

Abstract
Antimicrobial treatment failure threatens our ability to control infections. In addition to antimicrobial resistance, treatment failures are increasingly understood to derive from cells that survive antibiotic treatment without selection of genetically heritable mutations. Parasitic protozoa, such as Plasmodium species that cause malaria, Toxoplasma gondii and Kinetoplastid protozoa, including Trypanosoma cruzi and Leishmania spp., cause millions of deaths globally. These organisms can evolve drug resistance and they also exhibit phenotypic diversity, including the formation of quiescent or dormant forms that contribute to the establishment of long-term infections that are refractory to drug treatment, which we refer to as persister-like cells. In this Review, we discuss protozoan persister-like cells that have been linked to persistent infections and discuss their impact on therapeutic outcomes following drug treatment.
Table of content
Protozoa use various mechanisms to establish persistent infections. In the Review, Barrett and colleagues describe protozoan parasite ‘persister-like cells’, and they explore their possible role in persistent infections and drug treatment failure, and outline possible treatment options.
Introduction
Treatment failure of antimicrobial agents is a serious global health threat1. Drug resistance emerges from mutations in the microbial genome that enable microorganisms to survive in the presence of drugs2 and that are propagated in a genetically stable manner. Microorganisms can also be refractory to drugs without the selection of genetic mutations. In 1944, Irish physician Joseph Bigger3 noted that in a population of Staphylococcus aureus a small proportion of cells were refractory to penicillin cells within this subpopulation are termed persisters [G]. The formation of non-replicating but viable cells is common among bacteria4–7 and a fundamental and ancient evolutionary adaptive mechanism8, whereby a small sub-population of cells spontaneously enter dormancy or quiescence, terms we use interchangeably in this Review, given that different authors have used the terms arbitrarily in their studies describing cells that are non-replicating or replicating very slowly with associated metabolic alterations such as diminished DNA synthesis, and widespread downregulation of protein translation when compared to replicative cells. These cells are less vulnerable to a range of transient environmental stresses, including immunological attack and drug treatment8. Once those conditions pass, this population can return to a proliferative state and recolonizes the environment. Although the dormant state enables bacteria to survive stress, it is not necessarily triggered by stress, instead dormancy occurs spontaneously4–7 and recent preliminary data point to new approaches to identify and isolate low numbers of spontaneously arising Mycobacterium smegmatis persisters9; that is, a subpopulation of cells is prepared for the sudden onset of stress.
Eukaryotic cells also enter quiescent/dormant states that affect drug susceptibility -, and drug tolerant persister-like cell types have been described for both fungi10 and cancer cells11. Protozoan parasites are eukaryotes that cause millions of deaths each year. Treatment failure of anti-parasitic drugs is of concern, and drug resistance mechanisms in parasitic protozoa are increasingly understood and have recently been reviewed elsewhere12.
Protozoa use various mechanisms to establish persistent infections, including, for example, mechanisms to manipulate and avoid host immunity13,14. In this Review, we focus on parasite stages that are capable of entering a quiescent /dormant state and relate this phenomenon to drug treatment failure. In bacteria, metabolic divergence from proliferative cells is a hallmark of quiescence15, and a number of pathways have been implicated in metabolic adaptation have been identified, for example, toxin-antitoxin systems16 and stress response pathways4–7.
Several studies have shown that parasitic protozoa can generate various forms during their life cycle that have been linked to persistence. In particular, the hypnozoite [G] stages of Plasmodium vivax and related Plasmodium species have been shown to cause persistent infections and they have been linked to drug treatment failure. Moreover, it has been shown that Plasmodium falciparum parasites can enter a transient growth-arrested state as ring forms in erythrocytes, during which time they are refractory to the artemisinin-based drugs. In Toxoplasma gondii, the switch between the rapidly proliferative tachyzoite [G] form and the slow-replicating tissue cyst-bound bradyzoite [G] form underpins chronic infections that are refractory to treatment. In addition, it was recently shown that Trypanosoma cruzi can form dormant, transiently non-proliferating amastigote [G] forms17. Finally, evidence indicates Leishmania spp. form semi-quiescent amastigotes18. These protozoan parasites exhibit a loss of sensitivity to drugs that kill more rapidly proliferating cells because of phenotypic differences related to slow or arrested growth.
In this Review we describe these forms as ‘persister-like cells’. We describe the current knowledge of the observed forms, their possible role in persistent infections and treatment failure and outline possible treatment options. Table 1 compares key features of bacterial persister cells and their protozoan persister-like counterparts described in this Review.
Table 1:
Characteristics of bacterial persisters and protozoan persister-like cells
| Bacterial persister6 | Trypanosoma cruzi | Leishmania spp. | Plasmodium falciparum artemisinin refractory cells | Plasmodium vivax hypnozoites | Toxoplasma gondii bradyzoites in tissue cysts | |
|---|---|---|---|---|---|---|
| Cell growth | Sub-population of growth arrested or slow growing cells | Sub-population of growth-arrested cells | Sub-population of growth-arrested cells | Sub-population of growth-arrested cells | Sub-population of growth-arrested cells | Sub-population of semi-dormant cells |
| Drug sensitivity | Transient decreased sensitivity to drugs | Transient decreased sensitivity to drugs | Suspected transient sensitivity to drugs | Transient decreased sensitivity to artemisinin | Insensitive to most erythrocyte stage plasmodicidal drugs | Current therapies that are effective against tachyzoites are not effective against bradyzoites |
| Genetic background | Genetically identical to parent population | Presumed genetically identical to parent population | Presumed genetically identical to parent population | Mutations (for example, P. falciparum Kelch-13) confer increased survival possibly by increasing incidence of growth-arrested cells | Genetically identical to parent population | Genetically identical to tachyzoite population |
| Metabolism | Metabolically distinct from parent population | Presumed metabolically distinct from parent population | Presumed metabolically distinct from parent population | Presumed metabolically distinct from parent population | Metabolically distinct from parent population | Metabolically distinct from tachyzoite population |
| Reactivation | Recrudescence leads to the generation of a replicating population that can in turn generate low numbers of persisters | Recrudescence leads to the generation of a replicating population that can in turn generate low numbers of persister-like cells | Recrudescence presumed to lead to the generation of a replicating population | Recrudescence leads to the generation of a replicating population | A full life cycle needs to be completed before more hypnozoites can form | Reactivation of chronic infection occurs when bradyzoites differentiate back to the actively replicating tachyzoite stage |
| Persistent infection and treatment outcome | Contribute to persistent infections and treatment failure without resistance | Contribute to persistent infections and treatment failure without resistance | Contribute to persistent infections and treatment failure without resistance (for example, Post Kala azar dermal leishmaniasis leishmaniasis recidivans) | Contribute to persistent infections and recrudescence after artemisinin treatment | Contribute to persistent infections and treatment failure without resistance | Contribute to persistent infection treatment failure without resistance. |
Plasmodium species
Over 200 million people are infected with malaria-causing parasites of the genus Plasmodium, which cause the death of ~ half a million individuals annually19. The parasites have a complex life-cycle involving both an Anopheles mosquito vector and a vertebrate host. Infection begins when sporozoite [G] forms of human infectious Plasmodium species are injected by female Anopheles mosquitoes during a blood meal. Sporozoites rapidly invade liver cells, where the parasites undergo a proliferative stage, including substantial nuclear division without cell division to produce a schizont [G]. Subsequent cytokinesis results in the formation of multiple merozoites [G] that are released into the bloodstream and invade erythrocytes (developing into the erythrocytic stages). During infection with Plasmodium vivax in man, and Plasmodium cynomolgi in monkeys, a small number of liver stage parasites enter a different developmental stage, termed the hypnozoite, which can remain dormant for many months to years prior to entering the proliferative stage through schizogony. Within the erythrocyte, differentiation to a ring form, then a feeding trophozoite [G] that consumes haemoglobin precedes a further round of schizogony and the release of multiple merozoites. Depending on the species, this asexual erythrocyte cycle takes either two or three days. Some schizonts, instead of producing typical merozoites that proceed through further rounds of asexual reproduction, produce cells that upon reinfection of the next erythrocyte will form either male or female gametocytes. These non-proliferating gametocytes recombine when they enter a mosquito during a blood meal creating a zygote that further matures into a motile ookinete, which can migrate out of the mosquito midgut where it matures into an oocyst. Within the oocyst, sporozoite forms develop prior to bursting from the oocyst and these move to the salivary gland of the mosquito, from where the sporozoites are injected into the human host during the next blood meal of the mosquito.19 Sporozoites are a non-replicative form of the parasites. Persistent forms, outlined below, that affect therapeutic outcomes are known for some Plasmodium species that dwell in the liver and also during the erythrocytic stages. Other quiescent stages, for example the gametocytes, have also been considered as an important target for novel chemotherapy as they are refractory to most drugs used against replicative bloodstream forms20.
The hypnozoite.
Hypnozoites have been indicated in persistent infections with P. vivax, and they have been linked to drug treatment failure for some forms of malaria, particularly those caused by P. vivax and P. ovale in humans, and the monkey malaria parasite P. cynomolgi.
Hypnozoites can persist for weeks, months or even years before activation by unknown cues promotes schizogony21,22. These growth-arrested parasites arise spontaneously from a small number of the invading sporozoites, while most commence schizogony almost immediately [FIG.1A]. Interestingly, relapse arising from differentiation of hypnozoites into proliferative liver stage schizonts that in turn create merozoites may be more rapid in tropical strains, whereas in more temperate climates, hypnozoite-activation can occur a year or more after infection, to coincide with warm weather and high mosquito prevalence23. Morphologically, hypnozoites are uninucleate, non-growing stages first described in liver biopsies from P. vivax-infected chimpanzees and P. cynomolgi-infected Rhesus monkeys24,25. Hypnozoite-driven relapse is a major impediment to malaria elimination globally.
Figure 1. Persister-like cell types of Plasmodium spp.

A.The Plasmodium vivax hypnozoite. P. vivax sporozoites injected into the host by infected Anopheles mosquitoes rapidly infect hepatocytes. In the liver cells, the parasites undergo a proliferative stage, including substantial nuclear division without cell division to produce a schizont (schizogony). The schizont differentiates into merozoite forms that burst from hepatocytes and enter the bloodstream where they invade erythrocytes. Alternatively, some sporozoites will form hypnozoites that can persist in a growth-arrested state for weeks to many months before entering schizogony and completing the developmental cycle in the liver.
B. Artemisinin refractory dormant ring stages in P. falciparum
Exposure of synchronous ring stage P. falciparum parasites to dihydroartemisinin (DHA) induces cell-cycle arrest. These dormant ring stage forms are characterized morphologically by condensed chromatin and reduced cytoplasm. By contrast, pre-exposed ring stage forms have a relatively diffuse nucleus and large cytoplasmic area. Dormant ring stage parasites persist for several days before resuming normal growth. DHA-induced ring stage dormancy is observed in artemisinin-susceptible and artemisinin–resistant P. falciparum45, 47. The top panel shows Giemsa stained parasites (purple) inside erythrocytes. The lower panel provides a representation of the upper panel in which key structures visible in light microscopy are visible. The diameter of the erythrocyte is 6 micrometers.
Figure 1aadapted from Ref. 158. Fig 1b courtesy of Dennis E. Kyle (University of Georgia, Athens, USA).
The biology of hypnozoites is poorly understood, given the difficulties of studying a very rare parasite inside host liver cells and the lack of appropriate rodent models. However, several studies probing P. vivax or P. cynomolgi biology following sporozoite invasion of either immortalized hepatoma cells or primary hepatocytes have revealed key insights into hypnozoite biology. For example, histone acetyltransferase inhibitors can activate hypnozoites26 and the assessment of liver stage active drugs has been possible27,28. Transcriptome analysis of P. cynomolgi29 and P. vivax27 has revealed stage-specific gene expression, and comparison of P. cynomolgi liver stages at 9–10 days post infection30 with those at 6–7 days29 showed that schizonts express many genes and that most metabolic pathways are active. By contrast, hypnozoites, exhibit a marked decrease in the expression of genes for most biochemical pathways between days 6 and 9, which indicates progressive entry into a quiescent state. However, similar to other known persister cell types, quiescence does not involve the complete downregulation of metabolic pathways. Several pathways remain active, including those involved in protection against oxidative and other stresses, protein export, ATP maintenance and protein post-translational modifications, including those involved in genome structure (for example, histone methylation and acetylation)30, which suggests that epigenetic changes may contribute to the regulation of gene expression and heterochromatin structure in hypnozoites.
Fluorescent reporters have enabled the enrichment of P. cynomolgi liver stages31. A miniaturized culture system of P. vivax-infected primary human hepatocytes has offered new models to study liver stage infection28, 32 and can be used for high-content imaging-based assays to identify new hypnozoiticidal drugs. Immunodeficient mice, engrafted with human hepatocytes, have enabled the study of P. vivax liver stages33, including the elucidation of the hypnozoite maturation process, which enabled the testing of putative hypnozoiticidal drugs, and the identification of UIS4 (upregulated in infectious sporozoites protein 4) as a liver-specific marker for hypnozoites33,34 However, transcripts that are found only in hypnozoites and not other liver stages were not identified31.
Therapeutic options for hypnozoites are very limited. Most erythrocyte-stage active drugs are inactive against hypnozoites. This may be due to the absence of pathways such as heme degradation, which is targeted by chloroquine and the absence of pathways that are targeted by artemisinins. However, primaquine (PQ) and its newly approved analogue tafenoquine (TQ) are 8-aminoquinolines with proven efficacy against hypnozoites (and also liver stage schizonts). These drugs are most effective when given in combination with chloroquine or quinine that have some synergistic effect with PQ35,36. PQ is metabolized in the liver by the host cytochrome 2D6 into short-lived reactive metabolites. Different cytochrome 2D6 variants have been associated with reduced PQ metabolism in the host and diminished PQ efficacy37,38. However, reduced cytochrome 2D6 activity is not associated with decreased TQ efficacy39,40 even though TQ metabolites have been shown to eliminate liver stage parasites in rodents41.This suggests that unmetabolized TQ, or different metabolic routes may yield activity against liver stages for this drug. Unfortunately, 8-aminoquinoline metabolites can also cause serious side effects in glucose-6-phosphate dehydrogenase (G6PD)-deficient patients in whom hemolytic anemia [G] may occur. An estimated 35% of people at risk of malaria caused by P. vivax are therefore not eligible for PQ treatment either due to G6PD deficiency or to impaired 2D6 activity42. G6PD diagnostic tests will be used in conjunction with TQ following its approval in the US and Australia for human use, to exclude enzyme-deficient patients from treatment. New hypnozoiticidal drugs without G6PD liabilities are urgently needed, and the recent breakthroughs in hypnozoite models described above might expedite the discovery of novel avenues to target hypnozoites and thus enhance treatment success.
Recently discovered phosphatidylinositol 4-kinase (PI4K) inhibitors43 generated some excitement given their potency against liver stages of P. cynomolgi in vitro, yet treatment with such inhibitors failed to prevent relapses in Rhesus monkeys challenged with sporozoites of P. cynomolgi44. Despite this result, important insights into hypnozoite biology emerged from these studies; crucially, it was shown that hypnozoites mature by day 4–5 after infection of hepatocytes into PI4K inhibitor-refractory forms, whereas exposure to these inhibitors during early infection eliminates liver stage infection.
Artemisinin-induced dormant ring stages.
In addition to hypnozoites, recent evidence has pointed to another type of transiently growth-arrested form of Plasmodium falciparum that profoundly affects malaria therapy. Exposure of ring-stages of P. falciparum to artemisinin [G] derivatives (for example, dihydroartemisinin (DHA)) can induce a cell-cycle arrest that is time and dose dependent. These non-replicating ring-stage parasites persist in erythrocytes for days to weeks before they resume normal asexual growth45,46. This artemisinin-induced dormancy is characterized phenotypically with small parasites showing condensed nuclei (FIG. 1B). Whether such forms can also arise spontaneously is not known.
Artemisinin-induced dormancy has been confirmed in P. falciparum from lines that are refractory to the drug and also lines that are susceptible to the drug45,47, which suggests the involvement of an adaptive stress response in the formation of this dormant ring-form48. Dormant ring-stages are physiologically distinct from normal ring stage forms as shown by up-regulation of fatty acid and pyruvate metabolism49 and maintenance of mitochondrial membrane potential50. Interestingly, although artemisinin-based drugs induce dormancy during ring stage development, other current antimalarials, including quinolines, antifolates and atovaquone, do not seem to affect ring forms. However, the anti-folate pyrimethamine can induce ring stage arrest and dormant ring forms in the asexual cycle but not in the cycle where drug is applied, rather in the following cycle. Hence, parasites initially exposed to pyrimethamine develop into mature schizonts, some of which produce merozoites that invade new erythrocytes and only then arrest as dormant ring stages48. These observations explain the results of previous studies with pyrimethamine51,52, which found recrudescent parasites following drug exposure. This suggests the possibility of adaptive fitness advantages of dormancy (initiating at different parts of the life cycle) for parasite survival against a range of stresses, rather than a specific response to artemisinins. Additional drug or nutritional stressors can arrest development of intra-erythrocytic stages of P. falciparum. These include isoleucine starvation53, atovaquone exposure54, and difluoromethylornithine (DFMO)-induced depletion of polyamines55. However, these conditions block trophozoite development and thus differ from artemisinin-induced cell cycle arrest in ring stage parasites. Artemisinins (which are activated by cleavage of their endoperoxide bridge) alkylate many proteins in the parasite56,57, which leads to protein misfolding and ubiquitinylation and subsequent induction of the unfolded protein response involving protein degradation by the proteasome. However, treatment with artemisinins also diminishes proteasome activity58 and parasite death seems to be linked to the accumulation of damaged protein58. Inhibition of translation by the chemotherapeutic agent cyclophosphamide was shown to antagonize the antimalarial activity of dihydroartemisinin (DHA)58. Growth-arrested ring stages induced by artemisinin are likely to have diminished protein synthesis, which would explain why the drug loses activity against such forms.
Parasites isolated from patients failing artemisinin treatment in South East Asia were characterised by low clearance rates from blood. However, these parasites did not show alterations in artemisinin sensitivity in classic in vitro potency tests conducted over 72 hours60. The reasons for this have become clear in subsequent studies into understanding the nature of artemisinin treatment failure. In vitro selection of resistance in increasing doses of artemisinin61 revealed that these parasites also failed to yield discriminatory susceptibility to drug in classic assays, but did show an enhanced rate of dormant ring form formation. Mutated alleles of a gene, Pfk13, encoding the Kelch-13 protein (PfK13), were associated with parasites from artemisinin treatment failures in field studies as well as the in vitro selected line62. In each case, classic in vitro sensitivity assays could not detect the loss of response to artemisinins. However, a ring stage survival assay (RSA)63, where early rings (forming within the first three hours of erythrocyte invasion) from artemisinin refractory lines, exposed to 700 nM of artemisinin for 6 hours, show an enhanced ability to survive compared to sensitive lines. The same phenotype is found when various mutant alleles of Pfk13 are engineered into wild-type parasites64.
The function of PfK13 is not known although roles in protein ubiquitinylation have been predicted65. This includes ubiquitinylation of PfPI3K, and increased phosphatidylinositol-3-phosphate (PtdIns3P) levels have also been linked to diminished sensitivity to artemisinins66. It was recently shown that parasites with pfk13 mutant alleles have different kinetics of haemoglobin digestion67 which could also decrease the activation of drug. Recently, another gene, Pfcoronin, which like Pfk13 encodes a member of the WD40-propeller domain protein family, was identified in another P. falciparum line selected in vitro over a prolonged period for resistance to artemisinin68. Phenotypically, parasites with Pfcoronin mutant alleles mirror those of PfK13 in that the RSA is required to discriminate sensitive and artemisinin-refractory lines. The PfK13 and Pfcoronin mutations are, therefore, somewhat analogous to the so-called high persister (Hip) mutations that are known to enhance the rate at which persisters form in bacteria and yeast16. Many of the known Hip genes are part of toxin-antitoxin protein pairs, and given the predicted capacity of PfK13 to combine in protein-complex formations69, it will be of interest to determine whether analogous functions can be found for PfK13, PfCoronin and any other proteins that confer enhanced dormancy and artemisinin refractoriness as they are identified.
Recent studies evaluated the ability of antimalarial drugs to prevent the recrudescence of artemisinin-induced dormant rings stages. Mefloquine exposure reduced the rate of recrudescence, but did not completely abrogate activation and growth70. However, PI4K inhibitors and imidazolopiperazines did block recrudescence of dormant ring forms of artemisinin-susceptible and artemisinin-resistant P. falciparum71,72. A tetroxane-based compound was shown to eliminate parasites carrying PfK13 mutant alleles73 probably because of its long elimination half-life ensuring it remains at parasiticidal levels for over four days and thus can kill drug-induced dormant ring forms as they reactivate73.Interesting new data suggest that phytohormones74, including gibberellin and abscisic acid, activate DHA-induced dormant rings 24–48 hr earlier than controls treated with DHA alone and left to recover. This work offers a potential new route to drug discovery through re-activation of dormant forms, thereby re-sensitising them to current drugs. Reactivation is a more viable strategy for tropical infectious diseases than seeking molecules that sustain the growth-arrested state, as has been proposed for cancer cell persisters11, given economic considerations in long-term provision of therapy for diseases that most commonly occur in underprivileged areas. An additional study demonstrated the potential role of eIF2α phosphorylation as a mechanism promoting dormancy in ring stages in Plasmodium berghei75 which suggests that inhibition of eIF2α phosphorylation might prevent parasites entering dormancy20 and thus retaining sensitivity to artemisinins. The role of eIF2α phosphorylation in controlling dormancy was initially described in sporozoites76, and the phosphorylation status of eIF2α has since been proposed as a key regulator of different dormancy in apicomplexa20.
Toxoplasma gondii
Toxoplasma gondii, the causative agent of toxoplasmosis, is a tissue-cyst forming coccidian and a member of the Apicomplexa77,78. The ability to generate dormant (or semi-dormant forms that grow slowly as opposed to being growth arrested) yet highly infective forms as part of the developmental life cycle is critical to the persistent nature of these parasites. T. gondii is one of the most widespread protozoan parasites infecting a diverse range of warm-blooded vertebrate hosts, including humans79. Its complex, multi-host developmental life-cycle is defined by sexual and asexual stages, each with distinct, yet related end forms that enable the parasite to persist in nature. The natural life cycle for T. gondii involves small rodents that serve as intermediate hosts and cats that serve as the definitive host79. Within enterocytes of the cat gut, the parasite undergoes sexual development, culminating in shedding of oocysts in the feces79. After shedding, oocysts undergo meiosis to form eight haploid sporozoites, which remain dormant and do not divide, despite surviving in the environment for long periods of time79. Following oral infection, oocysts hatch, which enables sporozoites to invade the intestine and differentiate into replicating tachyzoites, which disseminate throughout the host, thus causing acute disease79. In response to immune pressure and environmental stress, the parasite differentiates into a chronic stage called the bradyzoite, which forms tissue cysts within host cells and give rise to persistent infection79. Bradyzoites are often referred to as dormant, quiescent, or latent forms – however, these terms are not strictly accurate as bradyzoites remain metabolically active, and do replicate, albeit slowly and asynchronously80. However, within a given tissue cysts, some bradyzoites seem to be non-replicative and may represent a truly dormant reservoir. Infections in the intermediate host are likely to persist for life in the host and are characterized by slow expansion of bradyzoites within tissue cysts, periodic rupture to release bradyzoites that can infect host cells and produce new tissue cysts, or alternatively convert back to actively replicating tachyzoites. This balance is controlled by immune surveillance that can suppress active parasite growth but not eliminate the tissue cyst reservoir81.
Dormant forms of Toxoplasma - the sporozoite and bradyzoite.
Outside of the feline, truly dormant oocyst forms of Toxoplasma can remain viable in the environment for many years, before being ingested by an intermediate host. Hatching of the oocyst in the gut releases highly invasive sporozoites that invade epithelial cells and then differentiate into proliferative tachyzoites which disseminate widely before differentiating into tissue cysts containing bradyzoites. The bradyzoite is the source of transmission between intermediate hosts, and to the definitive cat host. Tissue cysts are also the source of lifelong infections, having the potential to reactivate in a chronically infected host82. Humans serve as intermediate hosts and therefore the life cycle is similar to that described above for rodent. During the bradyzoite stage, infections are largely asymptomatic in immunocompetent individuals; pathology arising when these infections reactivate in immunocompromised patients. Individuals undergoing chemotherapy or organ transplantation, or patients with untreated HIV are at risk of reactivation when bradyzoites emerge from tissue cysts, and convert back to rapidly growing tachyzoites, which resume lytic growth and destroy their host cells. The process of reactivation is particularly severe when tissue cysts harbored within the central nervous system (CNS) undergo reactivation83,84. In the absence of appropriate antiparasitic treatment, the resulting syndrome of toxoplasmic encephalitis85 can become life-threatening.
The tachyzoite to bradyzoite transformation controls the transition between acute to chronic infection, while the reverse switch from semi-dormant bradyzoites to actively replicating tachyzoites is the key to disease reactivation. Gene expression analyses indicate that only a few hundred genes differ between tachyzoite and bradyzoite stages, whereas other stages (sporozoites and merozoites) show much greater differences86. However, the molecular basis of these life cycle transitions is not well defined. Slowing of the tachyzoite replication cycle87 coincides with the expression of a bradyzoite-specific transcriptional cascade88–90, which includes stage-specific roles for apicomplexan AP2 (ApiAP2) transcription factors91–95 that have also been implicated in transcriptional regulation of development in Plasmodium spp. (that is, ookinete, sporozoite and gametocyte stages)96–98. These tightly regulated developmental responses enable parasites to balance dissemination within the host and chronic infection that enables host-to-host transmission99. Previous studies have shown that stressors such as nutrient limitation100, high pH101 and nitric oxide exposure102 can induce bradyzoite differentiation. Infection of long-lived, differentiated cells such as skeletal muscle103 and neurons104 also induces bradyzoite formation and persistent infections105.106. Therefore, host immune pressure, environmental stressors and parasite-specific genetic programs can result in the formation of bradyzoite-containing tissue cysts. Triggering differentiation back to actively growing tachyzoites is also not fully understood, but might arise from abundance of nutrients or absence of immune pressure. Either model might explain why bradyzoites used to infect naive mice quickly revert to tachyzoite growth, before later developing into new bradyzoites and forming tissue cysts107. This switching ability is not understood, but it probably underlies the permissive nature of transmission between intermediate hosts, which include nearly all warm-blooded animals, which are usually infected by ingestion of semi-dormant tissue cysts108. This oral transmissibility of the asexual, tissue stages sets T. gondii apart from its close relatives that have more restricted modes of transmission.
Human infections arise either from ingestion of oocysts shed into the environment from the feline definitive host109 or consuming undercooked meat contaminated with cyst-bound bradyzoites110. Mothers first infected with T. gondii during pregnancy can transmit infection via tachyzoites crossing the placental barrier and infecting the fetus111. Although most infections are subclinical or only result in mild disease in healthy adults, hyper-virulent strains of T. gondii in South America cause an increased incidence of ocular toxoplasmosis in immunocompetent patients112.
Current therapies for toxoplasmosis based on pyrimethamine-sulphadiazine (PYR-Sulfa) or trimethoprim-sulfamethoxazole (TMP-SMX) target DNA synthesis by inhibiting the folate pathway and are effective at blocking replication and killing tachyzoites85. Adverse reactions are common and include low tolerance due to bone marrow toxicity and allergy to sulfa components while potential teratogenicity prevents use during the first trimester of pregnancy85. Clinical resistance to treatment (defined by the emergence of mutations rendering parasites stably insensitive to drug) is rare as human infections are zoonotic (and food animals are not routinely treated for toxoplasmosis) and human-to-human transmission is uncommon.
Although current therapies can control acute infection, owing to their mode of action, chronic infections are unaffected. Replicating tachyzoites are vulnerable, but slowly or asynchronously replicating bradyzoites are not85 (FIG.2). Typically, bradyzoite numbers expand as they replicate so tissue cysts contain 100–500 individual parasites over the course of 1–2 months (suggesting an average doubling time of several days), whereas tachyzoites double every 6–8 h80. Although PYR eliminates tachyzoites in culture overnight, a 4h exposure followed by drug wash out, does not inhibit parasite replication, hence continuous exposure of replicating tachyzoites over prolonged periods is required for effective treatment113. Neither the blood brain barrier85,114 nor the heavily glycosylated tissue cyst wall115,116 represent physical barriers to PYR, which suggest that the semi-dormant state of the bradyzoite is responsible for treatment failure. Alternative treatments such as clindamycin (which inhibits protein synthesis in the vital apicoplast organelle) also target actively replicating forms85.
Figure 2: Toxoplasma gondii tachyzoites and bradyzoites.

T. gondii, the causative agent of toxoplasmosis, undergoes asexual replication in nucleated host cells of intermediate hosts (such as rodents and humans). During initial infection tachyzoites disseminate throughout the host and cause acute disease. In response to immune pressure and environmental stress, the parasite differentiates into bradyzoites, which form tissue cysts within host cells and give rise to persistent infection (chronic stage). Shown are microscopy images of the tachyzoite (visualized using polyclonal rabbit anti- surface antigen 1 antibody with Alexa Fluor 488) and a tissue cyst containing bradyzoites (brightfield image). Current therapies are successful only against the acute phase, but fail to eliminate the chronic stage. See reference 80 for further detail on bradyzoite development. Image on the right courtesy of, L. David Sibley (Washington University School of Medicine, USA). Image on the left reproduced from Ref 81.
New drugs to treat toxoplasmosis are actively being investigated. The Malaria and Pathogen box compounds (https://www.pathogenbox.org/) selected in high throughput screens against malaria and other pathogens were screened to find compounds that hit biological targets shared among apicomplexans117. A number of other chemical libraries have been tested against T. gondii to possibly identify new therapeutic compounds118–121, including all FDA approved drugs seeking compounds for repurposing. In addition, screening of compounds that are active against malaria identified relatively few candidates that were effective against T. gondii122. Atovaquone123 and ELQ-300124 are effective against tachyzoites and both compounds reduced tissue cyst numbers in chronically infected mice, but complete cure was not achieved. Intriguingly, primaquine, used to treat P. vivax hypnozoites, has modest activity against T. gondii tachyzoites, but its therapeutic use against the bradyzoite has not been evaluated125. In separate studies, inhibitors of the calcium-dependent protein kinase 1 (CDPK1), an essential enzyme in T. gondii, have shown promise for eradicating infection in immune-compromised mice113. Learning more about essential processes in bradyzoites may lead to therapies that control or eliminate this stage.
Trypanosoma cruzi
Trypanosoma cruzi, which causes Chagas disease, afflicts millions of people mostly in Latin America126 and is vectored by blood-feeding insects of the Triatominae subfamily of reduviid bugs. Infections in humans and in many other mammals are often low level, but life-long and generally immune-controlled. The preferential persistence of T. cruzi in muscle, and the continued immune response to parasites in heart and other muscle tissues, probably accounts for the cardiac and gut pathology associated with Chagas disease. Infection with T. cruzi generally begins with invasion by flagellated (non-replicative) trypomastigotes (G) transmitted in bug faeces as they feed on their hosts. Trypomastigotes enter host cells and differentiate into amastigotes, which divide, notably in the host cell cytoplasm and not in a vacuole, by binary fission. Amastigotes convert back into trypomastigotes, which are released from the lysed host cell and invade new cells or are available in the blood for transmission. The intracellular replication cycle generally lasts for 4–5 days although it can be slower for some isolates127.
T. cruzi amastigotes are the primary targets of both immune control and drug treatment. Benznidazole and nifurtimox have rapid trypanocidal activity against T. cruzi and have been used therapeutically for decades. However, long treatment courses (30–60 days) are required and outcomes are unpredictable. These, and other observations, prompted the suggestion of drug-insensitive and potentially dormant subpopulations of T. cruzi in hosts128.
Dormant amastigote forms of T. cruzi.
Recently, non-replicating intracellular T. cruzi amastigotes were demonstrated in both mice and cultured host cells17. These dormant parasites occur spontaneously in the absence of drug pressure but resist killing by multiple trypanotoxic compounds that are highly active against replicating parasites. Entering the quiescent state seems not to be a response to a lack of nutrients or to the presence of other obvious stressors, as non-replicating amastigotes are frequently present in host cells alongside actively dividing amastigotes. Furthermore, these non-replicating amastigotes sense the signals that trigger amastigote-to -trypomastigote differentiation, as they differentiate synchronously with the actively dividing forms in the same host cell (FIG. 3). Trypomastigotes derived from non-replicating amastigotes can invade new host cells and re-establish both actively dividing and non-dividing progeny, a hallmark of phenotypic heterogeneity in bacterial persisters. Formerly dormant amastigotes, emerging after a 30 day continuous drug exposure in vitro are equally sensitive to drug as the parent (untreated) population17, hence drug exposure has not selected for genetically resistant parasites, instead suggesting that dormancy is the cause for drug refractoriness. Transient dormancy (that is, dormant cells that will re-activate) among a small subpopulation of intracellular T. cruzi amastigotes therefore underlies treatment failure of anti-T. cruzi compounds that kill actively dividing amastigotes. Although the metabolic state of non-replicating amastigotes has yet to be probed, dormant amastigotes have lower levels of expression of reporter proteins, which suggests decreased protein synthetic capacity and altered metabolism17.
Figure 3. Dormant amastigote forms of Trypanosoma cruzi.

Following invasion of host cells, T. cruzi trypomastigotes (shown in blue; indicating no or minimal proliferation) differentiate into non-replicating amastigotes. The presence or absence of dormant parasites determines treatment outcomes. Amastigotes may not replicate (bottom panel; blue amastigotes, dormant parasites), undergo minimal replication (top and middle panels, red amastigotes) before becoming dormant, or actively replicate (top, second round of invasion, red amastigotes). Replicating parasites are eliminated by drug treatment and under appropriate conditions (for example, long-treatment period), possibly resulting in sterile cure (top panel). However, parasites that are dormant during the time of drug exposure (middle and bottom, blue amastigotes) resist drug clearance and can revive the infection by resuming replication after drug treatment (bottom panel).
Many questions remain concerning dormancy in T. cruzi. Dormant amastigotes are not actively transmitted, instead trypomastigotes must reach the host bloodstream to be taken up by blood-feeding vectors. So what is the selective advantage of dormancy? Immune detection and control of T. cruzi is generally highly effective in keeping parasite load low. Host cells containing only dormant amastigotes probably evade surveillance by CD8+ T cells or other immune effectors and thus could function as reservoirs for the low but continuous production of trypomastigotes. This could extend the timeframe over which transmission is possible, and contribute to parasite persistence by reseeding tissue sites with new persister-like parasites. The occasional reactivation of these dormant parasites might also account for the dynamic nature of T. cruzi infection (for example, the appearance of new sites of parasite replication, appearing and disappearing in different anatomical locations)129.
Hosts with shorter length T. cruzi infections (for example, children) may be more responsive to drug treatment than those with longer term infections (decades in humans); of note this conclusion is still debated and is complicated by how cure is determined130–132. If true, longer term infections could result in increased seeding of tissues with dormant parasites, some of which may have exceptional longevity in the dormant state and thus avoid clearance by current, time-limited, treatment protocols.
As T. cruzi rarely undergoes sexual recombination, instead recombining and resorting existing genes to generate genetic and antigenic diversity133, dormant amastigotes might function as a stable ‘stem-like’ cell should the gene resorting yield compromised progeny in replicating forms.
It seems that the length of time that amastigotes stay dormant, or at least for the ability of an infection to persist under drug pressure, is finite. Sterile cure with drugs can be achieved, in some cases, with treatment courses as short 10–20 days134,135 and some, albeit not all, in vitro cultures clear parasites following a 30-day drug exposure17. For some T. cruzi strains, 16 days of in vitro exposure to benznidazole is sufficient to sterilize the culture136. However, 30 and even 60 day treatment regimens in humans and other animals often fail, and dormancy, in combination with other features of the infection (for example, the establishment of infection in a wide range of tissues which differ in drug penetration) are likely to be responsible. On the basis of this, currently available drugs might be more efficacious if used over longer periods (for example, several months) possibly at lower frequency of dosing to keep overall dose down. Indeed pulse-treating with benznidazole once every 5 days137 or even once every 7 days (Bustamante, J., unpublished observations)over several months can be as, or even more, efficacious than shorter, daily treatment protocols.
New trypanocidal compounds that can target dormant forms are clearly desirable. Nifurtimox and benznidazole both require metabolic activation by trypanosomal enzymes138, the activity of which is likely to be reduced in dormant forms. Therefore, establishing screens specific for these dormant forms may identify a new generation of drugs. The observation that non-dividing amastigotes can respond to the cues that trigger stage conversion to trypomastigotes also offers the possibility of finding molecules that might activate dormant parasites and sensitize parasites to existing drugs.
Leishmania species
The leishmaniases represent a series of conditions caused by over 20 different species of the genus Leishmania139 that belong to the same phylogenetic order, the Kinetoplastida as African and American trypanosomes. Leishmaniases span self-healing cutaneous disease to a frequently fatal visceral form. Around 1 million clinical cases are reported each year, with ~10 fold that number of people having sub-clinical infections in nearly 100 countries within the tropics and sub-tropics139. The parasites are transmitted by sandflies (FIG. 4). In the sandfly, a series of different flagellated promastigote [G] forms complete the life cycle, culminating in the formation of growth-arrested metacyclic promastigotes in the mouth parts ready for transmission to the vertebrate host. Here they enter phagocytes where they transform into amastigote forms within the phagolysosome to establish infection.
Figure 4. Leishmania life cycle showing impact of persisters.

Leishmania spp. alternate between motile promastigotes in the sand fly vector and intracellular amastigotes in the mammalian host. Following transmission of infective, non-dividing metacyclic promastigotes via the bite of a sandfly, promastigotes enter host phagocytic cells and differentiate within phagolysosomes into amastigote forms (with truncated flagellum) and undergo replication. Subsequently, host cells burst and the released amastigotes enter further host cells. Alternatively amastigotes may be taken up by a sandfly during a blood meal in which they differentiate into proliferating promastigotes (procyclic promastogotes) in the midgut
Studies have identified, discrete replicating and non-dividing intracellular amastigote populations and it is likely that growth-arrested leishmanial amastigote forms contribute to drug treatment failure and persistent infections (top right panel). Promastigotes have been shown to enter a non-proliferative but viable state in purine-depleted medium (bottom left panel). Figure reproduced with adaptation from Reference 159.
Successful treatment of leishmaniasis is notoriously difficult. Pentavalent antimony formulations were the mainstay of treatment for many years140 although toxicity and parasite drug-resistance increasingly limit use. Miltefosine and paromomycin are used in some settings140. Amphotericin B, which bind to ergosterol [G] found in both fungal and leishmanial membranes but not mammals, has become the drug of choice in many countries (its liposomal formulation, AmBisome, being safer than the non-encapsulated drug). Visceral leishmaniasis is caused by Leishmania donovani or Leishmania infantum in the old world and Leishmania chagasi in the new world. The cutaneous form is generally caused by L. major or L. tropica in the old world and a variety of different species in the new world. Mucocutaneous disease is caused by parasites of the Viannia group; L. (Viannia) braziliensis or L. (Viannia) guyenensis. Post Kala azar dermal leishmaniasis (PKDL) occurs in patients with visceral leishmaniasis, emerging as a tegumental condition several months to years after treatment141. For cutaneous leishmaniasis, a post-treatment relapsing form is known as leishmaniasis recidivans142. Both conditions are likely to arise from persister-type parasites; that is parasites that have survived drug treatment due to transient drug refractoriness during dormancy. They re-awaken into an immunologically altered environment, as the immune system has responded to the primary infection and in currently ill-understood ways, re-establish a pathological condition in the skin. Drug resistance mechanisms have been discovered143 and it is clear that drug treatment failure due to stably transmitted genetic mutation is an increasing problem. For example, treatment failure with antimony in the Indian sub-continent has been traced to mutations in the aquaporin protein, which is encoded by the AQP1 gene, that transports antimony into the cells144. A genetic locus associated with miltefosine resistance in field isolates has been identified145, and changes in genes involved in sterol metabolism have been implicated in amphotericin B resistance146, 147. However, treatment failures without classic genetic-based heritable resistance are also widespread (for example, PKDL and leishmaniasis recidivans), which indicates that Leishmania spp., like T. cruzi, produce persister-like cells.
Dormant forms of Leishmania species.
In mammals, Leishmania spp. dwell primarily within macrophages and dendritic cells. The growth rate of L. mexicana, in non-resolving infections in Balb/c mice148, was determined to be slow but constant, doubling every 12 days, and it was concluded that amastigotes are semi-quiescent in mice. Moreover, in mice, amastigotes (as an overall population) were shown to have a stringent metabolism, consuming relatively sparing quantities of metabolites as a source for carbon and energy and using them relatively efficiently compared to the cultured promastigote form149. The possibility of sub-populations of dividing and non-dividing was not taken into account in this study. However, discrete replicating and non-dividing intracellular amastigote populations were discriminated in a C57BL/6J mouse model of L. major cutaneous disease18. 4 months post-infection, two distinct populations could be distinguished based on their ability to incorporate the nucleotide analogue 5-bromo-2’-deoxyuridine (BrdU) into DNA: one actively dividing population that doubled around every 60 hours and a non-dividing, dormant population. In the early days following infection, amastigotes divide at a similar rate (around every 60 hours), but fewer dormant forms are found. Learning more about the comparative phenotypes of dormant and replicating Leishmania spp. amastigotes will shed more light on these persister-like cells.
Other Leishmania species also exhibit potential persister-like cellular states. Comparison of L. (Viannia) braziliensis promastigotes and amastigotes150 showed that amastigotes have diminished levels of protein, RNA, mitochondrial kDNA and ATP and exhibit no growth in macrophages in vitro. Untargeted metabolomics showed that axenic amastigotes also had diminished levels of multiple amino acids and polyamine pathway metabolites compared to promastigotes. Metabolomics offer a key route to understanding the persister phenotype (Box 1).
Box 1. Methods to study protozoan persister-like cells.
Fluorescent probes:
Fluorescent probes have become essential to visualize dormant protozoan cells. Labelling parasites using classic fluorescent reporters such as green fluorescent protein or ds red, Tmtomato are used to identify the parasites themselves. Labels that enter cells upon a pulsed exposure and are then either diluted out during cell division or remain at their initial pulse level in non-replicating cells enable the differentiation between replicating and non-replicating cells. Cell trace violet, carboxyfluorescein succinimidyl ester (CFSE) and Cell tracker red are examples of dyes used for this purpose. Fluorescence-activated cell sorters (FACS) can be used to sort differentially labelled populations for further analysis.
DNA replication probes:
Non-replicating cells do not incorporate new bases into their DNA, while replicating cells do. The thymidine analogues 5-ethynyl-2’-deoxyuridine (EdU) and 5-bromo-2’-deoxyuridine (BrDU) have both been shown to effectively incorporate into replicating DNA of Leishmania spp. and Trypanosoma cruzi, whereas non-replicating cells did not incorporate the activated derivatives of these analogues. To visualize their incorporation it is necessary to fix cells and use specific antibodies to detect them, precluding their use in sorting live cells. Toxoplasma gondii156and Plasmodium spp157 do not salvage thymidine and hence can only be labelled with BrdU and similar probes after stable transfection of thymidine kinase.
Sorting quiescent and replicating cells:
Cells that have been labelled with fluorescent dyes (see above)) can be separated using FACS. This approach can also be used to sort cells in which fluorescent reporters (for example, GFP) are regulated by stage-specific promoters, for example to separate tachyzoite from bradyzoites of T. gondii. Another simple way to enrich for persister-like cells is to treat a mixed population with varying doses of antimicrobials that kill replicating cells but not quiescent cells followed by the analysis of the viable cells remaining post treatment.
Metabolomics:
Untargeted metabolomics analysis enables the comprehensive profiling of metabolite levels in different cell types. As persister-like cells have altered metabolism when compared to proliferative cells, but genome sequences are unchanged, metabolomics offers a key route to profile the differences in biochemical physiology that distinguish cell types.
Transcriptomics and proteomics:
Also relating to the fact that the genomes of persister-like cells are unaltered, profiling the transcriptome and proteome of different cell types can report on adaptations to the quiescent cell phenotype and point to metabolic pathways that might be targeted in these cells.
Single cell transcriptomics:
Profiling transcription in single cells, rather than population averages, will enable a more detailed understanding of the changes at the transcriptional level that could be link to the formation of persister-like cells.
Non-replicating L. donovani were also found in in vitro cultivated THP-1 macrophages151 prompting speculation that these too represent persister-like cells. Although in their infancy, studying these growth-arrested leishmania forms will probably provide answers to the frequent drug treatment failures in the absence of genetically selected resistant parasites152.
Studying persister cells in vivo is challenging owing to low abundance and inaccessibility, being distributed across multiple host organs (Box1). As an evolutionarily ancient trait8, rather than an adaptation to mammalian parasitism, other life cycle stages of Leishmania spp. might be expected to form quiescent forms to enable population level survival during environmental stresses. Non-replicative forms also have important roles in the life cycle, for example, the transmitted metacyclic form is analogous to the non-proliferative Plasmodium spp. sporozoite. Leishmania donovani promastigotes enter a non-proliferative, but viable quiescent state in purine-depleted culture conditions153. Proteomics analysis154 revealed that purine transporters and purine salvage enzymes were upregulated within 24 hours of purine deprivation (presumably attempting to restore purine levels). However, after 48 hours, a profound remodeling of the proteome had occurred: enzymes involved in DNA replication and repair, and protein synthesis were diminished while oxidative stress response pathways were increased, all hallmarks of persister-like cells. In bacteria7 and fungi10 too, nutrient starvation responses have considerable overlap mechanistically with the formation of persister cells, hence the leishmanial promastigote model may offer a tractable means of probing mechanisms of persister-like cell formation and guide studies in less readily studied amastigotes (Box1).
Very recently155 insertion of a GFP gene into the ribosomal gene locus of both L. mexicana and L. braziliensis was reported in a preliminary study. Transformation of promastigotes to amastigotes in both species led to pronounced reduction in expression of the reporter gene, consistent with decreased translation in amastigotes. BrDU labelling showed that parasites with low GFP expression levels were also non-replicative. Axenic amastigotes were shown to form two populations (bright and dull with respect to GFP expression, and presumed to represent proliferative and non-replicating cells respectively). All macrophage resident amastigotes had dull expression. Using Cell tracker deep red staining it was seen that, in vivo, parasites retaining the dye (and thus not replicating) could be found up to 120 days after infection. GFP fluorescent parasites at this time lacked the dye indicating that this dual staining approach can distinguish non-proliferating (cell tracker positive) and proliferating (GFP positive, cell tracker negative) parasites. The tool kit necessary to probe Leishmania persisters more deeply is thus emerging.
To date, testing the susceptibility to drugs of dormant Leishmania spp. amastigotes has not been performed. However, by analogy to other systems – and given the widespread nature of drug treatment failure (including PKDL and leishmaniasis recidivans) it is probable that these parasite forms are refractory to treatment with anti-leishmanial drugs and by learning more about their biology we will be able to improve therapeutic interventions.
Concluding comments
Persister populations are increasingly appreciated as a problem leading to drug treatment failures in bacteria, fungi and cancer cells. Many parasitic protozoa also display the dormant/quiescent properties characteristic of persister phenotypes, ranging from discrete life-cycle stages (for example, non-dividing P. vivax hypnozoites and slow-growing T. gondii bradyzoites) to the stochastically arising, non-dividing amastigotes of Trypanosoma cruzi, to the slow-growing and non-replicating amastigotes of Leishmania spp. As in bacteria, these protozoan persister-like cells exhibit drug tolerance and are likely to be responsible for frequent drug treatment failure. Besides some advances, many challenges remain in studying protozoan persister-like cells: these cells are generally of low abundance in mammalian hosts and either non-replicating or only slowly growing; their tissue distribution is not well defined in mammalian hosts and they can be difficult to find; and finally, in vitro methods for their cultivation are not well developed
Understanding the mechanisms of dormancy, which involves reversible metabolic changes (FIG. 5), might lead to new treatment strategies. For example, screens that specifically target persister cells may lead to the discovery of compounds that facilitate sterile cure, either alone, or together with current compounds that eliminate actively replicating cells. It may also be possible to use current drugs in different ways; for example, pulsing therapy at intervals that mean reactivated persisters that emerge are confronted with drug to which they are sensitive. Alternatively, small molecules that trigger re-activation could resensitize the parasites to currently used drugs.
Figure 5. Metabolic changes in protozoan persisters.
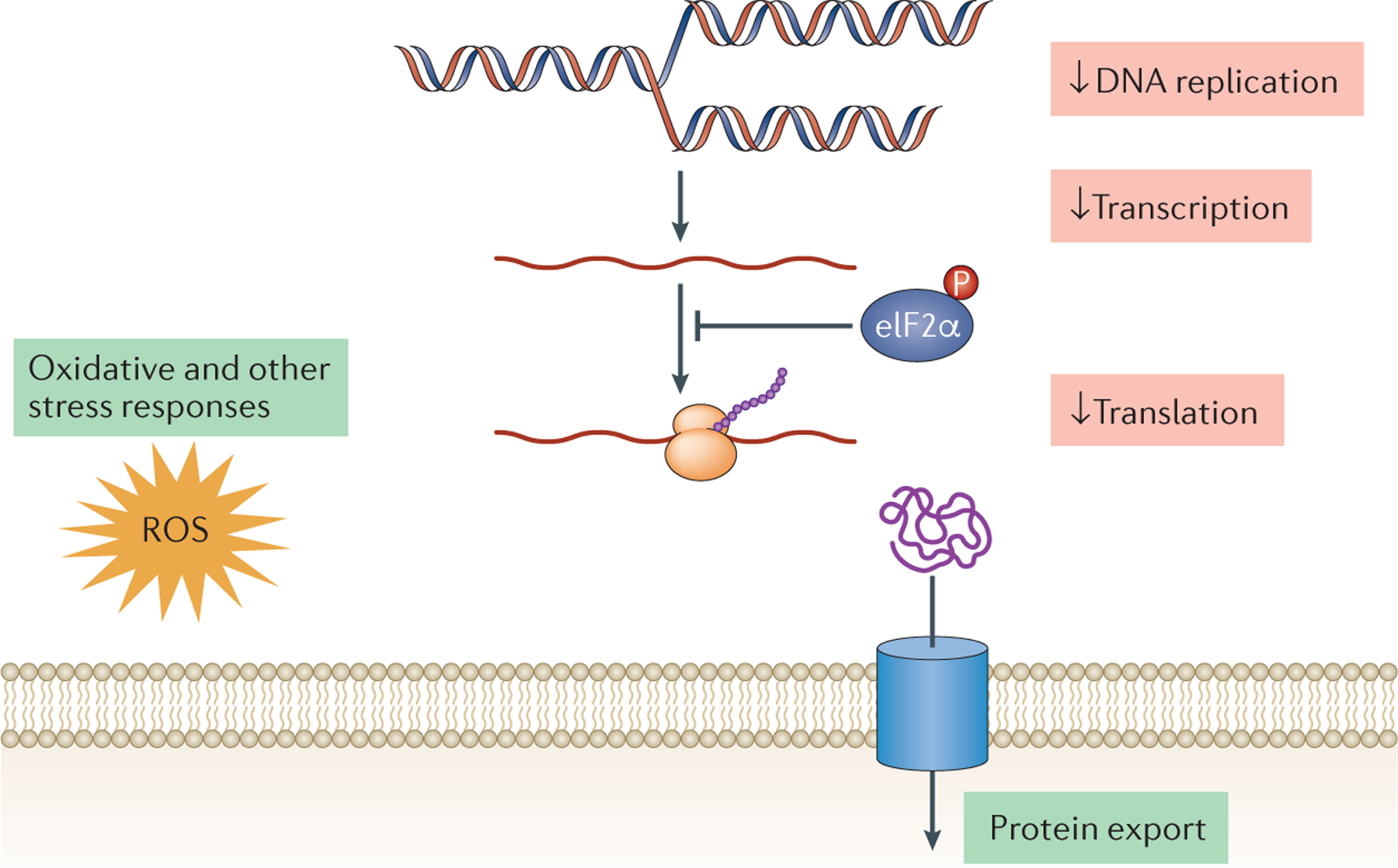
Currently, our knowledge is limited regarding metabolic changes across protozoan persister-like cells. Studies have shown that in Plasmodium spp. and Toxoplasma gondii DNA replication, general transcription and protein synthesis are decreased in persister-like cells. However, some biochemical pathways are sustained, such as protein export in Plasmodium cynomolgi hypnozoites forms and also oxidative and other stress responses in Plasmodium and T. gondii bradyzoites. In addition, in purine-depleted culture conditions, Leishmania donovani non-proliferative promastigotes exhibit decreased DNA replication and repair, and protein synthesis, whereas oxidative stress response pathways were increased. Finally, in the apicomplexan protozoa phosphorylation of parasite eukaryotic initiation factor-2α (eIF2α) has been implicated in the establishment of dormancy (possibly by inhibiting global protein synthesis), although the role of role eIF2α phosphorylation in the formation of persister-like cells in the kinetoplastid protozoa has yet to be investigated.
Finally, the identification of small molecules that modulate dormancy will also provide new tools for understanding the molecular basis of persister phenotypes.
Acknowledgements
M.P.B. is funded by the Wellcome Trust core grant to the Wellcome Centre for Integrative Parasitology (104111/Z/14/Z).
R.L.T. is supported by the U.S. National Institutes of Health grant R01 AI124692.
Glossary of terms
- Persisters
A subpopulation of slow-growing or growth-arrested cells that have a decreased susceptibility to killing by normally effective cytotoxic agents. Persisters survive treatment with drugs because of their altered metabolism during a temporary state of quiescence and are genetically identical to other, drug-susceptible cells in the population. Persisters may arise stochastically, or in responses to environmental cues such as nutrient starvation.
- Hypnozoite
A small dormant form of Plasmodium vivax and Plasmodium ovale (and Plasmodium cynomolgi in monkeys) that persists in host hepatocytes following infection of sporozoites.
- Tachyzoite
Rapidly growing intracellular form of Toxoplasma that divides asexually and undergoes successive round of lytic growth in a wide range of nucleated cells before being controlled by an efficient immune response. Cause of acute infection and disease manifestation in humans.
- Bradyzoite
Persistent form of Toxoplasma found in tissue cysts, which are intracellular and commonly found in skeletal muscle and neurons in the brain. Cysts are not cleared by active immunity or drug therapy. Reactivation can lead to serious disease, especially in those with compromised immune function.
- Amastigote
In Trypanosoma cruzi, an intracellular, replicative cell within the mammalian host cell cytoplasm (T. cruzi amastigotes reside in the cytoplasm of various of host cell types but shows preferential persistence in muscle and adipose tissues). In Leishmania, an intracellular form of Leishmania with a truncated flagellum that replicate in mammalian cells, including macrophages and dendritic cells, within the acidic phagolysome compartment of these cells.
- Sporozoite
In Plasmodium parasites a form forming in mosquitos where they migrate to salivary glands and are injected during a blood meal. They migrate to the liver and invade hepatocytes.In Toxoplasma, infectious form found in oocysts, the product of the sexual phase, which are shed into the environment and contaminate food and water leading to transmission. The sporozoite persists in a semi-dormant state within the oocysts, surviving for many months in the environment.
- Schizont
A multinucleated form of the Plasmodium parasite that forms through multiple nuclear divisions preceding cellular division to release merozoite forms. Can occur either in erythrocytes, or prior to the erythrocytic cycle in hepatocytes.
- Merozoites
Forms of Plasmodium parasites that emerges from liver cells after differentiation and invades red cells, where the asexual life cycle progression ultimately creates many more merozoites that burst from infected red cells and invade new ones.
- Trophozoite
A form in the Plasmodium parasite life cycle following the ring stage that consumes host hemoglobin before entering schizogony.
- hemolytic anemia
Anemia ensuing from lysis of red blood cells, for example due to oxidative stresses induced by primaquine in individuals that are deficient in glucose-6-phosphate dehydrogenase.
- Artemisinin
Anti-malarial drug derived from the sweet wormwood plant, Artemisia annua. The structure comprises a sesquiterpene lactone containing an endoperoxide responsible for activity.
- Trypomastigotes
In Trypanosoma cruzi, non-replicative, flagellated, extracellular cells that can invade host cells or be transmitted to reduviid vectors during a blood meal.
- Promastigote
A form of Leishmania, with an anterior flagellum, that replicate within the midgut of the sandfly vector that transmits these parasites. Several distinctive other forms exist in the sandfly vector too.
- Ergosterol
Major sterol of the Leishmania plasma membrane, also found in fungi. Binds to amphotericin B.
Footnotes
Further information
The Malaria and Pathogen box compounds: https://www.pathogenbox.org
There is NO Competing Interest.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peer review information
Nature Reviews Microbiology thanks P. Kaye, J. Kelly and the other anonymous reviewer(s) for their contribution to the peer review of this work.
REFS
- 1.O’Neill J Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Rev Antimicrob Resist. http://amr-review.org/Publications (2014). [Google Scholar]
- 2.Blair JM, Webber MA, Baylay AJ, Ogbolu DO & Piddock LJ Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol 13, 42–51 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Bigger JW The bactericidal action of penicillin on Staphylococcus pyogenes. Irish J. Med. Sci 19, 585–595 (1944). [Google Scholar]
- 4.Cohen NR, Lobritz MA & Collins JJ Microbial persistence and the road to drug resistance. Cell Host Microbe 13, 632–642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis K Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol 5, 48–56 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Fisher RA, Gollan B & Helaine S Persistent bacterial infections and persister cells. Nat. Rev. Microbiol 15, 453–464 (2017). [DOI] [PubMed] [Google Scholar]; Recent review summarizing knowledge of persister cells in bacteria.
- 7.Michiels JE, Van den Bergh B, Verstraeten N & Michiels J Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat 29, 76–89 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Van den Bergh B, Fauvart M & Michiels J Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev 41, 219–251 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Srinivas V, Arrieta-Ortiz M, Peterson ELR, & Baliga NS Characterization and elimination of stochastically generated persister subpopulation in mycobacteria. BioRxiv (2018). doi: 10.1101/463232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delarze E & Sanglard D Defining the frontiers between antifungal resistance, tolerance and the concept of persistence. Drug Resist. Updat 23, 12–19 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Vera-Ramirez L & Hunter KW Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Res. 6, 2134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fairlamb AH, Gow NA, Matthews KR & Waters AP Drug resistance in eukaryotic microorganisms. Nat Microbiol. 1,16092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaye P & Scott P Leishmaniasis: complexity at the host-pathogen interface. Nat. Rev. Microbiol 9, 604–615 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Yam XY & Preiser PR Host immune evasion strategies of malaria blood stage parasite. Mol Biosyst. 13, 2498–2508 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Cabral DJ, Wurster JI & Belenky P Antibiotic persistence as a metabolic adaptation: stress, metabolism, the host, and new directions. Pharmaceuticals 11, pii: E14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harms A, Brodersen DE, Mitarai N & Gerdes K Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell 70, 768–784 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Valdez FJ, Padilla A, Wang W, Orr D & Tarleton RL Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. Elife 7 pii: e34039(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first unequivocal demonstration of drug-resistant persister subpopulations in kinetoplastid parasites.
- 18.Mandell MA & Beverley SM Continual renewal and replication of persistent Leishmania major parasites in concomitantly immune hosts. Proc. Natl. Acad. Sci USA 114, E801–E810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Discovery of mixed populations of replicating and non-replicating sub-populations of L. major in persistent infections in mice.
- 19.Ashley EA, Pyae Phyo A & Woodrow CJ Malaria. Lancet 391, 1608–1621 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Holmes MJ, Augusto LDS, Zhang M, Wek RC & Sullivan WJ Jr. Translational control in the latency of apicomplexan parasites. Trends Parasitol. 33, 947–960 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imwong M Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J. Infect. Dis 195, 927–933 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Shanks GD & White NJ The activation of vivax malaria hypnozoites by infectious diseases. Lancet Infect. Dis 13, 900–906 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Battle KE et al. Geographical variation in Plasmodium vivax relapse. Malar J. 13, 144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krotoski WA et al. Observations on early and late post-sporozoite tissue stages in primate malaria. I. Discovery of a new latent form of Plasmodium cynomolgi (the hypnozoite), and failure to detect hepatic forms within the first 24 hours after infection. Am. J. Trop. Med. Hyg 31, 24–35 (1982). [PubMed] [Google Scholar]
- 25.Krotoski WA et al. Observations on early and late post-sporozoite tissue stages in primate malaria. IV. Pre-erythrocytic schizonts and/or hypnozoites of Chesson and North Korean strains of Plasmodium vivax in the chimpanzee. Am. J. Trop. Med. Hyg 35, 263–274 (1986). [DOI] [PubMed] [Google Scholar]
- 26.Dembele L et al. Persistence and activation of malaria hypnozoites in long-term primary hepatocyte cultures. Nat. Med 20, 307–312 (2014). [DOI] [PubMed] [Google Scholar]; Introduction of a model for P. cynomolgi hypnozoite characterization in cultured hepatocytes.
- 27.Gural N et al. In vitro culture, drug sensitivity, and transcriptome of Plasmodium vivax hypnozoites. Cell Host Microbe. 23, 395–406 e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Introduction of a model for P. vivax hypnozoite characterization in cultured hepatocytes
- 28.Roth A, et al. A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum. Nat Commun. 9, 1837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; In vitro hepatocyte system to enable screening for drugs against P. vivax hypnozoite and replicative liver stages and P. falciparum liver stages.
- 29.Voorberg-van der Wel A et al. , A comparative transcriptomic analysis of replicating and dormant liver stages of the relapsing malaria parasite Plasmodium cynomolgi. Elife 6 pii: e29605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertschi NL et al. Transcriptomic analysis reveals reduced transcriptional activity in the malaria parasite Plasmodium cynomolgi during progression into dormancy. Elife. 7, e41081 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Transcriptomic analysis demonstrating progressive changes in metabolism as hypnozoites form.
- 31.Voorberg-van der Wel A et al. Transgenic fluorescent Plasmodium cynomolgi liver stages enable live imaging and purification of Malaria hypnozoite-forms. PLoS One. 8, e54888 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maher SP et al. Microphysical space of a liver sinusoid device enables simplified long-term maintenance of chimeric mouse-expanded human hepatocytes. Biomed Microdevices. 16, 727–736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikolajczak SA Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe 17, 526–535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaefer C et al. A recombinant antibody against Plasmodium vivax UIS4 for distinguishing replicating from dormant liver stages. Malaria J 17, 370 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dow GS et al. Radical curative efficacy of tafenoquine combination regimens in Plasmodium cynomolgi-infected Rhesus monkeys (Macaca mulatta). Malar J. 10, 212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alving AS et al. Potentiation of the curative action of primaquine in vivax malaria by quinine and chloroquine. J. Lab. Clin. Med 46, 301–306 (1955). [PubMed] [Google Scholar]
- 37.Bennett JW et al. Primaquine failure and cytochrome P-450 2D6 in Plasmodium vivax malaria. N. Engl. J. Med 369, 1381–1382 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Pybus BS et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar J. 12, 212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milner EE et al. Cytochrome P450 2D-mediated metabolism is not necessary for tafenoquine and primaquine to eradicate the erythrocytic stages of Plasmodium berghei. Malar J. 15, 588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vuong C et al. Differential cytochrome P450 2D metabolism alters tafenoquine pharmacokinetics. Antimicrob. Agents Chemother 59, 3864–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcsisin SR, et al. Tafenoquine and NPC-1161B require CYP 2D metabolism for anti-malarial activity: implications for the 8-aminoquinoline class of anti-malarial compounds. Malar J. 13, 2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baird JK, Battle KE & Howes RE Primaquine ineligibility in anti-relapse therapy of Plasmodium vivax malaria: the problem of G6PD deficiency and cytochrome P-450 2D6 polymorphisms. Malar J. 17, 42 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McNamara CW et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 504, 248–253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeeman AM et al. PI4 Kinase Is a Prophylactic but not radical curative target in Plasmodium vivax-type malaria parasites. Antimicrob. Agents Chemother 60, 2858–2863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teuscher F, Chen N, Kyle DE, Gatton ML & Cheng Q Phenotypic changes in artemisinin-resistant Plasmodium falciparum lines in vitro: evidence for decreased sensitivity to dormancy and growth inhibition. Antimicrob. Agents Chemother 56, 428–431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nosten F Waking the sleeping beauty. J. Infect. Dis 202, 1300–1301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tucker MS, Mutka T, Sparks K, Patel J &Kyle, D.E. Phenotypic and genotypic analysis of in vitro-selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob. Agents Chemother 56, 302–314 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hott A, Tucker MS, Casandra D, Sparks K & Kyle DE Fitness of artemisinin-resistant Plasmodium falciparum in vitro. J. Antimicrob. Chemother 70, 2787–2796 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen N et al. Fatty acid synthesis and pyruvate metabolism pathways remain active in dihydroartemisinin-induced dormant ring stages of Plasmodium falciparum. Antimicrob. Agents Chemother 58, 4773–4781 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peatey CL et al. A small subset of artemisinin induced dormant P. falciparum parasites maintain mitochondrial membrane potential and resume growth in vitro. J. Infect. Dis 212, 426–434 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Nakazawa S, Kanbara H & Aikawa M Plasmodium falciparum: recrudescence of parasites in culture. Exp. Parasitol 81, 556–563 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Nakazawa S, Maoka T, Uemura H, Ito Y & Kanbara H Malaria parasites giving rise to recrudescence in vitro. Antimicrob. Agents Chemother 46, 958–965 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Babbitt SE et al. Plasmodium falciparum responds to amino acid starvation by entering into a hibernatory state. Proc. Natl. Acad. Sci. USA 109, E3278–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Painter HJ Mitochondrial electron transport inhibition and viability of intraerythrocytic Plasmodium falciparum. Antimicrob. Agents Chemother 54, 5281–5287 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Biljon R et al. Inducing controlled cell cycle arrest and re-entry during asexual proliferation of Plasmodium falciparum malaria parasites. Sci. Rep 8, 16581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ismail HM, et al. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc Natl Acad Sci USA. 113, 2080–2085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun. 6, 10111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bridgford JL et al. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat Commun. 9, 3801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noedl H et al. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med 359, 2619–2620 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Dondorp AM et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 361, 455–467 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witkowski B et al. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother 54, 1872–1877 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; After three years of selection in vitro, artemisinin refractory P. falciparum were isolated and refractoriness linked to ring-stage growth arrest.
- 62.Ariey F et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 505, 50–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Discovery of the link between Pfk13 mutations and artemisinin treatment failure in the field, which was shown to be related to ring-stage growth arrest.
- 63.Witkowski B et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect. Dis 2013 13, 1043–1049 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Straimer J et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347, 428–431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tilley L, Straimer J, Gnädig NF, Ralph SA & Fidock DA Artemisinin Action and Resistance in Plasmodium falciparum. Trends Parasitol. 32, 682–696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mbengue A A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature. 520, 683–687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heller LE. & Roepe P,D Quantification of freeferriprotoporphyrin IX heme and hemozoin for artemisinin sensitive versus delayed clearance phenotype Plasmodium falciparum malarial parasites. Biochemistry 57, 6927–6934 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Demas AR, et al. Mutations in Plasmodium falciparum actin-binding protein coronin confer reduced artemisinin susceptibility. Proc Natl Acad Sci U S A. 115, 12799–12804 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paloque L, Ramadani AP, Mercereau-Puijalon O, Augereau JM & Benoit-Vical F Plasmodium falciparum: multifaceted resistance to artemisinins. Malar J. 15, 149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Teuscher F et al. Artemisinin-induced dormancy in Plasmodium falciparum: duration, recovery rates, and implications in treatment failure. J. Infect. Dis 202, 1362–1368 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dembele L et al. The Plasmodium PI(4)K inhibitor KDU691 selectively inhibits dihydroartemisinin-pretreated Plasmodium falciparum ring-stage parasites. Sci Rep. 7, 2325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dembele L et al. Imidazolopiperazines Kill both rings and dormant rings in wild-type and K13 artemisinin-resistant Plasmodium falciparum in vitro. Antimicrob. Agents Chemother 62, pii: e02235–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Neill PM et al. A tetraoxane-based antimalarial drug candidate that overcomes PfK13-C580Y dependent artemisinin resistance. Nat Commun. 24, 15159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duvalsaint M & Kyle DE Phytohormones, isoprenoids, and role of the apicoplast in recovery from dihydroartemisinin-Induced dormancy of Plasmodium falciparum. Antimicrob. Agents Chemother 62, pii: e01771–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang M et al. Inhibiting the Plasmodium eIF2alpha kinase PK4 prevents artemisinin-Induced latency. Cell Host Microbe. 22, 766–776 e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang M et al. The Plasmodium eukaryotic initiation factor-2alpha kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. J. Exp. Med 207, 1465–1474 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dubey JP Foodborne and waterborne zoonotic sarcocystosis. Food and Waterborne Parasitology 1, 2–11 (2015). [Google Scholar]
- 78.Seeber F & Steinfelder S Recent advances in understanding apicomplexan parasites. F1000Res 5, pii: F1000 Faculty Rev-1369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dubey JP Toxoplasmosis of animals and humans. (CRC Press, 2010). [Google Scholar]
- 80.Watts E et al. Novel approaches reveal that Toxoplasma gondii bradyzoites within tissue cysts are dynamic and replicating entities in vivo. MBio 6, e01155–01115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrates that bradyzoites are not dormant but rather that they replicate slowly and asynchronously within the tissue cyst.
- 81.Hunter CA & Sibley LD Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol. 10, 766–778 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dubey JP Bradyzoite-induced murine toxoplasmosis: stage conversion pathogenesis, and tissue cyst formation in mice fed bradyzoites of different strains of Toxoplasma gondii. J. Euk. Microb 44, 592–602 (1997). [DOI] [PubMed] [Google Scholar]
- 83.Israelski DM & Remington JS Toxoplasmosis in the non-AIDS immunocompromised host. Curr. Clin. Top. Infect. Dis 13, 322–356 (1993). [PubMed] [Google Scholar]
- 84.Mariuz P & Steigbigel RT in Toxoplasmosis a comprehensive clinical guide (eds Joynson DHM & Wreghitt TG) 147–177 Cambridge Univ. Press; (2007). [Google Scholar]
- 85.Dunay IR, Gajurel K, Dhakal R, Liesenfeld O & Montoya JG Treatment of Toxoplasmosis: Historical perspective, animal models, and current clinical practice. Clin. Microbiol. Rev 31, pii: e00057–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Provides a current summary of treatment options and limitations for toxoplasmosis.
- 86.Behnke MS, Zhang TP, Dubey JP &, Sibley LD Toxoplasma gondii merozoite gene expression analysis with comparison to the life cycle discloses a unique expression state during enteric development. BMC Genomics. 15, 350 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Radke JR et al. Changes in the expression of human cell division autoantigen-1 influence Toxoplasma gondii growth and development. PLoS Pathog. 2, e105 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Behnke M, Radke J, Smith AT, Sullivan WJ & White MW The transcription of bradyzoite genes in Toxoplasma gondii is controlled by autonomous promoter elements. Mol. Microbiol 68, 1502–1518 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Behnke MS et al. Coordinated progression through two subtranscriptomes underlies the tachyzoite cycle of Toxoplasma gondii. PLoS One 5, e12354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Radke JR et al. The transcriptome of Toxoplasma gondii. BMC Biol. 3, 26 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hong DP, Radke JB & White MW Opposing transcriptional mechanisms regulate Toxoplasma development. mSphere 2, pii: e00347–16. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang S et al. Toxoplasma gondii AP2IX-4 Regulates Gene Expression during bradyzoite development. mSphere 2, pii: e00054–17. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Radke JB et al. ApiAP2 transcription factor restricts development of the Toxoplasma tissue cyst. Proc Natl Acad Sci USA 110, 6871–6876 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Radke JB et al. Transcriptional repression by ApiAP2 factors is central to chronic toxoplasmosis. PLoS Pathog. 14, e1007035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walker R et al. The Toxoplasma nuclear factor TgAP2XI-4 controls bradyzoite gene expression and cyst formation. Mol. Microbiol 87, 641–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Painter HJ, Campbell TL & Llinas M The Apicomplexan AP2 family: integral factors regulating Plasmodium development. Mol. Biochem. Parasitol 176, 1–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sinha A et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature 507, 253–257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yuda M, Iwanaga S, Shigenobu S, Kato T & Kaneko I Transcription factor AP2-Sp and its target genes in malarial sporozoites. Mol. Microbiol 75, 854–863 (2010). [DOI] [PubMed] [Google Scholar]
- 99.White MW, Radke JR & Radke JB Toxoplasma development - turn the switch on or off? Cell. Microbiol 16, 466–472 (2014). [DOI] [PubMed] [Google Scholar]; Summarizes how host and parasite-specific factors control stage differentiation of T. gondii.
- 100.Fox BA, Gigley JP, & Bzik DJ Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int J Parasitol. 34, 323–331 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Soête M, Camus D & Dubremetz JF Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp Parasitol. 78, 361–370 (1994). [DOI] [PubMed] [Google Scholar]
- 102.Bohne W, Heesemann J & Gross U Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect Immun. 62, 1761–1767 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ferreira-da-Silva Mda F, Takács AC, Barbosa HS, Gross U & Lüder CG Primary skeletal muscle cells trigger spontaneous Toxoplasma gondii tachyzoite-to-bradyzoite conversion at higher rates than fibroblasts. Int. J. Med. Microbiol 229, 381–388 (2009). [DOI] [PubMed] [Google Scholar]
- 104.Tanaka N, Ashour D, Dratz E & Halonen S Use of human induced pluripotent stem cell-derived neurons as a model for cerebral toxoplasmosis. Microbes Infect. 18, 496–504 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Blader IJ & Saeij JP Communication between Toxoplasma gondii and its host: impact on parasite growth, development, immune evasion, and virulence. APMIS 117, 458–476 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jeffers V, Tampaki Z, Kim K & Sullivan WJ Jr. A latent ability to persist: differentiation in Toxoplasma gondii. Cell. Mol. Life Sci 75, 2355–2373 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dubey JP Bradyzoite-induced murine toxoplasmosis: stage conversion, pathogenesis, and tissue cyst formation in mice fed bradyzoites of different strains of Toxoplasma gondii. J Eukaryot Microbiol. 44, 592–602 (1998). [DOI] [PubMed] [Google Scholar]
- 108.Su C,et al. Recent expansion of Toxoplasma through enhanced oral transmission. Science. 299, 414–416 (2003). [DOI] [PubMed] [Google Scholar]
- 109.Dubey JP, Miller NL & Frenkel JK The Toxoplasma gondii oocyst from cat feces. J. Exp. Med 132, 636–662 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jones JL & Dubey JP Foodborne toxoplasmosis. Clin. Infect. Dis 55, 864–851 (2012). [DOI] [PubMed] [Google Scholar]
- 111.Torgerson PR & Mastroiacovo P The global burden of congenital toxoplasmosis: a systematic review. Bull. World Health Organ 91, 501–508 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pfaff AW et al. New clinical and experimental insights into old world and neotropical ocular toxoplasmosis. Int. J. Parasitol 44, 99–107 (2013). [DOI] [PubMed] [Google Scholar]
- 113.Rutaganira FU et al. Inhibition of calcium dependent protein kinase 1 (CDPK1) by pyrazolopyrimidine analogs decreases establishment and reoccurrence of central nervous system disease by Toxoplasma gondii. J. Med. Chem 60, 9976–9989 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen M, Osman I & Orlow SJ Antifolate activity of pyrimethamine enhances temozolomide-induced cytotoxicity in melanoma cells. Mol. Cancer Res 7, 703–712 (2009). [DOI] [PubMed] [Google Scholar]
- 115.Tomita T et al. The Toxoplasma gondii cyst wall protein CST1 is critical for cyst wall integrity and promotes bradyzoite persistence. PLoS Pathog. 9, e1003823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lemgruber L, Lupetti P, Martins-Duarte ES, De Souza W & Vommaro RC The organization of the wall filaments and characterization of the matrix structures of Toxoplasma gondii cyst form. Cell. Microbiol 13, 1920–1932 (2011). [DOI] [PubMed] [Google Scholar]
- 117.Spalenka J et al. Discovery of new inhibitors of Toxoplasma gondii via the pathogen box. Antimicrob Agents Chemother. 62, e01640–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Adeyemi OS, Sugi T, Han Y & Kato K Screening of chemical compound libraries identified new anti-Toxoplasma gondii agents. Parasitol. Res 117, 355–363 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Benmerzouga I et al. Guanabenz repurposed as an antiparasitic with activity against acute and latent toxoplasmosis. Antimicrob. Agents Chemother 59, 6939–6945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dittmar AJ, Drozda AA & Blader IJ Drug repurposing screening identifies novel compounds that effectively inhibit Toxoplasma gondii growth. mSphere 1, pii: e00042–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Konrad C, Queener SF, Wek RC & Sullivan WJ Jr. Inhibitors of eIF2alpha dephosphorylation slow replication and stabilize latency in Toxoplasma gondii. Antimicrob. Agents Chemother 57, 1815–1822 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Radke JB, Burrows JN, Goldberg DE & Sibley LD Evaluation of current and emerging antimalarial medicines for inhibition of Toxoplasma gondii growth in vitro. ACS Infectious Diseases 4, 1264–1274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Evaluates sensitivity of T. gondii to current drugs and advanced leads for malaria treatment.
- 123.McFadden DC, Tomavo S, Berry EA & Boothroyd JC Characterization of cytochrome b from Toxoplasma gondii and Qo domain mutations as a mechanism of atovaquone-resistance. Mol. Biochem. Parasitol 108, 1–12 (2000). [DOI] [PubMed] [Google Scholar]
- 124.Alday PH et al. Genetic Evidence for Cytochrome b Qi Site Inhibition by 4(1H)-Quinolone-3-Diarylethers and Antimycin in Toxoplasma gondii. Antimicrob. Agents Chemother 61, pii: e01866–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tekwani BL & Walker LA 8-Aminoquinolines: future role as antiprotozoal drugs. Curr. Opin. Infect. Dis 19, 623–631 (2006). [DOI] [PubMed] [Google Scholar]
- 126.Pérez-Molina JA & Molina I Chagas disease. Lancet. 391, 82–94 (2017). [DOI] [PubMed] [Google Scholar]
- 127.Dumoulin PC & Burleigh BA Stress-induced proliferation and cell cycle plasticity of intracellular Trypanosoma cruzi Amastigotes. MBio. 9, e00673–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Francisco AF, et al. , Biological factors that impinge on Chagas disease drug development. Parasitology 144, 1871–1880 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lewis MD, et al. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell. Microbiol 16,1285–1300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sosa Estani S, et al. Efficacy of chemotherapy with benznidazole in children in the indeterminate phase of Chagas’ disease. Am. J. Trop. Med. Hyg 59, 526–529 (1998). [DOI] [PubMed] [Google Scholar]
- 131.Viotti R, Long-term I cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: a nonrandomized trial. Ann. Intern. Med 144,724–734 (2006). [DOI] [PubMed] [Google Scholar]; Demonstration in long-term follow-up studies of clinical benefits of treatment of chronic T. cruzi infection in humans
- 132.Bern C A new epoch in antitrypanosomal treatment for Chagas disease. J. Am. Coll. Cardiol 69, 948–950 (2017). [DOI] [PubMed] [Google Scholar]
- 133.Weatherly DB, Peng D & Tarleton RL Recombination-driven generation of the largest pathogen repository of antigen variants in the protozoan Trypanosoma cruzi. BMC Genomics 17, 729 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bustamante JM, et al. New, combined, and reduced dosing treatment protocols cure Trypanosoma cruzi infection in mice. J. Infect. Dis 209,150–162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alvarez MG, et al. Seronegative conversion after incomplete benznidazole treatment in chronic Chagas disease. Trans. R. Soc. Trop. Med. Hyg 106, 636–638 (2012). [DOI] [PubMed] [Google Scholar]
- 136.MacLean LM, et al. Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Negl. Trop. Dis, 12, e0006612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Alvarez MG, et al. New scheme of intermittent benznidazole administration in patients chronically infected with Trypanosoma cruzi: a pilot short-term follow-up study with adult patients. Antimicrob. Agents Chemother 60,833–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wilkinson SR, Taylor MC, Horn D, Kelly JM & Cheeseman I A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 105, 5022–5027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Burza S, Croft SL & Boelaert M Leishmaniasis. Lancet. 392, 951–970 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Sundar S & Chakravarty J Leishmaniasis: an update of current pharmacotherapy. Expert Opin. Pharmacother 14, 53–63. (2013) [DOI] [PubMed] [Google Scholar]
- 141.Mukhopadhyay D, Dalton JE, Kaye PM, Chatterjee M Post kala-azar dermal leishmaniasis: an unresolved mystery. Trends Parasitol. 30, 65–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Marovich MA et al. Leishmaniasis recidivans recurrence after 43 years: a clinical and immunologic report after successful treatment. Clin. Infect. Dis 33, 1076–1079 (2001). [DOI] [PubMed] [Google Scholar]
- 143.Ponte-Sucre A et al. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis 11, e0006052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Imamura H, et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. elife. 5, e12613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Carnielli JBT et al. A Leishmania infantum genetic marker associated with miltefosine treatment failure for visceral leishmaniasis. EBioMedicine. 36, 83–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pountain AW et al. Genomic instability at the locus of sterol C24-methyltransferase promotes amphotericin B resistance in Leishmania parasites. PLoS Negl Trop Dis. 13, e0007052 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Purkait B et al. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob. Agents Chemother 56, 1031–1041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kloehn J, Saunders EC, O’Callaghan S, Dagley MJ & McConville MJ Characterization of metabolically quiescent Leishmania parasites in murine lesions using heavy water labeling. PLoS Pathog. 11, e1004683 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Use of heavy water labelling incorporation into parasite biopolymers in vivo revealing very slow net replication rates of L. mexicana populations in mice.
- 149.Saunders EC et al. Induction of a stringent metabolic response in intracellular stages of Leishmania mexicana leads to increased dependence on mitochondrial metabolism. PLoS Pathog. 10, e1003888 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jara M et al. , Macromolecular biosynthetic parameters and metabolic profile in different life stages of Leishmania braziliensis: Amastigotes as a functionally less active stage. PLoS One. 12, e0180532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tegazzini D et al. Replicative in vitro assay for drug discovery against Leishmania donovani. Antimicrob. Agents Chemother 60, 3524–3532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wasunna M et al. , Efficacy and safety of AmBisome in combination with sodium stibogluconate or miltefosine and miltefosine monotherapy for African visceral leishmaniasis: Phase II Randomized Trial. PLoS Negl Trop Dis. 14, e0004880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Carter NS et al. Adaptive responses to purine starvation in Leishmania donovani. Mol Microbiol. 78, 92–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Martin JL et al. Metabolic reprogramming during purine stress in the protozoan pathogen Leishmania donovani. PLoS Pathog. 10, e1003938 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Proteomics and transcriptomics approach to probe changes in L. donovani non-proliferative promastigotes form selected in purine-free medium.
- 155.Jara M, et al. Tracking of quiescence in Leishmania by quantifying the expression of GFP in the ribosomal DNA locus. bioRxiv doi: 10.1101/641290 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]; Report of fluorescent dyes and GFP expression from ribosomal locus revealing mixed dividing versus non-dividing amastigote populations of Leishmania mexicana and L. braziliensis in vitro and in vivo.
- 156.Fox BA, Belperron AA & Bzik DJ Negative selection of herpes simplex virus thymidine kinase in Toxoplasma gondii. Mol. Biochem. Parasitol 116, 85–88 (2001). [DOI] [PubMed] [Google Scholar]
- 157.Merrick CJ Transfection with thymidine kinase permits bromodeoxyuridine labelling of DNA replication in the human malaria parasite Plasmodium falciparum. Malar. J 14, 490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wells TN, Burrows JN, Baird JK Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 26, 145–151 (2010). [DOI] [PubMed] [Google Scholar]
- 159.Handman E Leishmaniasis: current status of vaccine development. Clin. Microbiol. Rev 14, 229–243 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]