

Abstract
Lafora disease (LD) is both a fatal childhood epilepsy and a glycogen storage disease caused by recessive mutations in either the Epilepsy progressive myoclonus 2A (EPM2A) or EPM2B genes. Hallmarks of LD are aberrant, cytoplasmic carbohydrate aggregates called Lafora bodies (LBs) that are a disease driver. The 5th International Lafora Epilepsy Workshop was recently held in Alcala de Henares, Spain. The workshop brought together nearly 100 clinicians, academic and industry scientists, trainees, National Institutes of Health (NIH) representation, and friends and family members of patients with LD. The workshop covered aspects of LD ranging from defining basic scientific mechanisms to elucidating a LD therapy or cure and a recently launched LD natural history study.
Keywords: Lafora disease, Progressive myoclonus epilepsy, Neurodegeneration, Glycogen, Glycogen storage disease
Lafora disease (LD) is a fatal, autosomal recessive glycogen storage disease that results in progressive neurodegeneration with epilepsy, dementia, and myoclonus [1–3]. Patients present with an epileptic event in their early to mid-teen years, and these episodes along with the cognitive decline and myoclonus progressively increase until death approximately 10 years later because of respiratory complications, sudden unexpected death (SUDEP), or a massive epileptic event [4,5]. The National Institutes of Neurological Disease and Stroke (NINDS) of the National Institutes of Health (NIH) funded a program project grant in 2016 bringing together the top LD laboratories to define the basic mechanisms of LD and develop therapeutic options [2,6,7]. This grant established the Lafora Epilepsy Cure Initiative (LECI) that is directed by Dr. Matthew Gentry, Professor at the University of Kentucky College of Medicine (Fig. 1). The LECI is comprised of researchers at University of Kentucky, Indiana University, University of Texas-Southwestern, UC-San Diego, Fundación Jimenez Díaz (Madrid), Institute for Research in Biomedicine (Barcelona), and Institute of Biomedicine of Valencia (IBV-CSIC).
Fig. 1.
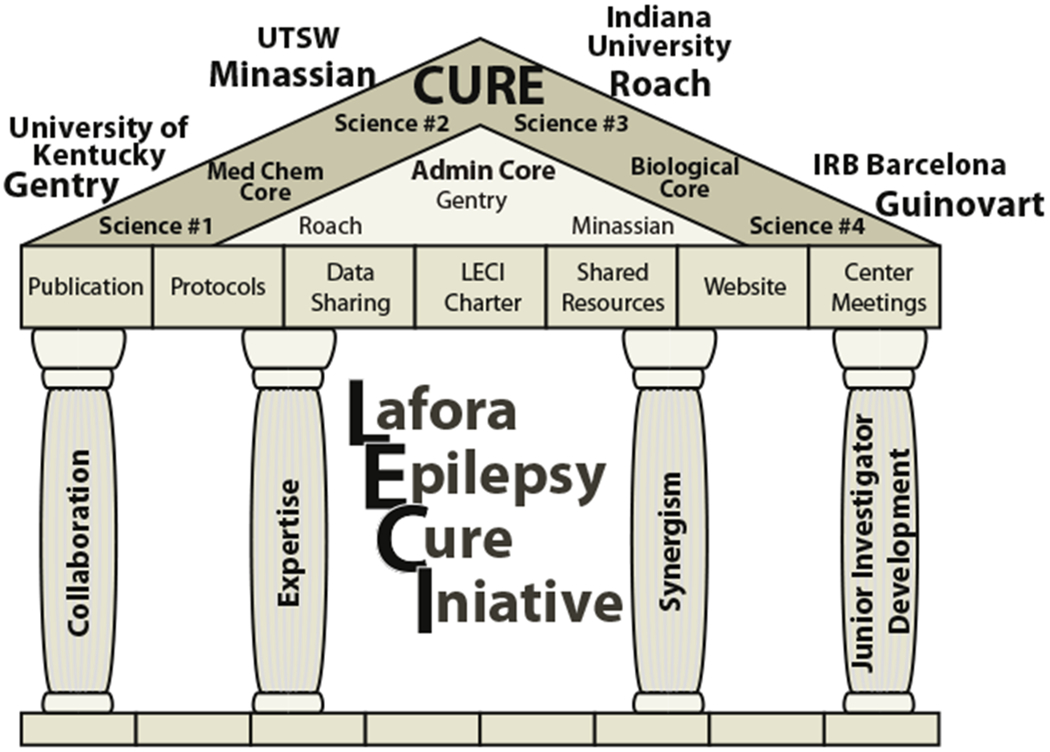
The Lafora Epilepsy Cure Initiative (LECI) is funded by NIH grant NS097197. The primary investigator’s and projects for the grant are Drs. Gentry — Science #1: Personalized diagnosis — defining how glycogen metabolism and proteostasis impact LD; Minassian — Science #2: Genome editing, mRNA suppression, and glycogen chain termination to inhibit glycogen storage as therapy for LD; Roach — Science #3: Suppressing glycogen storage with small molecule inhibitors as a therapeutic approach to LD; and Guinovart — Science #4: Defining the therapeutic window for the treatment of LD.
The 5th International Lafora Epilepsy Workshop was held at the Parador in Alcala de Henares, Spain on September 8–11, 2019. The workshop was organized and hosted by Drs. Jose Serratosa (Fundación Jimènez Díaz, Madrid, Spain) and Matthew Gentry; and it was sponsored by NINDS (P01 NS097197), Ionis Pharmaceuticals, Valerion Therapeutics, Maze Therapeutics, Chelsea’s Hope, and the University of Kentucky College of Medicine. The workshop is a unique blend of clinicians; scientists from academia, industry, and the NIH; trainees; friends and family members of patients with LD; and a patient with LD with approximately 100 representatives from the United States, Spain, Canada, Israel, Hungary, Slovakia, Russia, Honduras, Turkey, India, Italy, France, Australia, and the Netherlands (Fig. 2). The singular goal of this diverse group is to accelerate LD research to bring a LD therapy or cure to patients.
Fig. 2.

Over 100 scientists, clinicians, and family and friends of patients with LD attended the 5th International Lafora DiseaseWorkshop.
The workshop began on Monday morning, September 9, with a clinically focused meeting led by scientists from Ionis Pharmaceuticals and Valerion Therapeutics together with leading LD neurologists from around the world. This group discussed an ongoing Natural History and Functional Status Study that the two companies are jointly funding () [8]. The companies have established four clinical sites to monitor 30 patients with LD over a two-year period. The sites are led by Drs. Berge Minassian (UT-Southwestern), Antonio Delgado-Escueta (UCLA), Roberto Michelucci (Bologna), and Jose Serratosa (Madrid). The data from this patient cohort will be utilized to inform future clinical trial designs, including providing biomarker and historical control data. A robust poster session led by the trainees followed the Natural History Study meeting.
Serratosa welcomed the entire group to Alcala de Henares with a brief description of the historic city with its 15th century University of Alcala and as the birthplace of celebrated author Miguel de Cervantes. He then recounted how the Spanish neurologist Gonzalo Rodriguez Lafora left Madrid for Washington D.C. in 1910, and during this period, he discovered the intracellular, aberrant glycogen-like aggregates that are now called Lafora bodies (LBs) during an autopsy of a deceased patient with epilepsy [9–11]. Serratosa then highlighted the work by Delgado-Escueta from the 1980s hypothesizing that genetic mapping could be used to identify genes mutated in specific epilepsies [12]. Serratosa joined the Delgado-Escueta group in 1989 and they successfully mapped a LD gene locus to chromosome 6q [13]. Minassian later joined the Delgado-Escueta laboratory, and the trio independently identified the first LD gene known as Epilepsy progressive myoclonus 2A (EPM2A) that encodes the laforin phosphatase [14,15]. The Minassian and Delgado-Escueta laboratories collaborated to discover the second LD gene (EPM2B) that encodes the E3 ubiquitin ligase malin in 2003 [16]. Following the gene discoveries, multiple laboratories generated mouse and cell culture models as well as performed biochemical analyses of the malin and laforin proteins (Fig. 3). Importantly, multiple laboratories utilizing numerous model systems discovered that the LBs are the cause of LD and each group set out to remove, block, or ablate the LBs [17–20]. Serratosa closed by highlighting the five major therapeutic options that are being developed: small molecules, enzyme therapy, antisense oligonucleotides (ASOs), repurposing drugs, and gene therapy.
Fig. 3.
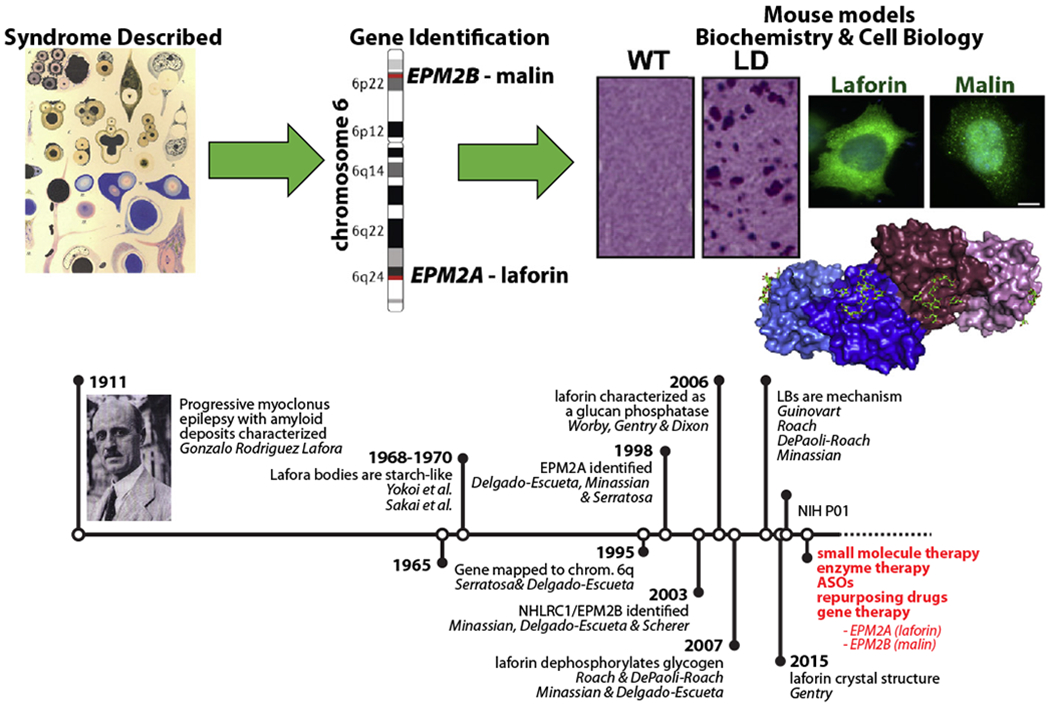
A timeline of LD-related research. In red are the five platforms that LECI scientists are pursuing as LD treatments. ASO — antisense oligonucleotides.
Dr. Frank Harris, President of the North American advocacy group for patients with LD, Chelsea’s Hope, followed with a welcome and a blunt call to arms: “What matters right now is moving a therapy into the clinic.” He pushed the basic scientists to further define cellular mechanisms and the translational and clinical scientists to do everything they can to move novel preclinical therapies into the clinic. Harris then showed a 5-minute video featuring a current teenage patient with LD from the Boston area. Harris then introduced Gentry as the opening speaker.
Gentry opened his talk emphasizing that like every great European football team, the LD community needs as many shots on goal as possible. He stated that the basic science has led to five therapeutic options that are currently being developed and that basic science could develop the next wave of potential cures. As he opened his lecture, Gentry stressed that each group focus on doing what they do best: the basic scientists define disease mechanisms, translational scientists complete preclinical mouse model experiments, and industry scientists partner with LD neurologists to rapidly advance therapies into the clinic. He then focused on recent work from his laboratory performed in collaboration with Valerion Therapeutics. They developed a novel antibody-enzyme fusion (AEF) that employs an antibody fragment with cell penetrating properties fused to an amylase that can degrade LBs [21–23]. Gentry showed that the AEF, termed VAL-0417, robustly degrades LBs in vitro releasing glucose and maltose. He went on to demonstrate that VAL-0417 degrades LBs in LD mouse models after both intramuscular and intravenous injections. However, removing LBs in astrocytes and neurons is the key to treating LD, and VAL-0417 likely does not cross the blood–brain barrier. The team utilized intracerebroventricular administration to deliver VAL-0417 into the brain of LD mouse models and Gentry showed that the drug ablated Periodic-acid Schiff positive (PAS+) LBs. He then highlighted a second AEF that the team has developed that also ablates PAS+ LBs and concluded with new mechanistic insights into how LBs drive disease progression.
Next, Dr. Paul Goldberg, VP of Clinical Development at Ionis Pharmaceuticals, presented the Ionis ASO platform in a talk geared for both scientists and friends and family members of patients with LD. After describing the ASO technology, Goldberg described the science that powers the amazing drug Spinraza® (Nusinersen) that is a collaborative program with Biogen and is now used successfully to treat patients with spinal muscular atrophy [24,25]. He then described how decreasing glycogen synthesis by 50% has been shown by multiple laboratories to prevent LD in preclinical mouse models. Ionis scientists, in collaboration with the Minassian laboratory, first identified ASOs against the mouse glycogen synthase gene (GYS1) in order to conduct initial proof-of-concept experiments in LD mouse models. Ionis screened numerous human ASOs and have now identified the development candidate human GYS1 ASO. They have initiated substantial safety and toxicity testing of this human ASO. At the same time, Ionis is preparing for a future clinical trial. Goldberg explained that since LD is an ultra-rare disorder, recruitment and powering for well-controlled studies will be very challenging. Ionis will explore novel trial designs and will work with the U.S. Food and Drug Administration (FDA) and other regulatory bodies to seek approval for a single clinical trial. He emphasized the importance of the Natural History Study and announced that Ionis is also funding a retrospective chart study. These natural history data will serve as comparator data when administering the drug and support evidence of endpoint improvement. He stressed that these data are key when talking to government regulators so that they can collaboratively design an appropriate clinical trial. The Natural History Study will allow the Ionis and Valerion teams to synthesize a coherent trajectory of disease and individual endpoint progression to support appropriate clinical trial endpoint selection. An additional important aspect of the study is to obtain serum and cerebrospinal fluid so that safety and efficacy biomarkers can be validated. Goldberg pointed out that these biomarkers may accelerate the approval process. To date, 15 patients have been enrolled in the study, and teams are working to identify and develop suitable biomarkers. During the question session, it became clear that scientists are triangulating their work so that the Natural History Study, retrospective chart study, and animal toxicity data are all completed as quickly as possible. Goldberg closed by passionately stating that Ionis is committed to the LD community and as efficiently as possible expediting the evaluation of the drug in the clinic.
Dr. Dustin Armstrong, Chief Scientific Officer for Valerion Therapeutics, then presented the history on how Valerion was founded and details regarding the Valerion drug platform. He focused much of his talk on the results of a recently completed Phase I/II clinical trial that treated patients with Pompe disease with the Valerion drug VAL-1221 and compared safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy versus the current standard of care, Myozyme® () [26]. Pompe disease is a glycogen storage disease caused by mutations in the lysosomal acid alpha-glucosidase [27]. Patients with Pompe disease primarily exhibit decreased functionality in skeletal muscle and the heart due to the accumulation of glycogen in lysosomes. Armstrong described how Myozyme® is a recombinant acid alpha-glucosidase and that it has a positive initial impact in patients with Pompe disease, but that the positive effect wanes over time because of the accumulation of cytoplasmic glycogen, which Myozyme® does not degrade because it targets the lysosome. VAL-1221 is an AEF that fuses the same Fab used in the VAL-0417 drug to acid alpha-glucosidase, thus incorporating both a cytoplasmic and lysosomal targeting region. VAL-1221 targets both the cytoplasm and lysosome and has shown great promise in preclinical mouse models [28]. Armstrong presented very encouraging data from the VAL-1221 Phase I/II clinical trial regarding both the safety and efficacy of VAL-1221 and stated that all of the patients from the clinical trial are now taking VAL-1221. Armstrong then reiterated the results presented by Gentry on VAL-0417, its ability to ablate PAS+LBs, and the commitment of Valerion to transitioning an AEF into a LD clinical trial. A key question was answered after his presentation regarding the Natural History Study and any future clinical trials. Ionis and Valerion agreed that a patient could be enrolled in a future clinical trial even if s/he did not participate in the Natural History Study. The evening closed with a meal at Restaurante de Desayunos where scientific discussions continued.
The second day consisted of three scientific sessions with two focused on basic/translational science and one on clinical science. Dr. Olga Varea from the Guinovart laboratory presented exciting data on defining the therapeutic window for treating LD. Varea presented data on multiple novel mouse models that they generated to answer the therapeutic timing question on when to intervene and when it was too late to intervene. Using Cre/lox technology, they generated a LD mouse model with a tamoxifen-inducible deletion of glycogen synthase (Gys1). Vera showed that suppression of Gys1 in either 4- or 6-month old LD animals dramatically reduced LB aggregates in the brain and also decreased neurodegeneration and inflammation markers. Varea presented data from a second project of a mouse model with a tamoxifen-dependent conditional expression of the malin gene (Epm2b). She showed that expression of malin in 4-month old malin knockout (KO) animals resulted in dramatically decreased LBs in the brain and near complete elimination of LBs in the muscle and heart. These data are very encouraging with respect to possible gene therapy as a treatment for LD.
Dr. Tom Hurley of Indiana University School of Medicine followed with an update on the Roach, DePaoli-Roach, and Hurley laboratories’ efforts to identify a small molecule inhibitor of glycogen synthase (GS). Hurley reminded the audience that a decrease in glycogen synthesis due to deficiency in GS or the glycogen synthesis-activating protein called Protein Targeting to Glycogen (PTG) normalized glycogen accumulation and neuronal function in LD mouse models [17–20]. Their work has focused on screening for compounds that compete with the binding of glucose-6-phosphate (G6P) since it is an allosteric inhibitor of GS. After screening numerous small molecule libraries, they identified a putative G6P competitor, but crystallographic studies demonstrated that the small molecule binds at the active site rather than the G6P binding site. In collaboration with the LECI Medicinal Chemistry Core, headed by Drs. David Watt (Kentucky) and Steve Johnson (Indiana), they screened analogs and identified a subset with >100-fold higher potency. They also reduced conformational flexibility of the compound and generated 60+ analogs with differential substitution patterns. Some of these compounds were markedly improved, and the group is continuing their work with these compounds. These and other efforts have yielded multiple compounds that the group is moving forward into animal testing.
The next speaker was Dr. Marina Sanchez (Fundación Jimenez Díaz) who presented new data on treating LD neonatal mice with metformin. Sanchez treated LD mice from conception to adulthood with metformin and observed decreases in myoclonic jerks, seizure reaction to convulsive doses of pentylenetetrazol (PTZ), decreased neuronal and astrocytic losses, and decreases in LBs. These findings serve as a good basic science model proving that early treatment provides improvement compared with later treatment. Sanchez also presented preclinical mouse data on magnetic resonance imaging (MRI) and positron-emission tomography (PET) analyses of LD mouse models. They are continuing to investigate these analyses as possible biomarkers. Dr. Tamar Grossman, Director of Translational Medicine at Ionis Pharmaceuticals, then presented the successes of bringing ASO drugs to the clinic, utilizing different chemistries that include ligand-conjugated ASO technology (LICA) that enriches ASO delivery to target tissues. She spoke of the seven ASOs that have been FDA-approved and highlighted the five Ionis drugs. Grossman then discussed the Ionis neurological drug pipeline that includes programs on Huntington’s disease, amyotrophic lateral sclerosis, and Alzheimer’s disease. She presented safety data as well as pharmacokinetics/pharmacodynamics data on ASOs that are intrathecally injected. The drugs achieve impressive biodistribution throughout the entire brain in nonhuman primate studies and in humans. She also highlighted that the half-life for most ASOs is long enough that patients could potentially receive quarterly injections.
Following a coffee break and poster session, Dr. Jordi Duran, senior scientist in the Guinovart laboratory, presented exciting new work on the role of p62 in LD and how LD is likely an astrocytic disease. Their work strongly suggests that p62 promotes the aggregation of abnormal glycogen and that malin and laforin may be involved in mechanisms driving glycogen autophagy. He then presented historical data suggesting that LD is a neuron-centric disease. However, the Guinovart laboratory has more recently demonstrated via elegant microscopy that far more astrocytes contain LBs compared with neurons [29]. He defined two subtypes of LBs: Corpora amylacea-like (CALs) that are astrocytic and neuronal LBs (nLBs). They have generated an astrocytic-specific Gys1 conditional KO mouse model, and the data are quite striking, showing that the CALs are the most abundant type of LB. The next talk was from Ms. Lyndsay Young, a graduate student in the Gentry laboratory, on the novel discovery of glycogen synthesis and glycogen degradation in the nucleus [30–32]. Young showed how normal lung tissue synthesizes glycogen in the nucleus and that the E3 ubiquitin ligase malin ubiquitinates glycogen phosphorylase (GP) to translocate it into the nucleus to degrade glycogen. The glucose-1-phosphate that is released by GP is converted via nuclear glycolytic enzymes into pyruvate and then acetate that is used to acetylate histones. She also described how non-small cell lung cancer cells downregulate malin levels, resulting in GP staying in the cytoplasm, and large LB-like aggregates form in the nucleus that results in decreased histone acetylation and increased cellular proliferation [30]. Dr. Carolyn Worby (UC-San Diego) then presented an update from the LECI biological core. Worby has generated a collection of unique and very useful reagents for the broader community. She has developed CRISPR-generated LD patient mutation cell lines as well as LD patient mutation mouse models for both laforin and malin mutations. Multiple groups are using these reagents in current experiments, and the groups are analyzing the new mouse models.
The last talk before lunch and poster viewing was presented by Dr. Felix Nitschke from the Miniassian laboratory. Nitschke presented on the important role of glycogen phosphate and branching in polyglucosan body (PGB) formation. His talk addressed a key unresolved aspect of LD, namely what is the biochemical and/or chemical mechanism underlying LB formation. Nitschke presented new data from multiple mouse models: laforin KO, malin KO, and glycogen branching enzyme Y329S. He has developed methods for separating the soluble from insoluble glucans so that they could be individually analyzed. The architecture of the soluble glycogen (i.e., chain length and branching) remains unchanged in the different models compared with wild-type. However, these parameters are altered for the insoluble glycogen in the diseased models. Nitschke stressed that glycogen is very heterogeneous in terms of chain length and its propensity to precipitate into PGBs.
After lunch and a poster session, Dr. Pascual Sanz presented work that followed his recent major discovery that in LD mice, >90% of astrocytes and only ~10% of neurons contain LBs [33]. The laboratory has recently focused on defining whether these accumulations cause changes in metabolism and/or inflammation. They performed RNA-sequencing on brain preparations from 3- and 16-month old laforin KO and malin KO mice brains compared with control wild-type animals. The number of genes downregulated in both models compared with wild-type was relatively small, and the number of upregulated genes was ~400. Strikingly, the classes of genes that were changed were very similar in both LD mouse models, suggesting similar disease mechanisms. Most of the genes encode for proinflammatory and phagocytosis proteins that are mainly expressed by reactive astrocytes and microglia. They focused on a subset of these genes and found that there is a clear upregulation in the LD animals as they age. Moving forward, they will address the following: What is the relative contribution of mature glial cells, microglial, astrocyte, or oligodendrocytes to the upregulation of gene expression? What is the relationship between metabolism and inflammatory gene expression? Sanz ended by stating that since they have established that there is inflammation in the LD brain, perhaps we should consider reducing neuroinflammation as a therapeutic strategy.
Following Sanz, Dr. Silvia Nitschke of the Minassian laboratory presented an exciting talk on a more recent addition to the family of brain PGB diseases. It is a complicated case linking PGBs with neuronal inflammation and immunodeficiency caused by mutations in the E3 ubiquitin ligase RBCK1 called PGB Myopathy 1 with or without immunodeficiency [34–37]. While the cellular mechanisms linking these clinical outcomes are currently undefined, it is clear that RBCK1 KO mice display dramatic central nervous system PGBs. Nitschke showed that Rbck1 −/− mice exhibit PGBs in the spinal cord, cortex, hippocampus, and cerebellum and that the PGBs exhibit longer glucose chains, aberrant branching, and increased C6-phosphorylation. Strikingly, she showed that crossing the Rbck1 −/− mice with mice lacking GS rescued the behavioral phenotypes as well as most of the PGBs in the brain. She highlighted how this model will allow further insights into how PGBs form and that a treatment for LD could be applicable for this disease as well.
Three short talks followed that were presented by Drs. Carlos Roma-Mateo (University of Valencia), Gino Cingolani (Thomas Jefferson University), and Mitchell Sullivan (Mater Research Institute — The University of Queensland). Roma-Mateo presented novel work on miRNA populations in laforin KO and malin KO animal models. He analyzed the brain tissue from both models and identified changes in a subset of miRNAs as LD mice aged. Cingolani updated the group on recent advances in cryo-electron microscopy (cryo-EM) and past work to determine the 3-dimensional structure of GS. While there is a structure of bacterial, yeast, and C. elegans GS, the structure of the human enzyme has been elusive. His laboratory has optimized the expression of GS using novel protocols to reduce the protein heterogeneity. Initial cryo-EM data are promising regarding a future structure of this important enzyme. Sullivan then gave an overview of diabetes, blood glucose, insulin, and glycogen metabolism. He then showed that diabetic liver glycogen is architecturally distinct from normal liver glycogen. Moving forward, his group is probing the location and structure of the abnormal glycogen that accumulates in diabetic kidneys.
The last basic science talk of the meeting was given by Dr. Eva Pérez-Jiménez of the Sanz laboratory. Pérez-Jiménez spoke on how the homeostasis of the astrocytic glutamate transporter Glt-1 is altered in LD mouse models [38,39]. She gave an excellent overview regarding signaling changes in the CA1–CA3 hippocampus regions of the epileptic brain and described how too much glutamate increases postsynaptic excitotoxicity. The glutamate transporter Glt-1 is normally responsible for mediating clearance of excess glutamate; however, Glt-1 levels are diminished at the cell surface of astrocytes in LD mice, resulting in increased synaptic glutamate levels. In cell culture experiments, laforin and malin direct the ubiquitination of Glt-1, but this ubiquitination does not result in Glt-1 degradation. Instead, this event stabilizes Glt-1 at the plasma membrane. Additionally, laforin and malin target an adapter protein of the E3 ubiquitin ligase Nedd4.2 for degradation. Since Nedd4.2 promotes endocytosis of Glt-1, the action of laforin and malin prevents endocytosis and retains Glt-1 at the cell surface. Therefore, the absence of laforin or malin results in a reduction of the Glt-1 transporter at the plasma membrane because of increased endocytosis.
The final session focused on clinical science from the world’s leading LD neurologists. Delgado-Escueta opened the session discussing the importance of enrolling all living patients with LD in a new registry that Serratosa launched. He advocated for using the Montreal Cognitive Assessment (MOCA) to assess patient cognitive progression. He then laid out a well-defined MOCA timeline of patient progression from first seizure to advanced dementia. Michelucci then discussed clinical data regarding 27 Italian patients with LD. He described how the electroencephalogram (EEG) from presymptomatic or early phase patients exhibit normal background activity with isolated generalized spike and wave or polyspike and wave discharges with photosensitivity, and no activation of the paroxysms during non-REM sleep. The EEG from mid- to late-phase patients display deterioration of background activity, subcontinuous fast spike and wave or polyspike and wave discharges with subcontinuous focal occipital and central spikes. Dr. Maria Machio from the Serratosa Epilepsy Unit then presented an analysis of the first symptoms of LD. Generalized tonic-clonic seizures, myoclonic seizures, and visual symptoms, were among the most common. Most patients were misdiagnosed at onset. Serratosa then presented EEG data from patients with the two main forms of LD: classic LD and slowly progressive LD. Patients with classic LD show a grossly abnormal EEG 2–3 years after onset, and their presymptomatic mutation-carrying siblings also show grossly abnormal EEGs. Background slowing and frequent epileptiform discharges were the rule in presymptomatic siblings of patients with LD. Patients with LD presenting with the slowly progressive form show less severe abnormalities and could be initially misdiagnosed as juvenile myoclonic epilepsy. In both forms, sleep is associated with a striking decrease in the amount of epileptiform discharges. Dr. Afawi presented information on families from Israel and his experience with genetic studies in epilepsies. Minassian then presented preliminary work on developing biomarkers from cerebrospinal fluid and stressed the importance of collecting patient serum and cerebrospinal fluid samples so that all aspects of the clinical trial are in place. He also discussed additional LD animal models, specifically canine and rat models. The canine model faithfully recapitulates the myoclonic and epileptic phenotypes of patients with LD, but these take years to develop. His laboratory has also established a LD rat model that they are currently evaluating. Serratosa concluded the clinical session by emphasizing that the community needs to define the early stages of LD in order to understand disease progression. His clinical laboratory has initiated some early stage behavioral and physiological assessment analyses to bring this picture into focus. He then restated the importance of the patient registry and having every patient enrolled through a qualified neurologist so that all patients are registered for any upcoming trials. Physicians can enter their patients by participating in the registry or by contacting Delgado-Escueta, Minassian, Michelucci, or Serratosa. The evening culminated with a dinner overlooking the Hosteria del Estudiante, a sixteenth century building built to host students and overlooking the Cervantes courtyard of the University of Alcala. On Wednesday, September 11, the LECI investigators met regarding the NIH P01 grant to discuss project details, exchange data and protocols, and set priorities for the upcoming year.
Acknowledgments
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award Numbers R01NS070899 (MSG) and P01NS097197 (MSG), the Spanish Ministry of Education and Science Award Number SAF2017-83151-R (PS), a grant from the Fundacion Ramon Areces CIVP18A3935 (PS), Spanish MINECO BFU2017-84345-P (JJG), Valerion Therapeutics (MSG), and Ionis Pharmaceuticals. We thank Ms. Cheylene Plummer at the University of Kentucky College of Medicine for logistical support and planning the event. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
Declaration of competing interest
Dr. Paul Goldberg is Vice President of Clinical Development at Ionis Pharmaceuticals, Dr. Tamar Grossman is Director of Translational Medicine at Ionis Pharmaceuticals, and Dustin Armstrong is Chief Scientific Officer of Valerion Therapeutics.
References
- [1].Turnbull J, Tiberia E, Striano P, Genton P, Carpenter S, Ackerley CA, et al. Lafora disease. Epileptic Disord 2016;18:38–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gentry MS, Guinovart JJ, Minassian BA, Roach PJ, Serratosa J. Lafora disease offers a unique window into neuronal glycogen metabolism. J Biol Chem 2018;293: 7117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nitschke F, Ahonen SJ, Nitschke S,Mitra S, Minassian BA. Lafora disease — from pathogenesis to treatment strategies. Nat Rev Neurol 2018;14:606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Minassian BA. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol 2001;25:21–9. [DOI] [PubMed] [Google Scholar]
- [5].Serratosa JM. Idiopathic epilepsies with a complex mode of inheritance. Epilepsia 1999;40(Suppl. 3):12–6. [DOI] [PubMed] [Google Scholar]
- [6].Brewer MK, Gentry MS. The 3rd International Lafora Epilepsy Workshop: evidence for a cure. Epilepsy Behav 2018;81:125–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brewer MK, Grossman TR, McKnight TR, Goldberg YP, Landy H, Gentry MS. The 4th International Lafora Epilepsy Workshop: shifting paradigms, paths to treatment, and hope for patients. Epilepsy Behav 2019;90:284–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Valerion_Therapeutics, and Ionis_Pharmaceuticals. (2019) Natural history and functional status study of patients with Lafora disease. . [Google Scholar]
- [9].Lafora GR. Uber des Vorkommen amyloider KJrperchen im innern der Ganglienzellen. Virchows Arch f Path Anat 1911;205:295. [Google Scholar]
- [10].Lafora GR, Gluck B. Beitrag zur histopathologie der myoklonischen epilepsie. Z Ges Neurol Psychiatr 1911;6:1–14. [Google Scholar]
- [11].Nanduri AS, Kaushal N, Clusmann H, Binder DK. The maestro don Gonzalo Rodriguez-Lafora. Epilepsia 2008;49:943–7. [DOI] [PubMed] [Google Scholar]
- [12].Delgado-Escueta AV, Greenberg D. The search for epilepsies ideal for clinical and molecular genetic studies. Ann Neurol 1984;16(Suppl):S1 [11]. [DOI] [PubMed] [Google Scholar]
- [13].Serratosa JM, Delgado-Escueta AV, Posada I, Shih S, Drury I, Berciano J, et al. The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum Mol Genet 1995;4:1657–63. [DOI] [PubMed] [Google Scholar]
- [14].Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S,Mungall AJ, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet 1998;20:171–4. [DOI] [PubMed] [Google Scholar]
- [15].Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, Lindhout D, et al. A novel protein tyrosine phosphatase gene ismutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet 1999;8:345–52. [DOI] [PubMed] [Google Scholar]
- [16].Chan EM, Young EJ, Ianzano L,Munteanu I, Zhao X, Christopoulos CC, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet 2003;35:125–7. [DOI] [PubMed] [Google Scholar]
- [17].Duran J, Gruart A, Garcia-Rocha M, Delgado-Garcia JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet 2014;23:3147–56. [DOI] [PubMed] [Google Scholar]
- [18].Pederson BA, Turnbull J, Epp JR,Weaver SA, Zhao X, Pencea N, et al. Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann Neurol 2013;74: 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Turnbull J, Depaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E, et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PloS Genet 2011;7:e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Turnbull J, Epp JR, Goldsmith D, Zhao X, Pencea N,Wang P, et al. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol 2014;75: 442–6. [DOI] [PubMed] [Google Scholar]
- [21].Austin GL, Simmons ZR, Klier JE, Rondon A, Hodges BL, Shaffer R, et al. Central nervous system delivery and biodistribution analysis of an antibody-enzyme fusion for the treatment of Lafora disease. Mol Pharm 2019;16:3791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brewer MK, Uittenbogaard A, Austin GL, Segvich DM, DePaoli-Roach A, Roach PJ, et al. Targeting pathogenic Lafora bodies in Lafora disease using an antibody-enzyme fusion. Cell Metab 2019;30:689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou Z, Austin GL, Shaffer R, Armstrong DD, Gentry MS. Antibody-mediated enzyme therapeutics and applications in glycogen storage diseases. Trends Mol Med 2019; 25:1094–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Finkel RS,Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 2017; 377:1723–32. [DOI] [PubMed] [Google Scholar]
- [25].Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 2018;378:625–35. [DOI] [PubMed] [Google Scholar]
- [26].Valerion_Therapeutics. (2016) VAL-1221 delivered intravenously in ambulatory and ventilator-free participants with late-onset Pompe disease. . [Google Scholar]
- [27].van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet 2008;372:1342–53. [DOI] [PubMed] [Google Scholar]
- [28].Yi H, Sun T, Armstrong D, Borneman S, Yang C, Austin S, et al. Antibody-mediated enzyme replacement therapy targeting both lysosomal and cytoplasmic glycogen in Pompe disease. J Mol Med (Berl) 2017;95:513–21. [DOI] [PubMed] [Google Scholar]
- [29].Auge E, Pelegri C, Manich G, Cabezon I, Guinovart JJ, Duran J, et al. Astrocytes and neurons produce distinct types of polyglucosan bodies in Lafora disease. Glia 2018;66:2094–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sun RC, Dukhande VV, Zhou Z, Young LEA, Emanuelle S, Brainson CF, et al. Nuclear glycogenolysis modulates histone acetylation in human non-small cell lung cancers. Cell Metab 2019;30:903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brewer MK, Gentry MS. Brain glycogen structure and its associated proteins: past, present and future. Adv Neurobiol 2019;23:17–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhou Z, Kinslow CJ, Hibshoosh H, Guo H, Cheng SK, He C, et al. Clinical features, survival and prognostic factors of glycogen-rich clear cell carcinoma (GRCC) of the breast in the U.S. population. J Clin Med 2019;8:246–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rubio-Villena C, Viana R, Bonet J, Garcia-Gimeno MA, Casado M, Heredia M, et al. Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum Mol Genet 2018;27:1290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med 2015;212:939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fanin M, Nascimbeni AC, Savarese M, Papa V, Cenacchi G, Nigro V, et al. Familial polyglucosan bodymyopathywith unusual phenotype. Neuropathol Appl Neurobiol 2015;41:385–90. [DOI] [PubMed] [Google Scholar]
- [36].Hedberg-Oldfors C, Oldfors A. Polyglucosan storage myopathies. Mol Aspects Med 2015;46:85–100. [DOI] [PubMed] [Google Scholar]
- [37].Nilsson J, Schoser B, Laforet P, Kalev O, Lindberg C, Romero NB, et al. Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann Neurol 2013;74: 914–9. [DOI] [PubMed] [Google Scholar]
- [38].Munoz-Ballester C, Berthier A, Viana R, Sanz P. Homeostasis of the astrocytic glutamate transporter GLT-1 is altered in mouse models of Lafora disease. Biochim Biophys Acta 2016;1862:1074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Munoz-Ballester C, Santana N, Perez-Jimenez E, Viana R, Artigas F, Sanz P. In vivo glutamate clearance defects in a mouse model of Lafora disease. Exp Neurol 2019; 320:112959. [DOI] [PMC free article] [PubMed] [Google Scholar]