modulated TKI treatment response in tumors other than glioblastoma. Hence, conclusions from studies on EGFR-TKIs/immunotherapies can only be drawn concerning this particular tumor. Finally, as Orellana et al. recently suggested the high probability that mutated ectodomain of EGFRvIII induces structural changes in the intracellular kinase domain [183], further research focused on detailed understanding of molecular aspects of EGFRvIII should be expected. On the other hand, considering current standard therapeutic GB regimen, EGFRvIII is associated with prolonged survival of GB patients treated with surgery and radio/chemotherapy [192]. It was clearly shown that cases of MGMT-methylated GB with endogenous EGFRvIII expression are significantly more sensitive to temozolomide, than their isogenic EGFRvIII -negative counterparts [193].
7. Immunotherapy in EGFRvIII-Positive Tumors
Apart from TKIs, antibodies constitute the most extensively analyzed group of EGFR-targeting compounds; however, their evident efficacy in GB has not been demonstrated so far. High molecular weight may be one of the factors limiting their applicability in treatment of this tumor type [194], but the integrity of blood-brain barrier may be compromised in case of tumors with high level of malignancy [180]. Cetuximab is a chimeric monoclonal IgG1 antibody directed against extracellular domain of EGFR that in clinical studies was demonstrated to exert anticancer effect and increase tumor cell sensitivity to radiotherapy in GB [92]. This molecule was approved by the Food and Drug Administration for the treatment of patients with head and neck cancer and advanced colon cancer. Interestingly, it may be used in case of increased expression of both EGFRWT and EGFRvIII [195, 196], as it was demonstrated that cetuximab may bind to domain III (L2) of EGFRvIII and reduce autophosphorylation of this mutated receptor [197]. Preclinical analyses indicate that following EGFRvIII binding cetuximab induces receptor internalization, resulting in 50% reduction of its active form [197]. Nevertheless, there is a lack of clinical studies evaluating the impact of cetuximab monotherapy on patients with primary glioblastoma [198, 199]. When tested in vitro on GB cell lines with EGFR overexpression or using in vivo GB models, cetuximab leads to decrease in proliferation rate and enhancement of apoptosis. Additionally, in the latter model, this antibody is able to significantly inhibit tumor growth and increase median survival rate [93]. During analyses conducted using stable cell lines as well as neurospheres, magnetic iron oxide particles (IONPs) were used to increase therapeutic availability of cetuximab and resulted in more effective binding of antibody to GB cells when compared to cetuximab alone, as evaluated by the inhibition of EGFR signaling pathway and increased receptor internalization [200]. There is also an ongoing research on the use of other antibodies in GB therapy, for example, panitumumab (humanized monoclonal IgG2 antibody) or nimotuzumab (humanized monoclonal IgG1 antibody), that are functionally similar to cetuximab [109, 201]. These antibodies also bind to L2 domain, preventing ligand binding and receptor dimerization [202]. Randomized phase III clinical trials demonstrated that nimotuzumab administration in adult GB patients increases overall survival when compared to standard treatment [109].
In order to achieve higher therapeutic response, it is also possible to conjugate antibodies with other drugs (antibody drug conjugates, ADC). So far, ABT-414 and AMG-595 were developed [114, 203] and the former conjugate was demonstrated to selectively induce apoptosis in cells with EGFRWT overexpression or EGFRvIII expression both in vitro and in vivo using xenograft models. ABT-414 conjugate consists of ABT-806 monoclonal antibody directed against EGFR and inhibitor of microtubule polymerization—monomethyl auristatin F. Despite the fact that ABT-806 was initially developed to specifically interact with EGFRvIII, it also binds to wild-type receptor, however, to a lesser extent [204]. Using xenograft GB models, it was demonstrated that combination of ABT-414 with standard chemo- and radiotherapy resulted in a significant decrease in cell proliferation and overall decrease in tumor growth [114]. Currently, there are ongoing phase I/II clinical trials aimed at evaluating the efficacy of ABT-414 administration in patients with newly diagnosed (NCT02573324) or recurring GB (NCT02343406). Analyses on an orthotopic mouse GB model showed that ind-111-labeled ABT-806 antibody can specifically recognize cancer cells [205].
Nowadays, one of the most promising immunotherapy-based approaches in GB treatment is the usage of autologous T lymphocytes with chimeric antigen receptor—CAR-T cells. These are T lymphocytes that have been modified ex vivo and able to recognize their molecular target irrespective of antigen presentation by the molecules of major histocompatibility complex [52, 206]. Structure of CAR-T cells makes them able to exhibit both activity of antibodies and toxicity of T lymphocytes [207]. CAR-T is a technology of interest in research on many cancer types; however, the prerequisite for its efficacy and lack of side effects is antigen expression specifically on cancer cells [208]. As EGFRvIII meets this requirement, it is possible to develop CAR-T recognizing mutated form of the receptor by antigen-specific, humanized single chain of variable fragment of antibody, conjugated with transmembrane and intracellular domains of T lymphocytes and NK cells. Similarly to modified T lymphocytes, NK cells with introduced CAR are able to exhibit cytotoxic activity in vitro [209]. CAR-T cells directed against EGFRvIII were able to effectively infiltrate tumor cells in brain in in vivo model [53]. Notably, EGFRvIII-targeting CAR-T therapies have currently reached phase I of clinical trials to treat GB patients (NCT03283631, NCT02209376). On the other hand, administration of CAR-T therapy in phase I and I/II clinical trials directed against antigens present on both normal and cancer cells (ErbB2, CD19) led to severe side effects and even patients death [210, 211]. It is still not clear whether antibodies designed to recognize EGFRvIII also detect wild-type EGFR [43, 205, 212, 213], but if so, it may lead to some serious side effects following administration of various immunotherapies, including CAR-T approach. Moreover, as glioblastoma tumor is highly heterogeneous, not all GB cells may respond to CAR-T therapy directed against EGFRvIII.
Apart from CAR-T technology, there is also an ongoing research on application of another immunotherapy-based approach in GB treatment in a form of bispecific antibodies activating T lymphocytes—bispecific T-cell engagers (BiTEs). BiTEs are recombined immunoglobulins composed of a single chain of variable fragments of two antibodies: one directed against antigen expressed on the surface of T lymphocytes and the second one against the antigen present on target cells [214, 215]. Heavy and light chains of variable antibody fragments are connected with a short, elastic linker, rich in glycine and serine residues. Extracellular EGFRvIII domain is small; hence, it may be efficiently bound by BiTEs [216], and specificity and cytotoxic activity of these molecules against this mutated receptor were demonstrated using in vitro and in vivo models. Properly designed bispecific antibodies are characterized with very high specificity, resulting in a minimal risk of induction of cross reactions in normal cells [55]. Using BiTEs, it was demonstrated that stimulated regulatory T lymphocytes secrete elevated levels of granzymes and perforins and that their activity is directed against EGFRvIII-positive cells [217, 218]. Currently, there is an ongoing phase I clinical trial on administration of AMG 596, drug containing BiTEs directed against EGFRVIII and CD3 surface protein in GB patients (NCT03296696).
It is worth to emphasize that results of our analyses, supported by the data gathered by other research teams, indicate that additional mutations within EGFRvIII that may have an impact on efficacy of antibodies or small molecules directed against EGFRvIII-characteristic protein fragments are rarely occurring, but if so, these are distant from EGFRvIII-specific parts [219, 220].
Vaccines constitute another therapeutic approach taking advantage of patient's immunological system to destroy EGFRvIII-positive cells. So far, only one peptide vaccine, rindopepimut, has been developed to induce humoral response leading to elimination of GB cells expressing mutated EGF receptor [221]. Rindopepimut (CDX-110) is based on 13-amino acid EGFRvIII-specific sequence conjugated with keyhole limpet hemocyanin (KLH; hemocyanin neoantigen) adjuvant [222]. In vivo preclinical analyses demonstrated that tumor volume significantly decreased in 70% of animals with subcutaneously injected cancer cells following CDX-110 administration, when compared to the control group. It was suggested that antibodies reactive against EGFRvIII-KLH are involved in triggering of antibody-dependent cell cytotoxicity (ADCC), regardless of antigen-specific T lymphocytes activity [223]. Median survival rate of GB patients treated with CDX-110 after surgical resection and chemotherapy was prolonged to 24 months, as demonstrated in 3 independent phase II clinical trials (ACTIVATE, ACT II, and ACT III). Moreover, EGFRvIII-expressing cells were not detected in 67% of patients receiving CDX-110 treatment for at least 3 months [222]. Nevertheless, phase III clinical trial (ACT IV), comparing the efficacy of temozolomide alone and temozolomide in combination with CDX-110 in GB, was terminated before the scheduled date, as despite premises from the previous stages of clinical trials, it failed to indicate the significant increase in patient survival (median survival for CDX-110-treated patients was 20.1 months, while in control group 20 months). Still, the researchers emphasized the relevance of research focused on determination of the type of immunological response induced by CDX-110 and highlighted the problem of the selection of the appropriate molecular target for immunotherapy approaches [58].
8. Anti-EGFRvIII Therapy Based on RNA Interference
Concerning the regulation of EGFRvIII expression, emphasis should be also put on noncoding RNAs, especially microRNAs (miRNAs). Aberrant expression of miRNAs has been implicated in various tumor types, including glioblastoma, and demonstrated to impact cancer cell proliferation, EGFR downstream signaling, as well as efficacy of several anti-EGFR-targeting therapeutic approaches. Unfortunately, the majority of data were focused on wild-type receptor and we can only speculate that similar mechanisms apply to EGFRvIII. Decrease in miR-137 level in glioblastoma tissue samples was found to be associated with poor prognosis and, consequently, overexpression of this miRNA in GB models resulted in elevated apoptosis and inhibition of tumor cell growth. It was suggested that miR-137 may act by decreasing translation of EGFR protein, hence decreasing proproliferative activity of this receptor in tumor cells [224]. Similarly, miR-615, miR-1231, or miR-133, also downregulated in glioblastoma, were found to inhibit EGFR levels [225–227]. On the other hand, upregulation of miR-21, often found in glioblastoma patients, promotes EGFR activity and supports tumor growth [228]. Yin et al. showed that miR-34a was often deleted in glioblastoma showing EGFR amplification; however, they did not evaluate the expression of EGFRvIII within analyzed samples. Nevertheless, considering the typical percentage of EGFRvIII-positive GB cases with EGFR amplification, it is very likely that miR-34a deletion coexists with EGFRvIII. Notably, Yin et al. indicated shorter mean survival rate of patients diagnosed with GB with EGFR amplification and miR-34a deletion compared to patients with only one of these alterations [229]. Moreover, EGFRvIII-mediated downstream signaling was found to be associated with inhibition of miR-9 expression, further promoting tumorigenicity in FOXP1-dependent manner [230]. Intriguingly, lncRNA EGFR-AS1 was found to act via miR-133b in regulation of glioblastoma cell migration, invasion, and apoptosis and knockout of this noncoding RNA negatively influenced tumor growth [231].
Besides protein-based therapeutic approaches, research focuses on targeting EGFRvIII at the mRNA level. RNA interference-based therapy relies on usage of ribozymes, antisense oligonucleotides, or siRNA molecules complementary to regions that silencing is beneficial from a clinical point of view [61]. Taking advantage of this technology enables to inhibit activity of EGFR signaling pathways, with relatively low toxicity and maintained high specificity against EGFRvIII [63, 232, 233]. As promising results were obtained in preclinical analyses with antisense oligonucleotides for the treatment of non-small-cell lung carcinoma and prostate cancer [234, 235], possibility to silence EGFR and EGFRvIII gained more attention. Sequence of mRNA nucleotides in junction site between introns 1 and 7 in EGFRvIII is highly specific and absent in any other human genes. Nevertheless, the majority of current literature data concerning siRNA is focused on EGFR in general, without distinguishing normal receptor from mutated one. Constructs with proper antisense RNA sequence were demonstrated to silence expression of mRNA encoding EGFRWT, both in vitro on GB cells with EGFRWT expression and in vivo on rat GB model. In the former model, significant decrease in level of EGFR mRNA and protein, decrease in proliferation rate, and induction of apoptosis were observed in cells with the expression of introduced construct, while in the latter model all rats with introduced antisense RNA were characterized with prolonged survival rate, when compared to animals with empty construct [236]. Comparison of the construct with antisense RNA complementary to 3′ end and to the whole EGFRWT mRNA encoding region demonstrated that inhibition is more effective in the first case, possibly as delivery of shorter construct may be much easier and efficient [236, 237]. First reports indicate that siRNA complementary to exon 1 and 8 junction site is able to inhibit EGFRvIII expression in human glioma cells, leading to decrease in AKT phosphorylation and inhibition of cell cycle in G2/M [238].
Gene therapy using ribozymes is based on the ability of antisense RNA to catalytically digest mRNA substrate within the specific nucleotide sequence [239]. Low-molecular hairpin-type ribozymes were able to specifically inhibit EGFR expression, as well as proliferation and clonogenicity of GB cells in vitro [60, 239]. In terms of gene-editing approaches, it is worth to mention that CRISPR-based technologies have only little chance of being successfully applied in case of EGFRvIII, as deletions within EGFR leading to the formation of this oncogenic variant are quite extensive and tend to differ between patients [240]. It is worth to mention that RNA interference can be achieved by miRNA upregulation. Moreover, one of the miRNAs, miRNA-34a, was demonstrated to enhance the antiproliferative effect of erlotinib [241].
9. In Vitro Models for EGFRvIII Analyses
One of the additional and still unresolved problems regarding development of an effective anti-EGFRvIII therapy is lack of the appropriate ex vivo/in vitro models reflecting heterogeneity of GB cell genotype and phenotype. Results obtained under in vitro conditions often tend to differ significantly from those obtained in clinical trials, as exemplified by results presented above. In primary GB cultures, EGFRvIII expression is quite stable in neurospheres, while in adherent cultures it tends to be lost as soon as after several passages. Analyses of SOX2 expression (marker of neural stem cells and factor crucial to maintain proliferation of GB cells) indicate that neurospheres and adherent cells differ in the state of differentiation—adherent cells gradually lose SOX2, while in spheroids expression of this marker remains at relatively constant level [242]. It is worth to emphasise that the majority of GB cells are SOX2-positive, as it is in contrary to the assumptions that cancer stem cells constitute only so-called side population or, it is possible that SOX2 is a marker not characteristic solely for stem cells [243, 244]. Apart from SOX2, glioblastoma cells also express GFAP, which can be considered quite surprising as GFAP for many years has been considered a marker of mature astrocytes. Nevertheless, GFAP-positive neural stem cells have been described in the literature [245] and these, similar to GB cells, were demonstrated to coexpress many other markers [163, 245].
Despite the fact that spheroid cultures maintain original phenotype of GB cells for a longer period (there is no stable GFAP+/SOX2+ adherent cell line), this approach is associated with various methodological difficulties. First of all, certain assays on 3D structure may be difficult to be performed. Moreover, cells maintained in medium containing serum are more resistant to the exposure to cytotoxic molecules than neurospheres cultures in serum-free media. Finally, not all primary GB cells are able to form spheroid structures [246]. Basically, in vitro culturing should promote survival and proliferation of cancer cells; however, it may lead to spontaneous senescence, mitotic catastrophe, or apoptosis. The occurrence of in vitro senescence described to play both pro- and antineoplastic role in vivo in primary GB cultures can be plausibly associated with failure in their stabilization [247]. Stable glioblastoma line may not only fail to reflect the heterogeneous nature of tumor cells observed in vivo, but also lack extrachromosomal amplicons encoding EGFRvIII [21, 150, 167]. However, the limited amount of tumor material derived from patients and its low stability force scientists to conduct research on commercially available stable cell lines with exogenously introduced EGFRvIII-encoding gene [153, 248]. Analyses on such models may be unreliable, as introduction of EGFRvIII cDNA via cell engineering methods may give biased results regarding such aspects as clonality (different results obtained depending on the analyzed clone) or neglect the dynamic regulation of amplicons released from EVs. Additionally, exogenously introduced EGFRvIII may not have an impact on the biology of already fully defined cancer cells, such as U87-MG cell line [147]. Therefore, biological differences observed between U87-MG clones may be easily confused and taken as the effect of EGFRvIII action. Hence, there is an ongoing search for the most appropriate model, reflecting nature of GB cells as precisely as possible.
10. Summary
Table 2 presents most important issues addressed in the article (except therapies in Table 1). EGFRvIII protein may be considered a suitable target in 28–30% of GB cases, as it is selectively expressed on cancer cells and structurally differs from wild-type receptor. Nevertheless, opinions on the role of EGFRvIII in GB biology are contradictory. This mutated receptor seems to play a key role in tumor cells, enhancing their proliferation, inhibiting apoptosis, or being considered a marker of CSCs. On the other hand, it is suggested that EGFRvIII is unnecessary for GB cells, especially at advanced stages of tumorigenesis, that may be considered a drawback in terms of therapeutic approaches directed against this mutated receptor. Despite many years of extensive research, EGFRvIII-specific inhibitors have not been developed yet. There are also many controversies regarding antibodies designed to specifically detect this oncogenic variant, which in turn may be negatively correlated with the efficacy of CAR-T and other immunotherapy-based approaches. Many factors hinder glioblastoma treatment, including heterogeneity of EGFRWT/EGFRvIII expression, the impact of receptor signaling on various cellular processes, mechanisms of cells resistance to treatment, or the presence of cancer stem cell populations. Undoubtedly, anti-EGFRvIII therapies constitute the important area of research, but the structure, mechanism of action, and the biological role of EGFRvIII need to be determined for their proper development. In particular, it is crucial to resolve whether EGFRvIII-negative glioblastoma cells are dependent on EGFRvIII-positive population or not.
Table 2.
Issues addressed in the article (except therapies in Table 1).
| EGFRvIII issue/process | Mechanism/way to address | Selected references |
|---|---|---|
| EGFRvIII presence in tumors/cancers | GB in about 40%, rarely in HNCSCC, lung prostate, colorectal cancer, breast cancer | [27, 33–41, 43] |
| EGFRvIII mechanism of mutation | Deletion of EGFR exons 2–7 | [26–29, 43] |
| EGFRvIII mechanism of action | Several models: (1) Heterodimerization with EGFRWT (2) Homodimerization (3) EGFRvIII and MET cooperation, FAK involved (4) OSMR mechanism Resistant to degradation important for all models |
[16, 17, 121–126, 142, 143] |
| EGFRvIII biological role | Extreme opinions: from lack of important role at advanced cancer (tumor) stages, to role in self-renewal, survival, and proliferation of cancer stem cells | [33, 147, 150, 152–158, 164, 165] |
| EGFRvIII cell culture models | 3D primary cell cancer cell models, DK-MG model, genetically modified cancer cell lines | [21, 150, 153, 167, 242–244, 246–248] |
Acknowledgments
This study was supported by the Medical University of Lodz (grant no. 503/0-166-01/503-01-001-19-00) and by National Science Center (grant no. 2016/21/D/NZ3/02616).
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- 1.Tzahar E., Waterman H., Chen X., et al. A hierarchical network of interreceptor interactions determines signal transduction by neu differentiation factor/neuregulin and epidermal growth factor. Molecular and Cellular Biology. 1996;16(10):5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weingaertner I. R., Koutnik S., Ammer H. Chronic morphine treatment attenuates cell growth of human BT474 breast cancer cells by rearrangement of the ErbB signalling network. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0053510.e53510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris R., Chung E., Coffey R. J. EGF receptor ligands. Experimental Cell Research. 2003;284(1):2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 4.Yu X., Sharma K. D., Takahashi T., Iwamoto R., Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Molecular Biology of the Cell. 2002;13(7):2547–2557. doi: 10.1091/mbc.01-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bessman N. J., Bagchi A., Ferguson K. M., Lemmon M. A. Complex relationship between ligand binding and dimerization in the epidermal growth factor receptor. Cell Reports. 2014;9(4):1306–1317. doi: 10.1016/j.celrep.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendoza M. C., Er E. E., Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends in Biochemical Sciences. 2011;36(6):320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones S., Rappoport J. Z. Interdependent epidermal growth factor receptor signalling and trafficking. The International Journal of Biochemistry & Cell Biology. 2014;51:23–28. doi: 10.1016/j.biocel.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 8.Treda C., Popeda M., Ksiazkiewicz M., et al. EGFR activation leads to cell death independent of PI3K/AKT/mTOR in an AD293 cell line. PLoS One. 2016;11(5) doi: 10.1371/journal.pone.0155230.e0155230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Water J. A. J. M., Bagci-Onder T., Agarwal A. S., et al. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proceedings of the National Academy of Sciences. 2012;109(41):16642–16647. doi: 10.1073/pnas.1202832109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigismund S., Argenzio E., Tosoni D., Cavallaro E., Polo S., Di Fiore P. P. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Developmental Cell. 2008;15(2):209–219. doi: 10.1016/j.devcel.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 11.Kang J. U. Characterization of amplification patterns and target genes on the short arm of chromosome 7 in early-stage lung adenocarcinoma. Molecular Medicine Reports. 2013;8(5):1373–1378. doi: 10.3892/mmr.2013.1686. [DOI] [PubMed] [Google Scholar]
- 12.Sato K.-I. Cellular functions regulated by phosphorylation of EGFR on Tyr845. International Journal of Molecular Sciences. 2013;14(6):10761–10790. doi: 10.3390/ijms140610761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omidfar K., Shirvani Z. Single domain antibodies: a new concept for epidermal growth factor receptor and EGFRvIII targeting. DNA and Cell Biology. 2012;31(6):1015–1026. doi: 10.1089/dna.2011.1529. [DOI] [PubMed] [Google Scholar]
- 14.Bradshaw R. A., Chalkley R. J., Biarc J., Burlingame A. L. Receptor tyrosine kinase signaling mechanisms: devolving TrkA responses with phosphoproteomics. Advances in Biological Regulation. 2013;53(1):87–96. doi: 10.1016/j.jbior.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tong J., Taylor P., Jovceva E., et al. Tandem immunoprecipitation of phosphotyrosine-mass spectrometry (TIPY-MS) indicates C19ORF19 becomes tyrosine-phosphorylated and associated with activated epidermal growth factor receptor. Journal of Proteome Research. 2008;7(3):1067–1077. doi: 10.1021/pr7006363. [DOI] [PubMed] [Google Scholar]
- 16.Purba E., Saita E.-I., Maruyama I. Activation of the EGF receptor by ligand binding and oncogenic mutations: the “rotation model”. Cells. 2017;6(2):p. 13. doi: 10.3390/cells6020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H.-J. S., Nagane M., Klingbeil C. K., et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. Journal of Biological Chemistry. 1997;272(5):2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 18.Shtiegman K., Kochupurakkal B. S., Zwang Y., et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene. 2007;26(49):6968–6978. doi: 10.1038/sj.onc.1210503. [DOI] [PubMed] [Google Scholar]
- 19.Sangar V., Funk C. C., Kusebauch U., Campbell D. S., Moritz R. L., Price N. D. Quantitative proteomic analysis reveals effects of Epidermal Growth Factor Receptor (EGFR) on invasion-promoting proteins secreted by glioblastoma cells. Molecular & Cellular Proteomics. 2014;13(10):2618–2631. doi: 10.1074/mcp.m114.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagane M., Coufal F., Lin H., Bögler O., Cavenee W. K., Huang H. J. S. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Research. 1996;56:5079–5086. [PubMed] [Google Scholar]
- 21.Francis J. M., Zhang C.-Z., Maire C. L., et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discovery. 2014;4(8):956–971. doi: 10.1158/2159-8290.CD-13-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong A. J., Ruppert J. M., Bigner S. H., et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proceedings of the National Academy of Sciences. 1992;89(7):2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillaudeau A., Durand K., Bessette B., et al. EGFR soluble isoforms and their transcripts are expressed in meningiomas. PLoS One. 2012;7(5) doi: 10.1371/journal.pone.0037204.e37204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho J., Pastorino S., Zeng Q., et al. Glioblastoma-derived epidermal growth factor receptor carboxyl-terminal deletion mutants are transforming and are sensitive to EGFR-directed therapies. Cancer Research. 2011;71(24):7587–7596. doi: 10.1158/0008-5472.CAN-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto I., Kenyon L. C., Emlet D. R., et al. Expression of constitutively activated EGFRvIII in non-small cell lung cancer. Cancer Science. 2003;94(1):50–56. doi: 10.1111/j.1349-7006.2003.tb01351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voldborg B. R., Damstrup L., Spang-Thomsen M., Poulsen H. S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Annals of Oncology. 1997;8(12):1197–1206. doi: 10.1023/a:1008209720526. [DOI] [PubMed] [Google Scholar]
- 27.Wikstrand C. J., Hale L. P., Batra S. K., et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Research. 1995;55:3140–3148. [PubMed] [Google Scholar]
- 28.Yamazaki H., Ohba Y., Tamaoki N., Shibuya M. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Japanese Journal of Cancer Research. 1990;81(8):773–779. doi: 10.1111/j.1349-7006.1990.tb02644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphrey P. A., Wong A. J., Vogelstein B., et al. Amplification and expression of the epidermal growth factor receptor gene in human glioma xenografts. Cancer Research. 1988;48:2231–2238. [PubMed] [Google Scholar]
- 30.Abou-Fayçal C., Hatat A. S., Gazzeri S., Eymin B. Splice variants of the RTK family: their role in tumour progression and response to targeted therapy. International Journal of Molecular Sciences. 2017;18(2) doi: 10.3390/ijms18020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Vecchio C. A., Giacomini C. P., Vogel H., et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene. 2013;32(21):2670–2681. doi: 10.1038/onc.2012.280. [DOI] [PubMed] [Google Scholar]
- 32.Snuderl M., Fazlollahi L., Le L. P., et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 33.Peciak J., Stec W. J., Treda C., et al. Low incidence along with low mRNA levels of EGFRvIII in prostate and colorectal cancers compared to glioblastoma. Journal of Cancer. 2017;8(1):146–151. doi: 10.7150/jca.16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moscatello D. K., Holgado-Madruga M., Godwin A. K., et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Research. 1995;55:5536–5539. [PubMed] [Google Scholar]
- 35.Olapade-Olaopa E. O., Moscatello D. K., MacKay E. H., et al. Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. British Journal of Cancer. 2000;82(1):186–194. doi: 10.1054/bjoc.1999.0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rae J. M., Scheys J. O., Clark K. M., Chadwick R. B., Kiefer M. C., Lippman M. E. EGFR and EGFRvIII expression in primary breast cancer and cell lines. Breast Cancer Research and Treatment. 2004;87(1):87–95. doi: 10.1023/B:BREA.0000041585.26734.f9. [DOI] [PubMed] [Google Scholar]
- 37.Sok J. C., Coppelli F. M., Thomas S. M., et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clinical Cancer Research. 2006;12(17):5064–5073. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 38.Chau N. G., Perez-Ordonez B., Zhang K., et al. The association between EGFR variant III, HPV, p16, c-MET, EGFR gene copy number and response to EGFR inhibitors in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Head & Neck Oncology. 2011;3(1):p. 11. doi: 10.1186/1758-3284-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohgaki H., Kleihues P. Genetic pathways to primary and secondary glioblastoma. The American Journal of Pathology. 2007;170(5):1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saikali S., Avril T., Collet B., et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Rα2, gp100 and TRP-2 for immunotherapy. Journal of Neuro-Oncology. 2006;81(2):139–148. doi: 10.1007/s11060-006-9220-3. [DOI] [PubMed] [Google Scholar]
- 41.Azuma M., Danenberg K. D., Iqbal S., et al. Epidermal growth factor receptor and epidermal growth factor receptor variant III gene expression in metastatic colorectal cancer. Clinical Colorectal Cancer. 2006;6(3):214–218. doi: 10.3816/ccc.2006.n.038. [DOI] [PubMed] [Google Scholar]
- 42.Steffensen K. D., Waldstrom M., Olsen D. A., et al. Mutant epidermal growth factor receptor in benign, borderline, and malignant ovarian tumors. Clinical Cancer Research. 2008;14(11):3278–3282. doi: 10.1158/1078-0432.CCR-07-4171. [DOI] [PubMed] [Google Scholar]
- 43.Jungbluth A. A., Stockert E., Huang H. J. S., et al. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proceedings of the National Academy of Sciences. 2003;100(2):639–644. doi: 10.1073/pnas.232686499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Graeff P., Crijns A. P. G., Ten Hoor K. A., et al. The ErbB signalling pathway: protein expression and prognostic value in epithelial ovarian cancer. British Journal of Cancer. 2008;99(2):341–349. doi: 10.1038/sj.bjc.6604471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spindler K.-L. G., Olsen D. A., Nielsen J. N., et al. Lack of the type III epidermal growth factor receptor mutation in colorectal cancer. Anticancer Research. 2006;26:4889–4893. [PubMed] [Google Scholar]
- 46.Cunningham M. P., Essapen S., Thomas H., et al. Coexpression, prognostic significance and predictive value of EGFR, EGFRvIII and phosphorylated EGFR in colorectal cancer. International Journal of Oncology. 2005;27:317–325. doi: 10.3892/ijo.27.2.317. [DOI] [PubMed] [Google Scholar]
- 47.Blehm K. N., Spiess P. E., Bondaruk J. E., et al. Mutations within the kinase domain and truncations of the epidermal growth factor receptor are rare events in bladder cancer: implications for therapy. Clinical Cancer Research. 2006;12(15):4671–4677. doi: 10.1158/1078-0432.CCR-06-0407. [DOI] [PubMed] [Google Scholar]
- 48.Ge H., Gong X., Tang C. K. Evidence of high incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. International Journal of Cancer. 2002;98(3):357–361. doi: 10.1002/ijc.10224. [DOI] [PubMed] [Google Scholar]
- 49.Nieto Y., Nawaz F., Jones R. B., Shpall E. J., Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. Journal of Clinical Oncology. 2007;25(28):4405–4413. doi: 10.1200/JCO.2006.09.8822. [DOI] [PubMed] [Google Scholar]
- 50.Tidow N., Boecker A., Schmidt H., et al. Distinct amplification of an untranslated regulatory sequence in the EGFR gene contributes to early steps in breast cancer development. Cancer Research. 2003;63:1172–1178. [PubMed] [Google Scholar]
- 51.Rosenthal M., Curry R., Reardon D. A., et al. Safety, tolerability, and pharmacokinetics of anti-EGFRvIII antibody-drug conjugate AMG 595 in patients with recurrent malignant glioma expressing EGFRvIII. Cancer Chemotherapy and Pharmacology. 2019;84(2):327–336. doi: 10.1007/s00280-019-03879-2. [DOI] [PubMed] [Google Scholar]
- 52.O’Rourke D. M., Nasrallah M. P., Desai A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science Translational Medicine. 2017;9(399) doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miao H., Choi B. D., Suryadevara C. M., et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One. 2014;9(4) doi: 10.1371/journal.pone.0094281.e94281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z., Jiang J., Wu X., et al. Chimeric antigen receptor T cell targeting EGFRvIII for metastatic lung cancer therapy. Frontiers of Medicine. 2019;13(1):57–68. doi: 10.1007/s11684-019-0683-y. [DOI] [PubMed] [Google Scholar]
- 55.Choi B. D., Kuan C.-T., Cai M., et al. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proceedings of the National Academy of Sciences. 2013;110(1):270–275. doi: 10.1073/pnas.1219817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellwanger K., Reusch U., Fucek I., et al. Highly specific and effective targeting of EGFRvIII-positive tumors with TandAb antibodies. Frontiers in Oncology. 2017;7:p. 100. doi: 10.3389/fonc.2017.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Study of AMG 596 in patients with EGFRvIII positive glioblastoma-full text view-ClinicalTrials.gov, 2017, https://clinicaltrials.gov/ct2/show/NCT03296696.
- 58.Weller M., Butowski N., Tran D. D., et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. The Lancet Oncology. 2017;18:1373–1385. doi: 10.1016/S1470-2045(17)30517-X. [DOI] [PubMed] [Google Scholar]
- 59.Luo X., Gong X., Tang C. K. Suppression of EGFRvIII-mediated proliferation and tumorigenesis of breast cancer cells by ribozyme. International Journal of Cancer. 2003;104(6):716–721. doi: 10.1002/ijc.11007. [DOI] [PubMed] [Google Scholar]
- 60.Halatsch M.-E., Schmidt U., Bötefür I. C., Holland J. F., Ohnuma T. Marked inhibition of glioblastoma target cell tumorigenicity in vitro by retrovirus-mediated transfer of a hairpin ribozyme against deletion-mutant epidermal growth factor messenger RNA. Journal of Neurosurgery. 2000;92(2):297–305. doi: 10.3171/jns.2000.92.2.0297. [DOI] [PubMed] [Google Scholar]
- 61.Kang C.-S., Zhang Z.-Y., Jia Z.-F., et al. Suppression of EGFR expression by antisense or small interference RNA inhibits U251 glioma cell growth in vitro and in vivo. Cancer Gene Therapy. 2006;13(5):530–538. doi: 10.1038/sj.cgt.7700932. [DOI] [PubMed] [Google Scholar]
- 62.Shir A., Levitzki A. Inhibition of glioma growth by tumor-specific activation of double-stranded RNA-dependent protein kinase PKR. Nature Biotechnology. 2002;20(9):895–900. doi: 10.1038/nbt730. [DOI] [PubMed] [Google Scholar]
- 63.Yamoutpour F., Bodempudi V., Park S. E., et al. Gene silencing for epidermal growth factor receptor variant III induces cell-specific cytotoxicity. Molecular Cancer Therapeutics. 2008;7(11):3586–3597. doi: 10.1158/1535-7163.MCT-08-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franceschi E., Cavallo G., Lonardi S., et al. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) British Journal of Cancer. 2007;96(7):1047–1051. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rich J. N., Reardon D. A., Peery T., et al. Phase II trial of gefitinib in recurrent glioblastoma. Journal of Clinical Oncology. 2004;22(1):133–142. doi: 10.1200/jco.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 66.Sim E. H. A., Yang I. A., Wood-Baker R., Bowman R. V., Fong K. M. Gefitinib for advanced non-small cell lung cancer. Cochrane Database of Systematic Reviews. 2018;2018 doi: 10.1002/14651858.cd006847.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao H., Fan Y., Ma S., et al. Final overall survival results from a phase III, randomized, placebo-controlled, parallel-group study of gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM; C-TONG 0804) Journal of Thoracic Oncology. 2015;10(4):655–664. doi: 10.1097/jto.0000000000000445. [DOI] [PubMed] [Google Scholar]
- 68.Jakob J. A., Kies M. S., Glisson B. S., et al. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head & Neck. 2015;37(5):644–649. doi: 10.1002/hed.23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalykaki A., Agelaki S., Kallergi G., Xyrafas A., Mavroudis D., Georgoulias V. Elimination of EGFR-expressing circulating tumor cells in patients with metastatic breast cancer treated with gefitinib. Cancer Chemotherapy and Pharmacology. 2014;73(4):685–693. doi: 10.1007/s00280-014-2387-y. [DOI] [PubMed] [Google Scholar]
- 70. Topotecan and gefitinib (iressa) for ovarian, peritoneal, or fallopian tube cancer-full text view-ClinicalTrials.gov, 2019, https://clinicaltrials.gov/ct2/show/NCT00317772?recrs=abdef&cond=gefitinib&draw=2&rank=21.
- 71. Adjuvant therapy of gefitinib (iressa, ZD1839) in patients with resectable hepatocellular carcinoma-full text view-ClinicalTrials.gov, 2019, https://clinicaltrials.gov/ct2/show/NCT00282100?recrs=abdef&cond=gefitinib&draw=2&rank=70.
- 72.Thiessen B., Stewart C., Tsao M., et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemotherapy and Pharmacology. 2010;65(2):353–361. doi: 10.1007/s00280-009-1041-6. [DOI] [PubMed] [Google Scholar]
- 73. Lapatinib with temozolomide and regional radiation therapy for patients with newly-diagnosed glioblastoma multiforme-full text view-ClinicalTrials.gov, 2019, https://clinicaltrials.gov/ct2/show/NCT01591577?term=lapatinib&cond=glioblastoma&rank=1.
- 74.Ryan Q., Ibrahim A., Cohen M. H., et al. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. The Oncologist. 2008;13(10):1114–1119. doi: 10.1634/theoncologist.2008-0816. [DOI] [PubMed] [Google Scholar]
- 75.Iqbal S., Goldman B., Fenoglio-Preiser C. M., et al. Southwest Oncology Group study S0413: a phase II trial of lapatinib (GW572016) as first-line therapy in patients with advanced or metastatic gastric cancer. Annals of Oncology. 2011;22(12):2610–2615. doi: 10.1093/annonc/mdr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Study of trastuzumab-emtansine in patients with HER2-positive metastatic colorectal cancer progressing after trastuzumab and lapatinib.-full text view-ClinicalTrials.gov, 2019, https://clinicaltrials.gov/ct2/show/study/NCT03418558?recrs=abdfh&cond=lapatinib&draw=4.
- 77.van den Bent M. J., Brandes A. A., Rampling R., et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. Journal of Clinical Oncology. 2009;27(8):1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raizer J. J., Abrey L. E., Lassman A. B., et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncology. 2010;12(1):95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Horowitz N. S., Olawaiye A. B., Borger D. R., et al. Phase II trial of erlotinib in women with squamous cell carcinoma of the vulva. Gynecologic Oncology. 2012;127(1):141–146. doi: 10.1016/j.ygyno.2012.06.028. [DOI] [PubMed] [Google Scholar]
- 80.Wang Y., Schmid-Bindert G., Zhou C. Erlotinib in the treatment of advanced non-small cell lung cancer: an update for clinicians. Therapeutic Advances in Medical Oncology. 2012;4(1):19–29. doi: 10.1177/1758834011427927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cicènas S., Geater S. L., Petrov P., et al. Maintenance erlotinib versus erlotinib at disease progression in patients with advanced non-small-cell lung cancer who have not progressed following platinum-based chemotherapy (IUNO study) Lung Cancer. 2016;102:30–37. doi: 10.1016/j.lungcan.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 82.Moore M. J., Goldstein D., Hamm J., et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. Journal of Clinical Oncology. 2007;25(15):1960–1966. doi: 10.1200/jco.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 83.William W. N., Tsao A. S., Feng L., et al. Single arm, phase II study of cisplatin, docetaxel, and erlotinib in patients with recurrent and/or metastatic head and neck squamous cell carcinomas. The Oncologist. 2018;23(5):526–e49. doi: 10.1634/theoncologist.2017-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bauman J. E., Duvvuri U., Gooding W. E., et al. Randomized, placebo-controlled window trial of EGFR, Src, or combined blockade in head and neck cancer. JCI Insight. 2017;2 doi: 10.1172/jci.insight.90449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. FDA Broadens Afatinib Indication to Previously Untreated, Metastatic NSCLC with Other Non-resistant EGFR Mutations, 2019, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-broadens-afatinib-indication-previously-untreated-metastatic-nsclc-other-non-resistant-egfr.
- 86.Cohen E. E. W., Licitra L. F., Burtness B., et al. Biomarkers predict enhanced clinical outcomes with afatinib versus methotrexate in patients with second-line recurrent and/or metastatic head and neck cancer. Annals of Oncology. 2017;28(10):2526–2532. doi: 10.1093/annonc/mdx344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vengoji R., Macha M. A., Nimmakayala R. K., et al. Afatinib and temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. Journal of Experimental & Clinical Cancer Research. 2019;38(1) doi: 10.1186/s13046-019-1264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reardon D. A., Nabors L. B., Mason W. P., et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro Oncol. 2015;17:430–439. doi: 10.1093/neuonc/nou160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goh G., Schmid R., Guiver K., et al. Clonal evolutionary analysis during HER2 blockade in HER2-positive inflammatory breast cancer: a phase II open-label clinical trial of afatinib ± vinorelbine. PLoS Medicine. 2016;13 doi: 10.1371/journal.pmed.1002136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hickish T., Cassidy J., Propper D., et al. A randomised, open-label phase II trial of afatinib versus cetuximab in patients with metastatic colorectal cancer. European Journal of Cancer. 2014;50(18):3136–3144. doi: 10.1016/j.ejca.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 91.Bonner J. A., Harari P. M., Giralt J., et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. New England Journal of Medicine. 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 92.Eller J. L., Longo S. L., Kyle M. M., Bassano D., Hicklin D. J., Canute G. W. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery. 2005;56(1):155–162. doi: 10.1227/01.neu.0000145865.25689.55. [DOI] [PubMed] [Google Scholar]
- 93.Eller J. L., Longo S. L., Hicklin D. J., Canute G. W. Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery. 2002;51:1004–1005. doi: 10.1227/00006123-200210000-00028. [DOI] [PubMed] [Google Scholar]
- 94.Hasselbalch B., Lassen U., Hansen S., et al. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncology. 2010;12:508–516. doi: 10.1200/jco.2008.26.15_suppl.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guren T. K., Thomsen M., Kure E. H., et al. Cetuximab in treatment of metastatic colorectal cancer: final survival analyses and extended RAS data from the NORDIC-VII study. British Journal of Cancer. 2017;116(10):1271–1278. doi: 10.1038/bjc.2017.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan J. A., Blaszkowsky L. S., Enzinger P. C., et al. A multicenter phase II trial of single-agent cetuximab in advanced esophageal and gastric adenocarcinoma. Annals of Oncology. 2011;22(6):1367–1373. doi: 10.1093/annonc/mdq604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jatoi A., Schild S. E., Foster N., et al. A phase II study of cetuximab and radiation in elderly and/or poor performance status patients with locally advanced non-small-cell lung cancer (N0422) Annals of Oncology. 2010;21(10):2040–2044. doi: 10.1093/annonc/mdq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carey L. A., Rugo H. S., Marcom P. K., et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. Journal of Clinical Oncology. 2012;30(21):2615–2623. doi: 10.1200/jco.2010.34.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fleming M. T., Sonpavde G., Kolodziej M., et al. Association of rash with outcomes in a randomized phase II trial evaluating cetuximab in combination with mitoxantrone plus prednisone after docetaxel for metastatic castration-resistant prostate cancer. Clinical Genitourinary Cancer. 2012;10(1):6–14. doi: 10.1016/j.clgc.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 100.Farley J., Sill M. W., Birrer M., et al. Phase II study of cisplatin plus cetuximab in advanced, recurrent, and previously treated cancers of the cervix and evaluation of epidermal growth factor receptor immunohistochemical expression: a gynecologic oncology group study. Gynecologic Oncology. 2011;121(2):303–308. doi: 10.1016/j.ygyno.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weber J., McCormack P. L. Panitumumab. BioDrugs. 2008;22(6):403–411. doi: 10.2165/0063030-200822060-00006. [DOI] [PubMed] [Google Scholar]
- 102.Asimakopoulou N., Souglakos J., Kentepozidis N., et al. Efficacy of panitumumab in older patients with metastatic colorectal cancer: a retrospective analysis using the database of the hellenic oncology research group (HORG) Journal of Geriatric Oncology. 2019;10(1):143–148. doi: 10.1016/j.jgo.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 103.Sohal D. P. S., Mykulowycz K., Uehara T., et al. A phase II trial of gemcitabine, irinotecan and panitumumab in advanced cholangiocarcinoma. Annals of Oncology. 2013;24(12):3061–3065. doi: 10.1093/annonc/mdt416. [DOI] [PubMed] [Google Scholar]
- 104.Mesía R., Henke M., Fortin A., et al. Chemoradiotherapy with or without panitumumab in patients with unresected, locally advanced squamous-cell carcinoma of the head and neck (CONCERT-1): a randomised, controlled, open-label phase 2 trial. The Lancet Oncology. 2015;16(2):208–220. doi: 10.1016/s1470-2045(14)71198-2. [DOI] [PubMed] [Google Scholar]
- 105.Rischin D., Spigel D. R., Adkins D., et al. PRISM: phase 2 trial with panitumumab monotherapy as second-line treatment in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Head & Neck. 2016;38(S1):E1756–E1761. doi: 10.1002/hed.24311. [DOI] [PubMed] [Google Scholar]
- 106.Pillay V., Allaf L., Wilding A. L., et al. The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia. 2009;11(5):448–IN2. doi: 10.1593/neo.09230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Panitumumab and Irinotecan for Malignant Gliomas-Full Text View-ClinicalTrials.Gov, 2019, https://clinicaltrials.gov/ct2/show/NCT01017653?cond=panitumumab+glioblastoma&draw=2&rank=2.
- 108.Matsuda N., Wang X., Lim B., et al. Safety and efficacy of panitumumab plus neoadjuvant chemotherapy in patients with primary HER2-negative inflammatory breast cancer. JAMA Oncology. 2018;4(9):1207–1213. doi: 10.1001/jamaoncol.2018.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Westphal M., Heese O., Steinbach J. P., et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. European Journal of Cancer. 2015;51(4):522–532. doi: 10.1016/j.ejca.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 110. Orphanet: Nimotuzumab, 2019, https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=82913.
- 111.Wang X., Gu J., Shao C., Han K., Meng J. Nimotuzumab plus chemotherapy with docetaxel, cisplatin, 5-fluorouracil for locally advanced head and neck squamous cell carcinoma: a clinical study. Journal of Cancer Research and Therapeutics. 2019;15(2):312–316. doi: 10.4103/jcrt.JCRT_889_17. [DOI] [PubMed] [Google Scholar]
- 112.Subramanian S., Sridharan N., Balasundaram V., Chaudhari S. Efficacy and safety of nimotuzumab in unresectable, recurrent, and/or metastatic squamous cell carcinoma of the head and neck: a hospital-based retrospective evidence. South Asian Journal of Cancer. 2018;7(3):p. 188. doi: 10.1007/978-1-4419-9464-6_47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Comp. committee for orphan medicinal products public summary of positive opinion for orphan designation of nimotuzumab for the treatment of pancreatic cancer, 2008, http://www.emea.europa.eu.
- 114.Phillips A. C., Boghaert E. R., Vaidya K. S., et al. ABT-414, an antibody-drug conjugate targeting a tumor-selective EGFR epitope. Molecular Cancer Therapeutics. 2016;15(4):661–669. doi: 10.1158/1535-7163.MCT-15-0901. [DOI] [PubMed] [Google Scholar]
- 115.van den Bent M., Gan H. K., Lassman A. B., et al. Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: results from a multi-center, international study. Cancer Chemotherapy and Pharmacology. 2017;80(6):1209–1217. doi: 10.1007/s00280-017-3451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goss G. D., Vokes E. E., Gordon M. S., et al. Efficacy and safety results of depatuxizumab mafodotin (ABT-414) in patients with advanced solid tumors likely to overexpress epidermal growth factor receptor. Cancer. 2018;124(10):2174–2183. doi: 10.1002/cncr.31304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Frederick L., Eley G., Wang Y., James C. D. Analysis of genomic rearrangements associated with EGFRvIII expression suggests involvement of Alu repeat elements. Neuro-Oncology. 2000;2(3):159–163. doi: 10.1093/neuonc/2.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Figueroa J. M., Skog J., Akers J., et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro-Oncology. 2017;19(11):1494–1502. doi: 10.1093/neuonc/nox085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Felsberg J., Hentschel B., Kaulich K., et al. Epidermal growth factor receptor variant III (EGFRvIII) positivity inEGFR-amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clinical Cancer Research. 2017;23(22):6846–6855. doi: 10.1158/1078-0432.CCR-17-0890. [DOI] [PubMed] [Google Scholar]
- 120.Turner K. M., Deshpande V., Beyter D., et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017;543(7643):122–125. doi: 10.1038/nature21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grandal M. V., Zandi R., Pedersen M. W., Willumsen B. M., van Deurs B., Poulsen H. S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis. 2007;28(7):1408–1417. doi: 10.1093/carcin/bgm058. [DOI] [PubMed] [Google Scholar]
- 122.Nishikawa R., Ji X. D., Harmon R. C., et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proceedings of the National Academy of Sciences. 1994;91(16):7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stec W., Rosiak K., Treda C., et al. Cyclic trans-phosphorylation in a homodimer as the predominant mechanism of EGFRvIII action and regulation. Oncotarget. 2018;9(9):8560–8572. doi: 10.18632/oncotarget.24058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Batra S. K., Castelino-Prabhu S., Wikstrand C. J., et al. Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth & Differentiation. 1995;6:1251–1259. [PubMed] [Google Scholar]
- 125.Hills D., Rowlinson-Busza G., Gullick W. J. Specific targeting of a mutant, activated EGF receptor found in glioblastoma using a monoclonal antibody. International Journal of Cancer. 1995;63(4):537–543. doi: 10.1002/ijc.2910630414. [DOI] [PubMed] [Google Scholar]
- 126.Sun L., Yu S., Xu H., et al. FHL2 interacts with EGFR to promote glioblastoma growth. Oncogene. 2018;37(10):1386–1398. doi: 10.1038/s41388-017-0068-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tan D. S. W., Chong F. T., Leong H. S., et al. Long noncoding RNA EGFR-AS1 mediates epidermal growth factor receptor addiction and modulates treatment response in squamous cell carcinoma. Nature Medicine. 2017;23(10):1167–1175. doi: 10.1038/nm.4401. [DOI] [PubMed] [Google Scholar]
- 128.Liccardi G., Hartley J. A., Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Research. 2011;71(3):1103–1114. doi: 10.1158/0008-5472.can-10-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dowlati A., Nethery D., Kern J. A. Combined inhibition of epidermal growth factor receptor and JAK/STAT pathways results in greater growth inhibition in vitro than single agent therapy. Molecular Cancer Therapeutics. 2004;3:459–463. [PubMed] [Google Scholar]
- 130.Narita Y., Nagane M., Mishima K., Huang H. J., Furnari F. B., Cavenee W. K. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Research. 2002;62:6764–6769. [PubMed] [Google Scholar]
- 131.Wiley H. Trafficking of the ErbB receptors and its influence on signaling. Experimental Cell Research. 2003;284(1):78–88. doi: 10.1016/s0014-4827(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 132.Huang F., Goh L. K., Sorkin A. EGF receptor ubiquitination is not necessary for its internalization. Proceedings of the National Academy of Sciences. 2007;104(43):16904–16909. doi: 10.1073/pnas.0707416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gajadhar A. S., Bogdanovic E., Munoz D. M., Guha A. In situ analysis of mutant EGFRs prevalent in glioblastoma multiforme reveals aberrant dimerization, activation, and differential response to anti-EGFR targeted therapy. Molecular Cancer Research. 2012;10(3):428–440. doi: 10.1158/1541-7786.MCR-11-0531. [DOI] [PubMed] [Google Scholar]
- 134.Hwang Y., Chumbalkar V., Latha K., Bogler O. Forced dimerization increases the activity of EGFR/EGFRvIII and enhances its oncogenicity. Molecular Cancer Research. 2011;9(9):1199–1208. doi: 10.1158/1541-7786.MCR-11-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kancha R., von Bubnoff N., Duyster J. Asymmetric kinase dimer formation is crucial for the activation of oncogenic EGFRvIII but not for ERBB3 phosphorylation. Cell Communication and Signaling. 2013;11(1):p. 39. doi: 10.1186/1478-811X-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fernandes H., Cohen S., Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association. Journal of Biological Chemistry. 2001;276(7):5375–5383. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- 137.Luwor R. B., Zhu H.-J., Walker F., et al. The tumor-specific de2–7 epidermal growth factor receptor (EGFR) promotes cells survival and heterodimerizes with the wild-type EGFR. Oncogene. 2004;23(36):6095–6104. doi: 10.1038/sj.onc.1207870. [DOI] [PubMed] [Google Scholar]
- 138.Fan Q.-W., Cheng C. K., Gustafson W. C., et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell. 2013;24(4):438–449. doi: 10.1016/j.ccr.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Greenall S. A., Donoghue J. F., Van Sinderen M., et al. EGFRvIII-mediated transactivation of receptor tyrosine kinases in glioma: mechanism and therapeutic implications. Oncogene. 2015;34(41):5277–5287. doi: 10.1038/onc.2014.448. [DOI] [PubMed] [Google Scholar]
- 140.Ymer S. I., Greenall S. A., Cvrljevic A., et al. Glioma specific extracellular missense mutations in the first cysteine rich region of epidermal growth factor receptor (EGFR) initiate ligand independent activation. Cancers. 2011;3(2):2032–2049. doi: 10.3390/cancers3022032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ruan Z., Kannan N. Mechanistic insights into R776H mediated activation of epidermal growth factor receptor kinase. Biochemistry. 2015;54(27):4216–4225. doi: 10.1021/acs.biochem.5b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chakravarty D., Pedraza A. M., Cotari J., et al. EGFR and PDGFRA co-expression and heterodimerization in glioblastoma tumor sphere lines. Scientific Reports. 2017;7(1):p. 9043. doi: 10.1038/s41598-017-08940-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Garnett J., Chumbalkar V., Vaillant B., et al. Regulation of HGF expression by ΔEGFR-mediated c-met activation in glioblastoma cells. Neoplasia. 2013;15(1):73–IN21. doi: 10.1593/neo.121536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Latha K., Li M., Chumbalkar V., et al. Nuclear EGFRvIII-STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl-XL promoter. International Journal of Cancer. 2013;132(3):509–520. doi: 10.1002/ijc.27690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gururaj A. E., Gibson L., Panchabhai S., et al. Access to the nucleus and functional association with c-myc is required for the full oncogenic potential of ΔEGFR/EGFRvIII. Journal of Biological Chemistry. 2013;288(5):3428–3438. doi: 10.1074/jbc.M112.399352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Newman J. P., Wang G. Y., Arima K., et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nature Communications. 2017;8(1):p. 1913. doi: 10.1038/s41467-017-01392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stec W. J., Rosiak K., Siejka P., et al. Cell line with endogenous EGFRvIII expression is a suitable model for research and drug development purposes. Oncotarget. 2016;7(22):31907–31925. doi: 10.18632/oncotarget.8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schmidt M. H. H., Furnari F. B., Cavenee W. K., Bogler O. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proceedings of the National Academy of Sciences. 2003;100(11):6505–6510. doi: 10.1073/pnas.1031790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Han W., Zhang T., Yu H., Foulke J. G., Tang C. K. Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII. Cancer Biology & Therapy. 2006;5(10):1361–1368. doi: 10.4161/cbt.5.10.3226. [DOI] [PubMed] [Google Scholar]
- 150.Inda M.-D.-M., Bonavia R., Mukasa A., et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes & Development. 2010;24(16):1731–1745. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zanca C., Villa G. R., Benitez J. A., et al. Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes & Development. 2017;31(12):1212–1227. doi: 10.1101/gad.300079.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liffers K., Kolbe K., Westphal M., Lamszus K., Schulte A. Histone deacetylase inhibitors resensitize EGFR/EGFRvIII-Overexpressing, erlotinib-resistant glioblastoma cells to tyrosine kinase inhibition. Targeted Oncology. 2016;11(1):29–40. doi: 10.1007/s11523-015-0372-y. [DOI] [PubMed] [Google Scholar]
- 153.Stockhausen M.-T., Kristoffersen K., Stobbe L., Poulsen H. S. Differentiation of glioblastoma multiforme stem-like cells leads to downregulation of EGFR and EGFRvIII and decreased tumorigenic and stem-like cell potential. Cancer Biology & Therapy. 2014;15(2):216–224. doi: 10.4161/cbt.26736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kristoffersen K., Villingshøj M., Poulsen H. S., Stockhausen M.-T. Level of Notch activation determines the effect on growth and stem cell-like features in glioblastoma multiforme neurosphere cultures. Cancer Biology & Therapy. 2013;14(7):625–637. doi: 10.4161/cbt.24595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Galli R., Binda E., Orfanelli U., et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Research. 2004;64(19):7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 156.Ignatova T. N., Kukekov V. G., Laywell E. D., Suslov O. N., Vrionis F. D., Steindler D. A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39(3):193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 157.Yuan X., Curtin J., Xiong Y., et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23(58):9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 158.Emlet D. R., Gupta P., Holgado-Madruga M., et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Research. 2014;74(4):1238–1249. doi: 10.1158/0008-5472.CAN-13-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ayuso-Sacido A., Moliterno J. A., Kratovac S., et al. Activated EGFR signaling increases proliferation, survival, and migration and blocks neuronal differentiation in post-natal neural stem cells. Journal of Neuro-Oncology. 2010;97(3):323–337. doi: 10.1007/s11060-009-0035-x. [DOI] [PubMed] [Google Scholar]
- 160.Bao S., Wu Q., Sathornsumetee S., et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Research. 2006;66(16):7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 161.Liu G., Yuan X., Zeng Z., et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular Cancer. 2006;5(1):p. 67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lathia J. D., Gallagher J., Myers J. T., et al. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLoS One. 2011;6(9) doi: 10.1371/journal.pone.0024807.e24807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rieske P., Golanska E., Zakrzewska M., et al. Arrested neural and advanced mesenchymal differentiation of glioblastoma cells-comparative study with neural progenitors. BMC Cancer. 2009;9(1):p. 54. doi: 10.1186/1471-2407-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen R., Pan Y., Gutmann D. H. The power of the few. Genes & Development. 2017;31(12):1177–1179. doi: 10.1101/gad.303453.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.van den Bent M. J., Gao Y., Kerkhof M., et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncology. 2015;17(7):935–941. doi: 10.1093/neuonc/nov013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ren P.-P., Li M., Li T.-F., Han S.-Y. Anti-EGFRvIII chimeric antigen receptor-modified T cells for adoptive cell therapy of glioblastoma. Current Pharmaceutical Design. 2017;23(14):2113–2116. doi: 10.2174/1381612823666170316125402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nathanson D. A., Gini B., Mottahedeh J., et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343(6166):72–76. doi: 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Read J., Ingram A., Al Saleh H. A., et al. Nuclear transportation of exogenous epidermal growth factor receptor and androgen receptor via extracellular vesicles. European Journal of Cancer. 2017;70:62–74. doi: 10.1016/j.ejca.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 169.Ricklefs F., Mineo M., Rooj A. K., et al. Extracellular vesicles from high-grade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer Research. 2016;76(10):2876–2881. doi: 10.1158/0008-5472.CAN-15-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lee D. W., Lee S.-Y., Doh I., Ryu G. H., Nam D.-H. High-Dose compound heat map for 3D-cultured glioblastoma multiforme cells in a micropillar and microwell chip platform. BioMed Research International. 2017;2017:7. doi: 10.1155/2017/7218707.7218707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Würstle S., Schneider F., Ringel F., et al. Temozolomide induces autophagy in primary and established glioblastoma cells in an EGFR independent manner. Oncology Letters. 2017;14(1):322–328. doi: 10.3892/ol.2017.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kummar S., Chen H. X., Wright J., et al. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nature Reviews Drug Discovery. 2010;9(11):843–856. doi: 10.1038/nrd3216. [DOI] [PubMed] [Google Scholar]
- 173.Levitzki A., Mishani E. Tyrphostins and other tyrosine kinase inhibitors. Annual Review of Biochemistry. 2006;75(1):93–109. doi: 10.1146/annurev.biochem.75.103004.142657. [DOI] [PubMed] [Google Scholar]
- 174.Park J. H., Liu Y., Lemmon M. A., Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochemical Journal. 2012;448(3):417–423. doi: 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vivanco I., Robins H. I., Rohle D., et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discovery. 2012;2(5):458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cross D. A. E., Ashton S. E., Ghiorghiu S., et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery. 2014;4(9):1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rich J. N., Bigner D. D. Development of novel targeted therapies in the treatment of malignant glioma. Nature Reviews Drug Discovery. 2004;3(5):430–446. doi: 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- 178.Mellinghoff I. K., Wang M. Y., Vivanco I., et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. New England Journal of Medicine. 2005;353(19):2012–2024. doi: 10.1056/nejmoa051918. [DOI] [PubMed] [Google Scholar]
- 179.Sepúlveda-Sánchez J. M., Vaz M. Á., Balañá C., et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro-Oncology. 2017;19(11):1522–1531. doi: 10.1093/neuonc/nox105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sarkaria J. N., Hu L. S., Parney I. F., et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology. 2018;20(2):184–191. doi: 10.1093/neuonc/nox175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Maemondo M., Inoue A., Kobayashi K., et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. New England Journal of Medicine. 2010;362(25):2380–2388. doi: 10.1056/nejmoa0909530. [DOI] [PubMed] [Google Scholar]
- 182.Mok T. S., Wu Y.-L., Thongprasert S., et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. New England Journal of Medicine. 2009;361(10):947–957. doi: 10.1056/nejmoa0810699. [DOI] [PubMed] [Google Scholar]
- 183.Orellana L., Thorne A. H., Lema R., et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proceedings of the National Academy of Sciences. 2019;116:10009–10018. doi: 10.1073/pnas.1821442116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Halatsch M.-E., Gehrke E. E., Vougioukas V. I., et al. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. Journal of Neurosurgery. 2004;100(3):523–533. doi: 10.3171/jns.2004.100.3.0523. [DOI] [PubMed] [Google Scholar]
- 185.Reardon D. A., Groves M. D., Wen P. Y., et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clinical Cancer Research. 2013;19(4):900–908. doi: 10.1158/1078-0432.ccr-12-1707. [DOI] [PubMed] [Google Scholar]
- 186.Brown P. D., Krishnan S., Sarkaria J. N., et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: north central cancer treatment group study N0177. Journal of Clinical Oncology. 2008;26(34):5603–5609. doi: 10.1200/jco.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Prados M. D., Chang S. M., Butowski N., et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. Journal of Clinical Oncology. 2009;27(4):579–584. doi: 10.1200/jco.2008.18.9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hegi M. E., Diserens A.-C., Bady P., et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—A phase II trial. Molecular Cancer Therapeutics. 2011;10(6):1102–1112. doi: 10.1158/1535-7163.mct-11-0048. [DOI] [PubMed] [Google Scholar]
- 189.Jutten B., Dubois L., Li Y., et al. Binding of cetuximab to the EGFRvIII deletion mutant and its biological consequences in malignant glioma cells. Radiotherapy and Oncology. 2009;92(3):393–398. doi: 10.1016/j.radonc.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 190.Fukai J., Nishio K., Itakura T., Koizumi F. Antitumor activity of cetuximab against malignant glioma cells overexpressing EGFR deletion mutant variant III. Cancer Science. 2008;99:2062–2069. doi: 10.1111/j.1349-7006.2008.00945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Neyns B., Sadones J., Joosens E., et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Annals of Oncology. 2009;20(9):1596–1603. doi: 10.1093/annonc/mdp032. [DOI] [PubMed] [Google Scholar]
- 192.Montano N., Cenci T., Martini M., et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia. 2011;13(12):p. 1113. doi: 10.1593/neo.111338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Struve N., Brend T., Stead L., et al. TMOD-09. EGFRvIII increases mismatch repair protein expression and is therefore a predictive marker for temozolomide response in O6-methylguanine-DNA methyltransferase negative glioblastoma cells and tumors. Neuro-Oncology. 2016;18(suppl_6):p. vi208. doi: 10.1093/neuonc/now212.879. [DOI] [Google Scholar]
- 194.Poduslo J. F., Curran G. L., Berg C. T. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proceedings of the National Academy of Sciences. 1994;91(12):5705–5709. doi: 10.1073/pnas.91.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rabney A., Baum K., Pitts D., et al. Cetuximab approved by FDA for treatment of head and neck squamous cell cancer. Cancer Biology & Therapy. 2006;5(4):339–348. doi: 10.4161/cbt.5.4.2666. [DOI] [PubMed] [Google Scholar]
- 196.Geng F., Wang Z., Yin H., Yu J., Cao B. Molecular targeted drugs and treatment of colorectal cancer: recent progress and future perspectives. Cancer Biotherapy and Radiopharmaceuticals. 2017;32(5):149–160. doi: 10.1089/cbr.2017.2210. [DOI] [PubMed] [Google Scholar]
- 197.Patel D., Lahiji A., Patel S., et al. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Research. 2007;27:3355–3366. [PubMed] [Google Scholar]
- 198.Patel D., Bassi R., Hooper A., Prewett M., Hicklin D. J., Kang X. Anti-epidermal growth factor receptor monoclonal antibody cetuximab inhibits EGFR/HER-2 heterodimerization and activation. International Journal of Oncology. 2009;34:25–32. doi: 10.3892/ijo_00000125. [DOI] [PubMed] [Google Scholar]
- 199.Chakraborty S., Filippi C. G., Wong T., et al. Superselective intraarterial cerebral infusion of cetuximab after osmotic blood/brain barrier disruption for recurrent malignant glioma: phase I study. Journal of Neuro-Oncology. 2016;128(3):405–415. doi: 10.1007/s11060-016-2099-8. [DOI] [PubMed] [Google Scholar]
- 200.Kaluzova M., Bouras A., Machaidze R., Hadjipanayis C. G. Targeted therapy of glioblastoma stem-like cells and tumor non-stem cells using cetuximab-conjugated iron-oxide nanoparticles. Oncotarget. 2015;6(11):8788–8806. doi: 10.18632/oncotarget.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Whittle J. R., Lickliter J. D., Gan H. K., et al. First in human nanotechnology doxorubicin delivery system to target epidermal growth factor receptors in recurrent glioblastoma. Journal of Clinical Neuroscience. 2015;22(12):1889–1894. doi: 10.1016/j.jocn.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 202.Schmitz K. R., Ferguson K. M. Interaction of antibodies with ErbB receptor extracellular regions. Experimental Cell Research. 2009;315(4):659–670. doi: 10.1016/j.yexcr.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hamblett K. J., Kozlosky C. J., Siu S., et al. AMG 595, an anti-EGFRvIII antibody-drug conjugate, induces potent antitumor activity against EGFRvIII-expressing glioblastoma. Molecular Cancer Therapeutics. 2015;14(7):1614–1624. doi: 10.1158/1535-7163.MCT-14-1078. [DOI] [PubMed] [Google Scholar]
- 204.Johns T. G., Stockert E., Ritter G., et al. Novel monoclonal antibody specific for the de2–7 epidermal growth factor receptor (EGFR) that also recognizes the EGFR expressed in cells containing amplification of the EGFR gene. International Journal of Cancer. 2002;98(3):398–408. doi: 10.1002/ijc.10189. [DOI] [PubMed] [Google Scholar]
- 205.Reilly E. B., Phillips A. C., Buchanan F. G., et al. Characterization of ABT-806, a humanized tumor-specific anti-EGFR monoclonal antibody. Molecular Cancer Therapeutics. 2015;14(5):1141–1151. doi: 10.1158/1535-7163.MCT-14-0820. [DOI] [PubMed] [Google Scholar]
- 206.Maus M. V., Plotkin J., Jakka G., et al. An MHC-restricted antibody-based chimeric antigen receptor requires TCR-like affinity to maintain antigen specificity. Molecular Therapy—Oncolytics. 2016;3:16023–16029. doi: 10.1038/mto.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Johnson L. A., Scholler J., Ohkuri T., et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Science Translational Medicine. 2015;7(275) doi: 10.1126/scitranslmed.aaa4963.275ra22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yong C. S. M., Dardalhon V., Devaud C., Taylor N., Darcy P. K., Kershaw M. H. CAR T-cell therapy of solid tumors. Immunology and Cell Biology. 2017;95(4):356–363. doi: 10.1038/icb.2016.128. [DOI] [PubMed] [Google Scholar]
- 209.Han J., Chu J., Keung Chan W., et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Scientific Reports. 2015;5(1):p. 11483. doi: 10.1038/srep11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Brentjens R., Yeh R., Bernal Y., Riviere I., Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Molecular Therapy. 2010;18(4):666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Morgan R. A., Yang J. C., Kitano M., Dudley M. E., Laurencot C. M., Rosenberg S. A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular Therapy. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Johns T. G., Mellman I., Cartwright G. A., et al. The antitumor monoclonal antibody 806 recognizes a high-mannose form of the EGF receptor that reaches the cell surface when cells over-express the receptor. The FASEB Journal. 2005;19(7):780–782. doi: 10.1096/fj.04-1766fje. [DOI] [PubMed] [Google Scholar]
- 213.Gupta P., Han S.-Y., Holgado-Madruga M., et al. Development of an EGFRvIII specific recombinant antibody. BMC Biotechnology. 2010;10(1) doi: 10.1186/1472-6750-10-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wu M.-R., Zhang T., Gacerez A. T., Coupet T. A., DeMars L. R., Sentman C. L. B7H6-Specific bispecific T cell Engagers lead to tumor elimination and host antitumor immunity. The Journal of Immunology. 2015;194(11):5305–5311. doi: 10.4049/jimmunol.1402517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Iwahori K., Kakarla S., Velasquez M. P., et al. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Molecular Therapy. 2015;23(1):171–178. doi: 10.1038/mt.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bluemel C., Hausmann S., Fluhr P., et al. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunology, Immunotherapy. 2010;59(8):1197–1209. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Choi B. D., Gedeon P. C., Herndon J. E., et al. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunology Research. 2013;1(3):163–167. doi: 10.1158/2326-6066.CIR-13-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Choi B. D., Gedeon P. C., Sanchez-Perez L., Bigner D. D., Sampson J. H. Regulatory T cells are redirected to kill glioblastoma by an EGFRvIII-targeted bispecific antibody. Oncoimmunology. 2013;2(12) doi: 10.4161/onci.26757.e26757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Banaszczyk M., Stoczynska-Fidelus E., Winiecka-Klimek M., et al. EGFRvIII—a stable target for anti-EGFRvIII therapy. Anticancer Research. 2013;33:5343–5348. [PubMed] [Google Scholar]
- 220.Idbaih A., Aimard J., Boisselier B., et al. Epidermal growth factor receptor extracellular domain mutations in primary glioblastoma. Neuropathology and Applied Neurobiology. 2009;35(2):208–213. doi: 10.1111/j.1365-2990.2008.00977.x. [DOI] [PubMed] [Google Scholar]
- 221.Binder D. C., Ladomersky E., Lenzen A., et al. Lessons learned from rindopepimut treatment in patients with EGFRvIII-expressing glioblastoma. Translational Cancer Research. 2018;7(S4):S510–S513. doi: 10.21037/tcr.2018.03.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Schuster J., Lai R. K., Recht L. D., et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-Oncology. 2015;17(6):854–861. doi: 10.1093/neuonc/nou348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Heimberger A. B., Crotty L. E., Archer G. E., et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clinical Cancer Research. 2003;9:4247–4254. [PubMed] [Google Scholar]
- 224.Zhang Z., Song X., Tian H., et al. MicroRNA-137 inhibits growth of glioblastoma through EGFR suppression. American Journal of Translational Research. 2017;9(3):1492–1499. [PMC free article] [PubMed] [Google Scholar]
- 225.Ji Y., Sun Q., Zhang J., Hu H. MiR-615 inhibits cell proliferation, migration and invasion by targeting EGFR in human glioblastoma. Biochemical and Biophysical Research Communications. 2018;499(3):719–726. doi: 10.1016/j.bbrc.2018.03.217. [DOI] [PubMed] [Google Scholar]
- 226.Xu F., Li F., Zhang W., Jia P. Growth of glioblastoma is inhibited by miR-133-mediated EGFR suppression. Tumor Biology. 2015;36(12):9553–9558. doi: 10.1007/s13277-015-3724-4. [DOI] [PubMed] [Google Scholar]
- 227.Zhang J., Zhang J., Qiu W., et al. MicroRNA-1231 exerts a tumor suppressor role through regulating the EGFR/PI3K/AKT axis in glioma. Journal of Neuro-Oncology. 2018;139(3):547–562. doi: 10.1007/s11060-018-2903-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhou X., Ren Y., Moore L., et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Laboratory Investigation. 2010;90(2):144–155. doi: 10.1038/labinvest.2009.126. [DOI] [PubMed] [Google Scholar]
- 229.Yin D., Ogawa S., Kawamata N., et al. MiR-34a functions as a tumor suppressor modulating EGFR in glioblastoma multiforme. Oncogene. 2013;32(9):1155–1163. doi: 10.1038/onc.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gomez G. G., Volinia S., Croce C. M., et al. Suppression of microRNA-9 by mutant EGFR signaling upregulates FOXP1 to enhance glioblastoma tumorigenicity. Cancer Research. 2014;74(5):1429–1439. doi: 10.1158/0008-5472.can-13-2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dong Z.-Q., Guo Z.-Y., Xie J. The lncRNA EGFR-AS1 is linked to migration, invasion and apoptosis in glioma cells by targeting miR-133b/RACK1. Biomedicine & Pharmacotherapy. 2019;118 doi: 10.1016/j.biopha.2019.109292. [DOI] [PubMed] [Google Scholar]
- 232.Davis M. E., Zuckerman J. E., Choi C. H. J., et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464(7291):1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shi Y., Jiang Y., Cao J., et al. Boosting RNAi therapy for orthotopic glioblastoma with nontoxic brain-targeting chimaeric polymersomes. Journal of Controlled Release. 2018;292:163–171. doi: 10.1016/j.jconrel.2018.10.034. [DOI] [PubMed] [Google Scholar]
- 234.Laskin J. J., Nicholas G., Lee C., et al. Phase I/II trial of custirsen (OGX-011), an inhibitor of clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. Journal of Thoracic Oncology. 2012;7(3):579–586. doi: 10.1097/JTO.0b013e31823f459c. [DOI] [PubMed] [Google Scholar]
- 235.Tannock I. F., Fizazi K., Ivanov S., et al. Aflibercept versus placebo in combination with docetaxel and prednisone for treatment of men with metastatic castration-resistant prostate cancer (VENICE): a phase 3, double-blind randomised trial. The Lancet Oncology. 2013;14(8):760–768. doi: 10.1016/S1470-2045(13)70184-0. [DOI] [PubMed] [Google Scholar]
- 236.Pu P., Liu X., Liu A., Cui J., Zhang Y. Inhibitory effect of antisense epidermal growth factor receptor RNA on the proliferation of rat C6 glioma cells in vitro and in vivo. Journal of Neurosurgery. 2000;92(1):132–139. doi: 10.3171/jns.2000.92.1.0132. [DOI] [PubMed] [Google Scholar]
- 237.Moroni M. C., Willingham M. C., Beguinot L. EGF-R antisense RNA blocks expression of the epidermal growth factor receptor and suppresses the transforming phenotype of a human carcinoma cell line. Journal of Biological Chemistry. 1992;267:2714–2722. [PubMed] [Google Scholar]
- 238.Fan Q.-W., Weiss W. A. RNA interference against a glioma-derived allele of EGFR induces blockade at G2M. Oncogene. 2005;24(5):829–837. doi: 10.1038/sj.onc.1208227. [DOI] [PubMed] [Google Scholar]
- 239.Yamazaki H., Kijima H., Abe Y., et al. Inhibition of tumor growth by ribozyme-mediated suppression of aberrant epidermal growth factor receptor gene expression. JNCI: Journal of the National Cancer Institute. 1998;90(8):581–587. doi: 10.1093/jnci/90.8.581. [DOI] [PubMed] [Google Scholar]
- 240.Koga T., Li B., Figueroa J. M., et al. Mapping of genomic EGFRvIII deletions in glioblastoma: insight into rearrangement mechanisms and biomarker development. Neuro-Oncology. 2018;20(10):1310–1320. doi: 10.1093/neuonc/noy058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Stahlhut C., Slack F. J. Combinatorial action of microRNAs let-7 and miR-34 effectively synergizes with erlotinib to suppress non-small cell lung cancer cell proliferation. Cell Cycle. 2015;14(13):2171–2180. doi: 10.1080/15384101.2014.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Witusik-Perkowska M., Rieske P., Hułas-Bigoszewska K., et al. Glioblastoma-derived spheroid cultures as an experimental model for analysis of EGFR anomalies. Journal of Neuro-Oncology. 2011;102(3):395–407. doi: 10.1007/s11060-010-0352-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Schmitz M., Temme A., Senner V., et al. Identification of sox2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. British Journal of Cancer. 2007;96(8):1293–1301. doi: 10.1038/sj.bjc.6603696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Berezovsky A. D., Poisson L. M., Cherba D., et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia. 2014;16(3):193–206. doi: 10.1016/J.NEO.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rieske P., Azizi S. A., Augelli B., Gaughan J., Krynska B. A population of human brain parenchymal cells express markers of glial, neuronal and early neural cells and differentiate into cells of neuronal and glial lineages. European Journal of Neuroscience. 2007;25(1):31–37. doi: 10.1111/j.1460-9568.2006.05254.x. [DOI] [PubMed] [Google Scholar]
- 246.Witusik-Perkowska M., Zakrzewska M., Sikorska B., et al. Glioblastoma-derived cells in vitro unveil the spectrum of drug resistance capability–comparative study of tumour chemosensitivity in different culture systems. Biosci Rep. 2017;37 doi: 10.1042/BSR20170058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Stoczynska-Fidelus E., Piaskowski S., Bienkowski M., et al. The failure in the stabilization of glioblastoma-derived cell lines: spontaneous in vitro senescence as the main culprit. PLoS One. 2014;9(1) doi: 10.1371/journal.pone.0087136.e87136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Huang P. H., Miraldi E. R., Xu A. M., et al. Phosphotyrosine signaling analysis of site-specific mutations on EGFRvIII identifies determinants governing glioblastoma cell growth. Molecular BioSystems. 2010;6(7):1227–1237. doi: 10.1039/c001196g. [DOI] [PMC free article] [PubMed] [Google Scholar]