

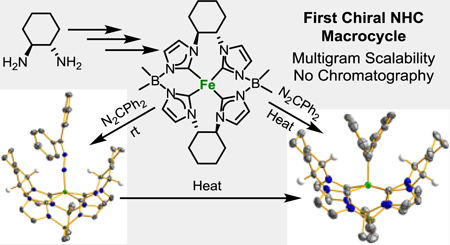
Abstract
The first chiral macrocyclic tetra-N-heterocyclic carbene (NHC) ligand has been synthesized. The macrocycle, prepared in high yield and large scale, was ligated onto palladium and iron to give divalent C2-symmetric square planar complexes. Multinuclear NMR and single crystal X-ray diffraction demonstrated that there are two distinct NHCs on each ligand, due to the bridging chiral cyclohexane. Oxidation of the iron(II) complex with trimethylamine N-oxide yielded a bridging oxo complex. Diazodiphenylmethane reacted with the iron(II) complex at room temperature to give a paramagnetic diazoalkane complex; the same reaction yielded the “all carbene” complex at elevated temperature. Electrochemical measurements support the assignment of the “all carbene” complex being as alkylidene. Notably, the diazoalkane complex can be directly transformed into the alkylidene complex, which had not been previously demonstrated on iron. Finally, a test catalytic reaction with a diazoalkane on the iron(II) complex does not yield the expected cyclopropane, but actually the azine compound.
Keywords: iron, NHCs, macrocycles, chiral ligands, alkylidene
Graphical Abstract
The first chiral NHC macrocycle and its associated complexes are reported. The macrocyclic ligand supports both diazoalkane and alkylidene complexes on iron.
Introduction
The ability to set stereochemistry during a catalytic reaction is a critical tool in synthetic chemistry, and three-membered ring formation is an increasingly active research topic.[1–4] The reason is that prochiral alkenes are widely abundant and can be combined with a variety of oxidants. The Jacobson-Katsuki epoxidation catalyst (Figure 1A) utilizes a tetradentate chiral salen ligand that promotes stabilization of the MnV=O intermediate that asymmetrically transfers oxygen to alkenes.[5–7] Research by Che established that chiral aziridines could be synthesized catalytically from single enantiomer Mn or Ru porphyrin (Figure 1B).[8, 9] More recently, Zhang has employed chiral D2-symmetric porphyrin cobalt complexes (Figure 1C) for enantioselective aziridination and cyclopropanation.[10–12] Nonetheless, there are several drawbacks for these catalyst designs. First, they often require strong oxidants or have a limited substrates scope to form the required group (O, NR, or CR2) that is to be selectively transferred to the alkene.[13, 14] Second, the synthesis of the ligands, particularly chiral porphyrins, is often cumbersome; many require challenging multistep syntheses as well as a chiral resolution step.[15, 16]
Figure 1.
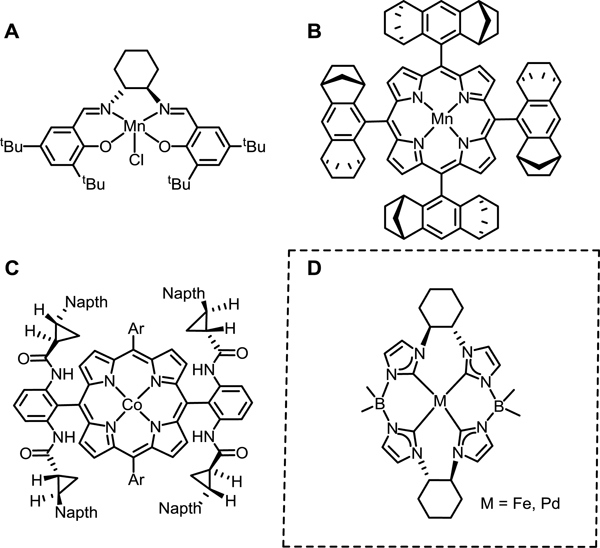
1A-1C: Examples of chiral tetradentate ligands and complexes used in previous asymmetric group transfer catalysis. 1D: New chiral ligand and complexes presented in this work.
The first drawback has been mitigated with the development of macrocyclic tetra-N-heterocyclic carbenes (NHCs) that have allowed for weaker oxidants to be effective in catalysis.[17–19] The increased electron donor strength of the NHCs stabilizes the required ligand multiple bond intermediates that are thought to be critical for these catalytic reactions, particularly on iron complexes.[20–24] For example, we have reported the synthesis of a neutral iron(II) complex utilizing a diborate tetra-NHC macrocyclic ligand that is able to activate completely aliphatic organic azides at room temperature, and perform catalytic aziridination to unactivated alkenes at elevated temperatures.[25] Furthermore, the Kühn group has demonstrated that iron macrocyclic tetra-NHC complexes are highly effective for epoxidation.[19]
Despite their benefits, chiral tetradentate NHC ligands have lagged behind significantly compared to other ligands, such as porphyrins.[26] The key obstacle to the synthesis of C2-symmetric bidentate NHC ligands is that the prototypical condensation reaction forms a 1:1 mixture of the imidazole amine species and the desired diimidazole. This mixture is difficult to purify, requires arduous column chromatography, and results in low yields.[27–29] While incomplete purification can be managed by alkylation bidentate NHCs, for tetra-NHC macrocycles the presence of an impurity can lead to a mixture of species that is intractable to separate. Indeed, to date, no chiral tetra-NHC ligands have been synthesized.
This manuscript explores the synthesis of the first chiral tetra-NHC macrocyclic ligand and its associated metal complexes. A scalable synthesis for a C2-symmetric chiral diimidazole is established that circumvents the typical byproduct complications. This key diimidazole is transformed into a chiral macrocycle that is then ligated to palladium(II) and iron(II). Initial oxidation reactions are explored with the iron complex which shows that an “all carbene” alkylidene complex can be formed from the addition of a diazoalkane with heat.
Results and Discussion
To develop a method for preparing a chiral macrocyclic tetraimidazolium, we desired to follow the route that we previously pioneered by reacting a diimidazole with a dielectrophile in a polar nitrile solvent.[30, 31] Crucially, to synthesize these NHC macrocycles, we require an unsubstituted C4 position on the imidazole, which renders some synthetic routes untenable.[32] Our approach to developing a robust and reliable route for preparing a chiral diimidazole, inspired by reports from Burgess and Kühn, would employ the formation of a bis-cyclic thiourea followed by reduction to yield the diimidazole (Scheme 1).[33, 34]
Scheme 1.
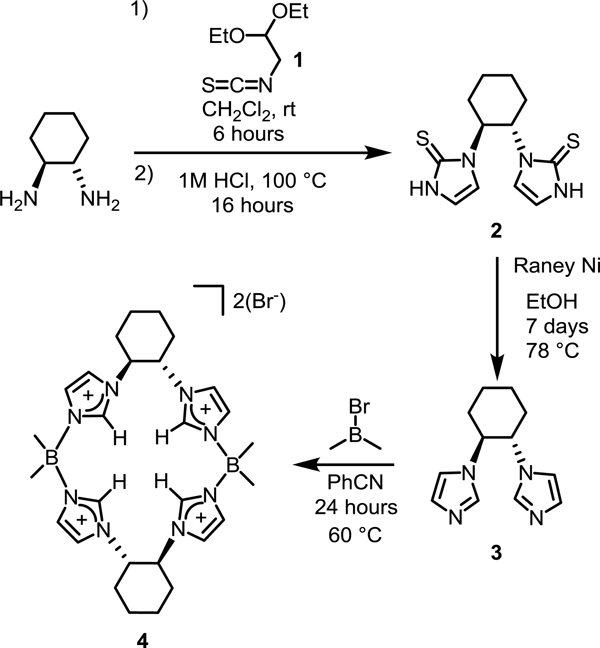
Synthesis of chiral macrocycle 4.
Addition of 1,1-diethoxy-2-isothiocyanatoethane (1) to (1S,2S)-1,2-diaminocyclohexane in dichloromethane led to the formation of the dithiourea 1,1’-((1S,2S)-cyclohexane-1,2-diyl)bis(3-(2,2-diethoxyethyl)thiourea), which was not isolated. The solvent was then removed and 1 M HCl was added and refluxed for 16 hours, leading to cyclization in 80% yield. Compound 2 formed as a white precipitate upon cooling and was collected by filtration. While some depictions of molecule 2 show it as the thiol tautomer,[34] we believe—based on IR evidence (Figure S9) of the broad N-H stretches at 3080 and 3016 cm−1 and no presence of a S-H stretch— that it is primarily the thione form.[35] The cyclic thiourea, 2, was reduced with excess Raney nickel in refluxing ethanol over seven days to yield (1S,2S)-1,2-di(imidazole)cyclohexane, 3, in 71% yield. Other desulfurization reagents such as hydrogen peroxide or benzoyl peroxide were ineffective.[33, 36] Both key steps for forming the chiral diimidazole can be completed on 10+ gram scales with no chromatography and the resulting product is consistent with similar compounds.[27]
The chiral macrocycle, ((S,S)−1,2-Cy,BMe2TCH)(Br)2, 4, was formed by reaction of 3 with bromodimethylborane in benzonitrile at 60 °C for 24 hours. The tan solid that was collected was washed with successively less polar solvents to yield a white powder in 81% yield. The absolute stereochemistry (S,S,S,S) of the macrocycle was confirmed by single crystal X-ray diffraction (Figure S1).
To assist with understanding the symmetry in solid state and solution, as well as its ability to support oxidative group transfer, we endeavored to synthesize palladium and iron complexes, respectively (Scheme 2). Based on recent research using a similar 16-atom macrocycle, we knew that it was necessary to fully deprotonate the ligand, 4, to prepare the metal complexes in high yield.[31] Therefore, we used a similar methodology of deprotonating the macrocycle 4 with nBuLi beginning at a low temperature (−35 °C) and then warming to rt. The clear solution of deprotonated 4 was then cooled to −35 °C and was added to the respective metal acetate salts. For Pd(OAc)2, this gave a white powder, [((S,S)−1,2-Cy,BMe2TCH)Pd], 5, in 72% yield, and for Fe(OAc)2 this gave a neon yellow powder, [((S,S)−1,2-Cy,BMe2TCH)Fe], 6, in 60% yield. Weaker lithium bases, such as lithium diisopropyl amide (LDA) or lithium bis(trimethylsilyl)amide (LiHMDS) were insufficiently strong to affect full deprotonation. Complex 5 is stable to atmospheric conditions while complex 6 is exceptionally air and moisture sensitive, consistent with other square planar tetra-NHCs we have prepared on these respective metals.[30, 31]
Scheme 2.
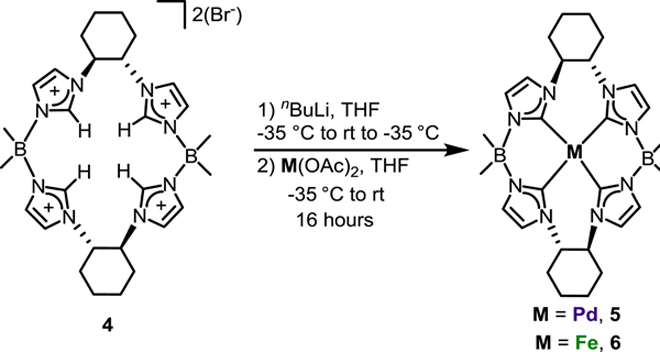
Synthesis of metal complexes 5 and 6.
Both the solution and solid state characterization of 5 and 6 demonstrate the rigid C2 symmetry that the chiral ligand imposes. 1H NMR of 5 in CDCl3 shows two separate resonances at 5.61 and 4.16 ppm for the protons off the chiral carbons in the cyclohexane ring (Figure S19). The 5.61 ppm resonance also does not shift appreciably in different polarity NMR solvents (Figure S52), which shows that the C-H by the metal center is strongly influenced by proximity to the metal. Even more conspicuous is the NHC resonances that are split in the 13C NMR as 176.9 and 165.6 ppm (Figure S20). All previous diamagnetic macrocyclic tetra-NHC complexes have only shown one resonance in solution implying that they are all equivalent on NMR time scale.[30, 37–40] The split resonances of both the 1H and 13C NMR for 5 is a common trait of constrained geometry C2-symmetric bidentate NHC complexes, but for 5 the difference between the 13C resonances are more pronounced.[27, 41, 42] The solid state structure for 5 confirms this absolute C2 symmetry and confirms that a single enantiomer is formed (Figure 2, top). Complex 5 has an average Pd-C bond distance of 2.01 Å which is consistent with its isostructural complexes and a τ4 value of 0.07 showing that is close to the square planar ideal.[37, 43]
Figure 2.
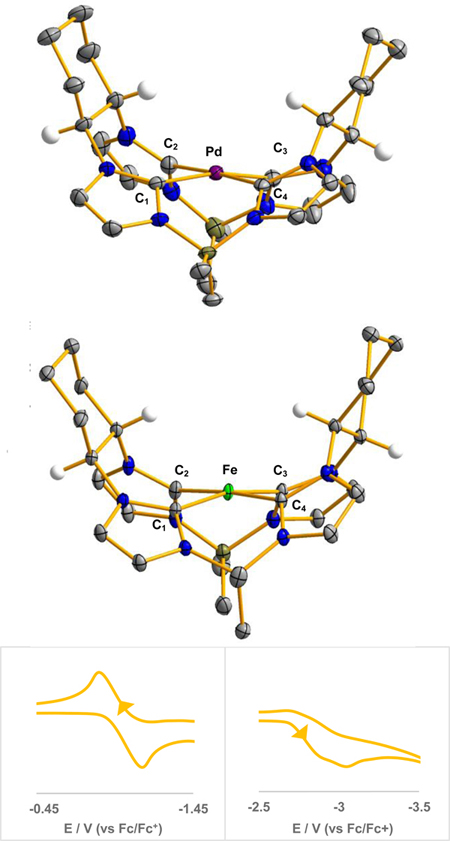
X-ray crystal structures of [((S,S)-1,2-Cy,BMe2TCH)Pd], 5 (top), and [((S,S)-1,2-Cy,BMe2TCH)Fe], 6 (middle). Purple, green, blue, gray, olive, and white ellipsoids (50% probability) represent Pd, Fe, N, C, B, and H atoms, respectively. Solvent molecules and H-atoms on non-stereogenic atoms are omitted for clarity; Selected bond lengths (Å) and angles (°) are as followed for 5: Pd-C1: 2.020(3), Pd-C2: 2.010(3), Pd-C3: 2.040(3), Pd-C4: 2.001(3), C1-Pd-C3: 167.88(12), C2-Pd-C4: 176.69(13); and 6: Fe-C1: 2.0057(15), Fe-C2: 1.9385(14), Fe-C3: 2.0049(14), Fe-C4: 2.0057(15), C1-Fe-C3: 166.64(6), C2-Fe-C4: 174.87(7). Bottom: Cyclic voltammogram traces of 6 in THF at 50 mV/s scan rate with 0.1 M (TBA)(PF6) as the supporting electrolyte, referenced to ferrocene/ferrocenium redox couple. Bottom, left: reversible Fe2+/Fe3+ oxidation wave at −0.98 V. Bottom, right: irreversible reduction wave at −3.09 V.
The iron complex, 6, exhibits many of the same characteristics as 5, but is complicated by its paramagnetism and extreme air sensitivity. Evans method measurements show two unpaired electrons, consistent with an isostructural iron complex, [(Et,BMe2TCH)Fe], that we reported previously.[25] All sixteen expected 1H resonances for a C2 symmetry are observed in a wide scan 1H NMR for 6 (Figure S23). As for [(Et,BMe2TCH)Fe] donor solvents such as CH3CN do not bind to the iron even at elevated temperature (Figure S25 and S53). The X-ray crystal structure of 6 shows a similar square planar geometry with slightly greater distortion (τ4 = 0.13) and Fe-C bond distances that average 1.97 Å (Figure 2, bottom). The iron(II) center in 6 is considerably more distorted from square planar than our previous isostructural iron(II) complexes, but has average Fe-C bonds (1.97 Å) in between the previously reported complexes.[25, 31]
Since divalent iron complexes supported by macrocyclic tetra-NHCs have been highly effective for oxidation catalysis,[18, 19, 25] we tested the reactivity of 6 with oxidants (Scheme 3). Reaction of complex 6 with trimethylamine N-oxide results in an immediate color change to red-brown. The X-ray crystal structure revealed that addition of oxygen led to a bridging oxo, [(((S,S)-1,2-Cy,BMe2TCH)Fe)2O] (7) (Figure 3). Complex 7 is similar to other bridging iron oxo complexes supported by tetracarbene ligands have been reported by Kühn, Meyer and our group.[31, 38, 44] The Fe-O bond lengths are each 1.816(2) Å and the Fe-O-Fe angle is nearly linear at 177.3(2)°. The 1H NMR of 7 showed a diamagnetic species similar to 5 (Figure S29). However, the broad peak at 6.59 ppm is highly shifted versus 5. This resonance is attributed to the protons off the chiral cyclohexane that are pointed toward the oxo ligand. These protons appear to have significant hydrogen bonding character with the oxo, shifting them downfield.
Scheme 3.
Reactions of 6 with Me3NO and Ph2CN2.
Figure 3.
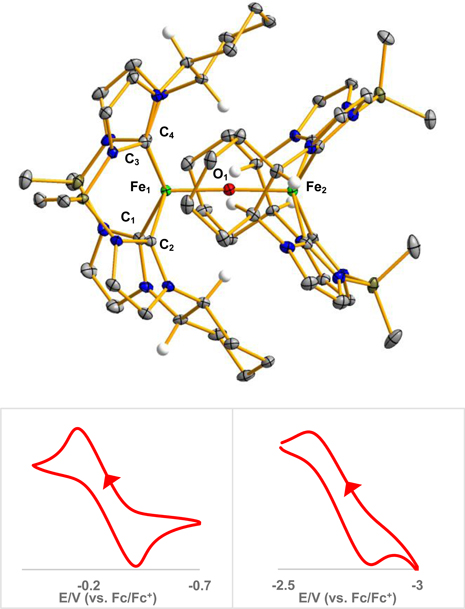
X-ray crystal structure of [(((S,S)-1,2-Cy,BMe2TCH)Fe)2O], 7 (top). Green, blue, grey, red, olive, and white ellipsoids (50% probability) represent Fe, N, C, O, B, and H atoms, respectively. Solvent molecules and H-atoms on non-stereogenic atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are as followed for 7: Fe1-C1: 2.070(3), Fe1-C2: 2.017(3), Fe1-C3: 2.068(3), Fe1-C4: 2.014(3), Fe1-O`: 1.816(2), Fe2-O: 1.816(2), C1-Fe1-C3: 142.37(13), C2-Fe1-C4: 152.07(14), Fe1-O1-Fe2: 177.33(15). Bottom: Cyclic voltammogram traces of 7 in THF at 100 mV/s scan rate with 0.1 M (TBA)(PF6) as the supporting electrolyte, referenced to ferrocene/ferrocenium redox couple. Bottom left: reversible Fe3+/Fe4+ oxidation wave at −0.28 V. Bottom, right: Quasi-reversible Fe3+/Fe2+ reduction wave −2.71 V
Addition of diazodiphenylmethane to 6 caused a color change to dark brown with no apparent N2 evolution. This paramagnetic product has a poorly resolved 1H NMR, but intriguingly has a peak in the IR spectrum at 2038 cm−1, suggesting that the N2 moiety is still present (Figure S35).[45, 46] Single crystal X-ray confirmed that the N2 from the diazoalkane had not been lost, and we had synthesized the diazoalkane complex, [((S,S)-1,2-Cy,BMe2TCH)Fe(N2CPh2)] (8) (Figure 4). Diazoalkane binding to transition metals is quite complex,[47] and few diazoalkane complexes have been prepared on iron.[48–51] Chirik and Albertin have synthesized linear diazoalkanes which bind as neutral adducts,[48, 50] while Betley and Holland have prepared complexes where the ligand binds as a diazoalkanyl radical.[49, 51] For complex 8, the key bond lengths (Fe-N, 1.779(4) Å; N-N, 1.215(5) Å; and N-C, 1.327(6) Å) are closer to the diazoalkanyl radical formulation (albeit with a slightly longer N-C bond). However, the ligand bond angles (Fe-N-N, 174.5(4)° and N-N-C, 140.4(5)°) are more consistent with a hydrazonido formulation.[51] Evans method measurements show a spin state of S = 1, which is consistent with either formulation (i.e. intermediate spin Fe(IV) or intermediate spin Fe(III) with antiferromagnetic coupling to diazoalkanyl radical, S = 3/2 −1/2). A final point of comparison is the average lengths of the Fe-NHC bonds since increasing the oxidation state in the same geometry should shorten the bonds. A comparison of 7, 8, and 9 (vide infra) indicates that the average Fe-CNHC distances are 2.042 Å, 2.008 Å, and 1.995 Å, respectively. We believe that the evidence is in favor of the hydrazonido formulation for 8, which is a similar spin state and oxidation state compared to our recently reported paramagnetic Fe(IV) imide complex, [(Me,BMe2TCH)Fe(NDipp)].[20]
Figure 4.
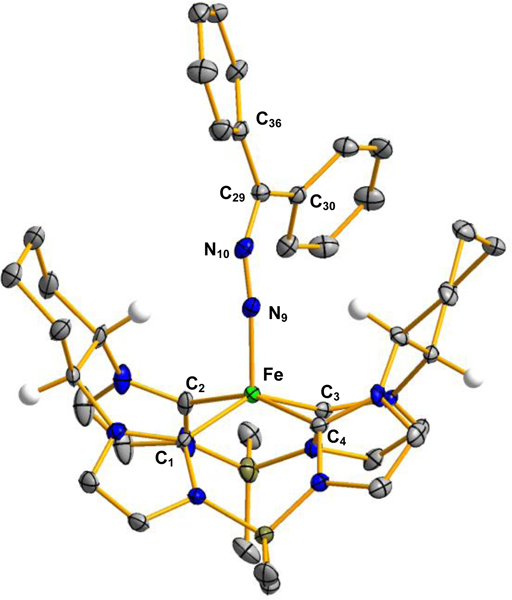
X-ray crystal structures of [((S,S)-1,2-Cy,BMe2TCH)Fe(N2CPh2)], 8 (top), and [((S,S)-1,2-Cy,BMe2TCH)Fe(CPh2)], 9 (bottom). Green, blue, gray, olive, and white ellipsoids (50% probability) represent Fe, N, C, B, and H atoms, respectively. Solvent molecules and H-atoms non-stereogenic atoms are omitted for clarity. Selected bond length (Å) and angles (°) are as followed for 8: Fe-C1: 2.019(5), Fe-C2: 1.900(5), Fe-C3: 2.032(5), Fe-C4: 1.978(5), Fe-N9: 1.779(4), N9-N10: 1.215(5), N10-C29: 1.327(6), C29-C30; 1.477(8), C29-C36: 1.471(7).
With the formation of the complex 8, we endeavored to form the alkylidene to determine if these species would be effective for chiral carbene transfer reactions. Increasing the temperature of the reaction on 8 with diazoalkane to 105 °C in toluene led to a diamagnetic product which was consistent with the carbene complex (Figure S38–S43), [((S,S)-1,2-Cy,BMe2TCH)Fe(CPh2)] (9). The 1H NMR of diamagnetic 9 has similarities to that of 5 with the addition of signals for the phenyl groups on the diazoalkane fragment. Of note, the peak at 7.58 ppm is much broader than expected. Due to the integration of 4H and the expected ortho C-H doublet missing, it was thought these H’s are fluxional in solution. Indeed, with the 2D-ROESY spectrum (S43) shows a cross peak to the C-H at 4.78 ppm. Along with other 2D NMR, this was absolutely assigned as the ortho C-H resonance on the phenyl ring. 13C NMR showed inequivalent NHC resonances at 188.8 and 172.4 ppm and, strikingly, a resonance at 309.3 ppm, attributed to the non-NHC carbene carbon which could only be definitively observed by an increase of the relaxation delay. Lapinte, Floriani, Che, and Wolczanski have reported 13C resonances for the carbene between 305 and 385 ppm, and are consistent with a diamagnetic NMR.[52–55] Only examples by Floriani[56, 57] and Chirik[45] have paramagnetic NMRs with this same diazoalkane fragment. A single crystal X-ray analysis of 9 showed the first “all carbene” iron complex (Figure 5). The Fe=C bond distance is 1.814(4) Å, making it slightly longer than examples with iron porphyrin complexes,[54, 58, 59] but considerably shorter than examples by Chirik and Wolczanski.[45, 55]
Figure 5:
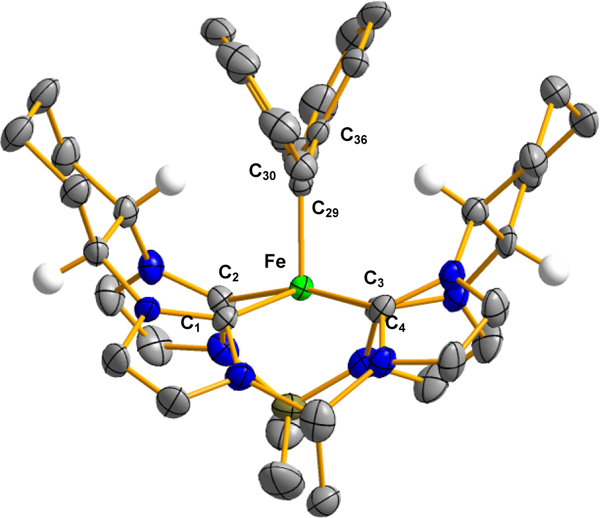
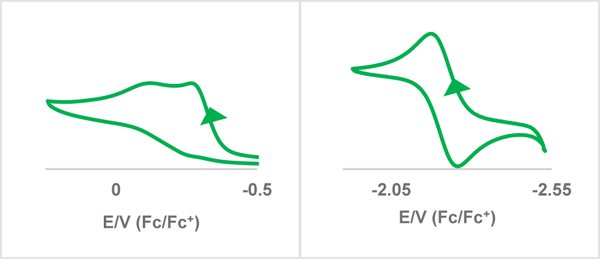
X-ray and [((S,S)-1,2-Cy,BMe2TCH)Fe(CPh2)], 9 (bottom). Green, blue, gray, olive, and white ellipsoids (50% probability) represent Fe, N, C, B, and H atoms, respectively. Solvent molecules and H-atoms non-stereogenic atoms are omitted for clarity. Selected bond length (Å) and angles (°) are as followed for 9: Fe-C1: 2.013(4), Fe-C2: 1.977(3), Fe-C3: 2.020(3), Fe-C4: 1.969(4), Fe-C29: 1.814(4), C29-C30: 1.508(5), C29-C36: 1.490(5). Bottom: Cyclic voltammogram traces of 9 in CH3CN at 100 mV/s scan rate with 0.1 M (TBA)(PF6) as the supporting electrolyte, referenced to ferrocene/ferrocenium redox couple. Bottom, left: irreversible oxidation wave at −0.28 V. Bottom, right: reversible Fe4+/Fe3+ wave at −2.21 V.
In recent years, the oxidation state systems similar to 9 has been heavily debated.[59, 60] The conventional knowledge was that in isostructural porphyrin compounds, the iron center goes from iron(II) to iron(IV) upon formation of alkylidene.[54] Spectroscopic evidence has corroborated this assignment. However, recently a combination of calculated properties by Zhang[60] and experimental studies by Li[59] has resulted in these complexes being re-evaluated as iron(II) singlet carbenes.
Recent reevaluation of literature discounts the possibility of iron(IV) for porphyrin complexes and other conjugated ligand platforms.[58–62] In contrast, for tetracarbene ligand platforms, we and others have shown that high oxidation states on iron are possible, with examples of isolated metallotetrazenes, imides, and oxos.[20, 38, 63] To help confirm the oxidation state on 9 we employed electrochemistry. Comparisons of the electrochemistry of molecules 6, 7 and 9 are shown below each crystal structure (Figures 2, 3 and 5, bottom). For complex 6, there is a reversible trace at −0.98 V and an irreversible trace at −3.09 V in THF versus the ferrocene/ferrocenium redox couple with (TBA)(PF6) as the supporting electrolyte. This reversible trace is consistent with an Fe2+/Fe3+ redox cycle. Furthermore, this oxidation potential is at a comparable voltage to our previously reported square planar iron(II) complex[31], and also consistent also with the extreme air sensitivity of 6. Comparing this to 7, which has two Fe(III) centers[38, 39, 44], there are reversible oxidation (−0.28 V) and quasi-reversible reduction (−2.71 V) potentials in THF. This result shows that the iron(II) and iron(IV) states are electrochemically obtainable with this iron complex. These measurements are in stark contrast to the electrochemistry of 9. Complex 9 has a reversible reduction at −2.21 V and an irreversible oxidation at −0.28 V in acetonitrile, which is the opposite of 6. This result supports that the iron center is high valent and can access a lower oxidation state (Fe(III)). Electrochemical studies on ruthenium salen carbene complexes do not show a similar reduction wave, only irreversible oxidation.[64] Reduction of a monomeric Fe(IV) is consistent with our previous reported result with an iron(IV) tetrazene.[63] Characterizing 9 as Fe(IV) is also consistent with the diamagnetic (S=0) 1H NMR.
Upon confirmation of the structure of 9, we set up a degradation reaction of 8 with heating to see if 9 could be formed directly from the diazoalkane complex (Scheme 3), which has never been observed on iron previously and rarely on other transition metals.[51, 65–68] The reaction in C6D6 at 85 °C is quite complex and the alkylidene complex 9 starts to appear after four hours; however, after one day, 6 is also being formed (Figures S54–S55). Five days were required to consume all of 8 and a mixture of 9 and 6 was still observed at that time. It was not clear what the organic byproduct was from the reaction, as no tetraphenylethylene was observed by NMR.
With the isolation of 9, we were optimistic that this complex would be effective for chiral cyclopropanation. Regrettably, stoichiometric reactions of 9 with both cyclooctene and α-methylstyrene yielded no transfer of the carbene ligand (Scheme 4). In addition, heating 9 in neat 1-decene at 170 °C did not result in cyclopropanation. Upon further examination, this is perhaps not too surprising since very few isolated iron alkylidene complexes transfer to give the cyclopropane, the notable exception being the example by Che.[54] An examination of a space-filling model of the X-ray structure of 9 (Figure S2) shows that there is relatively little space for disubstituted alkenes. However, a report by Zhu for intramolecular chiral cyclopropanation on iron inspired us to try a similar reaction to determine if 6 was an effective catalyst.[69] Reaction of 2-methylallyl 2-diazo-2-phenylacetate (10) with 10% 6 at 45 °C in acetonitrile did not form the expected intramolecular cyclopropane, but in fact formed the dimeric azine 11 in 81% yield. A control reaction of 10 without 6 showed no product after 16 hours. In addition, stoichiometric reaction of 10 with 6 in benzene formed the azine compound 11 in less than 15 minutes at room temperature. Sun found that Cp*RhCl2 catalyzed a similar reaction to form a similar azine from diphenyldiazomethane by diazo coupling, presumably from the Rh-carbene complex; we recently reported similar results with a less sterically encumbered tetra-NHC iron complex.[31, 70]
Scheme 4.
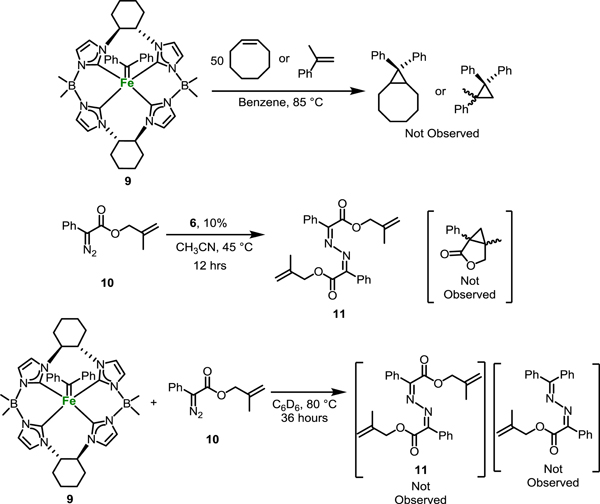
Attempts at cyclopropanation and cross reactivity with 9.
A variety of additional reactions were attempted to transfer the alkylidene group. We initially tried reacting 10 with 9 to synthesize a cross azine, but this did not result in any conversion of 9 after 24 hrs. In addition, reacting p-tolyl aldamine[71] with both 9 and in a stoichiometric reaction with 10 did not result in conversion or deviation from the same results, respectively. In the case of cyclopropanation, the mechanism is proceeds through a non-synchronous iron(II) mediated transfer to the alkene, and not through an iron(IV) radical-mediated pathway.[72] The lack of reactivity, therefore, gives an indication that the necessary electronics for compound 9 are incompatible for cyclopropanation.
Conclusion
In conclusion, we have synthesized the first chiral tetra-NHC macrocycle in three high yielding steps starting from (1S,2S)-1,2-diaminocyclohexane. This macrocycle was ligated onto palladium and iron in good yield through deprotonation with a strong base at low temperature. The diamagnetic palladium complex shows strict C2 symmetry in the NMR spectra as evidenced by the two distinct NHC resonances in 13C NMR. This C2 symmetry is also observed in the single crystal X-ray structures of both the palladium and iron complexes. Two different oxidation reactions were tested on the iron complex due to the effectiveness of similar macrocyclic tetracarbene complexes in catalysis. A reaction with trimethylamine N-oxide led to the bridging oxo complex which has hydrogen bonding to four of the cyclohexane protons from the macrocyclic ligands. The addition of diphenyldiazomethane to the iron(II) complex at room temperature led to a rare diazoalkane iron complex, which appears to bind in the hydrazonido formulation. Most notably, heating the same reaction with a slight excess of diazoalkane leads to the formation of an “all carbene” complex. The diazoalkane complex can also be transformed into the carbene complex and this is the first system where this has been demonstrated on iron. In addition, electrochemical studies of the starting iron(II) complex, bridging oxo complex, and carbene complex supports that the isolated “all carbene” complex is an iron(IV) alkylidene, which is different than other previously reported systems. While the iron(II) complex does not catalyze cyclopropanation, it does catalyze azine formation in a manner similar to Rh complexes. Subsequent stoichiometric reactions with the alkylidene do not transfer the carbene group, which is also in contrast to reported iron(II) carbenes. These results set the stage for further studies of chiral macrocyclic NHC complexes to complement porphyrin and salen complexes for oxidative group transfer reactions that are distinct from iron(II) species.
Experimental Section
All experimental details are described in the Supporting Information. Selected NMRs, IRs, MSs, UV-Vis, and X-ray structures are included (PDF). All X-ray crystallographic information is deposited as CIFs.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge the National Institute of Health (NIH-R15GM117494-01A) for financial support of this work. The University of Tennessee also provided additional financial support for this work via the X-ray facility. JFD graciously thanks Dr. Xian B. Carroll for valuable advice on X-ray crystallography.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Doyle MP, Angew. Chem. Int. Ed 2009, 48, 850–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pellissier H, Adv. Synth. Catal 2014, 356, 1899–1935. [Google Scholar]
- [3].Helene P, Lett. Org. Chem 2018, 15, 171. [Google Scholar]
- [4].Dalpozzo R, Lattanzi A, Pellissier H, Curr. Org. Chem 2017, 21, 1143–1191. [Google Scholar]
- [5].Zhang W, Loebach JL, Wilson SR, Jacobsen EN, J. Am. Chem. Soc 1990, 112, 2801–2803. [Google Scholar]
- [6].Irie R, Noda K, Ito Y, Matsumoto N, Katsuki T, Tetrahedron: Asymmetry 1991, 2, 481–494. [Google Scholar]
- [7].Linker T, Angew. Chem. Int. Ed 1997, 36, 2060–2062. [Google Scholar]
- [8].Lai T-S, Che C-M, Kwong H-L, Peng S-M, Chem. Comm 1997, 2373–2374. [Google Scholar]
- [9].Chan K-H, Guan X, Lo VK-Y, Che C-M, Angew. Chem. Int. Ed 2014, 53, 2982–2987. [DOI] [PubMed] [Google Scholar]
- [10].Jin L-M, Xu X, Lu H, Cui X, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2013, 52, 5309–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang Y, Wen X, Cui X, Wojtas L, Zhang XP, J. Am. Chem. Soc 2017, 139, 1049–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hu Y, Lang K, Tao J, Marshall MK, Cheng Q, Cui X, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2019, 58, 2670–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chang JWW, Ton TMU, Chan PWH, Chem. Rec 2011, 11, 331–357. [DOI] [PubMed] [Google Scholar]
- [14].Jenkins DM, Synlett 2012, 23, 1267–1270. [Google Scholar]
- [15].Halterman RL, Jan ST, J. Org. Chem 1991, 56, 5253–5254. [Google Scholar]
- [16].Marchon J-C, Ramasseul R, in:, The Porphyrin Handbook, ed. by Kadish KM, Smith KM and Guilard R, Academic Press, An Imprint of Elsevier Science: Amsterdam-Boston-London-Oxford-Paris-San Diego-San Francisco-Singapore-Sydney-Tokyo, 2003, 11, 75–132. [Google Scholar]
- [17].Keller CL, Kern JL, Terry BD, Roy S, Jenkins DM, Chem. Comm 2018, 54, 1429–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cramer SA, Jenkins DM, J. Am. Chem. Soc 2011, 133, 19342–19345. [DOI] [PubMed] [Google Scholar]
- [19].Kück JW, Anneser MR, Hofmann B, Pöthig A, Cokoja M, Kühn FE, ChemSusChem 2015, 8, 4056–4063. [DOI] [PubMed] [Google Scholar]
- [20].Anneser MR, Elpitiya GR, Townsend J, Johnson EJ, Powers XB, DeJesus JF, Vogiatzis KD, Jenkins DM, Angew. Chem. Int. Ed 2019, 58, 8115–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Searles K, Fortier S, Khusniyarov MM, Carroll PJ, Sutter J, Meyer K, Mindiola DJ, Caulton KG, Angew. Chem. Int. Ed 2014, 53, 14139–14143. [DOI] [PubMed] [Google Scholar]
- [22].Nieto I, Ding F, Bontchev RP, Wang H, Smith JM, J. Am. Chem. Soc 2008, 130, 2716–2717. [DOI] [PubMed] [Google Scholar]
- [23].Kropp H, King AE, Khusniyarov MM, Heinemann FW, Lancaster KM, DeBeer S, Bill E, Meyer K, J. Am. Chem. Soc 2012, 134, 15538–15544. [DOI] [PubMed] [Google Scholar]
- [24].Scepaniak JJ, Vogel CS, Khusniyarov MM, Heinemann FW, Meyer K, Smith JM, Science 2011, 331, 1049. [DOI] [PubMed] [Google Scholar]
- [25].Chandrachud PP, Bass HM, Jenkins DM, Organometallics 2016, 35, 1652–1657. [Google Scholar]
- [26].Janssen-Müller D, Schlepphorst C, Glorius F, Chem. Soc. Rev 2017, 46, 4845–4854. [DOI] [PubMed] [Google Scholar]
- [27].Bonnet LG, Douthwaite RE, Hodgson R, Organometallics 2003, 22, 4384–4386. [Google Scholar]
- [28].Lowry RJ, Veige MK, Clément O, Abboud KA, Ghiviriga I, Veige AS, Organometallics 2008, 27, 5184–5195. [Google Scholar]
- [29].Mechler M, Frey W, Peters R, Organometallics 2014, 33, 5492–5508. [Google Scholar]
- [30].Bass HM, Cramer SA, McCullough AS, Bernstein KJ, Murdock CR, Jenkins DM, Organometallics 2013, 32, 2160–2167. [Google Scholar]
- [31].Anneser MR, Elpitiya GR, Powers XB, Jenkins DM, Organometallics 2019, 38, 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mucha P, Mlostoń G, Jasiński M, Linden A, Heimgartner H, Tetrahedron: Asymmetry 2008, 19, 1600–1607. [Google Scholar]
- [33].Gigler P, Bechlars B, Herrmann WA, Kühn FE, J. Am. Chem. Soc 2011, 133, 1589–1596. [DOI] [PubMed] [Google Scholar]
- [34].Perry MC, Cui X, Powell MT, Hou D-R, Reibenspies JH, Burgess K, J. Am. Chem. Soc 2003, 125, 113–123. [DOI] [PubMed] [Google Scholar]
- [35].Bogatskii AV, Luk’yanenko NG, Kirichenko TI, Chem. Heterocyc. Compd 1983, 19, 577–589. [Google Scholar]
- [36].Wolfe DM, Schreiner PR, Synthesis 2007, 2007, 2002–2008. [Google Scholar]
- [37].Bass HM, Cramer SA, Price JL, Jenkins DM, Organometallics 2010, 29, 3235–3238. [Google Scholar]
- [38].Meyer S, Klawitter I, Demeshko S, Bill E, Meyer F, Angew. Chem. Int. Ed 2013, 52, 901–905. [DOI] [PubMed] [Google Scholar]
- [39].Anneser MR, Haslinger S, Pöthig A, Cokoja M, Basset J-M, Kühn FE, Inorg. Chem 2015, 54, 3797–3804. [DOI] [PubMed] [Google Scholar]
- [40].Fei F, Lu T, Chen X-T, Xue Z-L, New J Chem 2017, 41, 13442–13453. [Google Scholar]
- [41].Jeletic MS, Lower CE, Ghiviriga I, Veige AS, Organometallics 2011, 30, 6034–6043. [Google Scholar]
- [42].Tang J, He Y, Yu J, Zhang D, Organometallics 2017, 36, 1372–1382. [Google Scholar]
- [43].Yang L, Powell DR, Houser RP, Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- [44].Anneser MR, Haslinger S, Pöthig A, Cokoja M, D’Elia V, Högerl MP, Basset J-M, Kühn FE, Dalton Trans. 2016, 45, 6449–6455. [DOI] [PubMed] [Google Scholar]
- [45].Russell SK, Hoyt JM, Bart SC, Milsmann C, Stieber SCE, Semproni SP, DeBeer S, Chirik PJ, Chem. Sci 2014, 5, 1168–1174. [Google Scholar]
- [46].Reesbeck ME, Grubel K, Kim D, Brennessel WW, Mercado BQ, Holland PL, Inorg. Chem 2017, 56, 1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hillhouse GL, Haymore BL, J. Am. Chem. Soc 1982, 104, 1537–1548. [Google Scholar]
- [48].Bart SC, Bowman AC, Lobkovsky E, Chirik PJ, J. Am. Chem. Soc 2007, 129, 7212–7213. [DOI] [PubMed] [Google Scholar]
- [49].Sazama GT, Betley TA, Inorg. Chem 2014, 53, 269–281. [DOI] [PubMed] [Google Scholar]
- [50].Albertin G, Antoniutti S, Bortoluzzi M, Botter A, Castro J, Sibilla F, RSC Adv. 2016, 6, 97650–97658. [Google Scholar]
- [51].Bonyhady SJ, DeRosha DE, Vela J, Vinyard DJ, Cowley RE, Mercado BQ, Brennessel WW, Holland PL, Inorg. Chem 2018, 57, 5959–5972. [DOI] [PubMed] [Google Scholar]
- [52].Mahias V, Cron S, Toupet L, Lapinte C, Organometallics 1996, 15, 5399–5408. [Google Scholar]
- [53].Klose A, Solari E, Floriani C, Re N, Chiesi-Villa A, Rizzoli C, Chem. Comm 1997, 2297–2298. [Google Scholar]
- [54].Li Y, Huang J-S, Zhou Z-Y, Che C-M, You X-Z, J. Am. Chem. Soc 2002, 124, 13185–13193. [DOI] [PubMed] [Google Scholar]
- [55].Lindley BM, Jacobs BP, MacMillan SN, Wolczanski PT, Chem. Comm 2016, 52, 3891–3894. [DOI] [PubMed] [Google Scholar]
- [56].Giusti M, Solari E, Giannini L, Floriani C, Chiesi-Villa A, Rizzoli C, Organometallics 1997, 16, 5610–5612. [Google Scholar]
- [57].Esposito V, Solari E, Floriani C, Re N, Rizzoli C, Chiesi-Villa A, Inorg. Chem 2000, 39, 2604–2613. [DOI] [PubMed] [Google Scholar]
- [58].Wang H, Schulz CE, Wei X, Li J, Inorg. Chem 2019, 58, 143–151. [DOI] [PubMed] [Google Scholar]
- [59].Liu Y, Xu W, Zhang J, Fuller W, Schulz CE, Li J, J. Am. Chem. Soc 2017, 139, 5023–5026. [DOI] [PubMed] [Google Scholar]
- [60].Khade RL, Fan W, Ling Y, Yang L, Oldfield E, Zhang Y, Angew. Chem. Int. Ed 2014, 53, 7574–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lindley BM, Swidan A. a., Lobkovsky EB, Wolczanski PT, Adelhardt M, Sutter J, Meyer K, Chem. Sci 2015, 6, 4730–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jacobs BP, Agarwal RG, Wolczanski PT, Cundari TR, MacMillan SN, Polyhedron 2016, 116, 47–56. [Google Scholar]
- [63].Cramer SA, Hernández Sánchez R, Brakhage DF, Jenkins DM, Chem. Comm 2014, 50, 13967–13970. [DOI] [PubMed] [Google Scholar]
- [64].Li G-Y, Zhang J, Chan PWH, Xu Z-J, Zhu N, Che C-M, Organometallics 2006, 25, 1676–1688. [Google Scholar]
- [65].Messerle L, Curtis MD, J. Am. Chem. Soc 1980, 102, 7789–7791. [Google Scholar]
- [66].Mindiola DJ, Hillhouse GL, J. Am. Chem. Soc 2002, 124, 9976–9977. [DOI] [PubMed] [Google Scholar]
- [67].Cohen R, Rybtchinski B, Gandelman M, Rozenberg H, Martin JML, Milstein D, J. Am. Chem. Soc 2003, 125, 6532–6546. [DOI] [PubMed] [Google Scholar]
- [68].Iluc VM, Hillhouse GL, J. Am. Chem. Soc 2014, 136, 6479–6488. [DOI] [PubMed] [Google Scholar]
- [69].Shen J-J, Zhu S-F, Cai Y, Xu H, Xie X-L, Zhou Q-L, Angew. Chem. Int. Ed 2014, 53, 13188–13191. [DOI] [PubMed] [Google Scholar]
- [70].Qiu L, Huang D, Xu G, Dai Z, Sun J, Org. Lett 2015, 17, 1810–1813. [DOI] [PubMed] [Google Scholar]
- [71].Wang H, Wang C, Huang K, Liu L, Chang W, Li J, Org. Lett 2016, 18, 2367–2370. [DOI] [PubMed] [Google Scholar]
- [72].Wei Y, Tinoco A, Steck V, Fasan R, Zhang Y, J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.