

Abstract
Multiple type I interferons and interferon-γ (IFN-γ) are expressed under physiological conditions and are increased by stress and infections, and in autoinflammatory and autoimmune diseases. Interferons activate the Jak–STAT signaling pathway and induce overlapping patterns of expression, called ‘interferon signatures’, of canonical interferon-stimulated genes (ISGs) encoding molecules important for antiviral responses, antigen presentation, autoimmunity and inflammation. It has now become clear that interferons also induce an ‘interferon epigenomic signature’ by activating latent enhancers and ‘bookmarking’ chromatin, thus reprogramming cell responses to environmental cues. The interferon epigenomic signature affects ISGs and other gene sets, including canonical targets of the transcription factor NF-κB that encode inflammatory molecules, and is involved in the priming of immune cells, tolerance and the training of innate immune memory. Here we review the mechanisms through which interferon signatures and interferon epigenomic signatures are generated, as well as the expression and functional consequences of these signatures in homeostasis and autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis and systemic sclerosis.
Type I interferons (IFNs) and IFN-γ, the sole type II IFN, are secreted cytokines that are important regulators of immunity and inflammation. IFNs have been implicated in the dysregulation of immune responses in autoimmune diseases and more recently in the regulation of immune responsiveness and tissue integrity under homeostatic conditions1–4. IFNs have a key role in anti-tumor immunity, and activation of IFN-γ signaling has been implicated in the efficacy of checkpoint-blockade therapy (reviewed in ref.1); although checkpoint blockade has been associated with the emergence of autoimmunity, the role of IFNs in this phenomenon is unknown. Elevated production of IFNs during infection and in autoimmune diseases results in increased expression of target genes, most typically canonical interferon-stimulated genes (ISGs), in diseased tissues and often in circulating blood cells, in a pattern of expression defined as an IFN signature. Canonical ISGs are defined herein as genes transcriptionally activated by IFNs, as identified by transcriptomic analysis of IFN-stimulated cells, and they typically are directly activated by transcription factors of the STAT family. The presence of an IFN signature is often considered a hallmark of certain autoimmune diseases, and the ‘signature genes’ are inferred to have roles in pathogenesis.
Type I IFNs and IFN-γ bind specific cell-surface receptors expressed on most cell types and signal via pathways using the protein tyrosine kinases Jaks and STATs to activate gene expression1,5,6 (Fig. 1). Binding of type I IFNs to their heterodimeric receptor IFNAR activates the receptor-associated protein tyrosine kinases JAK1 and TYK2, which is followed by phosphorylation of STAT1 and STAT2 and their association with the transcription factor IRF9, thus forming the heterotrimeric complex ISGF3 (Fig. 1). ISGF3 binds DNA elements termed interferon-sensitive response element (ISREs) (with the consensus sequence TTTCNNTTTC) and subsequently activates ISGs, including genes encoding antiviral proteins such as Mx1 and OAS, and various transcription factors, including interferon-regulatory factors (IRFs). IFN-γ binding to its receptor activates JAK1 and JAK2, and predominantly STAT1 homodimers (Fig. 1). STAT1 binds a distinct DNA element termed a gamma-activated site (GAS; consensus sequence TTCNNNGGA) and directly activates a distinct set of ISGs, notably chemokines such as CXCL10 and transcription factors including IRFs.
Fig. 1 |. IFN-induced signaling and overlapping patterns of gene expression.
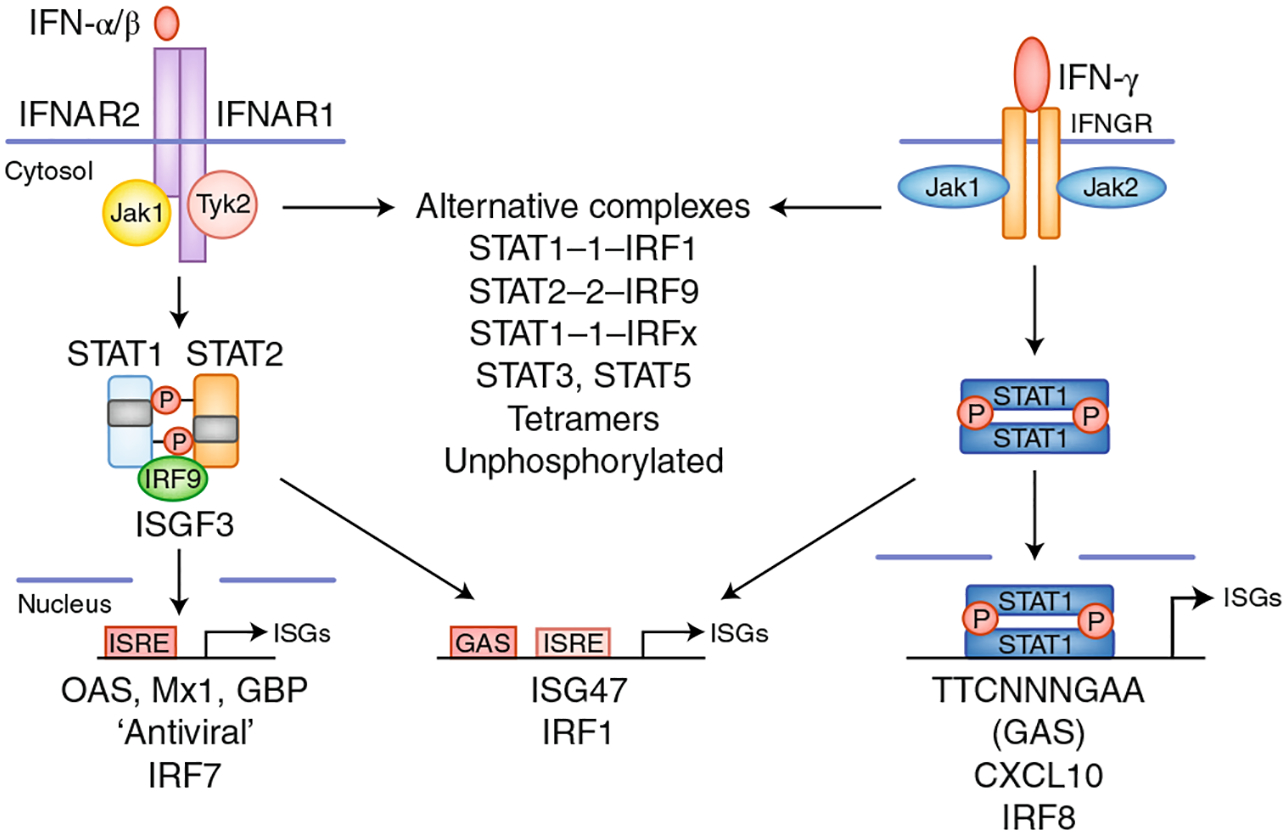
Type I and II IFNs activate distinct canonical signaling pathways leading to activation of ISGF3 and STAT1 homodimers, respectively, and downstream induction of ISRE- and GAS-driven target genes. The patterns of genes induced by type I and II IFNs overlap, partly because target genes can contain both ISRE and GAS elements, and overlap may be secondary to induction of transcription factors with shared target genes. This cascade of transcription factors, particularly IRF family members, which can interact with STATs and redirect their binding activity, can mediate the evolution of IFN signatures over time. Type I and II IFNs also activate noncanonical transcriptional complexes and additional STATs, and induce the expression of unphosphorylated STATs, thus contributing to the IFN signature.
Given their distinct core signaling pathways (Fig. 1), type I and type II IFN signatures might be predicted to be readily distinguishable, thus providing insight into which IFNs are driving gene expression and, by inference, disease pathogenesis. In practice, type I and type II IFN signatures greatly overlap and are difficult to distinguish1,3. Mechanistic explanations for such overlap include that many ISGs contain both ISREs and GAS elements and thus can be activated by both type I and II IFNs; both type I and type II IFNs can activate STAT–IRF complexes distinct from ISGF3, thus expanding the DNA binding profile, and IFNs can also activate STAT3 homodimers, STAT4, STAT5 and STAT6 in a context-dependent manner, can induce the expression and function of unphosphorylated STATs and can activate non-STAT pathways such as MAPK pathways; both type I and type II IFN induce a cascade of transcription factors, most notably IRFs, with overlapping DNA binding specificity, thus creating a dynamic IFN signature that can evolve over time; and the nature of the IFN response is context dependent, because IFN-induced gene expression is modulated by distinct environmental stimuli via signal-transduction cross-talk2 (Fig. 1).
IFN-induced signaling, gene expression and regulation of cellular responses to IFNs have recently been reviewed1,2,5,6, as have the functions of IFNs in normal immune responses (Box 1); these topics will not be further discussed herein. We describe recent developments in how IFN expression is regulated under physiological and stress conditions, and the functional consequences of IFN signatures, with a focus on their roles in maintaining tissue homeostasis and in the pathogenesis of autoimmune disease. We will cover emerging ideas about how IFN-mediated epigenomic regulation extends the concept of signatures to include chromatin accessibility, expression of non-ISGs and reprogrammed cell states, such as trained innate immunity and tolerance. We offer a detailed consideration of IFN signatures in three autoimmune diseases, including new insights obtained from single-cell genomics.
Box 1 |. Immune functions of type I and II IFNs.
Type I IFNs act on most cell types and induce an antiviral state; increase major histocompatibility complex expression, which in turn augments the lysis of infected cells; and induce the production of chemokines and cytokines that recruit immune and inflammatory cells and coordinate the immune response. Type I IFNs boost innate immunity by stimulating the differentiation and maturation of dendritic cells and the function of natural killer cells. These IFNs also augment adaptive immunity by promoting the activation and the differentiation of T and B cells and the development of immunological memory. Type I IFNs can also have suppressive effects, such as by inhibiting the responses of IL-17-producing helper T cells and inducing feedback inhibitory molecules such as IL-10 and PD-1 ligand in the setting of chronic infections.
IFN-γ also acts on most cell types; it has weaker antiviral effects than type I IFNs but potent effects on increasing major histocompatibility complex expression, antigen presentation and chemokine production, while suppressing cell proliferation. IFN-γ is the prototypic ‘macrophage-activating factor’ that augments cytokine and chemokine production, phagocytosis and intracellular killing of microbial pathogens. IFN-γ also promotes innate immunity by increasing ILC1 function. IFN-γ boosts type 1 adaptive immunity by promoting differentiation of type 1 helper T cells, generation of follicular helper T cells, B cell class switching, autoantibody production and generation of autoimmunity-associated B cells. IFN-γ can also have protective functions by suppressing responses mediated by type 2 and IL-17-producing helper T cells, inducing specialized regulatory T cells, and restraining tissue damage.
Induction of IFN expression
Type I IFNs can be induced in most cell types by microbial pathogen-associated and damage-associated molecular patterns. Type I IFNs were discovered as factors produced by virally infected cells, and many ISGs have anti-viral functions; conversely, viruses have evolved multiple mechanisms to evade IFN actions7. Nucleic acids are key inducers of type I IFNs; the recognition of nucleic acids by predominantly intracellular pattern recognition receptors (PRRs) induces inflammatory cytokines, including large amounts of type I IFNs (Fig. 2), and is essential to mounting effective immune responses to microbial pathogens8. Pathogens are constantly evolving, but nucleic acids are an intrinsic part of their structures, and many nucleic acid sensors have been described. The contribution of these sensors to the overall immune response is key and depends on their subcellular localization in the cytosol or in endosomes, and the nature of the nucleic acid that is recognized. Of the ten human Toll-like receptors (TLRs), four have a predominant endosomal localization and recognize nucleic acids: TLR3 (double-stranded RNA), TLR7 and TLR8 (single-stranded RNA) and TLR9 (double-stranded DNA)8. In addition to TLRs, cytosolic nucleic acid sensors, such as the RNA-specific RIG-I and MDA5, contribute to anti-viral responses. The response to DNA is driven by cytosolic sensors with distinct structures9, and cyclic GMP–AMP synthase (cGAS) is a key player in the induction of type I IFNs (Fig. 2). Notably, endogenous ligands that can be released from damaged tissues or apoptotic cells can activate nucleic-acid-sensing PRRs, and recognition of self nucleic acids appears to have critical roles in sterile inflammation and autoimmunity8. Important plasma-membrane receptors whose signaling induces type I IFN genes are TLR4 and the receptors for TNF family cytokines10,11. TNF activates an IRF1-dependent IFN-β-mediated autocrine loop that induces and sustains the expression of ISGs as part of the late-phase TNF response in macrophages. TLR4 senses microbial lipopolysaccharides and damage-associated molecular patterns, such as extracellular-matrix fragments generated by sterile injury. Signaling pathways used by nucleic-acid-sensing PRRs have been recently reviewed8 (Fig. 2). Their signaling pathways are often redundant, with commonalities including utilization of shared adaptors including MyD88, TRIF, MAVS and STING with downstream activation of IRF3, IRF5 and IRF7 (refs.10,12,13). These PRRs and signaling pathways are potential therapeutic targets in patients with autoimmune diseases characterized by an IFN signature.
Fig. 2 |. Nucleic acid sensors and downstream signaling pathways induce type I IFN production.
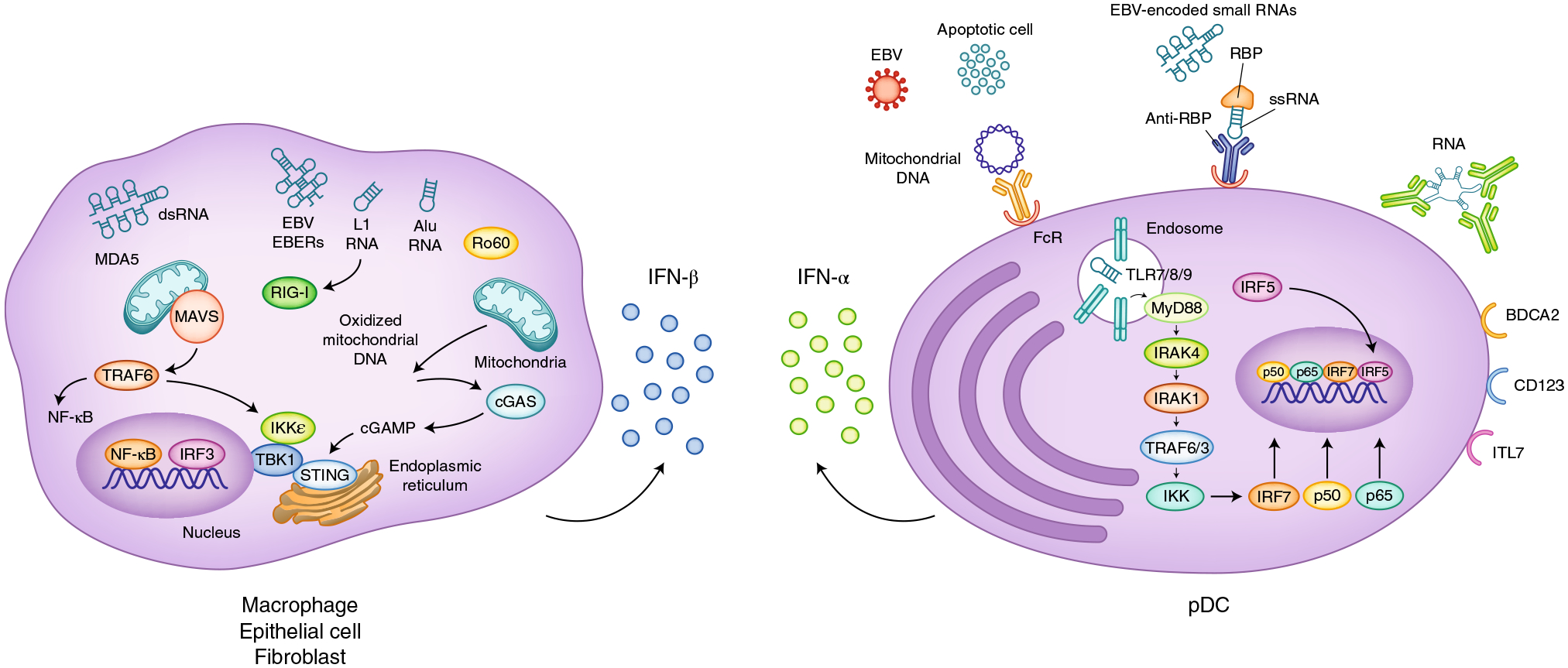
Cytosolic sensors of RNA (RIG-I and MDA5) and of DNA (cGAS) signal via the adaptors MAVS and STING, respectively, and activate the kinases TBK1 and IKKε and downstream IRF3 (left). IRF3 translocates to the nucleus, where it cooperates with NF-κB in driving Ifnb transcription in multiple cell types including macrophages, epithelial cells and fibroblasts. In pDCs that produce large amounts of IFN-α, nucleic-acid-containing immune complexes are endocytosed via Fc receptors (FcR) and delivered to endosomes, and then activate TLR sensors of RNA (TLR7, TLR8) or DNA (TLR9) (right). These endosomal TLRs signal via the adaptor MyD88 and activate IKK-kinase complexes, which in turn activate downstream transcription factors including IRF7 and the NF-κB subunits p50 and p65. IRF5 is activated by a yet-unknown mechanism. These transcription factors translocate to the nucleus and drive Ifna transcription. pDCs express cell-surface receptors including BDCA2, CD123 and ILT7, which inhibit IFN-α production through mechanisms that have not been fully clarified. EBV, Epstein–Barr virus; EBERs, EBV-encoded small RNAs; dsRNA, double-stranded RNA; ssRNA, single-stranded RNA; L1, LINE 1; TRAF, TNF receptor associated factor; RBP, RNA-binding protein.
A key issue in the biology of these nucleic acid sensors is their restricted expression in distinct cell types, which controls the nature of the response to specific nucleic acid agonists. In addition, the expression patterns of TLRs differ between mice and humans. For example, mouse TLR8 does not recognize viral RNA, and TLR7 and TLR9, whose expression is restricted to B cells and plasmacytoid dendritic cells (pDCs) in humans, are widely expressed in immune cells in mice14–16. Gaining a better understanding of the identities of the cells responsible for type I IFN production during normal immune responses and in disease situations is critical when thinking about ways to interfere with nucleic acid sensors in patients17.
Although type I IFNs, particularly IFN-β, can be produced by most cell types, production of type I IFNs by blood cells in response to viruses is for the most part dependent on nucleic acid sensing by pDCs via TLR7 and TLR9 (refs.18–20). pDCs have a plasma cell morphology and produce copious amounts of type I and type III IFNs, the main genes induced in these cells21,22. The contribution of pDCs to the overall type I IFN response in vivo in response to pathogens has been established in mouse models of viral infections19. However, because mouse pDCs can also produce large amounts of IL-12 and other proinflammatory cytokines21,23, a response not seen in human pDCs24, whether functions assigned to pDCs in mouse models can be fully extrapolated to human biology remains unclear.
IFN-γ is produced predominantly by immune cells, including innate natural killer cells, ILC1 and γδ T cells, and adaptive cells such as CD4+ type 1 helper T cells and CD8+ type 1 cytotoxic T cells (reviewed in ref.1). IFN-γ is induced primarily in response to stimulation of antigen receptors and by cytokines such as IL-12 and IL-18, which activate the transcription factors STAT4 and NF-κB, respectively. Given the different stimuli that induce their expression, and the distinct cell types that produce type I and II IFNs, their contributions to the IFN signature vary depending on the nature and timing of immune and inflammatory responses.
Homeostatic expression and function of IFNs
Both type I and II IFNs are expressed basally under physiological conditions and contribute to tissue homeostasis and ‘readiness’ to fight infection1,2. Basal IFN-β expression is maintained at least in part by the reactivity of cells at barrier tissues to commensal micro-flora, thus resulting in tonic IFN signaling that maintains expression of STAT1 and IRF9, and poises cells for robust responses to environmental challenges25. Basal IFN-β appears to have systemic effects and can act at a distance; for example, IFN induced by the gut microbiome maintains immune readiness at other sites such as the spleen26,27. In contrast, basal IFN-γ produced under homeostatic conditions in select tissues, such as lymph node lymphatics, uterine blood vessels, meninges, bone and skin, acts locally. The local functions of low concentrations of IFN-γ include remodeling of lymphatic and uterine arterial blood vessels, survival and connectivity of neurons important for social behavior, regulation of bone mass and immune cell trafficking in the skin28–32. The cell types and stimuli that maintain basal IFN-γ expression are not well understood but include tissue-resident memory T cells and stimulation by commensal microbes on barrier surfaces33,34. Overall, low-level basal IFN signatures have important physiological functions; although such signatures can be difficult to measure, they are clearly revealed when components of IFN signaling pathways, such as IFNAR or STAT1, are deficient25.
Genetic modulators of IFN expression
Although IFN signatures are induced by environmental cues, their amplitudes, time courses and patterns of gene expression are modulated by genetic factors. Complex multigenic autoimmune diseases exhibit a highly significant enrichment of allelic variation at gene loci encoding components of IFN–Jak–STAT–IRF signaling pathways35, in agreement with genetic modulation of IFN production36–38. Monogenic disorders, which are typically diagnosed in childhood and characterized by a type I IFN signature, have features of systemic autoimmunity and inflammation and are described as interferonopathies39,40. Those diseases are typically characterized by mutations that alter the regulation, degradation or sensing of endogenous RNA or DNA (described below). In contrast to the mutations associated with interferonopathies, which map to cytosolic pathways that induce type I IFN, single-nucleotide polymorphisms in IRF5, which encodes a protein primarily involved in signaling downstream of TLRs, are among the strongest genetic risk factors for systemic lupus erythematosus (SLE) and other systemic autoimmune diseases4,37. Dissecting the consequences of the genetic contributions to the risk of developing a systemic autoimmune disease becomes exceedingly complex, given that a heterozygous rare variant associated with a monogenic interferonopathy can amplify the risks conferred by common genetic variants41.
IFN gene expression signatures
IFN signatures commonly refer to sets of genes (ISGs) that can be upregulated by type I, II or III IFNs and were originally defined in in vitro culture systems3,42. Type I IFNs consist of 13 IFN-α subtypes, IFN-β, IFN-ω, IFN-κ and IFN-ε. There are four type III IFNs: IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B) and IFN-λ4 (ref.43). Structural studies of IFN–IFNAR complexes have revealed that type I IFNs can bind their common receptor IFNAR with different topologies and affinities, thus affecting downstream signaling and gene expression profiles44,45. Furthermore, structural studies of the IFN-γ–IFNγR complex have provided insights into signaling mechanisms that have enabled the development of partial agonists with differential gene expression profiles. These molecules include agonists that dissociate the induction of immunostimulatory and immunosuppressive genes, thereby paving the way to a new therapeutic strategy for boosting immune responses46. Whether the existence of multiple IFNs is due to strong evolutionary pressures or whether each IFN has distinct effects and can differentially modulate immune functions remains unclear. Although the three IFN types signal via distinct receptors43 (Fig. 1), the genes or signatures controlled by these IFNs overlap substantially. Differences between type I and II signatures have been described in mice47 and must be validated in humans. Being able to clearly define the differences between the type I and type II IFN signatures would clarify which IFNs and cell types are involved in disease situations and may aid in selecting the appropriate therapy. Distinctions between type I and type II signatures have been made in Sjogren syndrome48, lupus49,50 and infectious diseases51; determining how various therapeutic interventions that target the IFN pathway in patients affect these distinct signatures will be interesting.
IFN-induced epigenomic signatures
The epigenome or ‘epigenomic landscape’ is the genome-wide pattern of histone and DNA modifications, chromatin conformation and transcription-factor binding that determines cell-specific gene expression and responsiveness to environmental stimuli52. The epigenome regulates the access of signal-activated transcription factors and the general transcriptional machinery to gene-regulatory elements. The epigenomic landscape is shaped during development and is remodeled in response to environmental cues by transcription factors, histone- and DNA-modifying enzymes, nucleosome-remodeling complexes and factors that organize the three-dimensional structure of the genome. IFNs induce extensive remodeling of the epigenome, including the creation of new enhancers (termed latent enhancers), the disassembly of enhancers and the modulation of histone marks that regulate chromatin accessibility and the functions of enhancers and promoters53–61. This chromatin remodeling is mediated by IFN-activated STATs and by de novo–induced transcription factors, such as IRFs, which bind gene-regulatory elements and recruit chromatin-remodeling enzymes. Chromatin remodeling at ISG loci is associated with gene transcription, including sustained transcription at time points after IFN-induced proximal signaling has subsided.
IFN-induced transcription-factor binding, chromatin remodeling and changes in histone marks also occur at regulatory elements of non-ISGs (defined herein as genes whose transcription is not altered by IFN stimulation alone), including canonical targets of NF-κB that encode inflammatory molecules activated by prototypical inflammatory factors such as lipopolysaccharide53,57,59,61. In the case of IFN-γ, remodeling at non-ISGs is mediated in part by interactions of STAT1 with IRF1 and expansion of the STAT1 genomic binding profile to IRF-binding sites59, and by diminished occupancy of enhancers by IFN-γ-repressed transcription factors55 (reviewed in detail in ref.1). Stimulation with IFN-γ results in pervasive genome-wide changes in histone acetylation and chromatin accessibility at promoters and enhancers. In contrast, stimulation with type I IFNs increases the amount of trimethylated histone H3 Lys 4 (H3K4me3), a histone mark that promotes transcription at promoters of genes encoding inflammatory mediators. Furthermore, in an inflammatory context, type I IFNs, in cooperation with TNF, induce the tandem occupancy of regulatory elements at non-ISGs that encode inflammatory mediators by IRFs and NF-κB, a process associated with increased histone marks that facilitate transcription and mediate enhanced responsiveness to subsequent environmental challenges57 (Fig. 3). The binding of transcription factors and the presence of altered chromatin states that are stable over time can serve as ‘bookmarks’62 that mark genomic locations and mediate their responsiveness to subsequent environmental stimuli. Fully defining the functional role of such bookmarking of non-ISGs by IFNs in the absence of notable changes in transcription is an important area for future research.
Fig. 3 |. epigenomic regulation links type I IFN signaling to induction of inflammatory non-ISgs.
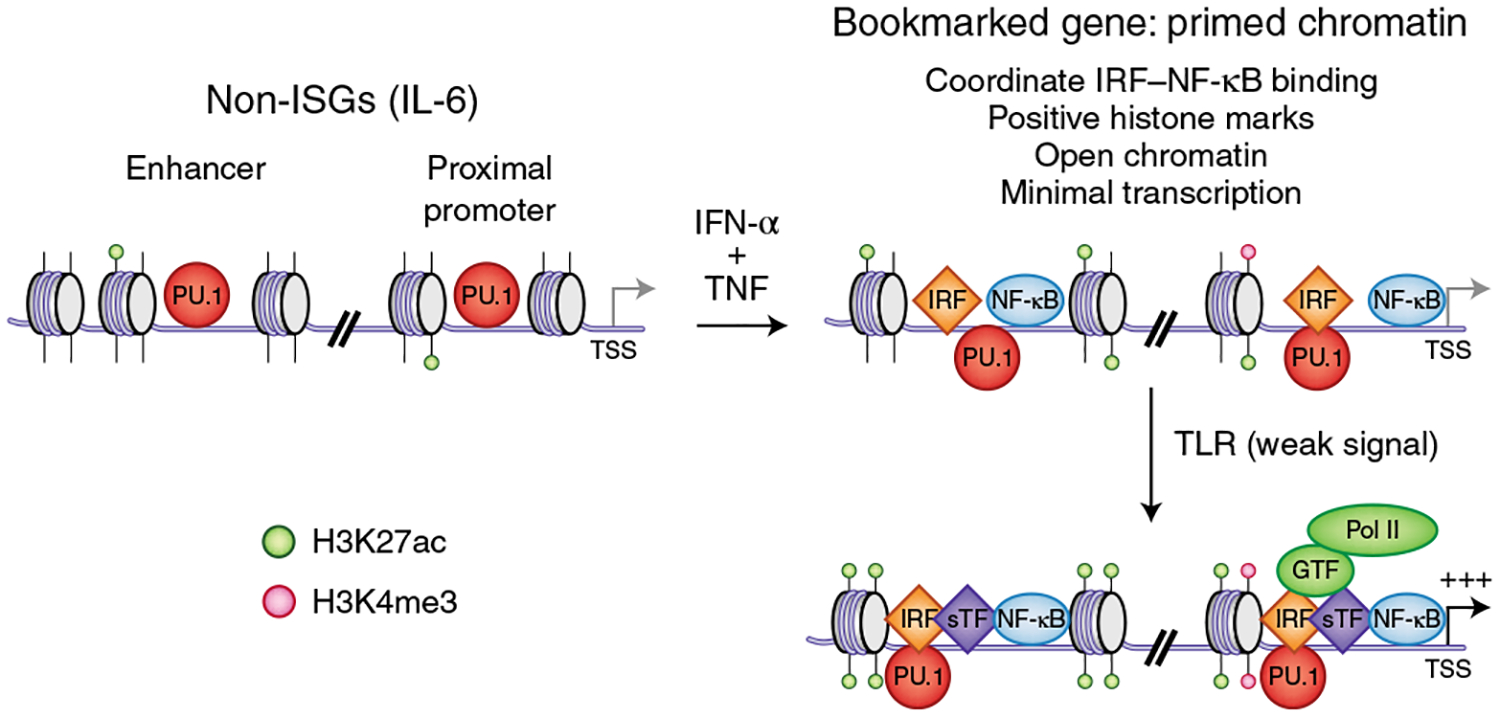
Stimulation of macrophages with TNF leads to transient expression of TNF-target genes encoding inflammatory mediators, such as IL6 and TNF, followed by a state of tolerance in which signaling responses to TLR ligands are strongly suppressed, and chromatin is not activated (not depicted). In contrast to tolerization with TNF alone, co-stimulation with TNF plus IFN-α results in coordinate binding of IRFs and NF-κB, increased chromatin accessibility and increased positive histone marks, most notably H3K4me3 (top right). These genes are thereby bookmarked with primed chromatin and subsequently exhibit a robust transcriptional response even to very weak proximal TLR-induced signals, such as those in TNF-tolerized cells on TLR stimulation (bottom right). TSS, transcription start site; ac, acetyl; Pol, polymerase.
Consequences of epigenomic remodeling by IFNs
IFN-induced epigenomic changes can last for days to weeks and thus persist beyond the period of IFN expression and upstream Jak–STAT signaling55–57,60,63. Such persistence can confer transcriptional memory and sustain the expression of ISGs. For non-ISGs, although IFN-induced epigenomic changes are often transcriptionally silent, the current model posits that such bookmarking alters how these genes respond to subsequent stimulation. For example, IFN-γ-induced marking of TNF and IL6 loci with IRF1–STAT1 and histone acetylation, or IFN-β-induced increases in H3K4me3, result in super-induction of the response to subsequent stimulation by TLR ligands, a phenomenon termed priming57,59. Thus, chromatin-mediated mechanisms link the IFNs to the induction of inflammatory genes beyond canonical ISGs; this linkage provides a potential explanation for the downregulation of genes encoding inflammatory mediators by the Jak inhibitors. IFN-induced chromatin changes can also make genes refractory to stimulation by suppressive factors such as glucocorticoids, IL-4 and IL-10, and resistant to tolerization53,57,58,60 (reviewed in refs.1,64). There is great interest in a phenomenon termed innate immune training, in which prior exposure to inflammation or infection results in a stronger secondary innate immune response that can be protective against an infectious challenge but also can result in increased inflammation and tissue damage65. In vitro, training can be induced with low-grade stimulation of macrophages with factors such as β-glucans, which initiate signaling mediated by immunoreceptor tyrosine-based activation motifs; in vivo, training is induced by various processes that elicit inflammation, including vaccination, bacillus Calmette–Guérin infection and high-fat-diet feeding. Innate immune training is mediated by epigenetic chromatin-based mechanisms65 that are similar to, and partially overlap with, those induced by IFNs. Emerging evidence indicates that IFN-γ signaling is important for the training of hematopoietic progenitors after induction with bacillus Calmette–Guérin66 and the viral-infection-induced training of alveolar macrophages67. Thus, IFNs are important for both priming and training, both of which are related by the utilization of overlapping epigenomic mechanisms; priming typically refers to a predominantly IFN-driven process, whereas training typically refers to phenomena driven by pathogens or pathogen-associated molecular patterns, to which IFNs can contribute.
The ability of an IFN epigenomic signature to augment and sustain immune responses suggests a potential role in the pathogenesis of autoimmune diseases. Indeed, IFN-induced epigenomic signatures, including increased histone acetylation, H3K4me3 modifications at the promoters of highly expressed genes, including ISGs, and evidence of resistance to endotoxin tolerance have been observed in monocytes from people with SLE57,68,69. These findings can be potentially explained on the basis of elevated IRF1 expression and occupancy of regulatory elements. Accordingly, assay for transposase-accessible chromatin using sequencing (ATAC–seq) analysis has revealed similarities in genome-wide chromatin-accessibility profiles between monocytes from people with SLE and in vitro IFN-primed monocytes that are resistant to tolerance. Digital footprinting under ATAC–seq peaks in these cells has revealed similar enrichment of tandem IRF1- and NF-κB-binding sites57. Although the effects of IFNs on DNA methylation are not well understood, several studies have reported altered DNA-methylation patterns, including hypomethylation of ISGs in SLE blood cells, which may augment gene expression and thus an IFN signature (reviewed in ref.70). These reports support further investigation of epigenomic signatures and the relationships among IFN signaling, chromatin changes and DNA methylation in autoimmune disease cells.
IFN signatures in SLE
The functions of IFNs and ISGs in normal immune responses have recently been reviewed1,2 (Box 1). Here we focus on three human autoimmune diseases in which IFN signatures have been extensively investigated, and we describe recent advances in understanding of the generation of an IFN signature, its utility as a biomarker and its role in pathogenesis. A contribution of IFN to the pathogenesis of SLE, the prototypic systemic autoimmune disease, was first suggested by studies in which induction of type I IFN in the NZB/NZW F1 mouse model accelerated disease71. Elevated expression of IFN in the blood in patients with SLE72,73 was corroborated by the elevated expression of hundreds of IFN-induced genes in microarray analyses of blood mononuclear cells from people with SLE, thus revealing that the IFN signature is the most prevalent molecular pathway activated in SLE74–77. The peripheral IFN signature is most consistent with induction by type I IFN78,79, although IFN-γ may contribute to the expression of some genes, for example, the gene encoding the chemokine CXCL10/IP-10 (refs.50,74,80; reviewed in ref.1).
The major cellular source of type I IFN in SLE is presumed to be pDCs. The depletion of these cells in mouse models of lupus leads to diminished disease81,82, and the administration of anti-BDCA2, a specific inhibitor of pDC function, induces a significant but partial decrease in the type I IFN signature in the blood in people with SLE83. IFN-α and the type I IFN–induced protein MxA have been found to be markedly decreased in the skin lesions in patients with SLE with cutaneous manifestations of the disease who were treated with anti-BDCA2 (ref.83), a finding that correlated with improved clinical scores, thus suggesting that the contribution of pDCs to SLE might be more dominant in the skin. Virtually all immune cell populations studied in the blood in people with SLE show a type I IFN response, and monocytes show a particularly prominent signature84. Synovial tissue collected from people with SLE with arthritis as well as renal biopsy tissue, particularly from people with class IV lupus nephritis, show strong type I IFN–induced gene expression85,86. Single-cell RNA sequencing (RNA-seq) of cells isolated from renal biopsies of people with SLE has detected an IFN signature in subsets (computationally defined cell clusters) of essentially all infiltrating leukocytes and tissue cells. This signature is especially prominent in subsets of B cells and CD4+ T cells; although target genes of type I IFN are expressed, the presence of natural killer cells and CD8+ T cells that can produce IFN-γ, and of autoimmunity-associated T-bet+ B cells, supports a contribution of IFN-γ to the IFN signatures87,88. Although cross-sectional studies of patients with lupus suggest that activation of the IFN pathway is associated with disease activity, type I IFN–stimulated gene expression is observed in cells from both inactive and active patients, and is often quite stable over time.
Characterization of the drivers and molecular pathways responsible for production of type I IFN in SLE has focused on both endosomal TLRs and cytosolic sensors (Fig. 2). Early studies demonstrated that the induction of IFN-α by immune complexes composed of necrotic or apoptotic cellular material, nucleic acids and autoantibodies targeting RNA-binding proteins, such as Ro or Sm, is strongly associated with an IFN signature89–91. These data, along with data from genome-wide association studies implicating IRF5 downstream of TLR7, and experiments using oligonucleotide inhibitors of TLR7, suggest that access of RNA-containing immune complexes to TLR7 is an important driver of the IFN pathway in SLE92. Neutrophil extracellular traps, DNA-containing microparticles and circulating mitochondrial DNA are additional candidate stimuli of endosomal TLRs93–97. The discovery of mutations in genes encoding cytosolic nucleic acid receptors for RNA and DNA and regulators of endogenous nucleic acids in interferonopathies associated with a type I IFN signature has drawn attention to these receptors for further study in SLE. Oxidized mitochondrial DNA and genome-derived DNA and RNA enriched in endogenous retroelement sequences have been proposed as candidate stimuli for cytosolic sensors and STING-dependent induction of type I IFN98,99. Additional investigations will be required to define the relative contributions of the endosomal TLR pathways and the cytosolic nucleic-acid-driven mechanisms in spontaneous SLE, as well as in the rare patients with SLE due to monogenic mutations.
The protean functional effects of type I IFN on the immune system4 are consistent with many of the features of altered immune-system function that characterize people with SLE. Augmented antigen-presenting-cell function, B cell differentiation, modulation of effector T cell function and promotion of inflammation by IFN-induced chemokines can be attributed at least partly to the induction of IFN-regulated genes. A major role for type I IFNs and the sustained activation of the type I IFN pathway in the pathogenesis of SLE, including its manifestations in the skin, arthritis, nephritis and premature atherosclerosis, is supported by extensive data in both mouse systems and human patients, and provides a rationale for drug-development programs targeting the upstream drivers and producers of type I IFN, the components of both endosomal TLR and cytosolic nucleic-acid-sensing pathways (Fig. 2). Potential stimuli for type I IFN production include RNA- or DNA-containing immune complexes, cytosolic nucleic acids enriched in endogenous retroelement sequences or mitochondrial DNA. These might be inhibited by RNases or DNases that degrade nucleic acids, or by inhibitors that suppress reverse transcription of retroelements. pDCs that robustly produce type I IFNs could be inhibited by targeting cell-surface receptors that are expressed by pDCs or that regulate pDC production of IFN, including BDCA2, CD123 or ILT7. Therapeutics targeting TLR7, TLR8 and possibly TLR9 or IRAK4 could decrease production of IFN by pDCs, and inhibitors of cGAS or STING might decrease the production of type I IFN triggered by cytosolic nucleic acids. Therapeutic targeting of IFN-α, IFNAR or the signaling components downstream of IFNAR (Fig. 1) has already shown promise. One approach is the blockade of IFNAR with a monoclonal antibody specific for IFNAR1, anifrolumab100, which significantly decreases the expression of type I IFN–induced transcripts101, but promising phase II studies have not been corroborated by phase III trials. The inhibition of gene transcription triggered by cytokines and IFNs and mediated by the Jak–STAT pathway is currently under study through use of Jak inhibitors102,103, and IFN-γ is also being targeted1,80. Assessment of the IFN signature has proven useful in the context of clinical-trial design, and its application is being extended to assessment of patients with pre-clinical SLE104,105. The type I IFN signature is being refined on the basis of the inclusion of transcripts representing distinct gene clusters derived from analysis of RNA-seq data106. Identifying those type I IFN–induced transcripts that fluctuate over time, either before or concurrently with flare-ups in disease activity, may provide new insights into the relevant molecular pathways driving and sustaining immune activation and clinical disease.
IFN signatures in rheumatoid arthritis
The expression of IFN-induced genes in inflamed rheumatoid arthritis (RA) synovium was described more than three decades ago107, and multiple studies including recent RNA-seq analysis have established the expression of IFN signatures in diseased synovial (joint) tissue108,109. The synovial IFN signature is sensitive to Jak inhibitors110, thus suggesting that the efficacy of these compounds may be related, at least in part, to inhibition of IFN signaling. Macrophages in the RA synovial fluid show a strong IFN-γ signature, elevated IRF1 and increased expression of genes associated with IFN-γ-induced latent enhancers55. These findings implicating the regulation of enhancers in RA synovial macrophages support further investigation into the IFN epigenomic signatures in RA synovial cells.
Single-cell RNA-seq and mass cytometry have identified four subsets of macrophages, four subsets of fibroblasts, four subsets of B cells and six subsets of T cells in the inflamed RA synovium111,112. In addition to identifying potentially novel pathogenic cell types, these studies show an IFN signature in one subset of HLAhi sublining fibroblasts that were proposed to be pathogenic on the basis of cytokine production, in two subsets of macrophages (one of which was defined by expression of IL1B and HBEGF and considered inflammatory) and in three subsets of B cells, including CD11c+ T-bet+ B cells, which produce high amounts of autoantibodies111. Synovial CD8+ T cells express IFNG at a higher frequency than CD4+ T cells, thus revealing a long-elusive source of synovial IFN-γ (ref.113) and a potentially pathogenic cell type that drives synovial IFN signatures. Characterization of the pathogenic functions of IFN-signature-expressing synovial cells is an important area of future research.
IFN signatures in systemic sclerosis
Systemic sclerosis (SSc) is a multisystem, fibrosing disorder in which vasculopathy, autoimmunity and inflammation lead to diverse clinical manifestations114. SSc is heterogeneous and life threatening, and is associated with the highest degree of morbidity and mortality among the rheumatic diseases, with a 10-year mortality rate of 23–45% (ref.115). Although evidence has linked the inflammatory response observed in patients and pro-fibrotic events, the roles of IFN and of key pathogenic cell types are only starting to emerge. The presence of an IFN signature and elevated expression of CXCL4, which is produced by pDCs in people with SSc116–118, suggested the possibility that pDCs may have a pathogenic role in skin disease. In agreement with this idea, pDCs infiltrate the skin after injury119,120, in ‘interface dermatitis’ skin inflammatory diseases121, and in a mouse model of the stiff skin syndrome122. pDCs directly promote skin fibrosis, and their depletion prevents and even reverts the fibrotic process in a mouse model of SSc123. Type I IFNs produced by pDCs might play a role, potentially by exacerbating the tissue-repair process or by inducing a cascade of IFN-related pro-fibrotic inflammatory events, as in other systems124. IFN produced by non-immune cells, such as keratinocytes, may contribute to the overall IFN response in the skin in people with SSc, similarly to cutaneous SLE125. One reason why mouse models of SSc only partially mimic the human disease is that human skin is very different from mouse skin. Although depleting pDCs can prevent fibrosis in mice, the pathogenic contributions of type I IFN or pDCs to SSc pathogenesis can be determined only in clinical trials using drugs that block IFN responses or deplete or attenuate pDC function.
IFN signatures in autoinflammatory diseases
Genetic analysis of children with features of inflammation refractory to immunosuppressive therapy has revealed novel single-gene mutations that affect the induction or regulation of type I or type II IFN and result in a variety of severe clinical syndromes. Insights gleaned from the identification of the molecular pathways affected by those mutations have the potential to inform understanding of disease mechanisms operative in polygenic complex systemic autoimmune and inflammatory diseases40,126. Aicardi–Goutières syndrome (AGS), chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome (CANDLE) and STING-associated vasculopathy with onset in infancy (SAVI) are among the so-called interferonopathies characterized by sustained high levels of type I IFN, particularly IFN-α, and an IFN signature39,40,126. AGS is based on mutations in TREX1, SAMHD1, RNASEH2A, RNASEH2B, RNASEH2C or ADAR1, which encode proteins that degrade or edit nucleic acids, or in IFIH1, which encodes a cytosolic RNA sensor. CANDLE is associated with mutations in genes encoding proteasome subunits127, and SAVI is due to a gain-of-function mutation in TMEM173, which encodes STING and promotes the transmission of signals initiated by cytosolic DNA128,129.
AGS presents with a distinct clinical picture with organ involvement different from that in SLE, possibly because of recognition of nucleic acids by cytosolic sensors in AGS, rather than recognition of nucleic-acid-containing immune complexes by endosomal TLRs in SLE. Although each of the gene mutations responsible for AGS has a distinct mechanism, the common consequence of the mutations is accumulation of cytosolic RNA, DNA or RNA–DNA hybrids that stimulate either cGAS or the RNA sensors RIG-I or MDA5. Mitochondrial DNA or RNA and cytosolic nucleic acids enriched in endogenous retroelement sequences have been proposed as candidate endogenous inducers of the type I IFN signature93,94,99,130. In contrast to AGS, CANDLE and SAVI have been described as ‘auto-inflammatory’ interferonopathies and are generally independent of the pathologic effects of autoantibodies40. The IFN signature in CANDLE is independent of STING and MAVS, thus suggesting that intracellular DNA and RNA may not be the relevant stimuli. Augmented responsiveness of CANDLE cells to IFN-γ has also been reported40. Clinical manifestations of CANDLE include fevers, nodular skin lesions with underlying panniculitis or lipodystrophy and arthritis with prominent joint contractures. The IFN signature in SAVI is likely to be driven by STING-mediated signaling and IRF3 translocation to the nucleus. Like people with CANDLE, people with SAVI have fever and rash, and often have interstitial lung disease and fibrosis. Although the current state of investigation does not allow for clear linkage of the distinct molecular pathways underlying each of these monogenic disorders to specific clinical manifestations of multigenic complex diseases, such as SLE, it is reasonable to propose that in addition to the immunopathologic effects of type I IFN, the pathogenesis of SLE may also require the production of autoantibodies and immune complexes that contribute to organ inflammation.
AGS, CANDLE or SAVI have been refractory to most of the immunosuppressive medications used in SLE, but promising data are emerging from studies of Jak inhibition131–133. The Jak inhibitor baricitinib results in particularly striking responses in patients with CANDLE133. A distinct and novel approach to therapy for patients with AGS is administration of a cocktail of reverse-transcriptase inhibitors, on the basis of the concept that reverse-transcribed DNA derived from endogenous retroelements may be enriched among the stimulatory nucleic acids driving the type I IFN pathway in those patients. The type I IFN signature shows an impressive transient inhibition in patients with AGS receiving a 12-month course of those agents134.
Hemophagocytic lymphohistiocytosis (HLH) is a potentially catastrophic syndrome affecting children and characterized by cytopenias, a sepsis-like presentation, hemophagocytosis by macrophages and abundant cytokine production by expanded CD8+ T cells135. A familial form is associated with single gene mutations that impair cytotoxic T cell function and appear to result in decreased clearance of APCs and infected cells, and the associated IFN-γ production that contributes to the characteristic macrophage activation and phagocytic function136,137. The effect of IFN-γ on macrophage function is reflected in the high expression of the IFN-γ-induced chemokines CXCL10 and CXCL9 and the IFN-γ-induced protein neopterin. A role for IFNs and IRFs in HLH and the related macrophage-activation syndrome is supported by findings in animal models138–141. Therapeutic targeting of IFN-γ with a specific monoclonal antibody (emapalumab) reverses clinical and laboratory manifestations of HLH in some patients142, and emapalumab has recently been approved by the US Food and Drug Administration for the treatment of patients with primary HLH with refractory, recurrent or progressive disease143.
Concluding remarks
The presence of IFN signatures at homeostasis and in autoimmune diseases is well established, and the characterization of IFN epigenomic signatures is emerging. Many important questions remain for future research. Although we have highlighted the potential pathogenic roles of IFN signatures in autoimmune diseases, both type I and II IFNs induce negative feedback and inhibitory pathways and have been implicated in suppressive effects, such as immune cell exhaustion1,2. Thus, similarly to infections and anti-tumor immune responses1,2, IFNs are likely to mediate both pathogenic and protective mechanisms in autoimmune diseases, and the effects of therapeutic IFN blockade are likely to be context and disease specific. Exogenous type I IFNs exhibit therapeutic efficacy in the autoimmune disease multiple sclerosis and are suppressive in the experimental autoimmune encephalitis animal model2,44. Given the panoply of cytokines that activate the Jak–STAT pathway, sorting out which IFNs and other cytokines contribute to gene expression patterns and disease pathogenesis will be important. Targeting therapies against individual cytokines will be helpful in this regard; defining STAT-specific gene expression signatures may also have great utility, especially if protective signatures can be identified (for example, genes mediating STAT3-dependent suppression of inflammatory responses in myeloid cells) and therapeutically augmented. The era of single-cell genomics is just beginning to be applied to the study of autoimmune diseases87,88,111,112 and holds great promise for identifying IFN-responsive cell types in vivo and defining their pathogenic roles and responses to therapy. In addition to autoimmune diseases and infections, IFN signatures may contribute to other pathologies, such as the increased inflammation and tissue dysfunction associated with aging, which is responsive to Jak inhibitors144.
Expanding investigations into IFN epigenomic signatures and the analysis of individual cell types will be important. Epigenomic analysis has the potential to provide insights into mechanisms of gene expression, identify chromatin- and DNA-modifying enzymes as new therapeutic targets, and reveal the effects of disease-associated allelic variants on the epigenome. Epigenomics-enabled measurement of the effects of environmental stimuli, which can be relatively stable over time, may provide a novel biomarker of environmental exposure and disease activity and, together with genetic analysis, can yield insights into the interplay between genetics and environment in disease pathogenesis.
Acknowledgements
This work was supported by grants from the NIH (to F.J.B. and L.B.I.) and from the Scleroderma Research Foundation (to F.J.B.).
Footnotes
Competing interests
F.J.B. has been acting as a consultant for Astra Zeneca, Janssen and EMD Serono but has no other conflicts. M.K.C. has served as a consultant for Astra Zeneca, Bristol-Myers Squibb, Janssen, Lilly and Novartis. L.B.I. serves as a consultant for Lilly but does not accept any personal compensation.
Peer review information: Ioana Visan was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ivashkiv LB IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol 18, 545–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ivashkiv LB & Donlin LT Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banchereau R, Cepika AM, Banchereau J & Pascual V Understanding human autoimmunity and autoinflammation through transcriptomics. Annu. Rev. Immunol 35, 337–370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crow MK, Olferiev M & Kirou KA Type I interferons in autoimmune disease. Annu. Rev. Pathol 14, 369–393 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Stark GR & Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity 36, 503–514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villarino AV, Kanno Y & O’Shea JJ Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol 18, 374–384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fensterl V, Chattopadhyay S & Sen GC No love lost between viruses and interferons. Annu. Rev. Virol 2, 549–572 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrat FJ, Elkon KB & Fitzgerald KA Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med 67, 323–336 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Dempsey A & Bowie AG Innate immune recognition of DNA: a recent history. Virology 479–480, 146–152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawai T & Akira S Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Yarilina A, Park-Min KH, Antoniv T, Hu X & Ivashkiv LB TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol 9, 378–387 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Guiducci C et al. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J. Exp. Med 205, 315–322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honda K et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Forsbach A et al. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol 180, 3729–3738 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Guiducci C et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med 210, 2903–2919 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janke M et al. Selective and direct activation of human neutrophils but not eosinophils by Toll-like receptor 8. J. Allergy Clin. Immunol 123, 1026–1033 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Kanzler H, Barrat FJ, Hessel EM & Coffman RL Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med 13, 552–559 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Guiducci C, Coffman RL & Barrat FJ Signalling pathways leading to IFN-alpha production in human plasmacytoid dendritic cell and the possible use of agonists or antagonists of TLR7 and TLR9 in clinical indications. J. Intern. Med 265, 43–57 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Reizis B Plasmacytoid dendritic cells: development, regulation, and function. Immunity 50, 37–50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swiecki M & Colonna M The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol 15, 471–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duramad O et al. IL-10 regulates plasmacytoid dendritic cell response to CpG-containing immunostimulatory sequences. Blood 102, 4487–4492 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Ito T, Kanzler H, Duramad O, Cao W & Liu YJ Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 107, 2423–2431 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Boonstra A et al. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J. Exp. Med 197, 101–109 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedberg JW et al. Combination immunotherapy with a CpG oligonucleotide (1018 ISS) and rituximab in patients with non-Hodgkin lymphoma: increased interferon-alpha/beta-inducible gene expression, without significant toxicity. Blood 105, 489–495 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Gough DJ, Messina NL, Clarke CJ, Johnstone RW & Levy DE Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36, 166–174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abt MC et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganal SC et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37, 171–186 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Filiano AJ et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 535, 425–429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao Y et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J. Clin. Invest 117, 122–132 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kataru RP et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity 34, 96–107 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Liu W et al. dNK derived IFN-γ mediates VSMC migration and apoptosis via the induction of lncRNA MEG3: a role in uterovascular transformation. Placenta 50, 32–39 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Nirschl CJ et al. IFNγ-dependent tissue-immune homeostasis is co-opted in the tumor microenvironment. Cell 170, 127–141.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison OJ et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science 363, eaat6280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanoue T et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langefeld CD et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun 8, 16021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niewold TB et al. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 58, 2481–2487 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sigurdsson S et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am. J. Hum. Genet 76, 528–537 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crow YJ Type I interferonopathies: a novel set of inborn errors of immunity. Ann. NY Acad. Sci 1238, 91–98 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Kim H, Sanchez GA & Goldbach-Mansky R Insights from Mendelian interferonopathies: comparison of CANDLE, SAVI with AGS, monogenic lupus. J. Mol. Med. (Berl.) 94, 1111–1127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Almlöf JC et al. Whole-genome sequencing identifies complex contributions to genetic risk by variants in genes causing monogenic systemic lupus erythematosus. Hum. Genet 138, 141–150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rusinova I et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wack A, Terczyńska-Dyla E & Hartmann R Guarding the frontiers: the biology of type III interferons. Nat. Immunol 16, 802–809 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ng CT, Mendoza JL, Garcia KC & Oldstone MB Alpha and beta type 1 interferon signaling: passage for diverse biologic outcomes. Cell 164, 349–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas C et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 146, 621–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mendoza JL et al. Structure of the IFNγ receptor complex guides design of biased agonists. Nature 567, 56–60 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mostafavi S et al. Parsing the interferon transcriptional network and its disease associations. Cell 164, 564–578 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hall JC et al. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc. Natl Acad. Sci. USA 109, 17609–17614 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banchereau R et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165, 551–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiche L et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 66, 1583–1595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suarez NM et al. Superiority of transcriptional profiling over procalcitonin for distinguishing bacterial from viral lower respiratory tract infections in hospitalized adults. J. Infect. Dis 212, 213–222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smale ST, Tarakhovsky A & Natoli G Chromatin contributions to the regulation of innate immunity. Annu. Rev. Immunol 32, 489–511 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Chen J & Ivashkiv LB IFN-γ abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proc. Natl Acad. Sci. USA 107, 19438–19443 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamada R et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl Acad. Sci. USA 115, e9162–e9171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang K et al. Interferon-γ represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor MAF. Immunity 47, 235–250.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ostuni R et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Park SH et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol 18, 1104–1116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piccolo V et al. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat. Immunol 18, 530–540 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qiao Y et al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity 39, 454–469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qiao Y, Kang K, Giannopoulou E, Fang C & Ivashkiv LB IFN-γ induces histone 3 lysine 27 trimethylation in a small subset of promoters to stably silence gene expression in human macrophages. Cell Rep. 16, 3121–3129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seeley JJ et al. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. Nature 559, 114–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daniel B et al. The nuclear receptor PPARγ controls progressive macrophage polarization as a ligand-insensitive epigenomic ratchet of transcriptional memory. Immunity 49, 615–626.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monticelli S & Natoli G Short-term memory of danger signals and environmental stimuli in immune cells. Nat. Immunol 14, 777–784 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Biswas SK & Lopez-Collazo E Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–487 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Netea MG et al. Trained immunity: a program of innate immune memory in health and disease. Science 352, aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaufmann E et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172, 176–190.e19 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Yao Y et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell 175, 1634–1650.e1617 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Leung YT et al. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics 10, 191–199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi L et al. The SLE transcriptome exhibits evidence of chronic endotoxin exposure and has widespread dysregulation of non-coding and coding RNAs. PLoS One 9, e93846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lanata CM, Chung SA & Criswell LA DNA methylation 101: what is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci. Med 5, e000285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steinberg AD, Baron S & Talal N The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinicpolycytidylic acid. Proc. Natl Acad. Sci. USA 63, 1102–1107 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hooks JJ et al. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med 301, 5–8 (1979). [DOI] [PubMed] [Google Scholar]
- 73.Skurkovich SV & Eremkina EI The probable role of interferon in allergy. Ann. Allergy 35, 356–360 (1975). [PubMed] [Google Scholar]
- 74.Baechler EC et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl Acad. Sci. USA 100, 2610–2615 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bennett L et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med 197, 711–723 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crow MK & Wohlgemuth J Microarray analysis of gene expression in lupus. Arthritis Res. Ther 5, 279–287 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Catalina MD, Bachali P, Geraci NS, Grammer AC & Lipsky PE Gene expression analysis delineates the potential roles of multiple interferons in systemic lupus erythematosus. Commun, Biol. 2, 140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kirou KA et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. 50, 3958–3967 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Rodero MP et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J. Exp. Med 214, 1547–1555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Welcher AA et al. Blockade of interferon-γ normalizes interferon-regulated gene expression and serum CXCL10 levels in patients with systemic lupus erythematosus. Arthritis Rheumatol. 67, 2713–2722 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rowland SL et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med 211, 1977–1991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sisirak V et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J. Exp. Med 211, 1969–1976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Furie R et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J. Clin. Invest 129, 1359–1371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Labonte AC et al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS One 13, e0208132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mavragani CP et al. Expression of long interspersed nuclear element 1 retroelements and induction of type I interferon in patients with systemic autoimmune disease. Arthritis Rheumatol. 68, 2686–2696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nzeusseu Toukap A et al. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 56, 1579–1588 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Arazi A et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol 20, 902–914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Der E et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol 20, 915–927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barrat FJ et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med 202, 1131–1139 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hua J, Kirou K, Lee C & Crow MK Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 54, 1906–1916 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Lövgren T, Eloranta ML, Båve U, Alm GV & Rönnblom L Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 50, 1861–1872 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Barrat FJ, Meeker T, Chan JH, Guiducci C & Coffman RL Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur. J. Immunol 37, 3582–3586 (2007). [DOI] [PubMed] [Google Scholar]
- 93.Caielli S et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med 213, 697–713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lood C et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med 22, 146–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ries M et al. Identification of novel oligonucleotides from mitochondrial DNA that spontaneously induce plasmacytoid dendritic cell activation. J. Leukoc. Biol 94, 123–135 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Sisirak V et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell 166, 88–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yasutomo K et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet 28, 313–314 (2001). [DOI] [PubMed] [Google Scholar]
- 98.Gehrke N et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495 (2013). [DOI] [PubMed] [Google Scholar]
- 99.Stetson DB, Ko JS, Heidmann T & Medzhitov R Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Furie R et al. Anifrolumab, an anti-interferon-α receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 69, 376–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Riggs JM et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci. Med 5, e000261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ladislau L et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain 141, 1609–1621 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Wallace DJ et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 392, 222–231 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Md Yusof MY et al. Prediction of autoimmune connective tissue disease in an at-risk cohort: prognostic value of a novel two-score system for interferon status. Ann. Rheum. Dis 77, 1432–1439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Munroe ME et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis 75, 2014–2021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.El-Sherbiny YM et al. A novel two-score system for interferon status segregates autoimmune diseases and correlates with clinical features. Sci. Rep 8, 5793 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burmester GR et al. Differential expression of Ia antigens by rheumatoid synovial lining cells. J. Clin. Invest 80, 595–604 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu X, Chakravarty SD & Ivashkiv LB Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev 226, 41–56 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Orange DE et al. Identification of three rheumatoid arthritis disease subtypes by machine learning integration of synovial histologic features and RNA sequencing data. Arthritis Rheumatol. 70, 690–701 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boyle DL et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis 74, 1311–1316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang F et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol 20, 928–942 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuo D et al. HBEGF+ macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci. Transl. Med 11, eaau8587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Firestein GS & Zvaifler NJ Peripheral blood and synovial fluid monocyte activation in inflammatory arthritis. II. Low levels of synovial fluid and synovial tissue interferon suggest that gamma-interferon is not the primary macrophage activating factor. Arthritis Rheum. 30, 864–871 (1987). [DOI] [PubMed] [Google Scholar]
- 114.Varga J & Abraham D Systemic sclerosis: a prototypic multisystem fibrotic disorder. J. Clin. Invest 117, 557–567 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mayes MD et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 48, 2246–2255 (2003). [DOI] [PubMed] [Google Scholar]
- 116.Rice LM et al. A longitudinal biomarker for the extent of skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 67, 3004–3015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Bon L et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med 370, 433–443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Volkmann ER et al. Changes in plasma CXCL4 levels are associated with improvements in lung function in patients receiving immunosuppressive therapy for systemic sclerosis-related interstitial lung disease. Arthritis Res. Ther 18, 305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gregorio J et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J. Exp. Med 207, 2921–2930 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Guiducci C et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J. Exp. Med 207, 2931–2942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wenzel J & Tüting T An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in “interface dermatitis”. J. Invest. Dermatol 128, 2392–2402 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Gerber EE et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature 503, 126–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ah Kioon MD et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med 10, eaam8458 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Triantafyllopoulou A et al. Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc. Natl Acad. Sci. USA 107, 3012–3017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sarkar MK et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann. Rheum. Dis 77, 1653–1664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Uggenti C, Lepelley A & Crow YJ Self-awareness: nucleic acid-driven inflammation and the type I interferonopathies. Annu. Rev. Immunol 37, 247–267 (2019). [DOI] [PubMed] [Google Scholar]
- 127.Liu Y et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 64, 895–907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jeremiah N et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Invest 124, 5516–5520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu Y et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dhir A et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Briand C et al. Efficacy of JAK1/2 inhibition in the treatment of chilblain lupus due to TREX1 deficiency. Ann. Rheum. Dis 78, 431–433 (2019). [DOI] [PubMed] [Google Scholar]
- 132.Meesilpavikkai K et al. Efficacy of baricitinib in the treatment of chilblains associated with Aicardi-Goutières syndrome, a type I interferonopathy. Arthritis Rheumatol. 71, 829–831 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sanchez GAM et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Invest 128, 3041–3052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rice GI et al. Reverse-transcriptase inhibitors in the Aicardi–Goutières syndrome. N. Engl. J. Med 379, 2275–2277 (2018). [DOI] [PubMed] [Google Scholar]
- 135.Schulert GS & Grom AA Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu. Rev. Med 66, 145–159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bracaglia C et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann. Rheum. Dis 76, 166–172 (2017). [DOI] [PubMed] [Google Scholar]
- 137.Canna SW Editorial: interferon-γ: friend or foe in systemic juvenile idiopathic arthritis and adult-onset Still’s Disease? Arthritis Rheumatol. 66, 1072–1076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Akilesh HM et al. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 363, eaao5213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jordan MB, Hildeman D, Kappler J & Marrack P An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 104, 735–743 (2004). [DOI] [PubMed] [Google Scholar]
- 140.Wang A et al. Specific sequences of infectious challenge lead to secondary hemophagocytic lymphohistiocytosis-like disease in mice. Proc. Natl Acad. Sci. USA 116, 2200–2209 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weaver LK, Chu N & Behrens EM Interferon-γ-mediated immunopathology potentiated by Toll-like receptor 9 activation in a murine model of macrophage activation syndrome. Arthritis Rheumatol. 71, 161–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lounder DT, Bin Q, de Min C & Jordan MB Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv. 3, 47–50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Al-Salama ZT Emapalumab: first global approval. Drugs 79, 99–103 (2019). [DOI] [PubMed] [Google Scholar]
- 144.Xu M, Tchkonia T & Kirkland JL Perspective: targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol. Res 111, 152–154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]