

Abstract
Human induced pluripotent stem cells (iPSCs) have emerged as a promising alternative to bone-marrow derived mesenchymal stem/stromal cells for cartilage tissue engineering. However, the effect of biochemical and mechanical cues on iPSC chondrogenesis remains understudied. This study evaluated chondrogenesis of induced pluripotent mesenchymal progenitor cells (iPS-MPs) encapsulated in a cartilage-mimetic hydrogel under different culture conditions: free swelling versus dynamic compressive loading and different growth factors (TGFβ3 and/or BMP2). Human iPSCs were differentiated into iPS-MPs and chondrogenesis was evaluated by gene expression (qPCR) and protein expression (immunohistochemistry) after three weeks. In pellet culture, both TGFβ3 and BMP2 were required to promote chondrogenesis. However, the hydrogel in growth factor-free conditions promoted chondrogenesis, but rapidly progressed to hypertrophy. Dynamic loading in growth factor-free conditions supported chondrogenesis, but delayed the transition to hypertrophy. Findings were similar with TGFβ3, BMP2, and TGFβ3+BMP2. Dynamic loading with TGFβ3, regardless of BMP2, was the only condition that promoted a stable chondrogenic phenotype (aggrecan+collagen II) accompanied by collagen X down-regulation. Positive TGFβRI expression with load-enhanced Smad2/3 signaling and low SMAD1/5/8 signaling was observed. In summary, this study reports a promising cartilage-mimetic hydrogel for iPS-MPs that when combined with appropriate biochemical and mechanical cues induces a stable chondrogenic phenotype.
Graphical Abstract
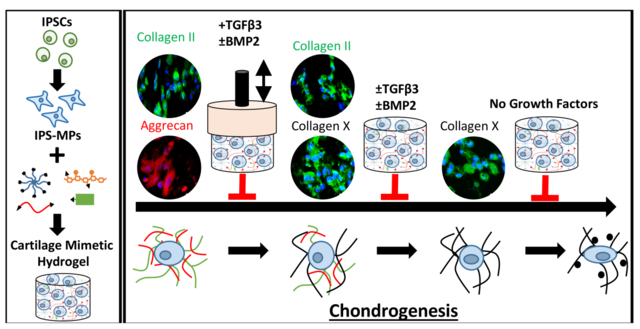
The chondrogenesis of iPSC-mesenchymal progenitor cells in a cartilage mimetic hydrogel is dependent on the combinatory effects of biophysical cues and exogenous growth factors.
Introduction
Articular cartilage when damaged due to injury or disease is unable to regenerate because of low cellular density and a lack of vascularity.1 If left untreated, damaged cartilage can lead to osteoarthritis. However, early treatment has the potential to stop disease progression.2 Cell-based therapies such as autologous chondrocyte implantation (ACI)3 and more recently, matrix assisted ACI (MACI)4–6 have shown improved patient-reported outcomes in young, healthy patients in the short-term, however repair tissue does not resemble hyaline cartilage.3,7–9 Bone marrow (BM)-derived mesenchymal stem/stromal cells (MSCs) (BM-MSCs) are a potential alternative to autologous chondrocytes, though they too have their own shortcomings including a loss in chondrogenic potential with age, age-related diseases, and higher passage number.10–13 Moreover, BM-MSCs can lead to fibrocartilage formation14,15 or undergo hypertrophic terminal differentiation, a precursor to endochondral ossification, during chondrogenesis.16,17 Consequently, achieving hyaline cartilage during cartilage repair remains a challenge and has led to further investigation into other cell sources.
Induced pluripotent stem cells (iPSCs) have emerged as a promising source of stem cells for tissue engineering.18 IPSCs are generated from adult somatic cells, such as skin fibroblasts, that are reprogrammed to an early state of differentiation similar to that of embryonic stem cells19. IPSCs have many of the benefits of embryonic stem cells including high proliferative capacity and pluripotency,20,21 but without the ethical concerns.22–24 Moreover, iPSCs offer an autologous cell source that when generated from older patients maintain pluripotency similar to that of younger patients, overcoming one of the major limitations of BM-MSCs.25
The chondrogenesis of iPSCs has been investigated in several studies using a variety of culturing conditions. In pellet culture, studies have reported improved chondrogenic differentiation of iPSCs following a mesenchymal progenitor intermediate step, with 70% of cell pellets revealing cartilage-matrix production.26 Collagen II and proteoglycan deposition by iPSC-mesenchymal progenitor cells was reported in another study, however, matrix production was heterogenous throughout the pellet.27 Only a limited number of studies have investigated iPSCs combined with scaffolds. One such study encapsulated human iPSCs from osteoarthritic chondrocytes in an alginate gel and reported that the cells underwent chondrogenesis but only when co-cultured with chondrocytes.28 In another study, murine iPSCs seeded onto polycaprolactone/gelatin electrospun fibrous scaffolds underwent chondrogenesis in the presence of TGFβ1 in vitro and revealed improvements in vivo over untreated osteochondral defects by histological assessments.29 Although these findings reveal the potential of iPSCs for cartilage tissue engineering, the role of external cues on iPSC fate in the context of chondrogenesis and differentiation towards hyaline, fibrous, and hypertrophic cartilage has yet to be fully elucidated.
Chondrogenic differentiation of MSCs has been shown to be highly dependent on the scaffold type, soluble growth factors and the presence of dynamic loading.30–32 Scaffolds influence cell-cell and cell-matrix interaction, ultimately impacting the response of MSCs to external cues which can vary their differentiation potential.32 For example, scaffolds that mimic aspects of the native cartilage environment have shown enhanced chondrogenesis and hyaline-like tissue deposition of encapsulated MSCs.33–36 A number of studies have used soluble cues, such as growth factors, to promote differentiation and extracellular matrix development.37 Members of the TGFβ family, for instance, are potent inducers of MSC chondrogenesis and regulators of tissue production38. Additionally, a variety of mechanical stimuli, including dynamic compression and shear stress, that recapitulate the natural joint loading environment have beneficial effects on MSC chondrogenesis, matrix elaboration, and the regulation of differentiation towards hyaline or hypertrophic cartilage.31 Interestingly, a synergistic effect of some or all of these factors (scaffold type, growth factors, and dynamic loading) has also been reported, where the benefit of one may rely on the presence of another.39–42 While there is extensive research on the role of external factors on MSC fate, further investigation into iPSC chondrogenesis as a function of their environment is still needed to realize their full potential in cartilage tissue engineering.
In this study, iPSCs were generated following an integration-free reprogramming method43,44 and then differentiated down the mesenchymal lineage, referred to as iPSC-mesenchymal progenitors (iPS-MPs). The specific goal for this study was to evaluate chondrogenesis of the iPS-MPs as a function of growth factors, dynamic compressive loading, and a combination thereof when encapsulated in a cartilage-mimetic hydrogel. We recently reported on a cartilage-mimetic hydrogel designed from a poly(ethylene glycol) (PEG) hydrogel containing extracellular matrix (ECM) analogs of chondroitin sulfate (ChS) and the adhesion peptide RGD that promoted stable chondrogenesis of BM-MSCs under dynamic compressive loading.33,39 Our prior studies identified that under exogenous TGFβ3 stimulation, the presence of ChS in the cartilage-mimetic hydrogel enhanced chondrogenesis of BM-MSCs and was necessary to suppress hypertrophy under loading.39 Given the promise of this cartilage-mimetic hydrogel for chondrogenesis of BM-MSCs, this study aimed to investigate if the hydrogel similarly could be promising for chondrogenesis of iPS-MPs. Moreover, this study investigates the role of the hydrogel itself under growth factor-free conditions and then when combined with exogenous growth factors, TGFβ3 and/or BMP2, on chondrogenesis of iPS-MPs. A stable version of the cartilage-mimetic hydrogel is used to investigate the effects of external cues in a controlled 3D culture environment. Chondrogenesis was first confirmed in pellet culture and then evaluated in the hydrogel for chondrogenic markers of hyaline cartilage, markers of fibrocartilage, and markers of hypertrophy using a combination of gene expression and immunohistochemistry methods. Moreover, TGFβRI expression and Smad signaling were probed to provide additional insight into the influence of external cues on the differentiation fate of iPS-MPs in the hydrogel. The study design is depicted in Figure 1. Overall, this study demonstrates that the cartilage-mimetic hydrogel under growth factor-free conditions is capable of inducing chondrogenesis of iPS-MPs under dynamic loading, but that the hydrogel combined with exogenous delivery of TGFβ3 and dynamic loading is required to achieve a stable hyaline cartilage phenotype that suppresses hypertrophy.
Figure 1.

Study design to evaluate the iPS-MP chondrogenesis. A) IPSCs were differentiated down the mesenchymal lineage into iPS-MPs prior to use in this study. B) In pellet culture, iPS-MPs were cultured with chondrogenic media containing no growth factor, TGFβ3 (10 ng ml−1), BMP2 (100 ng ml−1), or TGFβ3 (10 ng ml−1) and BMP2 (100 ng ml−1) and chondrogenesis assessed at day 21 by histology, IHC, and biochemical analysis. C) In 3D culture, iPS-MPs were photoencapsulated in a cartilage-mimetic hydrogel made from multi-arm PEG norbornene macromers, PEG dithiol crosslinkers, and ECM analogs of thiolated-chondroitin sulfate and CRGDS. The iPS-MP-laden hydrogels were cultured in chondrogenic media containing no growth factor, TGFβ3 (2.5 ng ml−1), BMP2 (25 ng ml−1), or TGFβ3 (2.5 ng ml−1) and BMP2 (25 ng ml−1) under free swelling culture or under a dynamic loading protocol. The latter consisted of one week free swelling followed by intermittent dynamic compressive loading for one hour per day. At day 21, iPS-MP differentiation was analyzed via qPCR, IHC and quantitative IHC.
Materials and methods
Macromer synthesis
Poly(ethylene glycol) (PEG) norbornene (8 arm, 10kDa) was synthesized from PEG amine as described previously.45 Briefly, 8-arm PEG amine (10kDa) (JenKem Technology) was dissolved in dimethyleformamide (DMF, Sigma Aldrich) and reacted under argon with 5-norbornene-2-carboxylic acid at 4 molar excess with n,n-diisopropylethylamine (DIEA, Sigma Aldrich) and 1-[Bis(dimethylamaino0methylene]-1H-1,2,3-triazolog[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU, Sigma Aldrich) overnight at room temperature. The 8-arm PEG norbornene product was precipitated in cold diethyl ether, vacuum filtered, purified by dialysis, and lyophilized to recover product. Conjugation of norbornene to each arm of the PEG amine molecule was ~100% determined by comparing the area under the peak for the allylic hydrogen closest to the norbornene hydrocarbon group (δ=3.1–3.2ppm) to the area under the peak for the methyl groups of the PEG backbone (δ=3.4–3.85 ppm) using 1H NMR.
Thiolated chondroitin sulfate (ChS-SH) was synthesized as described previously.46 Briefly, chondroitin sulfate (ChS) (Sigma Aldrich) was dissolved in water and reacted with two molar excess dithiobis(propanoic dihydrazide) (DTP) (2 moles DTP: 1 mole ChS repeat unit). The pH was adjusted to 4.75 by 1M HCl. Two molar excess of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI, Sigma Aldrich) was added per mole of ChS repeat unit and DTP solution and reacted overnight. The reaction was stopped by 1M NaOH to pH 7. Excess dithiothreitol (DTT, Sigma Aldrich) at 6.5 moles DTT: 1 mol ChS repeat unit was added to the solution and the pH was adjusted to 8.8 using 1M NaOH. The reaction was carried out for 24 hours to reduce the thiol groups of the ChS-conjugated DTP. The final product (ChS-SH) was purified and recovered by dialysis against 0.3 mM HCl, centrifuged to remove any particulates, and the supernatant was lyophilized. Conjugation of the thiol groups to ChS was determined to be 15% by 1H NMR by comparing the area under the peak representing the methylene groups associated with the ChS conjugated DTP (δ=2.5–2.6 and 2.6–2.8ppm) to that of the methyl protons of the acetyl amine on the ChS backbone (δ=1.8–2.0 ppm). The 15% conjugation of thiols to ChS indicates that there are approximately seven thiol groups per molecule of ChS assuming a molecular weight of 22kDa.47
Generation of human iPSCs
Human skin fibroblasts from a 50-year-old female (obtained commercially from American Type Culture Collection (ATCC)) were reprogrammed under low oxygen (5%) conditions by direct transfection of modified mRNA molecules encoding 6 human reprogramming factors, M3O (Myo-D-Oct4)48, SOX2, KLF4, cMYC, NANOG and LIN28A in combination with a cocktail of reprogramming embryonic stem cell-specific microRNAs, miRNA-367/302s (Qiagen, Hilden, Germany) as previously described.43,44 The reprogrammed cells were analyzed for pluripotency and underwent karyotype analysis before being used in this study.
Culture of human iPSCs
iPSCs were expanded on a feeder layer of neonatal human dermal fibroblasts (ATCC) and cultured in human embryonic stem cell medium (hESM) containing equal parts Dulbecco’s Modified Eagle Medium (DMEM)/F-12 (Life Technologies) and neurobasal medium (Life Technologies), supplemented with 1% N-2 supplement (Life Technologies), 2% B-27 supplement (Life Technologies), 50 μg/mL ascorbic acid (Fisher Scientific, Hampton, NH, USA), 1% nonessential amino acids (NEAA; Life Technologies), 1 mM L-glutamine (Life Technologies), 55 μM 2-mercaptoethanol (Fisher Scientific), 0.05% bovine serum albumin (Sigma-Aldrich), 1% penicillin-streptomycin (P/S; Life Technologies), 100 ng/mL recombinant human basic fibroblast growth factor (bFGF; Peprotech, Rocky Hill, NJ, USA), and 10 μM Y-27632 rho kinase inhibitor (Selleck Chemicals, Houston, TX, USA). iPSCs were passaged by enzymatic treatment with 4 mg/mL collagenase, type I (Life Technologies) in hESM. Colonies were then manually cut into squares using the StemPro EZPassage Disposable Stem Cell Passaging Tool (Life Technologies) and plated on new feeder layers at a 1:2 ratio. Cells were cultured in a humidified atmosphere at 37°C and 5% CO2.
Derivation of Mesenchymal Progenitors from Human iPSCs
iPSCs were induced to become mesenchymal progenitors using a previously published protocol.49 IPSCs underwent feeder depletion on 0.1% gelatin-coated dishes for 1 hour and were then plated on gelatin-coated tissue culture-treated flasks at 1×104 cells/cm2 in MSC induction medium consisting of DMEM-High Glucose (DMEM-HG; Life Technologies), 10% fetal bovine serum (FBS; Atlanta Biologicals), 1% NEAA, 1% P/S, and 5 ng/mL human bFGF. Cells were cultured in a humidified atmosphere at 37°C and 5% CO2, with media changes every 2–3 days. Cells were passaged when they reached 80% confluency using 0.25% trypsin/EDTA (Life Technologies) and subsequent passages were plated on tissue culture-treated flasks without gelatin at 1 × 104 cells/cm2 in MSC media.
Flow Cytometric Analysis of iPS-MPs
Mesenchymal differentiation was assessed by flow cytometry for mesenchymal cell surface markers CD90, CD73, and CD105. iPS-MPs were harvested using Accutase (Innovative Cell Technologies, San Diego, CA), washed in PBS, and resuspended in stain buffer consisting of 0.1% sodium azide and 5% FBS in PBS. Cell suspensions were mixed with FITC Mouse Anti-Human CD90, FITC Mouse Anti-Human CD105, or PE Mouse Anti-Human CD73. Nonspecific fluorescence was determined by incubation of cell samples with either PE Mouse IgG1, κ Isotype Control or FITC Mouse IgG1, κ Isotype Control. All antibodies were purchased from BD Biosciences, San Jose, CA, USA. A minimum of 10,000 events were analyzed using a Beckman Coulter Gallios in the Flow Cytometry Shared Resource at the University of Colorado Cancer Center.
Chondrogenic Differentiation of iPS-MPs in Pellet Culture
At passage 6, cells were treated with Accutase to dissociate, and 250,000 cells were added to each 15-mL conical polypropylene tube. Cells were centrifuged at 1500 RPM for 5 minutes and resuspended in 500 μL chondrogenic medium (CM) containing DMEM-HG, 50 μg/mL L-ascorbic acid 2-phosphate (Sigma-Aldrich), 40 μg/mL DL-proline (Sigma-Aldrich), 1% ITS+ Premix (Becton-Dickinson, Franklin Lakes, NJ), 1% P/S and with either no growth factors, or with 100 ng ml−1 BMP2 (Peprotech) and/or 10 ng ml−1 TGF-β3 (Peprotech). Cells were then pelleted at 500xg for 5 minutes. All pellets were incubated at 37°C in 5% CO2 for 21 days, with CM refreshed every 2–3 days.
Analysis of Chondrogenic Pellets
After 21 days, pellets were washed with PBS and fixed in 10% neutral buffered formalin followed by paraffin embedding. Cross sections (5 μm thick) were stained with Safranin O-Fast Green for sulfated glycosaminoglycans (sGAG) using standard protocols. Immunohistochemical analysis was performed using a Collagen Type II (Col II) antibody (Developmental Studies Hybridoma Bank), followed by a goat anti-mouse-HRP (Jackson ImmunoResearch). Slides were developed using the ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories) and counterstained with VECTOR Hematoxylin QS (Vector Laboratories) per manufacturer’s instructions. Slides were visualized and images taken using a Nikon Eclipse 90i microscope (Tokyo, Japan). To measure total sGAG content, pellets were washed with PBS and each dry pellet was frozen at −80°C until ready to be assayed. sGAG content per pellet was quantified via the dimethylmethylene blue (DMMB) assay as previously described (n=3 pellets/group).11
Hydrogel Formation, iPS-MP Encapsulation, and Culture
The hydrogel precursor solution was comprised of 9% (g/g) 8-arm PEG-norbornene, 1% (g/g) ChS-SH, 0.1 mM CRGDS (Genscript), 2.14% (g/g) PEG dithiol (1kDa) (Sigma-Aldrich), and 0.5% (g/g) Irgacure 2959 (I2959, BASF) in phosphate buffered saline (PBS, pH 7.4). The precursor solution was filter sterilized with a 0.22 μm filter. IPS-MPs were combined with the sterile precursor solution to a final concentration of 10 million cells per ml. The precursor solution with cells was photopolymerized with 352 nm light at 5 mW cm−2 for 8 minutes. The iPS-MP-laden constructs (5mm in diameter and 2 mm in height) were cultured in chondrogenic differentiation medium (50 μg ml−1 l-ascorbic acid 2-phosphate (Sigma), 40 μg ml−1 DL-proline (Sigma), 1% ITS+ Premix (Corning), 1% penicillin/streptomycin in DMEM-Glutamax (Gibco). Medium was supplemented with either no growth factor or with one of three growth factor conditions: 2.5 ng ml−1 TGF-β3 (TGF-β3, Peprotech), 25 ng ml−1 BMP2 (BMP2, Peprotech), or 2.5 ng ml−1 TGF-β3 plus 25 ng ml−1 BMP2 (TGF-β3+BMP2). The iPS-MP-laden hydrogels were cultured under standard cell conditions of 37°C with 5% CO2 under free swelling conditions for seven days in individual wells of a 24 well plate with two milliliters of chondrogenic media per well. After one week, a subset of hydrogels was continued under free swelling culture conditions. A different subset of hydrogels were transferred to a custom-built bioreactor50,51 and subjected to intermittent unconfined dynamic compressive strains applied at 5% peak to peak strain (2.5% amplitude strain) at 1 Hz in a sinusoidal waveform one hour daily and with 23 hours of rest under a tare strain of <0.1%. Medium was replaced every other day for the duration of the three-week study.
Gene expression
At day 21, iPS-MP-laden hydrogels (n=3) were removed from culture for RNA extraction. Briefly, hydrogels were homogenized (TissueLyzer II, Qiagen) at 30 Hz for 10 minutes in RNA lysis buffer. RNA was extracted from the hydrogels using MicroElute Total RNA Kit (Omega) per manufacturer instruction. RNA was transcribed to cDNA using a high capacity reverse transcription kit (Applied Biosystems) per manufacturer instruction. Quantitative PCR (qPCR) was performed using Fast SYBR Green (Applied Biosystems) and a 7500 Fast Real-time PCR Machine (Applied Biosystems). PCR efficiency for each set of primers was determined from the slope of Ct values of 15–25 from serial dilutions of cDNA following the methods described previously.52 Data are reported as normalized gene expression (NE)
which is determined from the delta Ct values (the difference in Ct values between the control and the sample) using true PCR efficiencies (E) of the gene of interest (GOI) to the housekeeping gene (HKG) L30 and normalizing to pre-encapsulated iPS-MPs (control). The genes of interest included the chondrogenic markers of SOX9, ACAN, COL2A1, and the hypertrophic and osteogenic markers of RUNX2, COL10A1, and COL1A1. Primer sequences and efficiencies are given in Table 1.
Table 1.
qPCR Primer Sequences
| Gene | Forward Sequence | Reverse Sequence | Efficiency |
|---|---|---|---|
| L30 | 5’-TTAGCGGCTGCTGTTGGTT-3’ | 5’-TCCAGCGACTTTTTCGTCTTC-3’ | 94% |
| SOX9 | 5’-TGACCTATCCAAGCGCATTACCA-3’ | 5’-ATCATCCTCCACGCTTGCTCTGAA-3’ | 95% |
| ACAN | 5’-AGTATCATCGTCCCAGAATCTAGCA-3’ | 5’-AATGCAGAGGTGGTTTCACTCA-3’ | 88% |
| COL2A1 | 5’-CAACACTGCCAACGTCCAGAT-3’ | 5’-TCTTGCAGTGGTAGGTGATGTTCT-3’ | 102% |
| RUNX2 | 5’-TTGGCCTGGTGGTGTCATTA-3’ | 5’-GAGTCCTTCTGTGGCATGCA-3’ | 98% |
| COL10A1 | 5’-TTTTGCTGCTAGTATCCTTGAACT-3’ | 5’-ACCTCTAGGGCCAGAAGGAC-3’ | 87% |
| COL1A1 | 5’-AGGAAGGGCCACGACAAAG-3’ | 5’-CAGTTACACAAGGAACAGAACAGTCTCT-3 | 99% |
Immunohistochemistry (IHC)
At day 21, iPS-MP-laden hydrogels (n=3) were removed from culture and processed for immunohistochemistry. Hydrogels constructs were fixed overnight in 4% paraformaldehyde at 4°C and transferred to a 30% sucrose solution at 4°C for storage. Constructs were subjected to a series of dehydration steps following standard methods, embedded in paraffin, and sectioned at 10 μm. Sections were stained for collagen II, collagen X, pSmad2/3, pSmad1/5/8, and TGFβ I receptor (TGFβRI). Sections were pretreated depending on antibody as follows: 2000 U ml−1 hyaluronidase for anti-collagen II, 1 mg ml−1 protease followed by 1 mg ml−1 pepsin, and lastly 0.25% trypsin in 1mM EDTA for anti-collagen X, and antigen retrieval using Retrievagen (BD Biosciences) for pSmad2/3, pSmad1/5/8, and TGβRI. Following permeabilization and blocking, sections were treated overnight at 4°C with the primary antibody: 1:50 anti-collagen II (US Biological, C7510–20F), 1:50 anti-collagen X (Abcam, ab49945), and 1:50 anti-pSmad2/3 (Santa Cruz Biotechnology, sc-11769), anti-pSmad1/5/8 (Santa Cruz Biotechnology, sc-12353) and anti-TGβRI (Abcam, ab31013) in blocking solution. Sections were subsequently treated for 2 hours with goat anti-mouse IgG or goat anti-rabbit IgG labelled with either Alexa Fluor 488 (Abcam) (1:100) or Alexa Flour 546 (Abcam) (1:100) and nuclei counterstained with DAPI (ThermoFischer).
Stained sections were further analyzed to quantify the fraction of cells that stained positive for the protein of interest or the relative intensity of staining for the protein of interest. Representative confocal microscopy images (n=3 images per hydrogel, n=3 hydrogels per condition) were selected and processed using NIH ImageJ. For collagen II and collagen X, the number of nuclei that were associated with cells that stained positive for the protein was counted per image and divided by the total number of nuclei in that image. The data are reported as a fraction of positively stained cells. For pSmad2/3, pSmad1/5/8, and TGFβR1, the intensity of protein stain per cell was determined, averaged for each image (n=8–15) per hydrogel (n=3) were averaged for each condition. The data are reported as average intensity per nuclei.
Statistical Analysis
For the study of chondrogenesis in pellet culture, a one-way ANalysis Of VAriance (ANOVA, α=0.05) with growth factor (TGFβ3, BMP2, TGFβ3+BMP2) as the factor was investigated. For the study investigating the effect of loading in the absence of growth factors, qPCR and quantitative IHC were analyzed using an unpaired two-sample t-test assuming unequal variances. For the study investigating the effect of growth factors concomitant with loading, qPCR and quantitative IHC were investigated using a two-way ANOVA (α=0.05) with growth factor (TGFb3, BMP2, TGFB3+BMP2) and culture condition (free swelling, loading) as factors. In cases of statistically significant interaction, data were re-analyzed using one-way ANOVAs for growth factors or using unpaired two-sample t-test assuming unequal variances for loading. For ANOVAs, a post-hoc Tukey’s HSD test was used for pairwise comparisons. Data were tested for normality using Shapiro-Wilk and normal distribution plots as well as equality of error variances by Levene’s test to meet criteria prior to ANOVA. Analyses were performed using IBM SPSS Statistics Software. Statistical significance was set at p<0.05.
Results
Mesenchymal Differentiation of Human IPSCs and their chondrogenesis in pellet culture
A two-stage differentiation protocol was used, where human iPSCs were first directed towards the mesenchymal lineage. After 1 week of culture in MSC induction medium, iPSCs transitioned from growing as round colonies to growing as single cells that displayed a fibroblast-like morphology. A population of spindle-shaped cells was obtained after 3–4 passages (Figure 2A). Flow cytometric analysis of iPS-MPs indicated that the mesenchymal cell surface markers CD73 and CD105 were expressed on greater than 95% of the cells, while CD90 was expressed on approximately 20% of the cells (Figure 2B). Chondrogenic differentiation of the iPS-MPs was confirmed using a pellet culture assay under different growth factor combinations. In the absence of exogenous growth factors, a pellet had formed but chondrogenesis was not observed by a lack of positive staining for sulfated glycosaminoglycans (sGAGs) (Figure 2C) and collagen II (Figure 2D). The addition of BMP2 (100 ng ml−1) to the culture medium led to pellets of larger size when compared to the no growth factor condition, and this was even more evident when BMP2 (100 ng ml−1) and TGFβ3 (10 ng ml−1) were both present. Positive sGAG staining was observed in pellet cultures stimulated with BMP2 alone, with greater staining when a combination of BMP2 and TGFβ3 was present (Figure 2C). Similar results were observed with the collagen II immunostaining, where the presence of BMP2 and TGFβ3 led to highest expression of collagen type II (Figure 2D). On the contrary, TGFβ3 led to slightly increased pellet size over the no growth factor control with minimal staining for sGAGs and no obvious staining for collagen II. Quantification of sGAG content per pellet confirmed that presence of BMP2 or BMP2 combined with TGFβ3 led to greater (p<0.05) sGAG content when compared to the no growth factor control and to the TGFβ3-only group (Figure 2E).
Figure 2.

A) The morphological transition of iPSCs cultured in MSC induction medium from round colonies to single spindle-shaped cells. B) Flow cytometry analysis for the cell surface markers CD73, CD105, and CD90. C) Sulfated glycosaminoglycans (red) and (D) Collagen II ( brown) were observed in pellet culture of chondrogenically differentiating iPS-MPs when cultured with TGFβ3 (10 ng ml−1), BMP2 (100 ng ml−1), or their combination. Scale bar is 100μm. E) Quantification of sGAG content per pellet in culture media without growth factors (CM) or with TGFβ3, BMP2, or their combination. Data are represented as mean with standard deviation (n=3).
The effect of dynamic compressive loading in the absence of growth factors on iPS-MP chondrogenesis in the 3D hydrogel
IPS-MPs were encapsulated in a cartilage-mimetic hydrogel (Figure 1B) and cultured in chemically defined chondrogenic differentiation media, but in the absence of any exogenous growth factors for 21 days. Differentiation of iPS-MPs was evaluated by gene expression for the chondrogenic markers SOX9, ACAN, and COL2A1, the hypertrophic markers RUNX2, COL10A1 and the fibrocartilage marker COL1A1. The latter is also present in MSCs prior to their differentiation. For all genes analyzed, with the exception of RUNX2, there was a 10–10,000-fold increase in expression over the pre-encapsulated iPS-MPs regardless of the culture condition (Figure 3A). RUNX2 expression levels remained below one indicating that its expression decreased in the hydrogels and over time. Under dynamic loading, SOX9 expression decreased (p=0.048) when compared to free swelling condition. For chondrogenic markers, loading reduced (p=0.045) COL2A1 expression, but had no effect on ACAN expression. Loading had no effect on the hypertrophic markers RUNX2 and COL10A1, or the fibrocartilage marker COL1A1.
Figure 3.

IPS-MP-laden hydrogels cultured without and with loading in the absence of growth factors. A) Gene expression data for the chondrogenic markers SOX9, ACAN, COL2A1, the fibrocartilage marker COL1A1, and the hypertrophic markers RUNX2 and COL10A1 at day 21 normalized to pre-encapsulated iPS-MPs cultured under free swelling (black) and loading (gray). B) Representative confocal microscopy images of ECM proteins collagen I (green), aggrecan (red), collagen II (green), collagen X (green) and nuclei (blue) and (C) quantitative analysis of the fraction of positively stained cells for each protein at day 21 of iPS-MPs cultured under free swelling (black) or loading (gray). D) Representative confocal microscopy images TGFβRI (green), pSmad2/3 (red), pSmad1/5/8 (red) and nuclei (blue) and quantitative analysis of the average intensity for each protein per nuclei (E) at day 21 of iPS-MPs cultured under free swelling (black) or loading (gray). Scale bar is 20 μm. Data presented as mean with standard deviation (n=3).
Chondrogenesis was evident by extracellular matrix (ECM) production (Figure 3B&C). Aggrecan and collagen II staining was evident, but faint with only 20% fraction of the cells staining positive for collagen II. However, there was strong staining for collagen X protein with 60% of the cells staining positive. Under dynamic loading, aggrecan and collagen II staining appeared more intense than under free swelling, but collagen X protein was also present. Quantitative analysis revealed an increase (p<0.001) from 20% to 90% in the fraction of cells that stained positive for collagen II under loading (Figure 3C). This result is contrary to the collagen II gene level, which suggests different temporal profiles of this gene and its protein. The fraction of cells that stained positive for collagen X protein under loading was similar to that under free swelling, with approximately 60% of cells staining positively for the protein. This result, however, is consistent with the gene expression, which was similar under both culture conditions.
The expression of TGFβR1 along with the cellular signaling proteins phosphorylated Smad2/3 (pSmad2/3) and phosphorylated Smad1/5/8 (pSmad1/5/8), which are downstream of the TGFβ-superfamily receptors were also examined (Figure 3D&E). IPS-MPs stained positively for TGFβR1, pSmad2/3, and pSmad1/5/8 regardless of culture condition. Loading of iPS-MP-laden hydrogels in the absence of growth factors did not alter the expression of TGFβR1 or pSmad1/5/8, as measured by the average intensity per nuclei levels (Figure 3E). The mean intensity for pSmad2/3 expression was higher under loading, but this was not statistically significant. Collectively, these results indicate that loading alone is capable of inducing chondrogenic differentiation of iPS-MPs when encapsulated in the cartilage mimetic hydrogel. The relative intensity of pSmad2/3 to pSmad1/5/8 may be playing a role, but its contribution appears to be subtle.
The effect of growth factors and dynamic compressive loading on iPS-MP chondrogenesis in the 3D hydrogel
The effects of exogenous growth factors, TGFβ3, BMP2, and a combination thereof, on iPS-MP chondrogenesis in the cartilage-mimetic hydrogels were examined concomitant with loading. Chondrogenic differentiation was evaluated by gene expression for the chondrogenic markers SOX9, ACAN, and COL2A1. SOX9 expression was not affected by growth factors or by loading (Figure 4A). For ACAN expression, a two-way ANOVA analysis revealed a significant interaction between growth factor and loading, but further analysis by one-way ANOVAs did not indicate any significant differences for the simple main effects. COL2A1 expression was affected (p=0.017) by loading, but not by growth factor. Post-hoc analysis did not reveal any significant pairwise differences with loading. These results indicate minimal differences between the effects of growth factors and/or loading on chondrogenic gene expression.
Figure 4.

IPS-MP-laden hydrogels cultured without and with loading and with TGFβ3, BMP2, and TGFβ3+BMP2 for 21 days. A.) Gene expression data for chondrogenic markers SOX9, ACAN, COL2A1 at day 21 normalized to pre-encapsulated iPS-MPs cultured under free swelling (black) and loading (gray). Representative confocal microscopy images of the ECM proteins (B) aggrecan (red), (C) collagen II (green) and nuclei (blue) and quantitative analysis of the fraction of collagen II positively stained cells of iPS-MPs cultured under free swelling (black) or loading (gray). Scale bar is 20 μm. Data are presented as mean with standard deviation (n=3).
At the protein level, aggrecan was expressed in all culture conditions regardless of growth factors or loading (Figure 4B). Similarly, collagen II protein was present in all culture conditions regardless of growth factors or loading, with 70–90% of cells staining positive for the protein (Figure 4C). A two-way ANOVA analysis revealed a significant interaction with growth factors and loading on the fraction of collagen II expressing cells. Follow-up analyses revealed that under free swelling there was a reduction in the number of positively stained cells when the cells were cultured with both TGFβ3 and BMP2 when compared to TGFβ3 (p=0.024) and to BMP2 (p=0.019). There was no significant effect from loading for any of the growth factors or combinations.
Collagen I was also evaluated by gene and protein expression as a fibrocartilage marker. COL1A1 expression was maintained at around 1000-fold higher than the iPS-MPs prior to encapsulation and differentiation regardless of growth factor and culture condition (Figure 5A). However, there was no significant effect between growth factors and/or loading on COL1A1 expression. At the protein level, collagen I protein was expressed in the free swelling cultures with BMP2 or BMP2+ TGFβ3, but not detectable in the TGFβ3-only culture (Figure 5B). Loading appeared to dampen collagen I protein expression in both of the BMP2 culture conditions, but it remained present in the TGFβ3+BMP2 condition (Figure 5B).
Figure 5.

IPS-MP-laden hydrogels cultured without and with loading with TGFβ3, BMP2, and TGFβ3+BMP2 for 21 days. A.) Gene expression data for fibrocartilage marker COL1A1 at day 21 normalized to pre-encapsulated iPS-MPs cultured under free swelling (black) and loading (gray). B.) Representative confocal microscopy images of the ECM protein collagen I (green) and nuclei (blue). Scale bar is 20 μm. Data are presented as mean with standard deviation (n=3).
The hypertrophic markers, RUNX2 and COL10A1, were not affected by loading or by growth factors (Figure 6A). Collagen X protein was present under free swelling culture conditions for all growth factor conditions, but its presence appeared reduced under loading when TGFβ3 was present (Figure 6B). Quantification of the fraction of positively stained cells revealed a significant interaction between growth factors and loading and follow-up statistical analyses were performed. Under free swelling culture, collagen X protein was expressed in all growth factor conditions with 90% of the cells staining positive for the protein (p=0.78). Loading down-regulated collagen X protein expression, resulting in 30% (p=0.002) and 50% (p=0.014) of the cells staining positive for the protein in the TGFβ3-only and the TGFβ3+BMP2 conditions, respectively (Figure 6B). Under loading, growth factor was a significant factor (p=0.011) in collagen X protein expression. TGFβ3-only condition with loading resulted in the lowest number of collagen X expressing cells. For example, there was 2.6-fold fewer (p=0.009) cells expressed collagen X in the TGFβ3-only condition compared to the BMP2-only condition.
Figure 6.

IPS-MP-laden hydrogels cultured without and with loading with TGFβ3, BMP2, and TGFβ3+BMP2 for 21 days. A.) Gene expression data for hypertrophic markers RUNX2 and COL10A1 at day 21 normalized to pre-encapsulated iPS-MPs cultured under free swelling (black) and loading (gray). B.) Representative confocal microscopy images of ECM protein collagen X (green) and nuclei (blue) and quantitative analysis of the fraction of collagen X positively stained cells at day 21 of iPS-MPs cultured under free swelling (black) or loading (gray). Scale bar is 20 μm. Data presented as mean with standard deviation (n=3).
Assessment of TGFβR1 and Smad Signaling During Chondrogenis of iPS-MP-laden Hydrogels
Protein expression of TGFβR1, pSmad2/3, and pSmad1/5/8 was evaluated by immunohistochemistry during chondrogenesis of the iPS-MPs when encapsulated in the cartilage mimetic hydrogels and cultured under different growth factor conditions and under loading. Quantification of TGFβR1 intensity staining per nuclei were affected by growth factors (p=0.008) and loading (p<0.001) following a two-way ANOVA. Under free swelling, TGFβR1 protein expression was highest in the BMP2-only condition over TGFβ3-only (p=0.025) and TGFβ3+BMP2 (p=0.028) condition. Loading did not affect TGFβR1 expression in the BMP2 condition. However, loading up-regulated TGFβR1 under TGFβ3-only condition (p=0.003) and the TGFβ3+BMP2 condition (p=0.02). Similarly, growth factors (p<0.001) and loading (p=0.027) had a significant effect on pSmad2/3 expression. Loading increased expression of pSmad2/3 when cultured under TGFβ3-only (p=0.025), BMP2-only (p=0.049), and TGFβ3+BMP2 (p=0.042) conditions. In addition, pSmad2/3 expression was greater (p=0.046) in the TGFβ3+BMP2 condition when compared to the TGFβ3-only condition. On the contrary, loading and growth factors did not have a significant effect on pSmad1/5/8, however, a load-induced reduction in mean pSmad1/5/8 was found when cultured with TGFβ3 only. These results indicate that exogenous growth factors and loading affect TGFβR1 expression and Smad signaling in iPS-MPs encapsulated in the cartilage mimetic hydrogel.
Discussion
In this study, chondrogenesis of iPS-MPs encapsulated in a cartilage mimetic hydrogel was investigated, with a focus on the effects of the growth factors, TGFβ3 and BMP2, in tandem with intermittent dynamic compressive loading. The key finding from this study is that dynamic compressive loading supported a stable chondrogenic phenotype with limited collagen I expression, an indicator of fibrocartilage, and collagen X expression, an indicator of hypertrophy, but only when TGFβ3 was present. On the contrary, the cartilage-mimetic hydrogel alone or when cultured with BMP2-only regardless of loading supported chondrogenesis but also promoted hypertrophy. These findings suggest that iPS-MPs when cultured in this biomimetic hydrogel and under standard culture conditions (e.g., normoxia) have a propensity towards hypertrophic chondrogenesis, but that biochemical and mechanical cues are able to maintain a hyaline cartilage phenotype and prevent further differentiation towards hypertrophy.
In the absence of exogenous growth factors, the cartilage mimetic hydrogel induced chondrogenesis of iPS-MPs. However, the iPS-MPs appeared to undergo hypertrophy regardless of loading. This observation is supported by elevated aggrecan, collagen II, and collagen X mRNA levels, which were accompanied by positive staining for collagen X. Interestingly, the free swelling culture condition exhibited weak staining for aggrecan and collagen II, while loading had more pronounced staining for the hyaline cartilage proteins, indicating different temporal rates of differentiation towards hypertrophy. While studies have reported that chondrogenically differentiating pluripotent stem cells (i.e., embryonic) resist hypertrophy53 of which iPSCs are similar, iPS-MPs has been shown to be more similar to BM-MSCs than their parent iPSCs.54 In addition, several studies have reported that pluripotent stem cells that are differentiated into mesenchymal progenitors and subsequently expanded give rise to growth plate-like chondrocytes displaying a hypertrophic tendency, regardless of their origin.55,56 Indeed, growth plate chondrocytes are considered transient and programmed to differentiate from hyaline cartilage-producing chondrocytes into hypertrophic chondrocytes that eventually turn into bone.57 We therefore surmise that the iPS-MPs in this study had a propensity to undergo hypertrophy prior to encapsulation in the hydrogel and this propensity led to the observed rapid transition towards hypertrophy in the hydrogel. Rapid induction of hypertrophic markers has been reported in chondrogenically differentiating BM-MSCs.16,58 Interestingly, dynamic loading appeared to delay this transition in the abence of growth factors, but loading was not sufficient to inhibit hypertrophy as collagen X was still present.
These findings indicate that cues within the cartilage-mimetic hydrogel were sufficient to induce iPS-MP chondrogenesis. This observation is supported by the pellet culture results which did not show evidence of chondrogenesis in the absence of exogenous growth factors, suggesting that the basal chondrogenic medium was insufficient. The hydrogel itself forces cells into a round morphology and cells are restricted from extending cellular processes due to the relatively tight mesh of the hydrogel. This morphology is maintained throughout the culture period in part due to the lack of degradable crosslinkers in the hydrogel and the small mesh size of the hydrogel. Thus, the differentiation potential towards other pathways by the iPS-MPs (e.g., osteoblast) may be limited simply by restricting cell morphology.59,60 Another potential contributing factor is the presence of insulin in the basal medium, which has been shown to activate IGF-1 signaling and enhance chondrogenesis.61,62 However, TGFβ signaling is essential to chondrogenesis38 and thus activation of IGF-1 signaling alone is likely insufficient. Positive staining for TGFβRI concomitant with pSmad2/3, indicating activated Smad2/3 signaling, suggests that TGFβ signaling was induced in the iPS-MPs without exogenous growth factors.63 This suggests that the hydrogel environment induced TGFβ up-regulation in the iPS-MPs. One possible explanation is hyperosmolarity, which has been reported to induce TGFβ secretion and TGFβRI expression in other cell types.64,65 We have previously shown that negatively charged chondroitin sulfate tethered into the hydrogel creates a hyperosmotic environment by attracting mobile cations from the media.66 We have also previously reported that BM-MSCs encapsulated in this cartilage-mimetic hydrogel containing chondroitin sulfate had elevated expressions of osmotic sensitive genes, indicating a responsiveness to an hyperosmotic environment.39 Once TGFβ signaling is induced, the ECM analogs within the cartilage-mimetic hydrogel can aid in amplifying TGFβ signaling. The RGD ligands tethered into the hydrogel can upregulate RGD-binding integrins on cells67,68, which can activate the latent form of TGFβ.69–71 In addition, the sulfation on chondroitin sulfate can bind soluble TGFβ3, prolonging its half-life and preventing its release from the hydrogel.68,72 Collectively, these observations lead us to hypothesize that the cartilage-mimetic hydrogel via hyperosmolarity from the ECM analogs, is sufficient to upregulate and amplify TGFβ signaling in the iPS-MPs and then induce chondrogenesis without the need for exogenous growth factors.
TGFβ3, BMP2, and a combination thereof, when exogenously delivered to the iPS-MPs in the cartilage-mimetic hydrogel and cultured under free swelling conditions, supported chondrogenesis. This finding is expected given that these growth factors are known to induce chondrogenesis.30 These growth factors, however, were also able to slow the progression of hypertrophy when compared to the growth factor-free condition. These results are in agreement with our previous reports with BM-MSCs in the cartilage-mimetic hydrogel, which showed simultaneous hyaline and hypertrophic protein expression under exogenous TGFβ3.39 Positive staining for TGFβRI and pSmad2/3 points to TGFβ signaling under all growth factor conditions. Studies have reported that BMP2 can stimulate Smad2/3 signaling;73 however, activation is through the BMP receptor ALK3 and independent of the receptors, such as TGFβRI, which is associated with pSmad2/3 signaling.74 Once Smad2/3 signaling is activated, cells can produce their own TGFβ to induce autocrine TGFβ signaling75. While BMP2 can initiate chondrogenesis of mesenchymal progenitors,76 it is also involved in hypertrophic differentiation77,78 and required for endochondral ossification through Smad1/5/8 activation.79 The pSmad1/5/8 expression was similar across all growth factors, but was overall lower than the pSmad2/3 expression. This result suggests that the cartilage-mimetic hydrogel under BMP2 stimulation may favor Smad2/3 over Smad1/5/8 signaling, similar to that under TGFβ3 stimulation. Collectively, these results indicate that under free swelling conditions, exogenous delivery of growth factors is necessary to slow progression towards hypertrophy of the encapsulated iPS-MPs.
Contrarily, the combination of dynamic loading and exogenous TGFβ3 supported chondrogenesis and limited hypertrophy. The addition of BMP2 along with TGFβ3 also showed a reduction in hypertrophy under loading, but was not as effective in reducing collagen X expressing cells as TGFβ3 alone. We recently reported that dynamic loading of BM-MSCs encapsulated in the cartilage-mimetic hydrogel inhibited hypertrophy in the cartilage-mimetic hydrogel through mechanisms mediated by p38, which were a direct result from the presence of chondroitin sulfate.39 In that study, we concluded that dynamic oscillations in osmolarity that result from loading a charged matrix was critical to load-induced inhibition of hypertrophy. Here, we showed that in the growth factor-free condition, collagen X was present with minimal staining for hyaline cartilage proteins, suggesting that the iPS-MPs may have already reached terminal differentiation. However, in this condition under loading, both hypertrophic and hyaline cartilage proteins were present, suggesting that loading slows the progression towards terminal differentiation. The addition of exogeonous TGFβ3 combined with loading is even more potent at inhibiting hypertrophy leading to the presence of hyaline cartilage proteins and minimal hypertrophic proteins. In studies with BM-MSCs, mechanical loading has been shown to induce chondrogenesis80–82 as a result of load-induced autocrine TGFβ signaling and subsequent amplification.83. We surmise that herein, loading up-regulated TGFβ signaling in addition to the hydrogel-alone, which contributed to the delay in the progression towards hypertrophy.
The high levels of collagen X mRNA in the iPS-MPs under all conditions, however, suggests that the propensity towards hypertrophy still remains; a finding that is consistent with our previous reports with BM-MSCs.33,39 It is interesting though that our previous report with BM-MSCs39 pointed to load-induced inhibition of Smad1/5/8 signaling leading to a shift in Smad2/3 dominant signaling, which is known to inhibit hypertrophy of BM-MSCs.84 In this work, loading upregulated Smad2/3 signaling, but had less of an effect on Smad1/5/8 signaling, which was relatively low in the iPS-MPs when compared to our previous study with BM-MSCs.39 It is worth noting, however, that the ratio of the intensity of the pSmad2/3 to pSmad1/5/8 staining was the highest at ~5 with TGFβ3 and loading, which led to the greatest inhibition in hypertrophy. With BMP2 and loading regardless of TGFβ3, the ratio was lower ~3, but hypertrophy was only reduced when TGFβ3 was present. This suggests that under BMP2, other signaling mechanisms beyond Smad1/5/8 signaling may contribute to collagen X protein expression. One potential explanation is differential regulation of Smad4, which forms a complex with pSmad2/3 and with pSmad1/5/8 and facilitates their translocation into the nucleus where Smad proteins regulate gene expression.85 In a recent study, Smad4 was reported to be necessary for cartilage formation86,87 and BM-MSC chondrogenesis.88 The abundance of Smad4 was also shown to play a role in determining Smad2/3 versus Smad1/5/8 dominated signaling.88 Thus, it is possible that Smad4 may be involved in regulating growth factor and/or load responses in the differentiation of iPS-MPs, however, the effects of loading on Smad4 remains to be elucidated. Moreover, other signaling mechanisms such as MAP kinase, WNT, and Notch, are known to be affected by mechanical loading and growth factor stimulation,31 and may also play a role in iPS-MP chondrogenesis and their progression towards hypertrophy.
Collectively findings from this study demonstrate that external cues in the form of dynamic loading and/or exogenous growth factors influence the temporal fate of iPS-MPs during chondrogenesis in the cartilage-mimetic hydrogel, which span from a stable chondrogenic phenotype towards a terminally differentiated phenotype (Figure 8). On one end, dynamic loading when combined with exogenous TGFβ3 supports stable chondrogenesis by inhibiting hypertrophy. We surmise that this occurs at least in part through load-enhanced Smad2/3 signaling accompanied by relatively low Smad 1/5/8 signaling. Hypertrophy was partially inhibited when BMP2 was present, suggesting that TGFβ3 with loading remained effective at slowing the progression to hypertrophy, but not to the same degree as when BMP2 was absent. IPS-MPs, however, were pushed towards hypertrophy under a range of culture conditions (see Figure 8). At low concentrations, TGFβ can bind not only to TGFβRI but also BMP receptors and activate both Smad2/3 and Smad1/5/8 signaling cascades,89,90 which could explain the hypertrophic phenotype observed under loading in the growth factor-free condition and that observed with TGFβ3 without loading. BMP signals through Smad1/5/8 signaling and thus it is not surprising that BMP2 promotes hypertrophy. Lastly, free swelling culture without loading or growth factors led to rapid induction of hypertrophy in iPS-MPs, which was evident by collagen X with minimal presence of hyaline cartilage proteins. This result suggests that cell-mediated degradation of the hyaline cartilage proteins occurred during the transition to hypertrophy or alternatively that transdifferentiation may have occurred. Studies have indicated that an immature chondrocyte, with limited hyaline cartilage matrix, can transdifferentiate into a pre-osteoblast.91 These findings indicate that the iPS-MPs when cultured in the cartilage-mimetic hydrogel rapidly undergo chondrogenesis and hypertrophy, but depending on the loading environment and the growth factors can slow this progression to achieve different stages of chondrogenesis.
Figure 8.

The proposed fate of iPS-MPS chondrogenesis in a cartilage mimetic hydrogel under different growth factor and loading conditions.
There are several limitations of this study. A stable, non-degradable hydrogel was chosen in order to maintain the local biochemical and mechanical cues within the hydrogel during the experiment without complications of a degradable hydrogel whose properties evolve over time. This limits the ECM proteins secreted by the encapsulated cells to the pericellular space. Nonetheless, this restricted matrix allows the type of ECM molecules to be identified. Further study is needed to assess chondrogenesis of iPS-MPs in a degradable hydrogel for cartilage tissue engineering. We investigated collagen X as an indicator of a hypertrophic phenotype in chondrogenesis.92 Although questions have been raised regarding collagen X as an appropriate marker at the gene level,58 collagen X protein is critical to the early events of cartilage hypertrophy that lead to endochondral ossification.93 The study was limited to assessment at day 21 and further analysis will need to include a time course to delineate the temporal progression of chondrogenesis. Although not a direct limitation of the study, the advantage of using iPS-MPs over BM-MSCs is that their chondrogenic differentiation capabilities are maintained in older and diseased patients. Future work will look to compare the chondrogenesis of iPS-MPs and BM-MSCs from the same patient in order to determine if iPS-MPs are a more effective cell source for chondrogenesis.
Conclusions
A photoclickable cartilage-mimetic hydrogel cultured in the presence of TGFβ3 and under dynamic mechanical stimulation was effective at promoting chondrogenesis of iPS-MPs while limiting hypertrophy. While the cartilage-mimetic hydrogel alone and when combined with mechanical loading promoted chondrogenesis of iPS-MPs cultured in the absence of growth factors, hypertrophy was only inhibited when mechanical loading was combined with TGFβ3. This result correlated with greater Smad2/3 signaling over pSmad1/5/8 signaling. However, our findings suggest that under BMP2 other signaling mechanisms are at play. Nonetheless, this study reports that a cartilage-mimetic hydrogel supports chondrogenesis of iPS-MPs and that when combined with the appropriate growth factors and loading can support a stable chondrogenic phenotype. Future efforts will examine the translatability of these findings into a degradable cartilage mimetic hydrogel where this hydrogel platform has clinical significance for its ability to be injected and photopolymerized into a chondral defect.
Figure 7.

Representative confocal microscopy images and quantitative analysis of (A) TGFβRI (green), (B) pSmad2/3 (red), (C) pSmad1/5/8 (red) and nuclei (blue) at day 21 of iPS-MPs cultured with TGFβ3, BMP2, and TGFβ3+BMP2 without (black) or with loading (gray). Scale bar is 20 μm. Data presented as mean with standard deviation (n=3).
Acknowledgments
This study was supported by the National Institutes of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases under the award #1R01AR069060. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors also acknowledge support from a National Science Foundation GFRP fellowship a Department of Education’s Graduate Assistantship in Areas of National Need fellowship to EAA and from the Gates Center for Regenerative Medicine.
References
- 1.Buckwalter JA, Mankin HJ and Grodzinsky AJ, Instr. Course Lect, 2005, 54, 465–480. [PubMed] [Google Scholar]
- 2.Buckwalter JA, Saltzman C and Brown T, Clin. Orthop, 2004, 427, S6. [DOI] [PubMed] [Google Scholar]
- 3.Dewan AK, Gibson MA, Elisseeff JH and Trice ME, Evolution of Autologous Chondrocyte Repair and Comparison to Other Cartilage Repair Techniques, https://www.hindawi.com/journals/bmri/2014/272481/, (accessed 13 September 2017). [DOI] [PMC free article] [PubMed]
- 4.Kon E, Filardo G, Di Matteo B, Perdisa F and Marcacci M, Bone Jt. Res, 2013, 2, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brittberg M, Am. J. Sports Med, 2010, 38, 1259–1271. [DOI] [PubMed] [Google Scholar]
- 6.Buckwalter JA, J. Orthop. Sports Phys. Ther, 1998, 28, 192–202. [DOI] [PubMed] [Google Scholar]
- 7.Gillogly SD and Myers TH, Orthop. Clin. North Am, 2005, 36, 433–446. [DOI] [PubMed] [Google Scholar]
- 8.Knutsen G, Engebretsen L, Ludvigsen TC, Drogset JO, Grøntvedt T, Solheim E, Strand T, Roberts S, Isaksen V and Johansen O, J. Bone Joint Surg. Am, 2004, 86-A, 455–464. [DOI] [PubMed] [Google Scholar]
- 9.Filardo G, Kon E, Andriolo L, Di Matteo B, Balboni F and Marcacci M, Am. J. Sports Med, 2014, 42, 898–905. [DOI] [PubMed] [Google Scholar]
- 10.Yoo JU, Barthel TS, Nishimura K, Solchaga L, Caplan AI, Goldberg VM and Johnstone B, J. Bone Joint Surg. Am, 1998, 80, 1745–1757. [DOI] [PubMed] [Google Scholar]
- 11.Payne KA, Didiano DM and Chu CR, Osteoarthritis Cartilage, 2010, 18, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brady K, Dickinson SC and Hollander AP, Cartilage, 2015, 6, 30S–35S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sethe S, Scutt A and Stolzing A, Ageing Res. Rev, 2006, 5, 91–116. [DOI] [PubMed] [Google Scholar]
- 14.Wakitani S, Mitsuoka T, Nakamura N, Toritsuka Y, Nakamura Y and Horibe S, Cell Transplant, 2004, 13, 595–600. [DOI] [PubMed] [Google Scholar]
- 15.Vinardell T, Sheehy EJ, Buckley CT and Kelly DJ, Tissue Eng. Part A, 2012, 18, 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelttari K, Winter A, Steck E, Goetzke K, Hennig T, Ochs BG, Aigner T and Richter W, Arthritis Rheum, 2006, 54, 3254–3266. [DOI] [PubMed] [Google Scholar]
- 17.Mackie EJ, Ahmed YA, Tatarczuch L, Chen K-S and Mirams M, Int. J. Biochem. Cell Biol, 2008, 40, 46–62. [DOI] [PubMed] [Google Scholar]
- 18.Yousefi A-M, James PF, Akbarzadeh R, Subramanian A, Flavin C and Oudadesse H, Stem Cells Int, 2016, 2016, 6180487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JS, Choi HW, Choi S and Do JT, Int. J. Stem Cells, 2011, 4, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong G, Ferrari D, Dealy CN and Kosher RA, J. Cell. Physiol, 2010, 224, 664–671. [DOI] [PubMed] [Google Scholar]
- 21.Lietman SA, World J Orthop, 2016, 7, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaspar-Maia A, Alajem A, Meshorer E and Ramalho-Santos M, Nat. Rev. Mol. Cell Biol, 2011, 12, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi K and Yamanaka S, Cell, 2006, 126, 663–676. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K and Yamanaka S, Cell, 2007, 131, 861–872. [DOI] [PubMed] [Google Scholar]
- 25.Rohani L, Johnson AA, Arnold A and Stolzing A, Aging Cell, 2014, 13, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koyama N, Miura M, Nakao K, Kondo E, Fujii T, Taura D, Kanamoto N, Sone M, Yasoda A, Arai H, Bessho K and Nakao K, Stem Cells Dev, 2012, 22, 102–113. [DOI] [PubMed] [Google Scholar]
- 27.Nejadnik H, Diecke S, Lenkov OD, Chapelin F, Donig J, Tong X, Derugin N, Chan RCF, Gaur A, Yang F, Wu JC and Daldrup-Link HE, Stem Cell Rev, 2015, 11, 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei Y, Zeng W, Wan R, Wang J, Zhou Q, Qiu S and Singh SR, Eur. Cell. Mater, 2012, 23, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Nie H, Xu Z, Niu X, Guo S, Yin J, Guo F, Li G, Wang Y and Zhang C, PLOS ONE, 2014, 9, e111566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tenney RM and Discher DE, Curr. Opin. Cell Biol, 2009, 21, 630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Conor CJ, Case N and Guilak F, Stem Cell Res. Ther, 2013, 4, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W and Chen CS, Cell Stem Cell, 2009, 5, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aisenbrey EA and Bryant SJ, J. Mater. Chem. B, 2016, 4, 3562–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coburn JM, Gibson M, Monagle S, Patterson Z and Elisseeff JH, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 10012–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamaddon M, Burrows M, Ferreira SA, Dazzi F, Apperley JF, Bradshaw A, Brand DD, Czernuszka J and Gentleman E, Sci. Rep,, DOI: 10.1038/srep43519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bian L, Guvendiren M, Mauck RL and Burdick JA, Proc. Natl. Acad. Sci, 2013, 110, 10117–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puetzer JL, Petitte JN and Loboa EG, Tissue Eng. Part B Rev, 2010, 16, 435–444. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Rigueur D and Lyons KM, Birth Defects Res. Part C Embryo Today Rev, 2014, 102, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aisenbrey EA and Bryant SJ, Biomaterials, 2019, 190–191, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park JS, Chu JS, Tsou AD, Diop R, Tang Z, Wang A and Li S, Biomaterials, 2011, 32, 3921–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kisiday JD, Frisbie DD, McIlwraith CW and Grodzinsky AJ, Tissue Eng. Part A, 2009, 15, 2817–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mauck RL, Byers BA, Yuan X and Tuan RS, Biomech. Model. Mechanobiol, 2007, 6, 113–125. [DOI] [PubMed] [Google Scholar]
- 43.McGrath PS, Diette N, Kogut I and Bilousova G, J. Vis. Exp. JoVE,, DOI: 10.3791/58687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kogut I, McCarthy SM, Pavlova M, Astling DP, Chen X, Jakimenko A, Jones KL, Getahun A, Cambier JC, Pasmooij AMG, Jonkman MF, Roop DR and Bilousova G, Nat. Commun, 2018, 9, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skaalure SC, Chu S and Bryant SJ, Adv. Healthc. Mater, 2015, 4, 420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shu XZ, Liu Y, Luo Y, Roberts MC and Prestwich GD, Biomacromolecules, 2002, 3, 1304–1311. [DOI] [PubMed] [Google Scholar]
- 47.da Cunha AL, de Oliveira LG, Maia LF, de Oliveira LFC, Michelacci YM and de Aguiar JAK, Carbohydr. Polym, 2015, 134, 300–308. [DOI] [PubMed] [Google Scholar]
- 48.Hirai H, Tani T, Katoku-Kikyo N, Kellner S, Karian P, Firpo M and Kikyo N, Stem Cells Dayt. Ohio, 2011, 29, 1349–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guzzo RM, Gibson J, Xu R-H, Lee FY and Drissi H, J. Cell. Biochem, 2013, 114, 480–490. [DOI] [PubMed] [Google Scholar]
- 50.Nicodemus GD and Bryant SJ, J. Biomech, 2008, 41, 1528–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villanueva I, Gladem SK, Kessler J and Bryant SJ, Matrix Biol. J. Int. Soc. Matrix Biol, 2010, 29, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfaffl MW, Nucleic Acids Res, 2001, 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toh WS, Guo X-M, Choo AB, Lu K, Lee EH and Cao T, J. Cell. Mol. Med, 2009, 13, 3570–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diederichs S and Tuan RS, Stem Cells Dev, 2014, 23, 1594–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JY, Matthias N, Pothiawala A, Ang BK, Lee M, Li J, Sun D, Pigeot S, Martin I, Huard J, Huang Y and Nakayama N, Stem Cell Rep, 2018, 11, 440–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Umeda K, Zhao J, Simmons P, Stanley E, Elefanty A and Nakayama N, Sci. Rep, 2012, 2, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsang KY, Chan D and Cheah KSE, Dev. Growth Differ, 2015, 57, 179–192. [DOI] [PubMed] [Google Scholar]
- 58.Mwale F, Stachura D, Roughley P and Antoniou J, J. Orthop. Res, 2006, 24, 1791–1798. [DOI] [PubMed] [Google Scholar]
- 59.Bilousova G, Jun DH, King KB, De Langhe S, Chick WS, Torchia EC, Chow KS, Klemm DJ, Roop DR and Majka SM, Stem Cells Dayt. Ohio, 2011, 29, 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McBeath R, Pirone DM, Nelson CM, Bhadriraju K and Chen CS, Dev. Cell, 2004, 6, 483–495. [DOI] [PubMed] [Google Scholar]
- 61.Mueller MB, Blunk T, Appel B, Maschke A, Goepferich A, Zellner J, Englert C, Prantl L, Kujat R, Nerlich M and Angele P, Int. Orthop, 2013, 37, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phornphutkul C, Wu KY and Gruppuso PA, Mol. Cell. Endocrinol, 2006, 249, 107–115. [DOI] [PubMed] [Google Scholar]
- 63.Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K and ten Dijke P, EMBO J, 1997, 16, 5353–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mozes MM, Szoleczky P, Rosivall L and Kokeny G, Bmc Nephrol, 2017, 18, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan Timur U, Caron M, van den Akker G, van der Windt A, Visser J, van Rhijn L, Weinans H, Welting T, Emans P and Jahr H, Int. J. Mol. Sci,, DOI: 10.3390/ijms20040795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Farnsworth NL, Mead BE, Antunez LR, Palmer AE and Bryant SJ, Matrix Biol, 2014, 40, 17–26. [DOI] [PubMed] [Google Scholar]
- 67.Salinas CN and Anseth KS, Biomaterials, 2008, 29, 2370–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Villanueva I, Bishop NL and Bryant SJ, Tissue Eng. Part A, 2009, 15, 3037–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Horiguchi M, Ota M and Rifkin DB, J. Biochem. (Tokyo), 2012, 152, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Munger JS and Sheppard D, Cold Spring Harb. Perspect. Biol, 2011, 3, a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wipff P-J, Rifkin DB, Meister J-J and Hinz B, J. Cell Biol, 2007, 179, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park JS, Woo DG, Yang HN, Na K and Park K-H, J. Biomed. Mater. Res. A, 2009, 91A, 408–415. [DOI] [PubMed] [Google Scholar]
- 73.Holtzhausen A, Golzio C, How T, Lee Y-H, Schiemann WP, Katsanis N and Blobe GC, FASEB J, 2014, 28, 1248–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Ho CC, Bang E, Rejon CA, Libasci V, Pertchenko P, Hébert TE and Bernard DJ, Endocrinology, 2014, 155, 1970–1981. [DOI] [PubMed] [Google Scholar]
- 75.Lee PT and Li W-J, J. Cell. Biochem, 2017, 118, 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmitt B, Ringe J, Häupl T, Notter M, Manz R, Burmester G-R, Sittinger M and Kaps C, Differ. Res. Biol. Divers, 2003, 71, 567–577. [DOI] [PubMed] [Google Scholar]
- 77.Caron MMJ, Emans PJ, Cremers A, Surtel DAM, Coolsen MME, van Rhijn LW and Welting TJM, Osteoarthritis Cartilage, 2013, 21, 604–613. [DOI] [PubMed] [Google Scholar]
- 78.Steinert AF, Proffen B, Kunz M, Hendrich C, Ghivizzani SC, Nöth U, Rethwilm A, Eulert J and Evans CH, Arthritis Res. Ther, 2009, 11, R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pogue R and Lyons K, in Current Topics in Developmental Biology, Academic Press, 2006, vol. 76, pp. 1–48. [DOI] [PubMed] [Google Scholar]
- 80.Huang C-YC, Reuben PM and Cheung HS, STEM CELLS, 2005, 23, 1113–1121. [DOI] [PubMed] [Google Scholar]
- 81.Huang C-YC, Hagar KL, Frost LE, Sun Y and Cheung HS, Stem Cells Dayt. Ohio, 2004, 22, 313–323. [DOI] [PubMed] [Google Scholar]
- 82.Li Z, Yao S-J, Alini M and Stoddart MJ, Tissue Eng. Part A, 2010, 16, 575–584. [DOI] [PubMed] [Google Scholar]
- 83.Li Z, Kupcsik L, Yao S-J, Alini M and Stoddart MJ, J. Cell. Mol. Med, 2010, 14, 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hellingman CA, Davidson ENB, Koevoet W, Vitters EL, van den Berg WB, van Osch GJVM and van der Kraan PM, Tissue Eng. Part A, 2011, 17, 1157–1167. [DOI] [PubMed] [Google Scholar]
- 85.Heldin CH, Miyazono K and ten Dijke P, Nature, 1997, 390, 465–471. [DOI] [PubMed] [Google Scholar]
- 86.Bénazet J-D, Pignatti E, Nugent A, Unal E, Laurent F and Zeller R, Dev. Camb. Engl, 2012, 139, 4250–4260. [DOI] [PubMed] [Google Scholar]
- 87.Lim J, Tu X, Choi K, Akiyama H, Mishina Y and Long F, Dev. Biol, 2015, 400, 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Kroon LMG, Narcisi R, van den Akker GGH, Vitters EL, Blaney Davidson EN, van Osch GJVM and van der Kraan PM, Sci. Rep, 2017, 7, 43164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goumans M-J, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P and ten Dijke P, EMBO J, 2002, 21, 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goumans M-J, Lebrin F and Valdimarsdottir G, Trends Cardiovasc. Med, 2003, 13, 301–307. [DOI] [PubMed] [Google Scholar]
- 91.Aghajanian P and Mohan S, Bone Res, 2018, 6, 19–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Joyce ME, Roberts AB, Sporn MB and Bolander ME, J. Cell Biol, 1990, 110, 2195–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chan D and Jacenko O, Matrix Biol, 1998, 17, 169–184. [DOI] [PubMed] [Google Scholar]