

Abstract
Cardiovascular disease is the leading cause of death worldwide. Despite overwhelming socioeconomic impact and mounting clinical needs, our understanding of the underlying pathophysiology remains incomplete. Multiple forms of cardiovascular disease involve an acute or chronic disturbance in cardiac myocytes, which may lead to potent activation of the Unfolded Protein Response (UPR), a cellular adaptive reaction to accommodate protein-folding stress. Accumulation of unfolded or misfolded proteins in the Endoplasmic Reticulum (ER) elicits three signaling branches of the UPR, which otherwise remain quiescent. This ER stress response then transiently suppresses global protein translation, augments production of protein-folding chaperones, and enhances ER-associated protein degradation, with an aim to restore cellular homeostasis. Ample evidence has established that the UPR is strongly induced in heart disease. Recently, the mechanisms of action and multiple pharmacological means to favorably modulate the UPR are emerging to curb the initiation and progression of cardiovascular disease. Here, we review the current understanding of the UPR in cardiovascular disease and discuss existing therapeutic explorations and future directions.
Keywords: Unfolded protein response, Endoplasmic reticulum, GRP78, PERK, IRE1, ATF6, XBP1s, Cardiovascular disease, Pathological cardiac remodeling, Ischemic heart disease
1. INTRODUCTION
Cardiovascular disease is the leading cause of mortality and morbidity worldwide [1]. Metabolic derangements, hypoxia, and inflammation constitute the majority of cardiovascular pathophysiology, most of which cause adverse disturbance of Endoplasmic Reticulum (ER) homeostasis [2]. The ER possesses multiple essential cellular functions, including protein folding, secretory/transmembrane protein translocation, calcium homeostasis, and lipid biosynthesis. Various physiological or pathological stimuli lead to luminal accumulation of misfolded and unfolded proteins, a condition known as ER stress. ER stress triggers the Unfolded Protein Response (UPR) to restore homeostasis of the ER through activating transcriptional and translational pathways [3]. While acute induction of the UPR may be adaptive, prolonged, persistent activation may cause dysfunction and cell death. Indeed, aberrant regulation of the UPR has been implicated in the pathogenesis of numerous diseases, including neurodegenerative disease, cancer, diabetes, etc. Recent emerging evidence indicates that UPR is involved in the initiation, progression, and development of the cardiovascular diseases, such as hypertensive heart disease, ischemic heart disease, and heart failure [4]. Precisely targeting the UPR may, therefore, represent a promising avenue to identify novel therapeutic interventions and to tackle the devastating cardiovascular disease.
2. UNFOLDED PROTEIN RESPONSE (UPR)
The ER is a major signal transduction organelle, which is sensitive to various cellular disturbances. Numerous extracellular stimuli such as heat, ischemia, gene mutation, hypoxia, and increase in protein synthesis can cause ER stress, which in turn triggers an adaptive response named UPR (Fig. (1) [5]. Under resting conditions, the master ER protein chaperone binding immunoglobulin protein (BiP)/glucose-regulated protein of 78 kDa (GRP78) interacts with the luminal domains of three UPR signaling transducers and secures them on the ER membrane [6]. As the first step of the UPR, GRP78 binds to the unfolded/misfolded client proteins, which in turn releases the three sequestered ER membrane signal transducers, including endoribonuclease Inositol-Requiring Enzyme 1 (IRE1), Protein Kinase-like ER kinase (PERK), and Activating Transcription Factor 6 (ATF6) [7]. Meanwhile, GRP78 binds to misfolded and unfolded proteins as a chaperone to help them refold correctly or guide for ER-associated Degradation (ERAD). As a consequence, the UPR leads to the downregulation of global protein translation, production of new chaperone proteins, enhancement of ER protein folding capacity, and elimination of terminally misfolded proteins.
Fig. (1). Endoplasmic Reticulum (ER) stress and the Unfolded Protein Response (UPR).
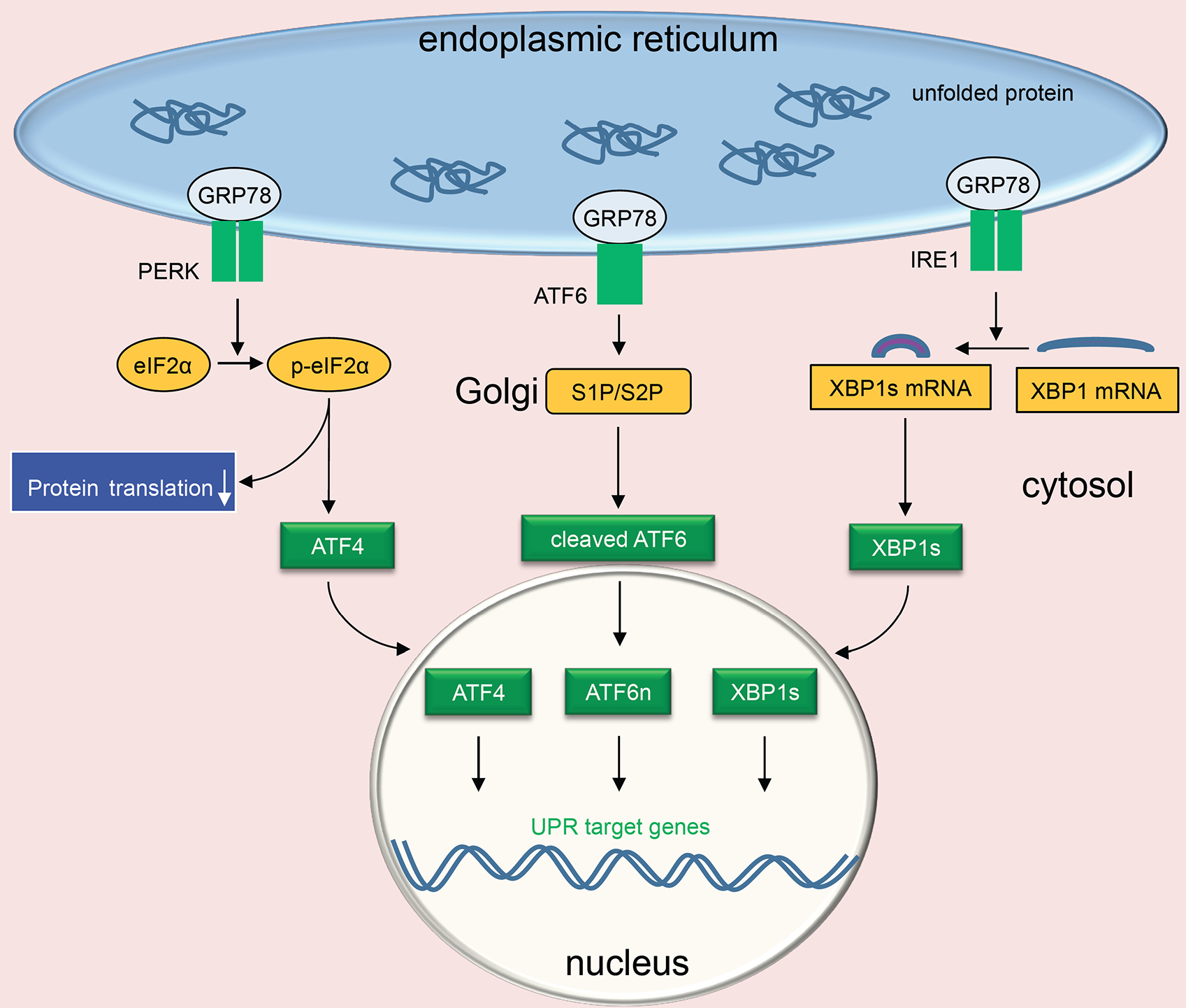
Under resting conditions, luminal domains of the three transducers of UPR bind ER master chaperone GRP78. ER stress ensues upon accumulation of misfolded and unfolded proteins. GRP78 then interacts with unfolded proteins, leading to activation of the three UPR branches. The initial response attempts to restore normal function by attenuation of protein synthesis and increasing chaperone production. PERK forms dimers and autophosphorylates, leading to downstream phosphorylation of eIF2α and a decrease in global translation. Phosphorylation of eIF2α also induces the translation of ATF4 and downstream targets such as CHOP. ATF6 is activated by site 1 and site 2 proteases (S1P and S2P) in the Golgi and transcriptionally competent nuclear ATF6 (ATF6n) is then translocated into the nucleus. IRE1 undergoes dimerization and autophosphorylation, which results in the cleavage of the XBP1 mRNA to form transcription factor XBP1s.
The three UPR branches are activated by distinct mechanisms and elicited different downstream signaling [8]. The liberation of IRE1 from GRP78 leads to autophosphorylation on serine 724 that triggers its endoribonuclease activity [9]. Cleavage of 26 base pairs from a cryptic intron of downstream target X-box binding protein 1 (XBP1) causes a frameshift and the resultant spliced XBP1 (XBP1s) functions as a strong transcriptional factor [10]. On the other hand, release of PERK and autophosphorylation lead to activation of its kinase activity, phosphorylation of downstream eukaryotic translation initiation factor 2α (eIF2α), and attenuation of global protein translation to decrease ER cargo load. Simultaneously, translation of activating transcriptional factor 4 (ATF4) is enhanced via preferable regulation of upstream small open reading frames to corroborate the protein-folding capacity of the ER. ATF6, after liberation from GRP78, is translocated from the ER to the Golgi where ATF6 is cleaved by site 1 protease and site 2 protease. The processed nuclear ATF6 (ATF6n) migrates to the nucleus and acts as a potent transcriptional factor. Various targets of ATF6n participate in ER protein folding, growth regulation, and cell survival [11].
All three branches stimulate UPR chaperone expression, but each transducer has special roles. The IRE1 effector XBP1s regulates multiple lipogenic enzymes and ERAD proteins [12, 13]. The PERK branch attenuates global protein synthesis through phosphorylation of eIF2α and induces CHOP (CCAAT/-enhancer-binding protein homologous protein)-mediated apoptosis under persistent ER stress [14]. ATF6 signaling is critical in maintaining cellular redox homeostasis and confers survival advantage [15, 16]. Furthermore, there is substantial crosstalk among the three branches. Although this interplay is essential for the cell to mount an adaptive action in response to complex intracellular and extracellular cues, it may make precise UPR targeting a daunting mission.
The UPR is a cellular stress response, highly conserved in evolution from yeast to mammals [17]. At the early phase, the three transducers activate their individual downstream pathways, resulting in elicitation of various pro-survival mechanisms including upregulation of ER chaperones such as GRP94, GRP78, and calreticulin and enhancement of ER homeostasis [18]. However, prolonged, sustained ER stress may turn the pro-survival action to pro-apoptotic signaling, mainly mediated by phosphorylation of c-Jun N-terminal kinase (p-JNK), upregulation of CHOP, cleavage of caspase-3, or a combination of these events [3]. Furthermore, a cell death response that involves cross-talk between ER and mitochondria may be triggered [19]. Taken together, optimal activation and execution of the UPR are critical for a pro-surviving adaptive response to restore cellular homeostasis, which may, however, lead to cell demise under persistent stress conditions.
3. CARDIOVASCULAR DISEASE AND UPR
Cardiovascular disease includes any conditions that affect blood vessels and internal structure or the morphology of heart, essentially contributing to the obstruction of continuous blood supply to the body. In cardiac myocytes, the sarco-ER (SER) plays a central role not only for general cellular function but also for myocyte contractility. SER stress that is common in heart disease can induce the UPR, which may reduce expression of essential proteins, adversely affect cell function, and even lead to cell death under uncontrolled stress conditions.
ER stress as an acute response has been found in many types of cardiovascular disease. For example, myocardial infarction is associated with oxygen and glucose deprivation in the myocardium, which disrupts ER homeostasis and generates a large amount of misfolded proteins, followed by the activation of UPR [20]. Hypertension or aortic stenosis can lead to pressure overload that is also associated with ER stress and stimulation of the UPR [21]. Additionally, congenital dilated cardiomyopathy is linked to a mutation in the KDEL receptor, which leads to buildup of misfolded proteins and prolonged ER stress in the heart [22]. Autoimmune cardiomyopathy is instigated by macrophage invasion to cardiac tissue and is associated with activation of the UPR in the heart [23]. In addition, atherosclerosis is a consequence of the accumulation of misfolded proteins due to excessive oxidation of lipids, elevation of homocysteines in vascular cells, and buildup of large amounts of cholesterol esters in macrophages [24]. Collectively, ER stress and cardiovascular disease are intimately linked together, and some of the key molecules of the UPR such as GRP78, IRE1, XBP1s, PERK, and ATF6 may play important roles in the pathogenesis of cardiovascular disease. Therefore, signaling pathways for coping with ER stress may present an important direction for the identification of therapeutic targets and design of novel treatment regimes.
Pharmacological agents that directly modulate the UPR are emerging as promising tools towards effective treatment of cardiovascular disease. Potential strategies to modulate the UPR include reducing unfolded proteins, preventing the UPR sensors from excessive activation (i.e., enhancing the binding between GRP78 and UPR transducers), suppressing the over-activated UPR sensors and effectors, etc. For example, it has been shown that salubrinal, an eIF2α phosphatase inhibitor, significantly increases GRP78 expression and protects the heart against ER stress-associated cardiomyocyte apoptosis in a rat myocardial infarction model [25]. However, the UPR participates in both pro-surviving and pro-apoptotic pathways and little is known about the biological consequences of switching from pro-survival to pro-apoptosis, or verse versa. As an example, the tyrosine kinase inhibitor sunitinib can directly stimulate IRE1 with the consequent activation of XBP1s and alleviation of ER stress [26]. However, in patients with previous clinical history of hypertension and heart disease, sunitinib was reported to increase the risk for cardiovascular disease [27].
Pharmacological alleviation of ER stress can also be achieved by stabilizing and rescuing misfolded proteins with chemical chaperones that mimic ER protein chaperones [28]. Tauroursodeoxycholic Acid (TUDCA) and 4-PBA have been approved for clinical use. Studies have shown that 4-PBA reduces ER stress and pressure overload-induced pathological cardiac remodeling in vivo [29]. Moreover, 4-PBA prevents doxorubicin - induced cardiac injury and isoprenaline-triggered cardiac fibrosis, highlighting its potential use as a cardioprotective drug [30]. Additionally, TUDCA treatment leads to improvement of contractile function in mouse cardiomyocytes under oxidative stress-induced UPR [31]. Taken together, by relief of ER stress, chemical chaperones may exert a protective role against pathological cardiac hypertrophy and, potentially, heart failure.
However, the UPR is an essential process in maintaining normal cellular function. Homozygous knockout mouse models of UPR sensors and effectors have shown various baseline detrimental effects [32]. For instance, whole body complete deficiency of either XBP1 or IRE1 is embryonic lethal with defective development of the heart and blood vessels [33, 34]. PERK knockout in mice leads to diabetes [35]. Moreover, ATF6 deficiency induces liver steatosis, hypoglycemia, and insulin resistance [36]. Collectively, targeting the UPR may be a double-edged sword, and more specific and temporary intervention of UPR sensors may be the safest strategy for modulating the UPR. Here, we review recent progresses that focus on the UPR as a therapy target in cardiovascular disease.
4. GRP78 IN CARDIOVASCULAR DISEASE
GRP78, also known as BiP, is a member of the Hsp70 family of protein chaperones [37]. Highly conserved across different species, GRP78 is localized primarily in the ER lumen, where it aids protein folding and regulates the UPR under stress conditions [6]. At the basal level, GRP78 binds the three signaling transducers PERK, IRE1, and ATF6 and prevents them from over-activation. Under the condition of accumulation of misfolded or unfolded proteins in the ER, GRP78 preferentially interacts with the exposed hydrophobic patches of misfolded proteins, releases the stress sensors, and activates the UPR.
Recently, accumulating studies have shown that GRP78 can be localized at the plasma membrane and may exert a cytoprotective role, especially in cancer cells [38–40]. Thapsigargin actively promotes cell surface localization of GRP78, as the increase of cell surface GRP78 is several-fold higher than the elevation in intracellular GRP78 [41]. GRP78 may be translocated and anchored to the cell surface membrane by binding to ER co-chaperones [42]. A recent study from Lee and colleagues showed that the activation of ER stress transducer IRE1 causes KDEL receptor dispersion from the Golgi and suppression of retrograde translocation of GRP78 [43]. Some GRP78, therefore, escapes to cell surface membrane where it counteracts the effects of transforming growth factor β and promotes cancer cell survival [43]. On the cell surface membrane, GRP78 may function as a signal transducing receptor or co-receptor for soluble ligands such as α2-macroglobulin (α2-M) [44], tumor differentiation factor [45], and vaspin [46]. Binding of GRP78 to most of these ligands activates the AKT/PI3K pro-survival pathway [47]. GRP78 is highly expressed on the cell surface membrane of a variety of cancer types owing to their inherently elevated ER stress level [46]. In contrast, expression of GRP78 in normal cells is modest and its membrane localization is therefore not prevalent. Consistently, global profiling of cell surface GRP78 is highly correlated with pathological states in cancers, making it a relevant target for therapeutic design.
Ample evidence suggests that GRP78 plays essential roles in development and cell survival [48]. Deletion of GRP78 causes defects in gestation and embryonic lethality at 3.5 days post coitum [49]. Moreover, tissue-specific elimination of GRP78 leads to cell death in respective cell types, including adipocytes and lung epithelial cells [50, 51]. Along these lines, GRP78 is also essential for physiological function and survival of cardiac myocytes. Our recent study highlighted the critical role of GRP78 in preventing cardiac cell death, maintaining contractile function and systolic performance, and survival [52]. We found that embryonic deficiency of GRP78 only in cardiomyocytes causes developmental defects in the heart and lethality before birth. Furthermore, inducible deletion of GRP78 in cardiomyocytes in adult animals leads to cardiomyocyte loss, severe heart failure, and accelerated death. Further work indicates that cell surface localized GRP78 may directly interact with PI3K and activate AKT, which can suppress overproduction of Reactive Oxygen Species (ROS) and promote survival [52]. Moreover, studies have shown that myocardial ischemia stimulates GRP78 expression in the heart [53]. We recently found that myocardial ischemia/reperfusion promotes GRP78 expression in cardiac myocytes [54] and overexpression GRP78 can mitigate the reperfusion injury [55]. The underlying mechanism may include GRP78 upregulation and translocation to cardiac myocyte surface membrane, where GRP78 interacts with PI3K and results in AKT activation to inhibit ROS accumulation [55]. In addition, our further study showed that GRP78 is also prominently upregulated in cardiac hypertrophy because of higher demand for protein synthesis/folding [56]. Interestingly, GRP78 overexpression leads to a higher degree of cardiac hypertrophic growth in response to pressure overload in vivo. Mechanistically, GRP78 may corroborate with GATA binding protein 4 (GATA4) and promote more profound hypertrophic growth under hemodynamic stress [56].
5. GRP78 AS A THERAPEUTIC TARGET
GRP78 overexpression can increase the binding probability of GRP78 to the UPR sensors and, therefore, prevent excessive activation. Meanwhile, GRP78 overexpression can augment the interaction of GRP78 to unfolded and misfolded proteins to help to refold or accelerate ERAD. Therefore, therapies targeting GRP78 may be beneficial for heart disease and other disorders with over-activated UPR (Fig. (2). As an example, overexpression of GRP78 has been reported to attenuate hypoxia-induced cardiomyocyte death [57].
Fig. (2). GRP78 as a therapeutic target in cardiovascular disease.
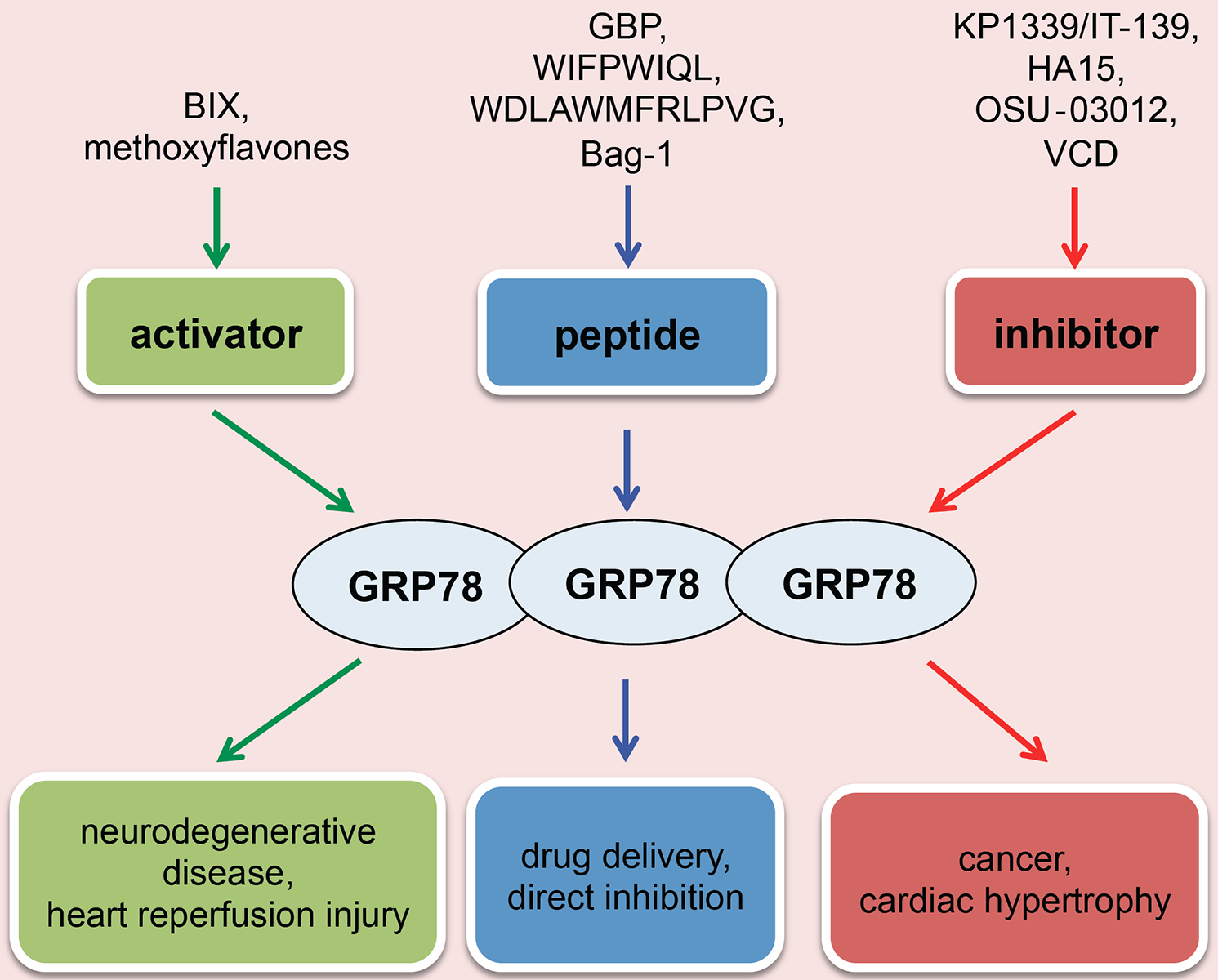
Different therapeutic approaches have been discovered for intervention of GRP78. The main approaches are as the following: (I) Direct activation of GRP78 can relieve neurodegenerative disorders and may be useful for cardiac ischemia/reperfusion injury; (II) Direct inhibition of GRP78 may suppress tumor growth and be useful to treat pathological cardiac remodeling; (III) GRP78 peptide can either directly modulate GRP78 activity or to be used as a precisely targeting vehicle to deliver therapeutic drugs.
6. GRP78 ACTIVATORS
Neurodegenerative disease is commonly associated with accumulation of misfolded and aggregated proteins, excessive oxidative stress, calcium dysregulation, and mitochondrial dysfunction [58]. A growing body of evidence indicates that the level and localization of GRP78 are altered, accompanied by accumulation and aggregation of misfolded proteins in neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s Disease (PD), Huntington’s Disease (HD), Amyotrophic Lateral Sclerosis (ALS), and prion protein disease [59–61]. Therefore, much research has been directed to validate GRP78 as a therapeutic target for the treatment of neurodegenerative disease. BiP Inducer X (BIX), originally identified to stimulate GRP78 mRNA expression, is a compound that preferentially induces GRP78 with modest augmentation of GRP94, calreticulin, and CHOP. BIX, however, does not modulate the signaling pathway downstream of IRE1 or the translational control branch from PERK [62]. Recently, BIX was tested in animal models with neurodegenerative disorders [63, 64]. It was shown that BIX administration significantly increases GRP78 expression, and ER stress-induced cell death is suppressed, leading to a reduction in the number of apoptotic cells. In addition, BIX was evaluated in ischemic animal models [65]. In global transient forebrain ischemic gerbils, BIX significantly upregulates the expression of GRP78. Moreover, pre-treatment with BIX reduces apoptosis cells in the hippocampus. In conclusion, BIX induces GRP78 to prevent neuronal death by ER stress, suggesting that BIX is a potential therapeutic agent for cerebral disease caused by excessive ER stress. Other GRP78 activators such as methoxyflavones also have a strong protective effect against detrimental ER stress [66]. Application of BIX and other GRP78 activators, however, has not been examined in heart disease. Using a transgenic mouse model, we recently showed that overexpression of GRP78 only in cardiac myocytes protects the heart from ischemia/reperfusion injury [55]. Further studies are warranted to investigate the pharmacokinetics of GRP78 activators, including its possible side effects, in pre-clinical tests in heart disease.
7. GRP78 INHIBITORS
The metabolic microenvironment of cancer cells is often acidic, hypoxic, and nutrient deprived, all of which are potent inducers of ER stress. GRP78 as the master regulator of ER stress is involved in cancer development, including tumor cell survival and proliferation, angiogenesis, and metastasis [67]. In addition, in different types of cancers, abnormally high GRP78 expression is correlated with anti-cancer drug resistance, greater risk for cancer recurrence, and an overall decrease in patient survival [68, 69]. Thus, small molecule agents that interfere with the synthesis, stability or activity of GRP78 in cancer cells can suppress its function and trigger cell death. Along with these lines, KP1339/IT-139 is one of the most sufficiently studied, potent GRP78 inhibitors [70, 71]. KP1339/IT-139 has already proven distinct anti-cancer activity while being extraordinarily well tolerated based on its dual prodrug nature: tumor site accumulation via albumin binding and tumor-specific activation by reduction [72, 73]. In a phase I clinical study designed to test KP1339, various tumors responded favorably to KP1339 treatment [74, 75].
Additionally, the thiazole benzenesulfonamide compound HA15 is a novel GRP78 inhibitor that has been shown to bind to and inhibit GRP78, leading to strong activation of ER stress pathways and subsequent cell death by apoptosis and autophagy [76, 77]. Recent studies have shown that treatment with HA15 can effectively kill melanoma cells and even to overcome BRAF inhibitor resistance. Recently, OSU-03012, a novel PDK-1 inhibitor, was discovered and the major mechanism is mediated through inhibition of GRP78, primarily by reducing the half-life of GRP78 protein [78–80]. Compared to the parent drug celecoxib (Celebrex), OSU-03012 has a greater level of bioavailability in preclinical large animal models and better efficacy at killing tumor cells [79, 80]. Verrucosidin (VCD) is a pyrone-type polyketide, which is produced by several species of the genus Penicillium. VCD was recently reported to be able to kill human HT29 colon carcinoma cells in culture under glucose-deprived conditions, which is mediated by downregulation of GRP78 [81, 82]. In addition, several purported GRP78 downregulators, for example, arctigenin [83], metformin [84], pyrvinium [85], and versipelostatin [86], have already shown anticancer activity in vivo. Pathological cardiac remodeling under chronic pressure overload is associated with elevation of GRP78. We showed that GRP78 potentiates cardiac hypertrophic growth through stabilization of a master cardiac transcriptional factor GATA4 [56]. On the other hand, siRNA-mediated knockdown of GRP78 in cardiomyocytes attenuates hypertrophic growth in response to hypertrophic stimuli. However, it remains to be determined whether GRP78 pharmacological inhibitors may regress pathological cardiac hypertrophy and improve heart failure. More work is therefore needed to evaluate the use of GRP78 inhibitors in this scenario.
8. GRP78-TARGETING PEPTIDES
Since GRP78 is strongly elevated in various cancer cells, it is a valid target to overcome tumor initiation and development. An additional advantage in targeting GRP78 is given by the observation that GRP78 translocates to the plasma membrane of malignant but not benign cells, offering the possibility of cancer cell-specific drug delivery with functionalized nanoparticles. Short peptides specifically binding GRP78 are therefore employed either for direct inhibition of GRP78 activity or to coat nanoparticles to deliver cytotoxic drugs.
One of the direct GRP78-targeting peptides is Bag-1, derived from a novel interaction site on the co-chaperone Bag-1 that binds the C-terminal domain of GRP78 [87]. Studies have shown that ectopic expression of Bag-1 peptide in several malignant but not benign prostate cancer cell lines as well as in prostate cancer xenograft models reduces tumor growth by inhibiting GRP78 protein-folding activity and inducing CHOP-mediated apoptosis. Similarly, intratumoral gene transfer of Mda-7, a tumor suppressor, was able to reduce the growth of several cancers. Importantly, M4, a peptide derived from Mda-7 and retaining the full functionality, specifically targets GRP78 [88, 89]. Taken together, these results indicate GRP78-targeting peptides may be exploited for therapeutic purposes.
Current antitumor chemotherapeutic drugs are often limited by severe side effects that leave cancer patients under extreme distress. To enhance delivery efficiency and decrease undesirable side effects, a large amount of efforts have been made in generating small molecules that can bind to cell surface GRP78 and activate the apoptotic pathway as an effective mode of anticancer therapy. GRP78 binding peptides are able to specifically bind to tumor cell surface GRP78, and to deliver the fused pro-apoptotic protein precisely into cancer cells to suppress tumor growth [90]. A successful example is given by the cyclic peptide Pep42 (CTVALPGGYVRVC) that can bind selectively to cell surface GRP78 and function as a cell-penetrating peptide [91].
In addition, the peptides WIFPWIQL and WDLAWM FRLPVG are able to bind GRP78 on the cell surface and have been employed successfully with in vivo models of prostate and breast cancers for the delivery of a cell death-inducing peptide [90]. Furthermore, GRP78 binding peptides can also be used in conjugation to other carriers like liposomes or nanoparticles. In fact, the peptide WIFPWIQL has been conjugated to liposomes for doxorubicin delivery to cancer endothelial cells and in an in vivo colon carcinoma mouse model [92]. Similarly, the peptide GIRLRG was employed for coating nanoparticles containing paclitaxel in irradiated breast carcinomas, which showed increased cell death compared to known chemotherapy approaches [93]. Based on these findings from the cancer studies, we propose that GRP78 targeted peptides may also be developed into a cardioprotective drug for reperfusion injury. The advantages may be twofold. These targeting peptides may themselves elicit pro-surviving actions in stressed but not dead cardiac myocytes through AKT activation. Moreover, targeted delivery of anti-apoptotic agents may provide another layer of cytoprotection. Exploration and validation of this peptide approach are currently underway in cardiac reperfusion models.
9. PERK IN CARDIOVASCULAR DISEASE
PERK is a protein kinase that belongs to the eIF2α kinase subfamily. PERK is composed of ER luminal and kinase domains. The former senses accumulation of unfolded and misfolded proteins in the ER lumen. After stimulation, PERK is activated by autophosphorylation of its kinase domain and acquires full catalytic activity to phosphorylate eIF2α at serine 51 [94]. Similar to most typical protein kinases, structure of the PERK kinase domain contains a C-terminal lobe (C-lobe) and an N-terminal lobe (N-lobe). There is a short hinge loop linking these two lobes. The N-lobe comprises three α-helices and five β-strands, whereas the C-lobe consists of two short β-strands, seven α-helices, and a long activation loop [95]. Located on the ER membrane, PERK is presented as monomers. Its ER luminal domain is bound by the master ER chaperone GRP78 under resting conditions. Initiation of ER stress facilitates disassociation of GRP78 from the PERK luminal domain. Then, the unfolded and misfolded proteins in the ER bind to MHC-like grooves of the PERK luminal domain, which subsequently triggers stacking of PERK homodimers. Along the polypeptide of unfolded and misfolded proteins, PERK dimers are lined up [95]. This steric configuration allows the activation loop of one dimer to reach the catalytic site of the other to trigger autophosphorylation. As a consequence, downstream molecules of PERK signaling are recruited and phosphorylated.
Recent studies by Liu et al. show that deficiency of PERK in the heart exacerbates the development of congestive heart failure in response to pressure overload, indicating that the PERK branch of UPR in cardiomyocytes is important in an adaptive response to high blood pressure [96]. Moreover, another work demonstrates that deletion of PERK in myocytes exhibits a strong protective effect against apoptosis induced by high glucose [97]. Interestingly, PERK in primary cardiomyocytes is found as a component of MAMs (mitochondria-associated endoplasmic reticulum membranes), which is the functional and physical contact site between ER and mitochondria. McAlpine et al. found that PERK is required for the activation of GSK3α/β by ER stress [98]. In mouse primary macrophages, PERK inhibition blocks ER stress-induced lipid accumulation, whereas constitutively active Ser9Ala-GSK3β promotes foam cell formation and CHOP elevation, even in cells treated with a PERK inhibitor. These data suggest that the ER stress/PERK-GSK3α/β signaling axis promotes pro-atherogenic macrophage lipid accumulation, highlighting a role in atherosclerosis and cardiovascular disease.
Studies have also shown that PERK may participate in the regulation of arrhythmias [57]. Gao et al. found that PERK activation in cardiomyocytes downregulates Na+ channel (Nav1.5) and cardiac rapidly activated K+ channel (Kv4.3) in human heart failure. The reduction of Nav1.5 leads to less Na+ current density and consequently decreased conduction velocity. On the other hand, the reduction of KCND3 encoding the α subunit of Kv4.3, which is the main contributor to the notch of phase 1 of the cardiac action potential, induces early repolarization, increases membrane resistance, and causes shortening of the cardiac action potential duration and phase 2 reentry. Moreover, blocking PERK prevents the decrease of these ion channels and reverses arrhythmogenic channel downregulation. Collectively, these results highlight a new paradigm of maintaining ion channel activity to prevent arrhythmia by modulating PERK.
10. PERK AS A THERAPEUTIC TARGET
Previous studies suggest that unresolved, chronic ER stress and sustained PERK activation have serious detrimental consequences in cell survival, which may be associated with cardiac disease. Acute stimulation of PERK, in contrast, may be beneficial for the heart to mount an adaptive response. This dichotomous impact of transient versus chronic PERK activation on normal physiology and disease pathogenesis is a subject of considerable interests and debates related to the therapeutic potential of PERK modulators (Fig. (3) [99].
Fig. (3). PERK as a therapeutic target in cardiovascular disease.
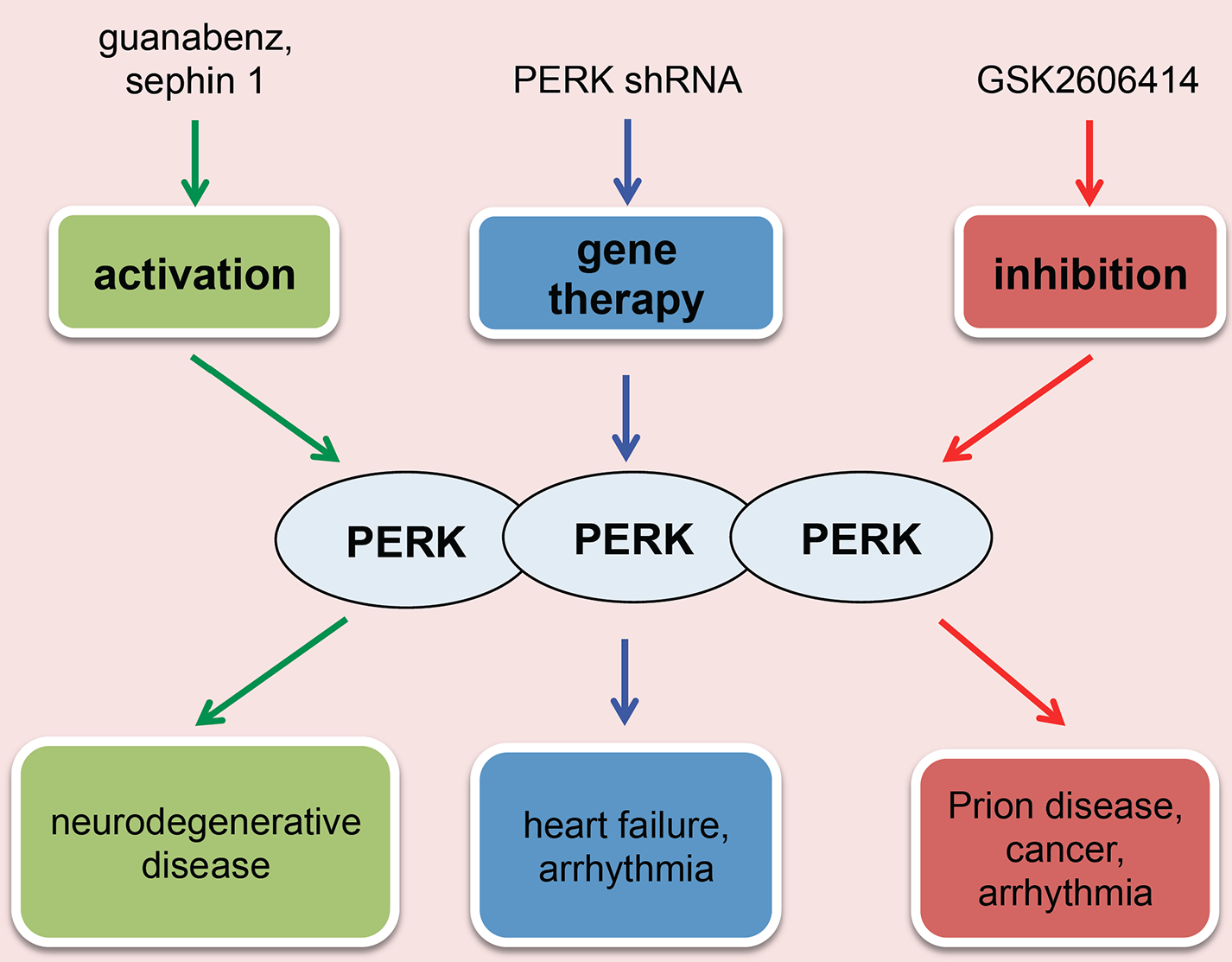
The main approaches to manipulate PERK are: (I) Direct activation of PERK can improve neurodegenerative disorders and suppress the development of cancer; (II) Direct inhibition of PERK can mitigate tumor growth, prion disease, and cardiac arrhythmia; (III) PERK targeting miRNA may be used to specifically suppress PERK for arrhythmia treatment.
A number of studies have shown that knockdown or genetic ablation of PERK can enhance tumor survival and growth and accelerate disease progression, indicating that activation of PERK may be a viable therapeutic approach [100]. Fv2E-PERK, an engineered PERK fusion protein with a modified luminal domain that can be dimerized and activated specifically with the FK506 analog AP20187, has proven to be a valuable tool to selectively activate PERK and precondition cells to tolerate following excessive ER stress, offer cytoprotective benefit, and induce or maintain a tumor cell quiescent state [101, 102]. However, more studies are required to be done in cardiac disease especially in congestive heart failure. Compared to direct PERK activation, increasing eIF2α phosphorylation by targeting the GADD34/PP1 eIF2α phosphatase with small molecule inhibitors such as guanabenz and Sephin1 has produced encouraging results in various models of neurodegenerative disease [103, 104].
On the other hand, PERK inhibition becomes increasingly attractive based on abundant preclinical and clinical data. These results reveal that PERK activation is pathogenic or associated with the promotion of tumor growth. Medicinal chemistry research on PERK inhibitors thus far is aiming at cancer therapy, using cell growth assays and models of tumor growth inhibition and angiogenesis [105, 106]. While some effect on tumor growth is observed in vivo with certain selective PERK inhibitors, no mechanistic insights have been revealed on cell proliferation in culture. Nonetheless, opportunities remain for the investigation of PERK inhibitors in models of metastasis, or in combination with existing chemotherapeutics, which is warranted based on findings that PERK inhibition can sensitize cancer cells to chemotherapy [107]. GSK2606414 was one of the most important discoveries of PERK inhibitors, which functions by targeting PERK in its inactive DFG conformation at the ATP-binding region. It was also reported as neuroprotective, and GSK2606414 prevents a progressive clinical and behavioral decline in a mouse model of prion disease [108]. Further evidence indicates that phosphorylation of tau is PERK dependent, and GSK2606414 administration can inhibit tau phosphorylation induced by metabolic stress, indicating a potential use in many tauopathies [109].
PERK inhibitors may also be used to benefit cardiovascular disease. Downregulation of Nav1.5 and Kv4.3 in heart failure has been shown to result from PERK activation. If a PERK inhibitor could be used to restore the channel protein levels, the arrhythmic risk might be improved. Alternatively, targeting the PERK branch of UPR might decrease cell apoptosis and improve cardiomyopathy. Recently human anti-PERK short hairpin RNA has been used in vitro to block PERK in human induced pluripotent stem cell-derived cardiomyocytes [57], which also highlights another means of PERK inhibition.
The therapeutic potential of PERK modulation is still under-defined. Studies supporting both inhibition and activation of PERK to ameliorate disease are confounding, which indicates significant gaps in our understanding of PERK in relation to disease treatment. In future studies, it will be important to characterize the timing and dynamics of PERK activation status in the context of different diseases, preferably combining both prophylactic and therapeutic intervention modalities with activators and inhibitors.
11. ATF6 IN CARDIOVASCULAR DISEASE
Mammals express two homologous ATF6 proteins, ATF6α (670 amino acids) and ATF6β (703 amino acids); the physiological characteristics of the former are much better understood than the latter. The C-terminal of ATF6 is inserted into the ER lumen, whereas the N-terminal is localized in the cytosol. The cytoplasmic part of ATF6 encompasses basic leucine zipper (bZIP) DNA binding and transcriptional activation domains, which are followed by a 20-amino acid transmembrane domain. ATF6α is a 90 kDa type II transmembrane glycoprotein and a member of the bZIP transcription factor family. ER stress induces the release of ATF6α from GRP78. Two Golgi localization sequences of ATF6α in the ER luminal domain are then exposed, evoking ATF6α translocation to the Golgi apparatus and cleavage by two proteases therein [110]. As a consequence, the soluble cytoplasmic region of 400 amino acids is translocated to the nucleus. This nuclear ATF6 (ATF6n) possesses both DNA binding and transcriptional activation domains, which contributes to the upregulation of multiple ER chaperones to enhance the ER folding capacity and restore cellular homeostasis.
Recent emerging evidence suggests important and unique roles of ATF6 in the heart. Primary cardiomyocytes manifest ATF6 elevation under hypoxia and nutrient-deprivation conditions [111]. Importantly, induction of ATF6 expression shows cardioprotection in the heart by reperfusion injury [112]. Under these conditions, knockdown of GRP78 partially mitigates the protective effect of ATF6. Moreover, in mice after myocardial infarction, inhibition of ATF6 impairs cardiac function and increases mortality [15]. The Glembotski lab found that ectopic expression of activated ATF6 decreases reperfusion damage in the heart [113]. Further, they determined that ATF6 acts as a transcriptional factor of several antioxidant genes [15]. Interestingly, it was further proposed that compensatory cardiac hypertrophy under pressure overload activates ATF6, which induces Rheb and activates mTORC1 [114]. Thus, ATF6 is critical in coupling growth stimulation and mTORC1-mediated cardiac growth. However, another study points out that ATF6 signaling plays an important role via calcium-mediated NFAT nuclear translocation mechanism in the process of diabetic cardiomyopathy, which was characterized by loss of cardiac compliance and function caused by cardiac fibrosis [115]. The precise role of ATF6 in the heart is probably dependent on disease conditions and various pathological contexts.
12. ATF6 AS A THERAPEUTIC TARGET
Since ATF6 plays a cardioprotective role in heart disease, approaches that target ATF6 might be employed for therapeutic gain (Fig. (4). Several drugs that act on ATF6 have been independently identified by multiple groups. N-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (147) is one of the most promising compounds, which was confirmed to preferentially activate ATF6 [116]. But the underlying mechanism by which 147 stimulates ATF6 remains unclear. A recent study by Blackwood et al. showed that 147 treatment confers strong cytoprotection against reperfusion damage in various tissues, highlighting a general mechanism of reprogramming cellular proteostasis [16]. Another study points out that ATF6 possesses two activation mechanisms: lipotoxic activation and proteotoxic activation. Tam et al. found that ATF6 containing a luminal achromatopsia eye disease mutation, unresponsive to proteotoxic stress, can be activated by fenretinide, a drug that upregulates dihydroceramide, suggesting a potential therapy for ATF6-related diseases including heart disease and stroke [117]. Recently, Jin et al. used the method of AAV9-mediated ATF6 cardiac overexpression to reverse the damage and to improve the decreased function in ATF6 knockout mice under ischemia/reperfusion [15]. These studies highlight the potential use of AAV-mediated gene transfer as a therapeutic strategy in ER stress-related cardiomyopathy.
Fig. (4). ATF6 as a therapeutic target in cardiovascular disease.
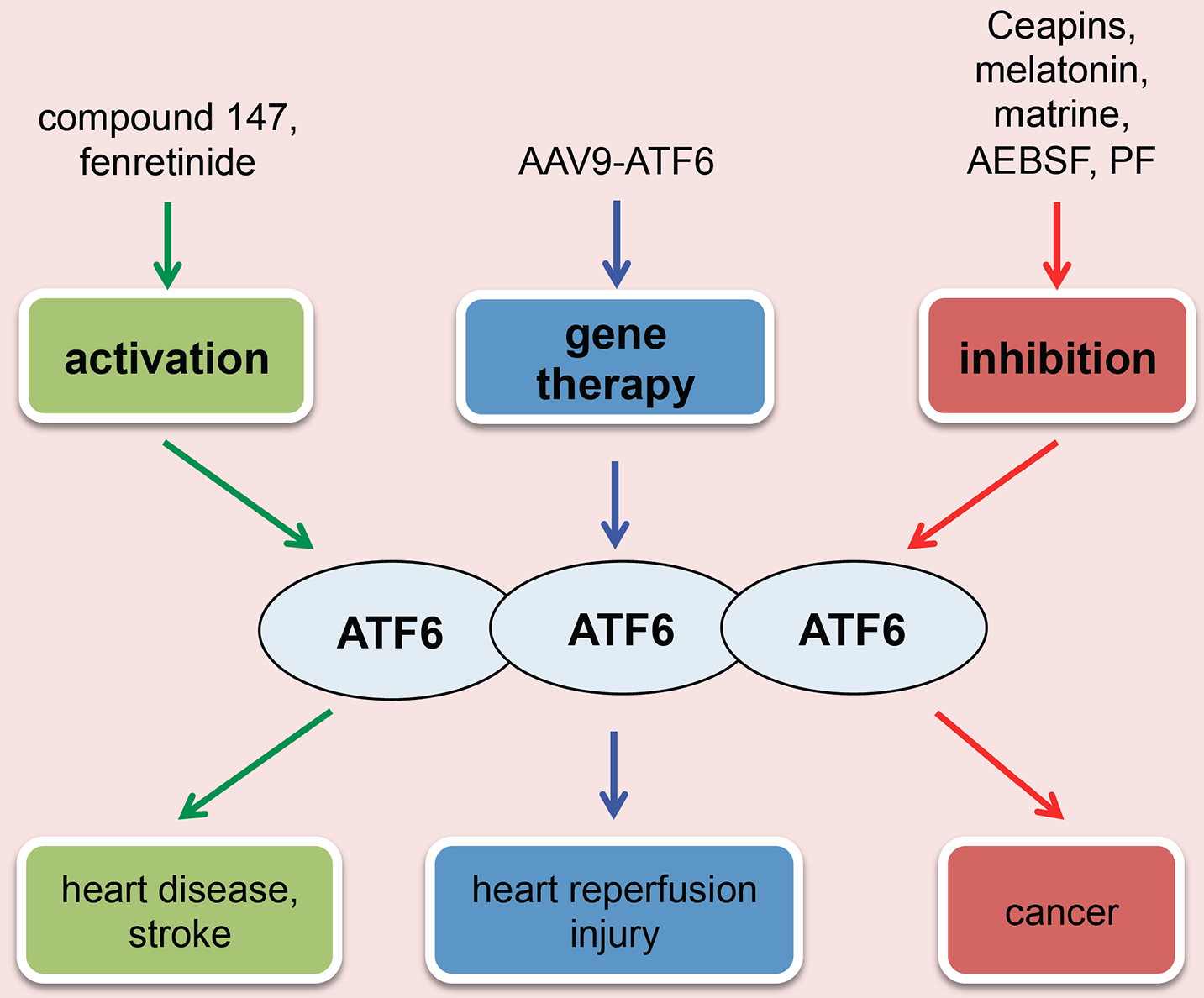
Different therapeutic approaches aiming to intervene ATF6 include: (I) Activation of ATF6 may relieve ER stress-related heart disease and stroke; (II) Inhibition of ATF6 may be exploited to treat cancer; (III) AAV9-mediated gene therapy can improve the outcome of cardiac ischemia/reperfusion.
On the other hand, ATF6 inhibitors that can reduce the activity of ATF6 are also attractive to researchers. Ceapins are first-in-class inhibitors that are used to explore both the mechanism of ATF6 activation and its role in multiple pathological settings. Gallagher et al. found that the small molecules of Ceapins can selectively block the activity of ATF6 under ER stress by inducing rapid, reversible clustering of ATF6α and preventing the exit of ATF6α from ER, but have no effect on other proteins involved in UPR [118]. The discovery of Ceapins now enables pharmacological modulation of the ATF6 branch individually, without perturbation of the other two. Other inhibitors such as melatonin, matrine, and 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride are all in the process to be tested in vitro and in vivo. Site 1 protease inhibitor, such as PF-429242, can also attenuate ATF6 expression by blocking proteolytic processing in the Golgi apparatus. But the effect may also be associated with the induction of IRE1 and increased phosphorylation of PERK [119]. Further studies are needed to delineate the action of ATF6 inhibitors in cardiac disease, particularly in diabetic cardiomyopathy and heart failure.
13. IRE1/XBP1S IN CARDIOVASCULAR DISEASE
As one of the three branches of the UPR, IRE1 is the most conserved, from yeast to mammals. IRE1 is a type I transmembrane glycoprotein of the ER that possesses both serine or threonine protein kinase and endoribonuclease functions. Under resting conditions, IRE1 luminal domain binds to the master chaperone GRP78. Upon accumulation of misfolded and unfolded proteins in the ER, GRP78 is dislocated from IRE1 and the latter autophosphorylates, leading to activation of the endoribonuclease function. Activated IRE1 then triggers unconventional cytoplasmic splicing of XBP1 mRNA. IRE1 removes 26 nucleotides from the unspliced XBP1 (XBP1u) mRNA, thereby leading to a frameshift and the generation of XBP1s [120]. XBP1u does not contain a transactivation domain in contrast to the potent transcriptional factor XBP1s. Because of the extremely short half-life of XBP1u, little is known about its biological functions. XBP1s, on the other hand, attracts the majority of attention due to its inducibility by the UPR and versatile roles in physiology and pathophysiology [13]. Indeed, XBP1s exerts multiple functions and is involved in both classical UPR and non-UPR pathways. During ER stress, XBP1s protein is translocated into the nucleus to initiate transcriptional programs that upregulate a large amount of UPR-associated genes involved in protein folding, ERAD, and ER biogenesis.
Accumulating evidence has shown that IRE1/XBP1s plays an important role in cardiovascular disease [121]. Recently, IRE1α is proposed to play a protective role against pressure overload-induced pathological remodeling in the heart [122]. Cardiac-specific overexpression of IREα exhibits preserved function and reduced fibrotic area, associated with increased adaptive UPR signaling and blunted inflammatory and pathological gene expression. In addition, IRE1 was reported to play an important role in atherosclerosis by promoting macrophage-derived foam cell formation [123].
As a direct downstream target of IRE1, XBP1s also plays critical roles in cardiovascular disease [124]. A recent study suggests that XBP1s regulates Vascular Endothelial Growth Factor (VEGF) - mediated cardiac angiogenesis and contributes to the progression of adaptive hypertrophy [125]. Using a cardiac-specific XBP1 knockout animal model, we observed an increase in cardiac myocyte death and more profound pathological remodeling during ischemia/reperfusion, suggesting that XBP1s protects the heart from reperfusion damage in vivo [54]. Consistently, using an inducible transgenic mouse model, we showed dramatic protection against reperfusion injury by XBP1s overexpression [54]. The underlying mechanism may involve transcriptional augmentation of the hexosamine biosynthetic pathway [126, 127]. Another study showed that transient activation of XBP1s increases endothelial cell proliferation, while sustained activation can lead to endothelial cell apoptosis, endothelium denudation, and atherosclerotic lesion development via multiple caspase activation and down-regulation of VE-cadherin transcription and MMP-mediated degradation [128]. These findings suggest that optimal levels of XBP1s are required in maintaining endothelial integrity.
14. IRE1/XBP1S AS A THERAPEUTIC TARGET
Due to its critical role in cell growth and survival, various approaches have been developed to target IRE1 (Fig. 5). Recent studies showed that IRE1 inhibitors, 4μ8c and STF-083010, which selectively inhibit IRE1’s RNase function, uncouple lipid-induced ER stress from inflammasome activation in both rodent and human macrophages [129]. At the in vivo level, these IRE1 inhibitors lead to a significant decrease in hyperlipidemia-induced IL-1β and IL-18 production, less T-helper type-1 immune response, and reduced atherosclerotic plaque formation, without altering the plasma lipid profiles in apolipoprotein E-deficient mice. These data suggest that pharmacologic modulation of IRE1 counteracts metaflammation and alleviates atherosclerosis development. Other IRE1 inhibitors such as salicylaldehydes [130], MKC-3946 [131], toyocamycin [132], and Hydroxylaryl-Aldehydes (HAA) [133] also show potent inhibition of IRE1 but have not yet been tested in cardiovascular disease. Although specific compounds that only activate XBP1s splicing have not been discovered, flavonol quercetin was reported to activate the endoribonuclease activity of IRE1, which may be used for this purpose. A recent study revealed that Ginkgolide K (GK) can significantly enhance IRE1/XBP1s activity and decrease ER stress-induced cell death in both in vitro and in vivo models [134]. GK may, therefore, be a promising therapeutic agent to ameliorate ER stress for cardiovascular disease treatment.
Fig. (5). IRE1/XBP1s as a therapeutic target in cardiovascular disease.
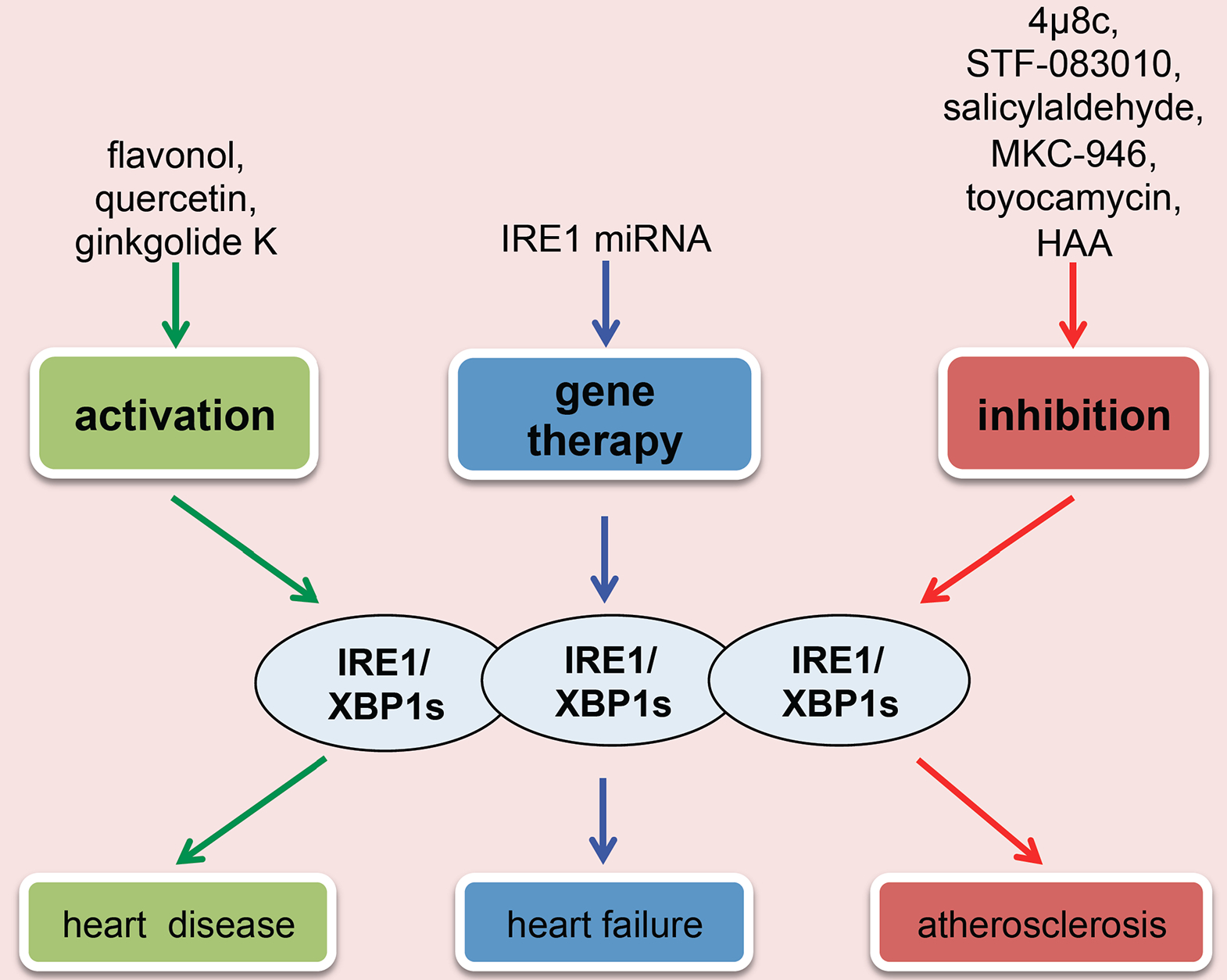
The main approaches are as the following: (I) Direct activation of IRE1/XBP1s may relieve ER stress-related cardiovascular disease; (II) Inhibition of IRE1/XBP1s can suppress atherosclerosis development; (III) miRNA-mediated gene therapy can be used to improve cardiac dysfunction of heart failure.
Recently, studies from Duan et al. established that XBP1s is an important angiogenic factor to maintain normal cardiac function in the early stage of hypertrophy growth [135]. Dysregulation of miR-214 and miR-30 in the hypertrophic and failing hearts inhibits XBP1s and XBP1s-induced angiogenesis, which leads to the pathological transition from hypertrophy to heart failure. Thus, modulation of miRNAs may be a valid therapeutic approach to stimulate XBP1s and to prevent heart failure.
15. ERAD IN CARDIOVASCULAR DISEASE
Beyond the three main branches of UPR, the ER also employs a mechanism termed ERAD to clear aggregated misfolded or unfolded proteins [136]. ERAD, a quality control process, can remove terminally misfolded proteins from the ER by the cytosolic ubiquitin-proteasome system. ER transmembrane and luminal misfolded proteins are first transported out of the ER into the cytosol. Then, the misfolded proteins are ubiquitinated by the ER transmembrane E3 ubiquitin (Ub) ligases. This process targets them for degradation by cytosolic proteasomes. ERAD has multiple components such as HRD1, EDEM, SEL1, and OS-9 [137]. Each of them has a different function in ERAD. A study from the Glembotski lab found that overexpressing Derl3, which can enhance ERAD, attenuates chronic ER stress response and cell death in response to simulated ischemia/reperfusion, suggesting that enhancing the ERAD machinery in the heart is cardioprotective [138]. Furthermore, another study showed that HRD1 contributes to the adaptive ER stress response in cultured cardiac myocytes and that HRD1 preserves cardiac function in a mouse model of pathological cardiac hypertrophy [139]. It is hypothesized that under certain conditions, sufficient levels of HRD1 facilitate the degradation of misfolded proteins, which adaptively enhances myocyte viability. However, when HRD1 is not enough, such as in the adult heart, pathology-driven maladaptive accumulation of misfolded proteins threatens myocyte viability and cardiac function. Along these lines, AAV9-mediated HRD1 overexpression directed to ventricular myocytes preserves heart function and reduces cardiac hypertrophy in mice with pressure overload-induced cardiac pathology. Taken together, the aforementioned findings suggest that intervention of ERAD components may be a viable approach to prevent cardiac cell death and to improve heart function under pathological conditions.
CONCLUSION AND PERSPECTIVES
Cardiovascular disease is the leading cause of death globally. Current therapies are incapable to arrest disease progression and as a result, socioeconomic burdens are ever climbing. Various heart diseases involve moderate to disastrous disturbances of cellular homeostasis in cardiac myocytes. The UPR as an acute, adaptive response has been found activated in multiple forms of heart diseases. From the past decades, our understanding of the UPR and ER stress response has been flourishing. The three signaling branches of ER stress are stimulated via distinct mechanisms. Downstream actions may differ but orchestrate in nature to regain cellular homeostasis. Only upon unsolved, persistent stress, the UPR may turn from adaptive to detrimental. The timing and duration of activation of the three branches therefore together determine the final outcome in cardiac myocytes. Indeed, the UPR has shown pro-survival and pro-apoptotic actions under different circumstances. While there is still a long way before modulating the UPR in clinic, novel discoveries are underway and exciting progresses have been made. In conclusion, the UPR plays a crucial role in the pathological processes of cardiovascular disease. A better understanding of the underlying mechanisms will greatly help us identify novel targets and define more effective approaches to tackle the devastating cardiovascular disease.
ACKNOWLEDGEMENTS
We thank members of the Wang lab for valuable discussions.
FUNDING
This work was supported by grants from the American Heart Association (14SDG18440002 to ZVW, 17IRG3346-0191 to ZVW), American Diabetes Association (1-17-IBS-120 to ZVW), and NIH (R01-HL137723 to ZVW).
Footnotes
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- [1].Benjamin EJ; Virani SS; Callaway CW; Chamberlain AM; Chang AR; Cheng S; Chiuve SE; Cushman M; Delling FN; Deo R; de Ferranti SD; Ferguson JF; Fornage M; Gillespie C; Isasi CR; Jiménez MC; Jordan LC; Judd SE; Lackland D; Lichtman JH; Lisabeth L; Liu S; Longenecker CT; Lutsey PL; Mackey JS; Matchar DB; Matsushita K; Mussolino ME; Nasir K; O’Flaherty M; Palaniappan LP; Pandey A; Pandey DK; Reeves MJ; Ritchey MD; Rodriguez CJ; Roth GA; Rosamond WD; Sampson UKA; Satou GM; Shah SH; Spartano NL; Tirschwell DL; Tsao CW; Voeks JH; Willey JZ; Wilkins JT; Wu JH; Alger HM; Wong SS; Muntner P Heart disease and stroke statistics-2018 update: A report from the american heart association. Circulation, 2018, 137(12), e67–e492. [ 10.1161/CIR.0000000000000558] [DOI] [PubMed] [Google Scholar]
- [2].Arrieta A; Blackwood EA; Glembotski CC ER Protein quality control and the unfolded protein response in the heart. Curr. Top. Microbiol. Immunol, 2018, 414, 193–213. [ 10.1007/82_2017_54] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ron D; Walter P Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol, 2007, 8(7), 519–529. [ 10.1038/nrm2199] [DOI] [PubMed] [Google Scholar]
- [4].Groenendyk J; Sreenivasaiah PK; Kim DH; Agellon LB; Michalak M Biology of endoplasmic reticulum stress in the heart. Circ. Res, 2010, 107(10), 1185–1197. [ 10.1161/CIRCRESAHA.110.227033] [DOI] [PubMed] [Google Scholar]
- [5].Schröder M; Kaufman RJ The mammalian unfolded protein response. Annu. Rev. Biochem, 2005, 74, 739–789. [ 10.1146/annurev.biochem.73.011303.074134] [DOI] [PubMed] [Google Scholar]
- [6].Lee AS The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci, 2001, 26(8), 504–510. [ 10.1016/S0968-0004(01)01908-9] [DOI] [PubMed] [Google Scholar]
- [7].Groenendyk J; Sreenivasaiah PK; Kim DH; Agellon LB; Michalak M Biology of endoplasmic reticulum stress in the heart. Circ. Res, 2010, 107(10), 1185–1197. [ 10.1161/CIRCRESAHA.110.227033] [DOI] [PubMed] [Google Scholar]
- [8].Walter P; Ron D The unfolded protein response: from stress pathway to homeostatic regulation. Science, 2011, 334(6059), 1081–1086. [ 10.1126/science.1209038] [DOI] [PubMed] [Google Scholar]
- [9].Hetz C; Martinon F; Rodriguez D; Glimcher LH The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol. Rev, 2011, 91(4), 1219–1243. [ 10.1152/physrev.00001.2011] [DOI] [PubMed] [Google Scholar]
- [10].Yoshida H; Matsui T; Yamamoto A; Okada T; Mori K XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell, 2001, 107(7), 881–891. [ 10.1016/S0092-8674(01)00611-0] [DOI] [PubMed] [Google Scholar]
- [11].Glembotski CC Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J. Mol. Cell. Cardiol, 2014, 71, 11–15. [ 10.1016/j.yjmcc.2013.09.018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lee AH; Scapa EF; Cohen DE; Glimcher LH Regulation of hepatic lipogenesis by the transcription factor XBP1. Science, 2008, 320(5882), 1492–1496. [ 10.1126/science.1158042] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Glimcher LH XBP1: The last two decades. Ann. Rheum. Dis, 2010, 69(Suppl. 1), i67–i71. [ 10.1136/ard.2009.119388] [DOI] [PubMed] [Google Scholar]
- [14].Wang S; Kaufman RJ The impact of the unfolded protein response on human disease. J. Cell Biol, 2012, 197(7), 857–867. [ 10.1083/jcb.201110131] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jin JK; Blackwood EA; Azizi K; Thuerauf DJ; Fahem AG; Hofmann C; Kaufman RJ; Doroudgar S; Glembotski CC ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ. Res, 2017, 120(5), 862–875. [ 10.1161/CIRCRESAHA.116.310266] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Blackwood EA; Azizi K; Thuerauf DJ; Paxman RJ; Plate L; Kelly JW; Wiseman RL; Glembotski CC Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun, 2019, 10(1), 187 [ 10.1038/s41467-018-08129-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tabas I; Ron D Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol, 2011, 13(3), 184–190. [ 10.1038/ncb0311-184] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ferri KF; Kroemer G Organelle-specific initiation of cell death pathways. Nat. Cell Biol, 2001, 3(11), E255–E263. [ 10.1038/ncb1101-e255] [DOI] [PubMed] [Google Scholar]
- [19].Kim DY; Kim HR; Kim KK; Park JW; Lee BJ NELL2 function in the protection of cells against endoplasmic reticulum stress. Mol. Cells, 2015, 38(2), 145–150. [ 10.1007/s10059-013-0117-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Thuerauf DJ; Marcinko M; Gude N; Rubio M; Sussman MA; Glembotski CC Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res, 2006, 99(3), 275–282. [ 10.1161/01.RES.0000233317.70421.03] [DOI] [PubMed] [Google Scholar]
- [21].Okada K; Minamino T; Tsukamoto Y; Liao Y; Tsukamoto O; Takashima S; Hirata A; Fujita M; Nagamachi Y; Nakatani T; Yutani C; Ozawa K; Ogawa S; Tomoike H; Hori M; Kitakaze M Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation, 2004, 110(6), 705–712. [ 10.1161/01.CIR.0000137836.95625.D4] [DOI] [PubMed] [Google Scholar]
- [22].Hamada H; Suzuki M; Yuasa S; Mimura N; Shinozuka N; Takada Y; Suzuki M; Nishino T; Nakaya H; Koseki H; Aoe T Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol. Cell. Biol, 2004, 24(18), 8007–8017. [ 10.1128/MCB.24.18.8007-8017.2004] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gargalovic PS; Gharavi NM; Clark MJ; Pagnon J; Yang WP; He A; Truong A; Baruch-Oren T; Berliner JA; Kirchgessner TG; Lusis AJ The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler. Thromb. Vasc. Biol, 2006, 26(11), 2490–2496. [ 10.1161/01.ATV.0000242903.41158.a1] [DOI] [PubMed] [Google Scholar]
- [24].Tabas I The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res, 2010, 107(7), 839–850. [ 10.1161/CIRCRESAHA.110.224766] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li RJ; He KL; Li X; Wang LL; Liu CL; He YY Salubrinal protects cardiomyocytes against apoptosis in a rat myocardial infarction model via suppressing the dephosphorylation of eukaryotic translation initiation factor 2α. Mol. Med. Rep, 2015, 12(1), 1043–1049. [ 10.3892/mmr.2015.3508] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Korennykh AV; Egea PF; Korostelev AA; Finer-Moore J; Zhang C; Shokat KM; Stroud RM; Walter P The unfolded protein response signals through high-order assembly of Ire 1. Nature, 2009, 457(7230), 687–693. [ 10.1038/nature07661] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chu TF; Rupnick MA; Kerkela R; Dallabrida SM; Zurakowski D; Nguyen L; Woulfe K; Pravda E; Cassiola F; Desai J; George S; Morgan JA; Harris DM; Ismail NS; Chen JH; Schoen FJ; Van den Abbeele AD; Demetri GD; Force T; Chen MH Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet, 2007, 370(9604), 2011–2019. [ 10.1016/S0140-6736(07)61865-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Perlmutter DH Chemical chaperones: A pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res, 2002, 52(6), 832–836. [ 10.1203/00006450-200212000-00004] [DOI] [PubMed] [Google Scholar]
- [29].Park CS; Cha H; Kwon EJ; Sreenivasaiah PK; Kim DH The chemical chaperone 4-phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem. Biophys. Res. Commun, 2012, 421(3), 578–584. [http://dx.doi.org/10.1016Zj.bbrc.2012.04.048] [DOI] [PubMed] [Google Scholar]
- [30].Ayala P; Montenegro J; Vivar R; Letelier A; Urroz PA; Copaja M; Pivet D; Humeres C; Troncoso R; Vicencio JM; Lavandero S; Diaz-Araya G Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp. Mol. Pathol, 2012, 92(1), 97–104. [ 10.1016/j.yexmp.2011.10.012] [DOI] [PubMed] [Google Scholar]
- [31].Guo R; Ma H; Gao F; Zhong L; Ren J Metallothionein alleviates oxidative stress-induced endoplasmic reticulum stress and myocardial dysfunction. J. Mol. Cell. Cardiol, 2009, 47(2), 228–237. [ 10.1016/j.yjmcc.2009.03.018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cornejo VH; Pihan P; Vidal RL; Hetz C Role of the unfolded protein response in organ physiology: Lessons from mouse models. IUBMB Life, 2013, 65(12), 962–975. [ 10.1002/iub.1224] [DOI] [PubMed] [Google Scholar]
- [33].Masaki T; Yoshida M; Noguchi S Targeted disruption of CRE-binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem. Biophys. Res. Commun, 1999, 261(2), 350–356. [ 10.1006/bbrc.1999.0972] [DOI] [PubMed] [Google Scholar]
- [34].Chen Y; Brandizzi F IRE1: ER stress sensor and cell fate executor. Trends Cell Biol, 2013, 23(11), 547–555. [ 10.1016/j.tcb.2013.06.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gao Y; Sartori DJ; Li C; Yu QC; Kushner JA; Simon MC; Diehl JA PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol. Cell. Biol, 2012, 32(24), 5129–5139. [ 10.1128/MCB.01009-12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen X; Zhang F; Gong Q; Cui A; Zhuo S; Hu Z; Han Y; Gao J; Sun Y; Liu Z; Yang Z; Le Y; Gao X; Dong LQ; Gao X; Li Y Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosis in mice through peroxisome proliferator-activated receptor α. Diabetes, 2016, 65(7), 1904–1915. [ 10.2337/db15-1637] [DOI] [PubMed] [Google Scholar]
- [37].Little E; Ramakrishnan M; Roy B; Gazit G; Lee AS The glucose-regulated proteins (GRP78 and GRP94): Functions, gene regulation, and applications. Crit. Rev. Eukaryot. Gene Expr, 1994, 4(1), 1–18. [ 10.1615/CritRevEukarGeneExpr.v4.i1.10] [DOI] [PubMed] [Google Scholar]
- [38].Koumenis C ER stress, hypoxia tolerance and tumor progression. Curr. Mol. Med, 2006, 6(1), 55–69. [ 10.2174/156652406775574604] [DOI] [PubMed] [Google Scholar]
- [39].Kao C; Chandna R; Ghode A; Dsouza C; Chen M; Larsson A; Lim SH; Wang M; Cao Z; Zhu Y; Anand GS; Ge R Proapoptotic cyclic peptide BC71 targets cell-surface GRP78 and functions as an anticancer therapeutic in mice. EBioMedicine, 2018, 33, 22–32. [ 10.1016/j.ebiom.2018.06.004] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Araujo N; Hebbar N; Rangnekar VM GRP78 is a targetable receptor on cancer and stromal cells. EBioMedicine, 2018, 33, 2–3. [ 10.1016/j.ebiom.2018.06.030] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang Y; Liu R; Ni M; Gill P; Lee AS Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem, 2010, 285(20), 15065–15075. [ 10.1074/jbc.M109.087445] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Birukova AA; Singleton PA; Gawlak G; Tian X; Mirzapoiazova T; Mambetsariev B; Dubrovskyi O; Oskolkova OV; Bochkov VN; Birukov KG GRP78 is a novel receptor initiating a vascular barrier protective response to oxidized phospholipids. Mol. Biol. Cell, 2014, 25(13), 2006–2016. [ 10.1091/mbc.e13-12-0743] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tsai YL; Ha DP; Zhao H; Carlos AJ; Wei S; Pun TK; Wu K; Zandi E; Kelly K; Lee AS Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-β signaling. Proc. Natl. Acad. Sci. USA, 2018, 115(18), E4245–E4254. [ 10.1073/pnas.1714866115] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Misra UK; Deedwania R; Pizzo SV Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J. Biol. Chem, 2005, 280(28), 26278–26286. [ 10.1074/jbc.M414467200] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sokolowska I; Woods AG; Gawinowicz MA; Roy U; Darie CC Identification of a potential tumor differentiation factor receptor candidate in prostate cancer cells. FEBS J, 2012, 279(14), 2579–2594. [ 10.1111/j.1742-4658.2012.08641.x] [DOI] [PubMed] [Google Scholar]
- [46].Ni M; Zhang Y; Lee AS Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J, 2011, 434(2), 181–188. [ 10.1042/BJ20101569] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Philippova M; Ivanov D; Joshi MB; Kyriakakis E; Rupp K; Afonyushkin T; Bochkov V; Erne P; Resink TJ Identification of proteins associating with glycosylphosphatidylinositol- anchored T-cadherin on the surface of vascular endothelial cells: Role for Grp78/BiP in T-cadherin-dependent cell survival. Mol. Cell. Biol, 2008, 28(12), 4004–4017. [ 10.1128/MCB.00157-08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhu G; Lee AS Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol, 2015, 230(7), 1413–1420. [ 10.1002/jcp.24923] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Luo S; Mao C; Lee B; Lee AS GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol, 2006, 26(15), 5688–5697. [ 10.1128/MCB.00779-06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhu G; Ye R; Jung DY; Barron E; Friedline RH; Benoit VM; Hinton DR; Kim JK; Lee AS GRP78 plays an essential role in adipogenesis and postnatal growth in mice. FASEB J, 2013, 27(3), 955–964. [ 10.1096/fj.12-213330] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Flodby P; Li C; Liu Y; Wang H; Marconett CN; Laird-Offringa IA; Minoo P; Lee AS; Zhou B The 78-kD Glucose-regulated protein regulates endoplasmic reticulum homeostasis and distal epithelial cell survival during lung development. Am. J. Respir. Cell Mol. Biol, 2016, 55(1), 135–149. [ 10.1165/rcmb.2015-03270C] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang X; Bi X; Zhang G; Deng Y; Luo X; Xu L; Scherer PE; Ferdous A; Fu G; Gillette TG; Lee AS; Jiang X; Wang ZV Glucose-regulated protein 78 is essential for cardiac myocyte survival. Cell Death Differ, 2018, 25(12), 2181–2194. [ 10.1038/s41418-018-0109-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Thuerauf DJ; Marcinko M; Gude N; Rubio M; Sussman MA; Glembotski CC Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res, 2006, 99(3), 275–282. [ 10.1161/01.RES.0000233317.70421.03] [DOI] [PubMed] [Google Scholar]
- [54].Wang ZV; Deng Y; Gao N; Pedrozo Z; Li DL; Morales CR; Criollo A; Luo X; Tan W; Jiang N; Lehrman MA; Rothermel BA; Lee AH; Lavandero S; Mammen PPA; Ferdous A; Gillette TG; Scherer PE; Hill JA Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell, 2014, 156(6), 1179–1192. [ 10.1016/j.cell.2014.01.014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bi X; Zhang G; Wang X; Nguyen C; May HI; Li X; Al-Hashimi AA; Austin RC; Gillette TG; Fu G; Wang ZV; Hill JA Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation. Circ. Res, 2018, 122(11), 1545–1554. [ 10.1161/CIRCRESAHA.117.312641] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang G; Wang X; Bi X; Li C; Deng Y; Al-Hashimi AA; Luo X; Gillette TG; Austin RC; Wang Y; Wang ZV GRP78 (Glucose-regulated protein of 78 kDa) promotes cardiomyocyte growth through activation of GATA4 (GATA-binding protein 4). Hypertension, 2019, 73(2), 390–398. [ 10.1161/HYPERTENSI0NAHA.118.12084] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gao G; Xie A; Zhang J; Herman AM; Jeong EM; Gu L; Liu M; Yang KC; Kamp TJ; Dudley SC Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ Arrhythm Electrophysiol, 2013, 6(5), 1018–1024. [ 10.1161/CIRCEP.113.000274] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ross CA; Poirier MA Protein aggregation and neurodegenerative disease. Nat. Med, 2004, 10(Suppl), S10–17. [ 10.1038/nm1066] [DOI] [PubMed] [Google Scholar]
- [59].Selkoe DJ Folding proteins in fatal ways. Nature, 2003, 426(6968), 900–904. [ 10.1038/nature02264] [DOI] [PubMed] [Google Scholar]
- [60].Taylor JP; Hardy J; Fischbeck KH Toxic proteins in neurodegenerative disease. Science, 2002, 296(5575), 1991–1995. [ 10.1126/science.1067122] [DOI] [PubMed] [Google Scholar]
- [61].Kopito RR; Ron D Conformational disease. Nat. Cell Biol, 2000, 2(11), E207–E209. [ 10.1038/35041139] [DOI] [PubMed] [Google Scholar]
- [62].Kudo T; Kanemoto S; Hara H; Morimoto N; Morihara T; Kimura R; Tabira T; Imaizumi K; Takeda M A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ, 2008, 15(2), 364–375. [ 10.1038/sj.cdd.4402276] [DOI] [PubMed] [Google Scholar]
- [63].Nakanishi T; Shimazawa M; Sugitani S; Kudo T; Imai S; Inokuchi Y; Tsuruma K; Hara H Role of endoplasmic reticulum stress in light-induced photoreceptor degeneration in mice. J. Neurochem, 2013, 125(1), 111–124. [ 10.1111/jnc.12116] [DOI] [PubMed] [Google Scholar]
- [64].Shimazawa M; Inokuchi Y; Okuno T; Nakajima Y; Sakaguchi G; Kato A; Oku H; Sugiyama T; Kudo T; Ikeda T; Takeda M; Hara H Reduced retinal function in amyloid precursor protein-over-expressing transgenic mice via attenuating glutamate-N-methyl-d-aspartate receptor signaling. J. Neurochem, 2008, 107(1), 279–290. [ 10.1111/j.1471-4159.2008.05606.x] [DOI] [PubMed] [Google Scholar]
- [65].Oida Y; Hamanaka J; Hyakkoku K; Shimazawa M; Kudo T; Imaizumi K; Yasuda T; Hara H Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neurosci. Lett, 2010, 484(1), 43–46. [ 10.1016/j.neulet.2010.08.015] [DOI] [PubMed] [Google Scholar]
- [66].Takano K; Tabata Y; Kitao Y; Murakami R; Suzuki H; Yamada M; Iinuma M; Yoneda Y; Ogawa S; Hori O Methoxy-flavones protect cells against endoplasmic reticulum stress and neurotoxin. Am. J. Physiol. Cell Physiol, 2007, 292(1), C353–C361. [ 10.1152/ajpcell.00388.2006] [DOI] [PubMed] [Google Scholar]
- [67].Lee AS Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer, 2014, 14(4), 263–276. [ 10.1038/nrc3701] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang M; Wey S; Zhang Y; Ye R; Lee AS Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal, 2009, 11(9), 2307–2316. [ 10.1089/ars.2009.2485] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Casas C GRP78 at the centre of the stage in cancer and neuroprotection. Front. Neurosci, 2017, 11, 177 [ 10.3389/fnins.2017.00177] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bytzek AK; Koellensperger G; Keppler BK; G Hartinger C Biodistribution of the novel anticancer drug sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)] KP-1339/IT139 in nude BALB/c mice and implications on its mode of action. J. Inorg. Biochem, 2016, 160, 250–255. [ 10.1016/jjinorgbio.2016.02.037] [DOI] [PubMed] [Google Scholar]
- [71].Burris HA; Bakewell S; Bendell JC; Infante J; Jones SF; Spigel DR; Weiss GJ; Ramanathan RK; Ogden A; Von Hoff D Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: a first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open, 2017, 1(6), e000154 [ 10.1136/esmoopen-2016-000154] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chang SW; Lewis AR; Prosser KE; Thompson JR; Gladkikh M; Bally MB; Warren JJ; Walsby CJ CF3 derivatives of the anticancer Ru(III) complexes KP1019, NKP-1339, and their imidazole and pyridine analogues show enhanced lipophilicity, albumin interactions, and cytotoxicity. Inorg. Chem, 2016, 55(10), 4850–4863. [ 10.1021/acs.inorgchem.6b00359] [DOI] [PubMed] [Google Scholar]
- [73].Dömötör O; Hartinger CG; Bytzek AK; Kiss T; Keppler BK; Enyedy EA Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. J. Biol. Inorg. Chem, 2013, 18(1), 9–17. [ 10.1007/s00775-012-0944-6] [DOI] [PubMed] [Google Scholar]
- [74].Schonhacker-Alte B; Baier D; Mohr T; Pirker C; Buck A; Hofmann T; Keppler B; Berger W; Heffeter P Update on NKP-1339/IT-139, a ruthenium-based GRP78 inhibitor in clinical development. Oncol. Res. Treat, 2018, 41, 47–48.29402861 [Google Scholar]
- [75].Lentz F; Drescher A; Lindauer A; Henke M; Hilger RA; Hartinger CG; Scheulen ME; Dittrich C; Keppler BK; Jaehde U Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs, 2009, 20(2), 97–103. [ 10.1097/CAD.0b013e328322fbc5] [DOI] [PubMed] [Google Scholar]
- [76].Cerezo M; Lehraiki A; Millet A; Rouaud F; Plaisant M; Jaune E; Botton T; Ronco C; Abbe P; Amdouni H; Passeron T; Hofman V; Mograbi B; Dabert-Gay AS; Debayle D; Alcor D; Rabhi N; Annicotte JS; Héliot L; Gonzalez-Pisfil M; Robert C; Moréra S; Vigouroux A; Gual P; Ali MMU; Bertolotto C; Hofman P; Ballotti R; Benhida R; Rocchi S compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell, 2016, 29(6), 805–819. [ 10.1016/j.ccell.2016.04.013] [DOI] [PubMed] [Google Scholar]
- [77].Ronco C; Millet A; Plaisant M; Abbe P; Hamouda-Tekaya N; Rocchi S; Benhida R Structure activity relationship and optimization of N-(3-(2-aminothiazol-4-yl)aryl)benzenesulfonamides as anti-cancer compounds against sensitive and resistant cells. Bioorg. Med. Chem. Lett, 2017, 27(10), 2192–2196. [ 10.1016/j.bmcl.2017.03.054] [DOI] [PubMed] [Google Scholar]
- [78].Bhattacharjee R; Devi A; Mishra S Molecular docking and molecular dynamics studies reveal structural basis of inhibition and selectivity of inhibitors EGCG and OSU-03012 toward glucose regulated protein-78 (GRP78) overexpressed in glioblastoma. J. Mol. Model, 2015, 21(10), 272 [ 10.1007/s00894-015-2801-3] [DOI] [PubMed] [Google Scholar]
- [79].Booth L; Cazanave SC; Hamed HA; Yacoub A; Ogretmen B; Chen CS; Grant S; Dent P OSU-03012 suppresses GRP78/BiP expression that causes PERK-dependent increases in tumor cell killing. Cancer Biol. Ther, 2012, 13(4), 224–236. [ 10.4161/cbt.13.4.18877] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Park MA; Yacoub A; Rahmani M; Zhang G; Hart L; Hagan MP; Calderwood SK; Sherman MY; Koumenis C; Spiegel S; Chen CS; Graf M; Curiel DT; Fisher PB; Grant S; Dent P OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells. Mol. Pharmacol, 2008, 73(4), 1168–1184. [ 10.1124/mol.107.042697] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Park HR; Ryoo IJ; Choo SJ; Hwang JH; Kim JY; Cha MR; Shin-Ya K; Yoo ID Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 downregulator. Toxicology, 2007, 229(3), 253–261. [ 10.1016/j.tox.2006.11.049] [DOI] [PubMed] [Google Scholar]
- [82].Thomas S; Sharma N; Gonzalez R; Pao PW; Hofman FM; Chen TC; Louie SG; Pirrung MC; Schönthal AH Repositioning of Verrucosidin, a purported inhibitor of chaperone protein GRP78, as an inhibitor of mitochondrial electron transport chain complex I. PLoS One, 2013, 8(6), e65695 [ 10.1371/journal.pone.0065695] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kim JY; Hwang JH; Cha MR; Yoon MY; Son ES; Tomida A; Ko B; Song SW; Shin-ya K; Hwang YI; Park HR Arctigenin blocks the unfolded protein response and shows therapeutic antitumor activity. J. Cell. Physiol, 2010, 224(1), 33–40. [ 10.1002/jcp.22085] [DOI] [PubMed] [Google Scholar]
- [84].Kato K; Gong J; Iwama H; Kitanaka A; Tani J; Miyoshi H; Nomura K; Mimura S; Kobayashi M; Aritomo Y; Kobara H; Mori H; Himoto T; Okano K; Suzuki Y; Murao K; Masaki T The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol. Cancer Ther, 2012, 11(3), 549–560. [ 10.1158/1535-7163.MCT-11-0594] [DOI] [PubMed] [Google Scholar]
- [85].Yu DH; Macdonald J; Liu G; Lee AS; Ly M; Davis T; Ke N; Zhou D; Wong-Staal F; Li QX Pyrvinium targets the unfolded protein response to hypoglycemia and its anti-tumor activity is enhanced by combination therapy. PLoS One, 2008, 3(12), e3951 [ 10.1371/journal.pone.0003951] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Park HR; Tomida A; Sato S; Tsukumo Y; Yun J; Yamori T; Hayakawa Y; Tsuruo T; Shin-ya K Effect on tumor cells of blocking survival response to glucose deprivation. J. Natl. Cancer Inst, 2004, 96(17), 1300–1310. [ 10.1093/jnci/djh243] [DOI] [PubMed] [Google Scholar]
- [87].Maddalo D; Neeb A; Jehle K; Schmitz K; Muhle-Goll C; Shatkina L; Walther TV; Bruchmann A; Gopal SM; Wenzel W; Ulrich AS; Cato AC A peptidic unconjugated GRP78/BiP ligand modulates the unfolded protein response and induces prostate cancer cell death. PLoS One, 2012, 7(10), e45690 [ 10.1371/journal.pone.0045690] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cunningham CC; Chada S; Merritt JA; Tong A; Senzer N; Zhang Y; Mhashilkar A; Parker K; Vukelja S; Richards D; Hood J; Coffee K; Nemunaitis J Clinical and local biological effects of an intratumoral injection of mda-7 (IL24; INGN 241) in patients with advanced carcinoma: a phase I study. Mol. Ther, 2005, 11(1), 149–159. [ 10.1016/j.ymthe.2004.09.019] [DOI] [PubMed] [Google Scholar]
- [89].Gupta P; Walter MR; Su ZZ; Lebedeva IV; Emdad L; Randolph A; Valerie K; Sarkar D; Fisher PB BiP/GRP78 is an intracellular target for MDA-7/IL-24 induction of cancer-specific apoptosis. Cancer Res, 2006, 66(16), 8182–8191. [ 10.1158/0008-5472.CAN-06-0577] [DOI] [PubMed] [Google Scholar]
- [90].Arap MA; Lahdenranta J; Mintz PJ; Hajitou A; Sarkis AS; Arap W; Pasqualini R Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell, 2004, 6(3), 275–284. [ 10.1016/j.ccr.2004.08.018] [DOI] [PubMed] [Google Scholar]
- [91].Yoneda Y; Steiniger SCJ; Capkova K; Mee JM; Liu Y; Kaufmann GF; Janda KD A cell-penetrating peptidic GRP78 ligand for tumor cell-specific prodrug therapy. Bioorg. Med. Chem. Lett, 2008, 18(5), 1632–1636. [ 10.1016/j.bmcl.2008.01.060] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Katanasaka Y; Ishii T; Asai T; Naitou H; Maeda N; Koizumi F; Miyagawa S; Ohashi N; Oku N Cancer antineovascular therapy with liposome drug delivery systems targeted to BiP/GRP78. Int. J. Cancer, 2010, 127(11), 2685–2698. [ 10.1002/ijc.25276] [DOI] [PubMed] [Google Scholar]
- [93].Passarella RJ; Spratt DE; van der Ende AE; Phillips JG; Wu H; Sathiyakumar V; Zhou L; Hallahan DE; Harth E; Diaz R Targeted nanoparticles that deliver a sustained, specific release of Paclitaxel to irradiated tumors. Cancer Res, 2010, 70(11), 4550–4559. [ 10.1158/0008-5472.CAN-10-0339] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Saito A; Ochiai K; Kondo S; Tsumagari K; Murakami T; Cavener DR; Imaizumi K Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J. Biol. Chem, 2011, 286(6), 4809–4818. [ 10.1074/jbc.M110.152900] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Cui W; Li J; Ron D; Sha B The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D Biol. Crystallogr, 2011, 67(Pt 5), 423–428. [ 10.1107/S0907444911006445] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Liu X; Kwak D; Lu Z; Xu X; Fassett J; Wang H; Wei Y; Cavener DR; Hu X; Hall J; Bache RJ; Chen Y Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension, 2014, 64(4), 738–744. [ 10.1161/HYPERTENSIONAHA.114.03811] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu ZW; Zhu HT; Chen KL; Dong X; Wei J; Qiu C; Xue JH Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc. Diabetol, 2013, 12, 158 [ 10.1186/1475-2840-12-158] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].McAlpine CS; Werstuck GH Protein kinase R-like endoplasmic reticulum kinase and glycogen synthase kinase-3α/β regulate foam cell formation. J. Lipid Res, 2014, 55(11), 2320–2333. [ 10.1194/jlr.M051094] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Axten JM Protein kinase R(PKR)-like endoplasmic reticulum kinase (PERK) inhibitors: a patent review (2010–2015). Expert Opin. Ther. Pat, 2017, 27(1), 37–48. [ 10.1080/13543776.2017.1238072] [DOI] [PubMed] [Google Scholar]
- [100].Han J; Back SH; Hur J; Lin YH; Gildersleeve R; Shan J; Yuan CL; Krokowski D; Wang S; Hatzoglou M; Kilberg MS; Sartor MA; Kaufman RJ ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol, 2013, 15(5), 481–490. [ 10.1038/ncb2738] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lu PD; Jousse C; Marciniak SJ; Zhang Y; Novoa I; Scheuner D; Kaufman RJ; Ron D; Harding HP Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J, 2004, 23(1), 169–179. [ 10.1038/sj.emboj.7600030] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ranganathan AC; Ojha S; Kourtidis A; Conklin DS; Aguirre-Ghiso JA Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res, 2008, 68(9), 3260–3268. [ 10.1158/0008-5472.CAN-07-6215] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Wang L; Popko B; Tixier E; Roos RP Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis, 2014, 71, 317–324. [ 10.1016/j.nbd.2014.08.010] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Way SW; Popko B Harnessing the integrated stress response for the treatment of multiple sclerosis. Lancet Neurol, 2016, 15(4), 434–443. [ 10.1016/S1474-4422(15)00381-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Smith AL; Andrews KL; Beckmann H; Bellon SF; Beltran PJ; Booker S; Chen H; Chung YA; D’Angelo ND; Dao J; Dellamaggiore KR; Jaeckel P; Kendall R; Labitzke K; Long AM; Materna-Reichelt S; Mitchell P; Norman MH; Powers D; Rose M; Shaffer PL; Wu MM; Lipford JR Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J. Med. Chem, 2015, 58(3), 1426–1441. [ 10.1021/jm5017494] [DOI] [PubMed] [Google Scholar]
- [106].Wang H; Blais J; Ron D; Cardozo T Structural determinants of PERK inhibitor potency and selectivity. Chem. Biol. Drug Des, 2010, 76(6), 480–495. [ 10.1111/j.1747-0285.2010.01048.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Pytel D; Majsterek I; Diehl JA Tumor progression and the different faces of the PERK kinase. Oncogene, 2016, 35(10), 1207–1215. [ 10.1038/onc.2015.178] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Moreno JA; Halliday M; Molloy C; Radford H; Verity N; Axten JM; Ortori CA; Willis AE; Fischer PM; Barrett DA; Mallucci GR Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med, 2013, 5(206), 206ra138 [ 10.1126/scitranslmed.3006767] [DOI] [PubMed] [Google Scholar]
- [109].Nijholt DAT; van Haastert ES; Rozemuller AJM; Scheper W; Hoozemans JJM The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol, 2012, 226(5), 693–702. [ 10.1002/path.3969] [DOI] [PubMed] [Google Scholar]
- [110].Shen J; Chen X; Hendershot L; Prywes R ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell, 2002, 3(1), 99–111. [ 10.1016/S1534-5807(02)00203-4] [DOI] [PubMed] [Google Scholar]
- [111].Doroudgar S; Thuerauf DJ; Marcinko MM; Glembotski CC Simulated .ischemia activates the ATF6 branch of the endoplasmic reticulum stress response in cultured cardiac myocytes. Circ.n Res, 2008, 103(5), E69–E70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Doroudgar S; Thuerauf DJ; Marcinko MC; Belmont PJ; Glembotski CC Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem, 2009, 284(43), 29735–29745. [ 10.1074/jbc.M109.018036] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Martindale JJ; Fernandez R; Thuerauf D; Whittaker R; Gude N; Sussman MA; Glembotski CC Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res, 2006, 98(9), 1186–1193. [ 10.1161/01.RES.0000220643.65941.8d] [DOI] [PubMed] [Google Scholar]
- [114].Blackwood EA; Hofmann C; Santo Domingo M; Bilal AS; Sarakki A; Stauffer W; Arrieta A; Thuerauf DJ; Kolkhorst FW; Müller OJ; Jakobi T; Dieterich C; Katus HA; Doroudgar S; Glembotski CC ATF6 regulates cardiac hypertrophy by transcriptional induction of the mTORC1 activator, Rheb. Circ. Res, 2019, 124(1), 79–93. [ 10.1161/CIRCRESAHA.118.313854] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Liu Z; Zhang Y; Tang Z; Xu J; Ma M; Pan S; Qiu C; Guan G; Wang J Matrine attenuates cardiac fibrosis by affecting ATF6 signaling pathway in diabetic cardiomyopathy. Eur. J. Pharmacol, 2017, 804, 21–30. [ 10.1016/j.ejphar.2017.03.061] [DOI] [PubMed] [Google Scholar]
- [116].Paxman R; Plate L; Blackwood EA; Glembotski C; Powers ET; Wiseman RL; Kelly JW Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. eLife, 2018, 7, 7 [ 10.7554/eLife.37168] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Tam AB; Roberts LS; Chandra V; Rivera IG; Nomura DK; Forbes DJ; Niwa M The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev. Cell, 2018, 46(3), 327–343.e7. [ 10.1016/j.devcel.2018.04.023] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Gallagher CM; Garri C; Cain EL; Ang KKH; Wilson CG; Chen S; Hearn BR; Jaishankar P; Aranda-Diaz A; Arkin MR; Renslo AR; Walter P Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6 alpha branch. eLife, 2016, 5, e11878 [DOI: 10.7554/eLife.11878] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Lebeau P; Byun JH; Yousof T; Austin RC Pharmacologic inhibition of S1P attenuates ATF6 expression, causes ER stress and contributes to apoptotic cell death. Toxicol. Appl. Pharmacol, 2018, 349, 1–7. [ 10.1016/j.taap.2018.04.020] [DOI] [PubMed] [Google Scholar]
- [120].Calfon M; Zeng H; Urano F; Till JH; Hubbard SR; Harding HP; Clark SG; Ron D IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature, 2002, 415(6867), 92–96. [ 10.1038/415092a] [DOI] [PubMed] [Google Scholar]
- [121].Wang ZV; Hill JA Protein quality control and metabolism: bidirectional control in the heart. Cell Metab, 2015, 21(2), 215–226. [ 10.1016/j.cmet.2015.01.016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Steiger D; Yokota T; Li J; Ren S; Minamisawa S; Wang Y The serine/threonine-protein kinase/endoribonuclease IRE1α protects the heart against pressure overload-induced heart failure. J. Biol. Chem, 2018, 293(25), 9652–9661. [ 10.1074/jbc.RA118.003448] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Yao S; Miao C; Tian H; Sang H; Yang N; Jiao P; Han J; Zong C; Qin S Endoplasmic reticulum stress promotes macrophage-derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression. J. Biol. Chem, 2014, 289(7), 4032–4042. [ 10.1074/jbc.M113.524512] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Wang X; Xu L; Gillette TG; Jiang X; Wang ZV The unfolded protein response in ischemic heart disease. J. Mol. Cell. Cardiol, 2018, 117, 19–25. [ 10.1016/j.yjmcc.2018.02.013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Duan Q; Ni L; Wang P; Chen C; Yang L; Ma B; Gong W; Cai Z; Zou MH; Wang DW Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell, 2016, 15(4), 625–633. [ 10.1111/acel.12460] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Vincenz L; Hartl FU Sugarcoating ER stress. Cell, 2014, 156(6), 1125–1127. [ 10.1016/j.cell.2014.02.035] [DOI] [PubMed] [Google Scholar]
- [127].Glembotski CC Finding the missing link between the unfolded protein response and O-GlcNAcylation in the heart. Circ. Res, 2014, 115(6), 546–548. [ 10.1161/CIRCRESAHA.114.304855] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Zeng L; Zampetaki A; Margariti A; Pepe AE; Alam S; Martin D; Xiao Q; Wang W; Jin ZG; Cockerill G; Mori K; Li YS; Hu Y; Chien S; Xu Q Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA, 2009, 106(20), 8326–8331. [ 10.1073/pnas.0903197106] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Tufanli O; Telkoparan Akillilar P; Acosta-Alvear D; Kocaturk B; Onat UI; Hamid SM; Çimen I; Walter P; Weber C; Erbay E Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA, 2017, 114(8), E1395–E1404. [ 10.1073/pnas.1621188114] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Volkmann K; Lucas JL; Vuga D; Wang X; Brumm D; Stiles C; Kriebel D; Der-Sarkissian A; Krishnan K; Schweitzer C; Liu Z; Malyankar UM; Chiovitti D; Canny M; Durocher D; Sicheri F; Patterson JB Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem, 2011, 286(14), 12743–12755. [ 10.1074/jbc.M110.199737] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mimura N; Fulciniti M; Gorgun G; Tai YT; Cirstea D; Santo L; Hu Y; Fabre C; Minami J; Ohguchi H; Kiziltepe T; Ikeda H; Kawano Y; French M; Blumenthal M; Tam V; Kertesz NL; Malyankar UM; Hokenson M; Pham T; Zeng Q; Patterson JB; Richardson PG; Munshi NC; Anderson KC Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood, 2012, 119(24), 5772–5781. [ 10.1182/blood-2011-07-366633] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ri M; Tashiro E; Oikawa D; Shinjo S; Tokuda M; Yokouchi Y; Narita T; Masaki A; Ito A; Ding J; Kusumoto S; Ishida T; Komatsu H; Shiotsu Y; Ueda R; Iwawaki T; Imoto M; Iida S Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J, 2012, 2(7), e79 [ 10.1038/bcj.2012.26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Sanches M; Duffy NM; Talukdar M; Thevakumaran N; Chiovitti D; Canny MD; Lee K; Kurinov I; Uehling D; Alawar R; Poda G; Prakesch M; Wilson B; Tam V; Schweitzer C; Toro A; Lucas JL; Vuga D; Lehmann L; Durocher D; Zeng Q; Patterson JB; Sicheri F Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat. Commun, 2014, 5, 4202 [ 10.1038/ncomms5202] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wang SB; Wang ZZ; Fan QR; Guo J; Galli G; Du GH; Wang X; Xiao W Ginkgolide K protects the heart against ER stress injury by activating the IRE1 alpha/XBP1 pathway. Acta Pharmacol. Sin, 2017, 38(7), 1075–1075. [DOI: 10.1111/bph.13516] [DOI] [Google Scholar]
- [135].Duan Q; Chen C; Yang L; Li N; Gong W; Li S; Wang DW MicroRNA regulation of unfolded protein response transcription factor XBP1 in the progression of cardiac hypertrophy and heart failure in vivo. J. Transl. Med, 2015, 13, 363 [ 10.1186/s12967-015-0725-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Lederkremer GZ; Glickman MH A window of opportunity: timing protein degradation by trimming of sugars and ubiquitins. Trends Biochem. Sci, 2005, 30(6), 297–303. [ 10.1016/j.tibs.2005.04.010] [DOI] [PubMed] [Google Scholar]
- [137].Hwang J; Qi L Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci, 2018, 43(8), 593–605. [ 10.1016/j.tibs.2018.06.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Belmont PJ; Chen WJ; San Pedro MN; Thuerauf DJ; Gellings Lowe N; Gude N; Hilton B; Wolkowicz R; Sussman MA; Glembotski CC Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ. Res, 2010, 106(2), 307–316. [ 10.1161/CIRCRESAHA.109.203901] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Doroudgar S; Völkers M; Thuerauf DJ; Khan M; Mohsin S; Respress JL; Wang W; Gude N; Müller OJ; Wehrens XH; Sussman MA; Glembotski CC Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ. Res, 2015, 117(6), 536–546. [ 10.1161/CIRCRESAHA.115.306993] [DOI] [PMC free article] [PubMed] [Google Scholar]