

Abstract
Zoonotic monkey B virus (Macacine alphaherpesvirus 1; BV) infections are extremely serious and usually fatal. Drugs currently used for treatment were developed for the treatment of herpes simplex virus but are less effective against BV. Effective suppression of viral replication in the skin could prevent the virus from invading the nervous system. To test this hypothesis, the efficacy of topical administration of several drugs against lethal BV infection was evaluated in female BALB/c mice that were infected by scarification. Drugs were then applied to the site of inoculation. As 3% preparations, most drugs were only minimally effective or ineffective. In contrast, ganciclovir and cidofovir were very effective. The ED50 for cidofovir was 0.007%, compared with 1.1% for ganciclovir. At 0.5%, cidofovir protected against both death and neurologic signs, whereas 5% ganciclovir only protected against death but not neurologic involvement. All genotypes of BV were equally susceptible to cidofovir and ganciclovir. For maximal effectiveness, treatment with both cidofovir and ganciclovir had to be initiated within 8 h of infection. Cidofovir was completely protective when administered only on the day of infection, whereas a minimum of 5 d of treatment was required for maximal ganciclovir efficacy. These studies showed that topical cidofovir treatment started soon after BV exposure was very effective in preventing BV from invading the nervous system, whereas ganciclovir treatment was only partially effective. In addition, cidofovir was protective against a ganciclovir-resistant BV mutant, whereas ganciclovir was not. These studies showed that topical cidofovir treatment started soon after BV exposure is more effective than ganciclovir in preventing BV from invading the CNS.
Abbreviations: BV, Macacine alphaherpesvirus 1; HSV, herpes simplex viruses; dpi, days after inoculation; DRG, dorsal root ganglion; PLO, pluronic lecithin organogel; TK, thymidine kinase
Macaque monkeys are an invaluable resource for biomedical research. Monkey B virus (Macacine alphaherpesvirus 1; BV) is an α-herpesvirus indigenous in macaques (Macaca spp.) and is closely related to herpes simplex viruses (HSV) of humans. Like HSV, BV rarely causes serious disease in its natural macaque host. However, when transmitted to humans through bites or scratches from macaques, BV readily invades the CNS, resulting in a fatality rate of approximately 80%.13,24,32,49 With only a single exception, all documented zoonotic BV infections to date have occurred in research, veterinary, or animal care personnel working with macaque tissues or NHP. Although zoonotic BV infections are uncommon, their high fatality rate (approximately 80%) makes BV the single most important zoonotic agent of concern for persons having contact with macaques.16,24 In addition, the increasing popularity of monkey temple and monkey forest visitation in southeast Asia raises a public health concern that tourists may acquire BV infections through direct contact with infected wild monkeys.17,25,39
Although all exposure incidents (through bites, scratches, ocular mucous membranes, and so forth) have the potential for developing into an active BV infection, the probability of actual BV transmission likely varies in different situations. The dose of infectious BV necessary to successfully infect humans is unknown, and exposure incidents can range from the transmission of high doses of BV (for example, a bite from a monkey actively shedding infectious BV) to very low or no infectious BV. Furthermore, serologic diagnosis of BV zoonotic infections is problematic due to the extensive antigenic crossreactivity between BV and HSV, coupled with the high incidence of HSV in humans. Consequently, it is virtually impossible to discriminate among patients that were actually infected with BV and were successfully treated, patients that were infected with a sufficiently low dose of virus that they were capable of controlling the infection (through innate or acquired immune responses) regardless of drug treatment, and patients that were ‘exposed’ but did not actually acquire infectious BV. The current inability to predict the outcome of an exposure incident means that if prophylactic treatment is pursued, many patients likely will be treated unnecessarily.
Because BV is closely related to HSV, most genes encoding enzymes involved in DNA replication are conserved to a high degree between the 2 viruses.19,27,30,33 As a result, many antiviral drugs that inhibit HSV replication are also active against BV, although BV is typically less sensitive to them than is HSV.4,8,19,27,51 The comparative rarity of zoonotic BV infections has not provided a financial incentive for the development of drugs specifically directed against BV. Consequently, drugs used for the treatment of HSV infections are currently used—despite their suboptimal efficacy—to treat patients with or suspected of having a BV infection.11,37 Current recommendations for prophylactic treatment after a BV exposure incident (possible infection) are oral acyclovir or its prodrug, valacyclovir; when neurologic symptoms are apparent, ganciclovir is recommended.11,37 Although there are reports of persons infected with BV that were treated with antiviral drugs and survived the infection, there are also reports of drug-treated patients who have died and of untreated patients that have survived BV infection.2,9,10,12,13,18,22,29 Thus, the true efficacies of acyclovir and ganciclovir for treatment of BV infections in exposed people is not really known.
Although valacyclovir and acyclovir are relatively safe drugs, with few side effects in most patients, experiments in animals indicate that acyclovir is actually not very effective (in rabbits) or completely ineffective (in mice) against BV infection.4,5,8,51 The use of acyclovir for prophylactic treatment after BV exposure incidents could be problematic if acyclovir is as poorly effective against BV in humans as it is in mice and rabbits, in that ineffective therapy could allow BV to replicate at the site of inoculation to levels sufficient to invade the CNS.26 Although ganciclovir is the most effective drug currently available for the treatment of zoonotic BV infections, even ganciclovir is not completely effective and has significant toxicity when administered systemically.10,34
Given the infrequency of zoonotic BV cases relative to the number of exposure incidents, whether prophylactic drug therapy after an exposure incident is appropriate is unclear, especially given the uncertain efficacy and potential toxicity of some drugs currently used when administered systemically. Many, if not the vast majority, of BV exposure incidents do not result in clinically apparent infections; consequently prophylactic treatment probably is unnecessary in many cases. However, the extremely poor prognosis of BV cases, once neurologic symptoms are evident, makes prevention of possible BV infections of paramount importance. Ideally, after an exposure incident, all persons would be treated prophylactically with a highly efficacious and minimally toxic drug that would prevent the replication of BV to levels sufficient to become established in the patient and, especially, to invade the CNS.
Topical drug application has several major advantages over systemic drug delivery. Drug toxicity and adverse side effects are typically much lower with topical rather than systemic administration. Topical drug treatment is also less costly since it can generally be self-administered on an out-patient basis (with a reasonable expectation of good compliance in the case of BV, given the potentially severe consequences of noncompliance). Several viral infections of the skin have been treated successfully with topical drug formulations.20,44,50 In addition, new vehicle compounds have been developed that improve drug permeation through the skin, allowing accumulation of the drug in the skin at the site of the infection, thereby increasing treatment efficacy.1,36
If topical drug treatment after a BV exposure effectively prevented BV from replicating at the initial site of infection in the skin, this outcome would prevent the accumulation of levels of infectious virus necessary for invasion of the CNS through unmyelinated sensory nerve endings present in the dermis. Here we present a series of experiments assessing the efficacy of topical drug treatment for the prevention and control of BV infections in a mouse model.
Materials and Methods
Humane care guidelines.
All animal experiments were reviewed and approved by the Oklahoma State University IACUC, and mice were maintained in an AAALAC-accredited facility. The mouse model of BV infection was previously described.8,15,40,41 Briefly, the left flank of female Balb/c mice (weight, 10 to 12 g; Charles River, Wilmington, MA) was shaved and lightly scarified by scratching with a 20-gauge needle in a 6×6 checkerboard pattern, approximately 10 LD50 of BV in 10 μL was applied to the scarified area, and the inoculum was gently rubbed in by using the side of a micropipet tip. Because some mice that received drug treatment survived with neurologic symptoms, acetaminophen was included in drinking water (2 mg/mL) in all experiments.
Mouse procedures.
Mice euthanized after 10 dpi were bled, and their serum was tested by ELISA for antiBV IgG as described.31 Histopathology and immunohistochemical staining were performed as previously described.6,7 In addition to using mortality as a measure of protection, neurologic disease was also assessed. A scoring system based on the highly reproducible progression of neurologic symptoms in untreated infected mice was used (Figure 1).8 The initial sign of neurologic involvement in mice is an abnormal reflex in abduction of the ipsilateral hindleg, with marked flexion of the foot, when a mouse is lifted by the tail. This lesion rapidly evolves into paresis of the ipsilateral foot followed by spastic then flaccid paralysis of the leg. Mice progressively become immobile, show a decrease in body temperature, and develop bilateral paralysis of the hindlegs, tremors, ataxia, and urinary and fecal retention. When the infection reaches this point, mice are euthanized. Although skin lesions were often evident, their presence was not used in the scoring system because not all mice developed cutaneous lesions, lesions were inconsistent in severity, some mice regrew hair very quickly thus making detection of lesions problematic, and lesions appeared both soon after infection (as a result of primary infection) and days later when virus traveled retrograde from dorsal root ganglia (DRG) back to the skin.
Figure 1.

Neurologic scoring system for mice inoculated with BV. Criteria for euthanasia: score of 4 plus signs including tremors, pale extremities, decreased consciousness, or poor response to stimuli.
Unless otherwise stated, groups of 8 mice were used in all experiments. The standard drug treatment protocol used was initiated on day 0 at 4 h after inoculation, with a second dose administered at 8 h after inoculation. For the next 6 d, drugs (or vehicle control) were administered 3 times daily at 5-h intervals. Approximately 4 to 5 mm of the emulsion containing pluronic lecithin organogel (PLO) and drug was extruded from the syringe onto a Dacron-tipped swab and rubbed into the hindflank, covering the entire inoculation site and surrounding area. Any variation from this standard protocol during experiments is noted in the text.
Viruses and cells.
BV strain E90-136 originally was isolated from a cynomolgus (long-tailed) macaque (M. fasicularis);45 the other BV strains were isolated from rhesus macaques (M. mulatta).23,38 All work with infectious BV was performed under biocontainment conditions approved by the Oklahoma State University Institutional Biosafety Committee and the US Centers for Disease Control and Prevention.
A ganciclovir-resistant mutant of BV E90-136, similar to a penciclovir-resistant mutant described previously,3 was identified when testing in vitro sensitivity to 40 µg/mL of ganciclovir. This ganciclovir-resistant mutant was plaque-purified through multiple rounds of limiting dilution. PCR amplification and sequencing, performed as described,15 identified a mutation in the UL23 thymidine kinase (TK) gene. The new ganciclovir-resistant mutant had a single-nucleotide deletion approximately 167 nucleotides from the 3′ end of the UL23 ORF, resulting in replacement of the terminal 59 amino acids with 10 codons of unrelated sequence (Genbank accession MH512907).
Drugs.
Analytical grade reagents were used in all experiments. Drugs used included acyclovir, penciclovir, ganciclovir, cidofovir, 5-iodo-2ʹ-deoxyuridine, 5-trifluoromethyl-2ʹ-deoxyuridine, and 5-ethyl-2ʹ-deoxyuridine. Drugs were purchased from Sigma Chemical (St Louis, MO; acyclovir, 5-ethyl-2ʹ-deoxyuridine, 5-iodo-2ʹ-deoxyuridine, 5-trifluoromethyl-2ʹ-deoxyuridine) or Gemini Biologicals (West Sacramento, CA; cidofovir, ganciclovir). An experimental inhibitor of the HSV ribonucleotide reductase has been reported to be effective against HSV and to augment the inhibitory activity of acyclovir;28,46-48 one of the less toxic variants (BW348U87)48 was synthesized by GLSynthesis (Worchester, MA). A commercially available product for treatment of HSV cold sores (Abreva) was purchased locally.
PLO was purchased from Pharmedica Enterprise (Selangor, Malaysia) and used as a vehicle for topical application of drugs. PLO–drug preparations (3 mL) were prepared the day before mice were infected, and emulsions were stored at room temperature in the dark for the duration of the experiment. Briefly, a slurry of drug was prepared in 300 µL of ethoxydiglycol (all drugs for the initial experiment) or 0.02 N HCl (cidofovir and ganciclovir for all subsequent experiments) and mixed with 700 µL of the organic phase solution. This slurry was placed in a 3-mL syringe and mixed through a female–female luer-lock connecter with 2.1 mL of the aqueous phase solution (in a 3-mL syringe) until a stable emulsion formed. When more than 3 mL of drug was required for an experiment, the preparation was scaled up by using 10-mL syringes and the emulsion then divided among 3-mL syringes for use.
Statistical analyses.
For drugs that appeared to successfully prevent mortality due to BV inoculation, a range of doses were administered to groups of 4 to 8 mice over multiple experiments. Survival in each dose group was regressed against the log10 of drug dose (% drug in topical PLO) by using the Hill equation (Kinetica version 5.0, Thermo Fisher Scientific, Philadelphia, PA) to estimate the drug's ED50 against BV. The mean of curves from individual experiments was used to estimate the mean ED50. The maximal neurologic score recorded after BV inoculation was compared between dose rates by using nonparametric one-way, Kruskall–Wallis ANOVA with the Dunn test (SigmaPlot version 11.0, Systat Software, San Jose, CA), to determine whether treatment groups differed from vehicle controls. Statistical significance was defined as α of less than 0.05.
Results
Many zoonotic BV infections result from bites or scratches on the skin. Unless very high amounts of infectious virus are transmitted, the virus must first replicate locally in the skin to attain levels of infectious virus sufficient to invade unmyelinated sensory nerve endings located in the dermis. We therefore reasoned that topical application of antiviral drugs to a wound site might suppress viral replication at the site of infection, thereby preventing BV from ever accessing the CNS where drugs are less effective. To test this hypothesis, we prepared 8 drugs that have been used to treat HSV and, in some cases, zoonotic BV infections as 3% concentrations in a PLO topical vehicle. Mice were infected with 10 LD50 of BV, and drugs were applied at 4 and 8 h after inoculation on the day of infection (0 dpi) and 3 times daily on 1 through 6 dpi. The efficacy of the drugs tested varied (Figure 2). Both Abreva and penciclovir were completely ineffective, providing less protection than vehicle alone. Acyclovir was only marginally better than vehicle alone, whereas 5-trifluoromethyl-2ʹ-deoxyuridine and 5-ethyl-2ʹ-deoxyuridine were somewhat effective, providing 40% to 50% protection against lethal infection. However, 5-trifluoromethyl-2ʹ-deoxyuridine demonstrated toxicity, as evidenced by weight loss, depression, and inactivity in treated mice. In contrast, ganciclovir, cidofovir, and the experimental ribonucleotide reductase inhibitor BW348U87 were 90% to 100% effective in protecting against lethal BV infection. These results indicate that topical drug application can provide effective protection against lethal BV infection.
Figure 2.
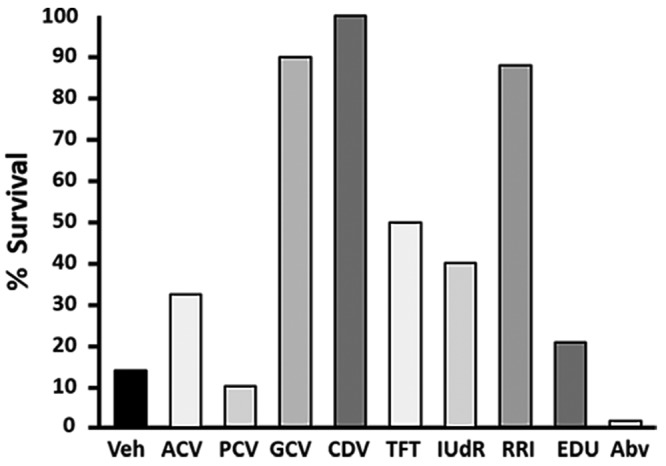
Screening of antiherpetic drugs for topical efficacy. Drugs were prepared at a concentration of 3% in PLO vehicle and applied to the area of skin where virus had been applied. Mice were treated at 4 and 8 h after inoculation on the day of infection (0 dpi) and 3 times daily at 5-h intervals on 2 through 6 dpi. Survival through 14 dpi is shown.
Because ganciclovir is currently the most effective drug recommended for treatment of zoonotic BV infections, we determined its topical ED50. Concentrations ranging from 5.0% to 0.1% were tested by using the 7 d treatment regimen described earlier (Figure 3 A). No signs of toxicity were noted for any dose of ganciclovir or cidofovir. Three experiments were performed that yielded ED50 values of 1.1%, 1.8%, and 0.3%, for an average ganciclovir ED50 of 1.1% ± 0.7% (Figure 4 A). Because previous studies8 with the BV–mouse model using systemic drug administration found cidofovir to be more effective than ganciclovir, we also determined the ED50 of cidofovir (Figures 3 B and 4 B). Duplicate experiments gave ED50 values of 0.006% and 0.007%, yielding an average of 0.007% ± 0.0001%, approximately 150-fold lower than the ED50 of ganciclovir. Consistent with this difference in ED50 values, neurologic symptoms were much more prevalent and more severe in mice treated with ganciclovir than in cidofovir-treated mice. Although variability between experiments was considerably less with cidofovir as compared with ganciclovir, the Hill coefficients were similar (4.0 ± 1.9 and 3.0 ± 0.1, respectively; P > 0.05).
Figure 3.
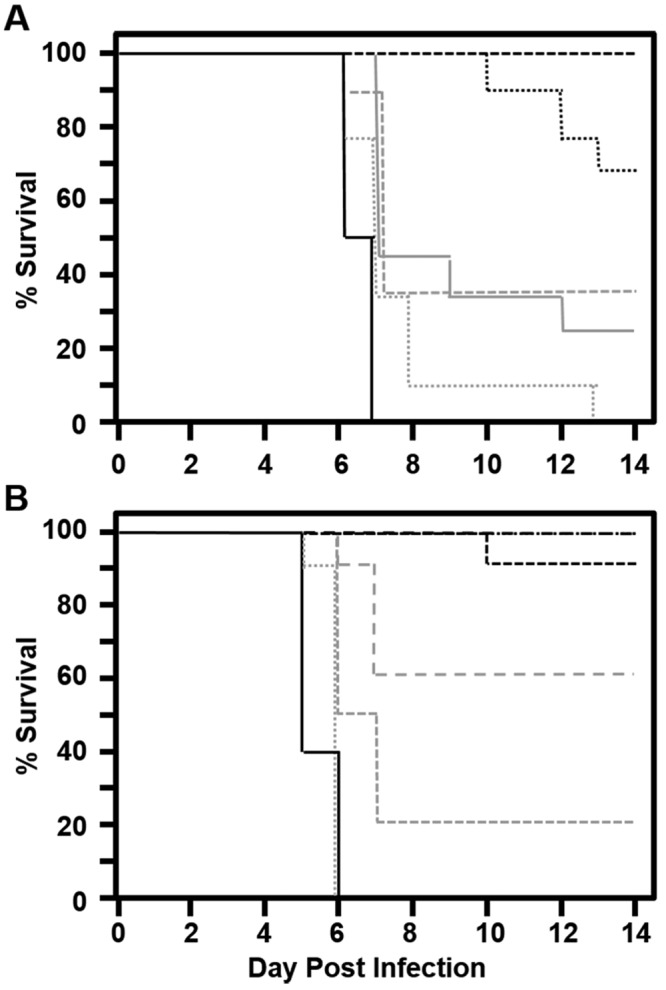
Survival curves after topical application of increasing concentrations of (A) ganciclovir or (B) cidofovir by using the standard treatment regimen after BV inoculation through skin scarification. For ganciclovir: black long dash, 5%; black short dash, 3%; black dot, 1%; gray long dash, 0.5%; gray short dash, 0.3%; gray dot, 0.1%; solid black, vehicle control. For cidofovir: black long dash, 0.3%; black short dash, 0.1%; black dot, 0.03%; gray long dash, 0.01%; gray short dash, 0.003%; gray dot, 0.001%; solid black, vehicle control.
Figure 4.
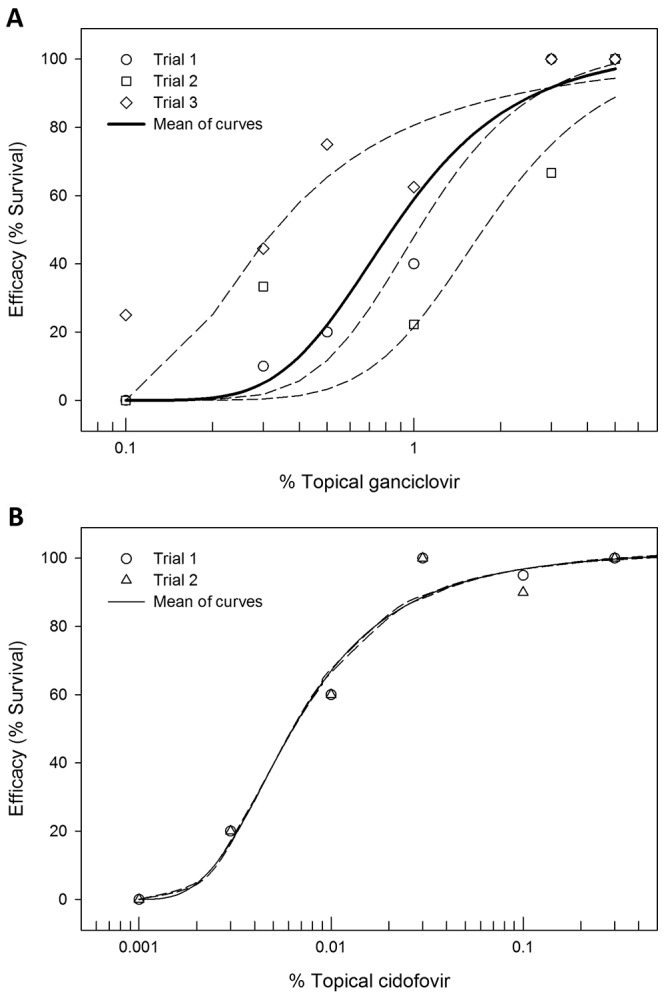
Dose–response curves for the survival of mice after topical application of increasing concentrations (A) ganciclovir or (B) cidofovir by using the standard treatment regimen after inoculation with BV through skin scarification.
Neurologic signs generally paralleled the ED50 results, in that the drug dose groups with the highest survival had correspondingly low neurologic scores, both during and after the study (Table 1). The highest dose groups for both ganciclovir and cidofovir showed few neurologic signs. However, at the highest tested concentrations of cidofovir, mice had little to no neurologic signs, whereas even at 5% ganciclovir, some mice had neurologic signs during the study but stabilized and survived through 14 dpi. At a ganciclovir concentration of 3%, all 28 mice studied survived to 14 dpi, but neurologic deficits were noted and decreased during the course of the study. At 0.5% to 1% ganciclovir, neurologic deficits persisted in surviving mice at 14 dpi. At lower ganciclovir concentrations, the majority of mice showed severe neurologic signs that culminated in death. With rare exception (2 mice treated with 0.01% and one with 0.05%), cidofovir treatment resulted in an ‘all-or-none’ response, with mice either developing encephalitis and dying or not developing any neurologic signs and surviving to 14 dpi.
Table 1.
Neurologic scores of BV-inoculated treated with topical ganciclovir or cidofovir
| Neurologic score |
|||
| Maximum | Survivors at 14 dpi | ||
| Topical ganciclovir | |||
| 0% (control) | 5 (5, 5); n = 23 | no survivors | |
| 0.1% | 5 (5, 5); n = 28 | 0 (0, 0); n = 6 | |
| 0.3% | 5 (5, 5); n = 28 | 0 (0, 0.5); n = 6 | |
| 0.5% | 5 (2, 5); n = 28 | 1.5 (0, 2); n = 11 | |
| 1% | a3 (1, 5); n = 28 | 0.5 (0, 2); n = 19 | |
| 3% | a1.75 (0, 2.9); n = 28 | 0 (0, 1); n = 28 | |
| 5% | *0 (0, 1); n = 28 | 0 (0, 0); n = 28 | |
| % cidofovir | |||
| 0% (control) | 5 (5, 5); n = 15 | no survivors | |
| 0.001% | 5 (5, 5); n = 20 | no survivors | |
| 0.003% | 5 (5, 5); n = 20 | 0 (0, 0); n = 4 | |
| 0.005% | 5 (5, 5); n = 10 | no survivors | |
| 0.01% | 0.5 (0, 5); n = 20 | 0 (0, 0); n = 13 | |
| 0.03% | a0 (0, 0); n = 20 | 0 (0, 0); n = 18 | |
| 0.05% | a0 (0, 0); n = 10 | 0 (0, 0); n = 10 | |
| 0.1% | a0 (0, 0.75); n = 20 | 0 (0, 0); n = 16 | |
| 0.3% | a0 (0, 0); n = 20 | 0 (0, 0); n = 20 | |
| 1% | a0 (0, 0); n = 9 | 0 (0, 0); n = 9 | |
| 3% | a0 (0, 0); n = 9 | 0 (0, 0); n = 9 | |
| 5% | a0 (0, 0); n = 9 | 0 (0, 0); n = 9 | |
Data are presented as the median (lower quartile, upper quartile); number of mice in group.
Value is significantly (P < 0.05) different from maximal neurologic score of control mice.
The ED50 values noted earlier indicated that topical cidofovir was much more potent than ganciclovir in protecting mice from mortality and neurologic signs after BV inoculation. Therefore, we hypothesized that cidofovir may be similarly effective at a shorter duration of treatment than the full 7-d regimen. To test this hypothesis, we inoculated groups of mice with 10 LD50 of BV, and treatment (vehicle, 0.5% cidofovir, or 5.0% ganciclovir) was initiated at 4 and 8 h after inoculation on the day of infection and 3 times daily for the next 6 d (standard 7-d regimen). Treatment ceased for 1 group each day, resulting in groups treated for 1 to 7 d (0 to 6 dpi). Resulting mortality was 0% to 37.5% for mice treated with ganciclovir for less than 5 d (Figure 5 A); ganciclovir provided 100% protection from death or euthanasia only when the drug was administered for 6 or 7 d. Most (13 of 19) of the ganciclovir-treated mice that survived infection after various lengths of treatment developed signs of neurologic involvement. Although neurologic symptoms in most of these survivors diminished over time, neurologic signs in several mice slowly increased in severity over time but did not reach a point that required euthanasia by the end of the experiment (14 dpi). In contrast, all mice treated with cidofovir—even those that received only 2 doses of cidofovir (at 4 and 8 h after inoculation on day 0)—survived infection. Furthermore, none of the cidofovir-treated mice developed any clinical signs of neurologic involvement.
Figure 5.

Effect of varying the drug treatment regimen. Data shown indicate the effect of (A) decreasing the duration of ganciclovir or cidofovir, (B) delaying the start of ganciclovir or cidofovir treatment, or (C) varying the concentration of cidofovir on the survival of mice to 14 dpi. Cidofovir was 100% protective when administered only on the day of infection, whereas ganciclovir had to be administered for at least 5 d to be 100% protective. Delaying the initiation of cidofovir and ganciclovir treatment only 24 h after infection resulted in a decrease in efficacy. In panels A and B: gray, 5% ganciclovir; black, 0.5% cidofovir. In panel C: black, 3% cidofovir; gray, 1% cidofovir; white, 0.5% cidofovir.
If the efficacy of cidofovir is due to rapid and efficient shutdown of viral replication at the site of inoculation, delaying the start of treatment could be a more critical factor in treatment efficacy for cidofovir than for ganciclovir. To assess the importance of timing in treatment initiation, groups of mice were infected and treated with ganciclovir (5.0%) or cidofovir (0.5%). Delaying the start of cidofovir treatment just 4 h (that is, starting treatment at 8 h after inoculation instead of at 4 h) resulted in only 87.5% protection, and delaying the start of cidofovir treatment until 24 h after inoculation resulted in no protection at all (Figure 5 B). Delaying the time of initiation of ganciclovir treatment had a similar effect, albeit not as severe as for cidofovir. In addition, starting ganciclovir treatment at 8 h after inoculation was 100% protective against mortality, and starting treatment as late as 24 h after inoculation still protected 62.5% of mice; any longer delay in initiating treatment did not provide effective protection. As previously, surviving cidofovir-treated mice did not develop any clinical signs of neurologic involvement, whereas most (22 of 33) ganciclovir-treated survivors did. Even beginning ganciclovir treatment at 4 or 8 h after inoculation did not prevent the development of some signs of neurologic involvement. These results clearly demonstrate that initiating topical drug treatment very soon after infection is essential to provide effective protection, especially for cidofovir.
Despite its seemingly high efficacy, the inability of cidofovir to provide 100% protection when started as soon as 8 h after inoculation was unexpected. To confirm the critical importance of starting cidofovir treatment within 4 h of infection and to determine whether higher doses of cidofovir provided better protection if started later than 4 h after inoculation, 3% and 1% cidofovir were compared with 0.5% cidofovir. Whereas 3% cidofovir provided 100% protection when treatment was initiated at 8 h after inoculation, 1% cidofovir did not (Figure 5 C). Initiating treatment at 12 h after inoculation provided some protection with all 3 dosages, but starting treatment at 24 h after inoculation was completely ineffective for all dosages. Furthermore, several mice surviving treatment initiated at 8 or 12 h after inoculation developed clinical signs of neurologic involvement.
The comparatively low EC50 of cidofovir suggests that this drug is very effective against BV. To further assess cidofovir's effectiveness, mice were infected with increased doses of virus. At 0.5% cidofovir, mice were 100% protected against both 10 and 100 LD50 but only 50% against 1000 LD50 of BV. Ganciclovir (5%) provided 100% protection against 10, 100, and 1000 LD50 BV. None of the surviving mice treated with cidofovir developed any clinical signs of neurologic involvement, whereas 80% of ganciclovir-treated mice infected with 10 LD50 and 100% of those infected with 100 or 1000 LD50 developed signs of neurologic involvement.
All experiments above were conducted by using a BV strain isolated from a cynomolgus macaque. However, rhesus monkeys are more widely used in biomedical research, and BV exposure incidents are thus more likely to involve a rhesus-adapted BV genotype virus. Therefore, the efficacies of ganciclovir and cidofovir were assessed against 5 rhesus-adapted BV strains (E2490, 20620, 16293, 26896-G, and 32425-O). Mice were infected with 10 LD50 of virus and treated for 3 d (0.5% cidofovir) or 7 d (vehicle, 5.0% ganciclovir) beginning at 4 h after inoculation on the day of infection. Both ganciclovir and cidofovir provided 100% protection against death due to all 5 rhesus-adapted BV virus strains. As we observed for the cynomolgus-adapted BV virus, approximately 50% of the mice that survived infection with rhesus-adapted BV and that were treated with ganciclovir developed various neurologic symptoms whereas cidofovir-treated mice failed to develop any clinical signs of neurologic involvement.
The short treatment duration needed for protection and the lack of any neurologic symptoms in cidofovir-treated survivors suggests that the efficacy of cidofovir rests in its immediate and complete suppression of viral replication in the skin. If so, infected mice treated with cidofovir may never experience sufficient exposure to the virus to stimulate an adaptive immune response. In contrast, ganciclovir efficacy may depend more on reducing viral replication to a level that allows the host to survive long enough to mount an adaptive immune response that ultimately controls the infection. In mice treated from 4 h after inoculation through 6 dpi and that survived through 14 dpi, serum antiBV IgG (as a measure of an adaptive immune response) was detected in only 1 of the 26 mice treated with 0.5% cidofovir (OD range, 0.000 to 0.163; mean, 0.022; Figure 6), whereas 26 of the 31 mice treated with 5% ganciclovir were seropositive (OD range, 0.000 to 0.748; mean, 0.252). These results support our supposition that cidofovir very quickly and effectively suppresses BV replication in the skin to the point that the virus does not effectively spread and the host never mounts an adaptive immune response. In contrast, ganciclovir likely sufficiently reduces viral replication to enable the host to mount an adaptive immune response that can result in protection from lethality.
Figure 6.
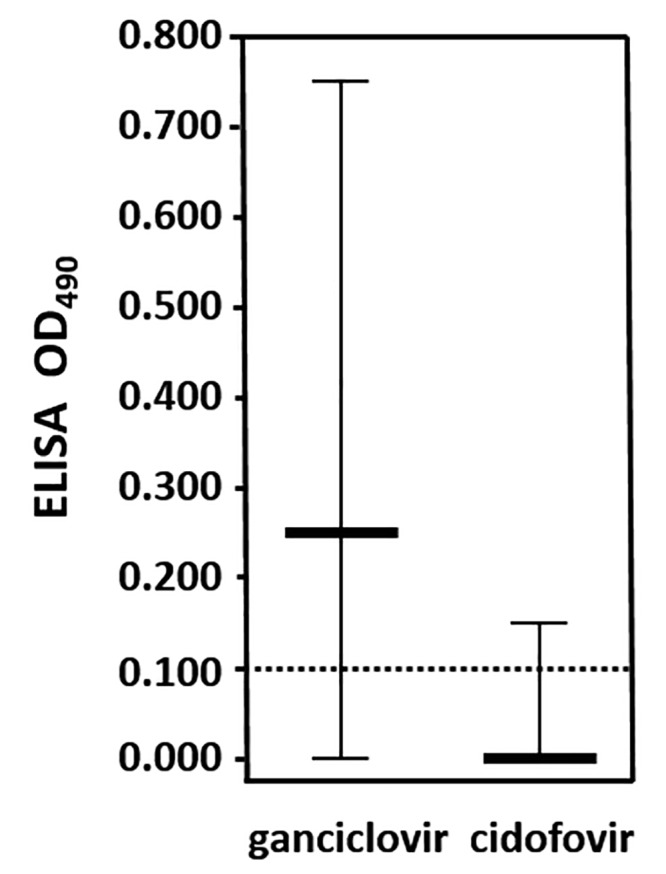
Immune response of mice treated with cidofovir compared with ganciclovir. Median OD data are from mice that survived through 14 dpi after treatment with cidofovir (0.5%; n = 16) or ganciclovir (5.0%; n = 17) by using the standard regimen were bled and their serum tested by ELISA for IgG to BV.
To further assess the effect of cidofovir compared with ganciclovir on viral replication, we followed the histologic progression of the infection over time in 5 vehicle control mice and in groups of 6 mice that were treated with either topical ganciclovir (5%) or cidofovir (0.5%). Mice were infected and euthanized at 0.5, 1, 2, 3, 4, and 5 dpi. Skin from the site of inoculation, the spinal cord, and dorsal root ganglia were examined for histologic evidence of viral replication. In untreated mice as well as in mice treated with cidofovir and ganciclovir, skin lesions at 0.5 and 1 dpi were similar in all groups and were characterized by multifocal epidermal erosion with neutrophilic dermatitis and were presumed to represent lesions of scarification at the inoculation site (Table 2). By 2 dpi and continuing at 3 dpi, untreated mice had regionally extensive epidermal erosion to ulceration with scattered vesicle formation in which necrotic keratinocytes often had discernible intranuclear inclusion bodies. For the ganciclovir treatment group, all but one mouse in each of the 2- and 3-dpi groups had intact epidermis beneath neutrophilic crusts and no evidence of keratinocyte degeneration. One ganciclovir-treated mouse in each of the 2- and 3-dpi groups had a focal epidermal lesion similar to those in untreated mice, characterized by a few degenerate keratinocytes containing intranuclear inclusion bodies. At 2 and 3 dpi, all cidofovir-treated mice had multifocal neutrophilic epidermal crusts overlying intact and often hyperplastic epidermis without any evidence of intranuclear inclusion bodies. No abnormalities were seen in the dorsal root ganglia (DRG) or spinal cord of any mice at 2 dpi or earlier. By 3 dpi, untreated mice had subtle neuronal degeneration and early necrosis in unilateral lumbar DRG with occasional discernible intranuclear inclusion bodies. No lesions were seen in the DRG or spinal cords of any ganciclovir- or cidofovir-treated mice at 3 dpi.
Table 2.
Lesions and BV antigen (detected by immunohistochemistry) in the skin and dorsal root ganglia of mice inoculated with BV and treated topically with vehicle (control), 5% ganciclovir, or 0.5% cidofovir
| Skin (at time point [dpi]) |
Dorsal root ganglion (at time point [dpi]) |
|||||||||||
| Lesion |
Immunohistochemistry |
Lesion |
Immunohistochemistry |
|||||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 1 dpi | 2 dpi | 3 dpi | 1 dpi | 2 dpi | 3 dpi | |
| Control | +* | +++ | +++ | + | +++ | +++ | — | — | +++ | — | — | +++ |
| Ganciclovir | +* | + | + | — | + | + | — | — | — | — | — | — |
| Cidofovir | +* | — | — | — | — | — | — | — | — | — | — | — |
+, mild lesions that are not virus-specific or lesions present in only 1 individual per group; +++, virus-specific lesions with discernible intranuclear inclusion bodies in the majority of animals per group; *, lesions consistent with skin scarification procedure; –, no lesions
Immunohistochemistry for viral antigen was done to definitively correlate microscopic lesions in skin and DRG with viral infection. In untreated mice, viral antigen was first detected in the skin at 1 dpi within nuclei of a few foci of keratinocytes (Table 2). By 2 and 3 dpi, viral antigen was detected within keratinocytes with intranuclear inclusion bodies and other changes previously described in histologic sections (Figure 7 A). In ganciclovir-treated mice, viral antigen was detected in a small focus of keratinocytes in the skin of one mouse each in the 2- and 3-dpi groups and corresponded with previously described microscopic epidermal lesions. No viral antigen was detected in the skin of cidofovir-treated mice. No viral antigen was detected in DRG or spinal cords of any mice up to 2 dpi. By 3 dpi, many neurons in unilateral lumbar DRG of untreated mice had strong immunohistochemical signals for viral antigen that corresponded with subtler neuronal changes visible in sections stained with hematoxylin and eosin (Figure 7 B). No immunohistochemical positivity for viral antigen was seen in DRG or spinal cord of any ganciclovir or cidofovir-treated mice at 3 dpi.
Figure 7.

Photomicrographs of (A) skin (magnification, 200×) and (B) lumbar DRG (magnification, 400×) from an untreated mouse at 3 dpi. Immunohistochemical stain for BV antigen shows a focus of keratinocytes with abundant intranuclear positive signal within the epidermis (skin from inoculation site). A few cells within sebaceous glands near the center of the photo have intranuclear viral antigen. Immunohistochemical staining for BV antigen also shows strong positive signal within the nuclei (arrowheads) of several neurons within the DRG.
A major problem seen with acyclovir treatment of HSV infections is the occurrence of acyclovir-resistant virus. A spontaneous penciclovir-resistant mutant of BV has been reported previously; this mutant was resistant to acyclovir and ganciclovir also.3 Unlike ganciclovir, cidofovir does not require phosphorylation by viral thymidine kinase enyzmes to have antiviral activity.50 Consequently, cidofovir is used to treat acyclovir-resistant HSV infections42,43,50 and could be similarly effective for treatment of infections with ganciclovir-resistant BV mutants. To test this hypothesis, mice were infected with a dose of a ganciclovir-resistant BV mutant equivalent to 10 or 100 LD50 of the parental wildtype virus (2 × 105 and 2 × 106 pfu, respectively). Mice infected with either dose of the ganciclovir-resistant mutant developed only mild neurologic signs when untreated or treated with vehicle (Figure 8 A through C). A similar level of neurologic involvement was evident in mice treated with a standard 7-d regimen of 5% ganciclovir. In contrast, treatment with a 3-d regimen of 0.5% cidofovir resulted in no signs of neurologic involvement. Skin lesions in mice treated with vehicle and ganciclovir were much more frequent and of greater severity than were observed in ganciclovir-treated mice infected with the parental wildtype virus (Figure 8 D through F). A significant percentage of vehicle-treated mice infected with 2 × 105 pfu and all mice infected with 2 × 106 pfu of the ganciclovir-resistant BV mutant developed skin lesions. Similar incidences of skin lesions of similar severity were apparent in mice treated with 5% ganciclovir. No mice infected with either wildtype or ganciclovir-resistant virus that were treated with a 3-d regimen of 0.5% cidofovir developed any skin lesions.
Figure 8.

Efficacy of ganciclovir and cidofovir against ganciclovir-resistant BV. Mice were infected with (A and D) 10 LD50 (2 × 105 pfu) of wildtype BV or (B and E) 2 × 105 pfu or (C and F) 2 × 106 pfu of a ganciclovir-resistant BV mutant. Mice were treated with vehicle only (circles with solid line), 5% ganciclovir (squares with dashed line), or 0.5% cidofovir (triangles with dotted line). The (A through C) group neurologic score and (D through F) percentage of mice showing skin lesions are shown.
Discussion
Given that the development of new drugs specifically directed against BV is unlikely, this study focused on assessing the efficacy of existing antiherpetic drugs when applied topically. It is well known that the in vitro efficacy of antiviral drugs does not always accurately reflect their efficacy in vivo. In vitro efficacy testing of several drugs administered systemically against BV found 5-iodo-2ʹ-deoxyuridine, penciclovir, 5-trifluoromethyl-2ʹ-deoxyuridine, and 5-ethyl-2ʹ-deoxyuridine to be about as effective as ganciclovir, with acyclovir and cidofovir being somewhat less effective.19 However, when this drug series was tested in vivo by using a mouse model, systemically administered cidofovir and ganciclovir were most effective.8 Even so, once BV had invaded the CNS, neither ganciclovir nor cidofovir was effective.8 Given the poor prognosis for BV infections once neurologic involvement is evident, preventing BV from ever getting into the nervous system is the most effective approach for dealing with potential BV exposures.
To test the potential of this approach, we used a mouse model involving infection with a lethal BV dose by scarification of the skin followed by topical application of drugs. By using 3% concentrations of various drugs utilized for treating HSV infections, topical efficacy closely mirrored that of their efficacy when administered parenterally.8 Notably, penciclovir was not at all effective, and acyclovir was only marginally protective, whereas ganciclovir and cidofovir provided the best protection. Previous studies in which drugs were administered systemically found cidofovir to be more effective than ganciclovir against BV.8 Given that ganciclovir is currently deemed the most effective drug against BV and is recommended for treatment of zoonotic BV infections,11,37 we further examined the comparative efficacy of cidofovir and ganciclovir.
Cidofovir is currently used in human patients to treat several infections but has not been used to treat BV infections. Cidofovir is a synthetic, acyclic monophosphate nucleotide analog of deoxycytidine.14,50 Cidofovir and ganciclovir compete with dCTP or dGTP, respectively, for viral DNA polymerase and are incorporated into replicating DNA strands. Once incorporated into the growing DNA strands, both drugs cause chain termination, preventing further DNA polymerization. However, unlike ganciclovir, cidofovir does not need to be phosphorylated to the monophosphate form by viral TK to have antiviral activity; rather, cidofovir is phosphorylated by cellular pyruvate kinases to its active diphosphate form. To date, both cidofovir and ganciclovir have primarily been used for the therapy of cytomegalovirus infections in immunocompromised patients. However, topical cidofovir has found some use in the treatment of epithelial HSV infections that are resistant to acyclovir.35,42,43
High doses of ganciclovir do prevent lethality when administered to BV-infected mice either parenterally or topically, but many to most surviving mice still develop symptoms of neurologic involvement and generate a strong adaptive antiBV immune response.8 Similarly, treatment of a case of symptomatic, zoonotic BV infection with ganciclovir failed to halt advancement of the infection.10 Taken together, these results indicate that ganciclovir does not completely suppress BV replication or prevent it from accessing the nervous system; instead, ganciclovir likely suppresses viral replication sufficiently to allow the host time to mount an adaptive immune response that eventually clears the infection. In contrast, topically applied cidofovir rapidly and very effectively terminates BV replication in the skin, thereby preventing the virus from ever entering the CNS. The termination of BV infection in the skin by cidofovir is so effective that it obviates the development of an adaptive immune response to the infection in most cases. Furthermore, cidofovir exhibited this high efficacy at a much lower concentration than ganciclovir (0.5% cidofovir compared with 5.0% ganciclovir, which was still not completely protective) and required a much shorter minimal treatment duration for maximal efficacy (1 d for cidofovir compared with 5 d for ganciclovir).
For treatment with either ganciclovir or cidofovir to be effective, therapy needed to be initiated very soon after infection. Once BV invaded the nervous system, neither ganciclovir nor cidofovir was effective at preventing lethality. As seen previously when drug was administered systemically, efficacy of both drugs dropped to near zero once virus was detectable in sensory ganglia serving the site of infection.11 This finding suggests that either these drugs do not efficiently enter the nervous system or that, once the virus is in the CNS, replication is so rapid and destructive that the infection cannot be overcome.
The high efficacy of cidofovir when applied topically (low concentration and rapidity of action) suggests that, with the PLO organogel vehicle, the drug readily penetrates the skin to effectively inhibit local viral replication in the dermis and epidermis, thereby preventing the virus from entering unmyelinated sensory nerve endings. Indeed, histopathology showed an absence of viral skin and DRG lesions and viral antigen in mice treated topically with 0.5% cidofovir. By comparison, ganciclovir either does not penetrate the skin as effectively as cidofovir or is not as effective at inhibiting viral replication as was cidofovir, because histopathology and immunohistochemistry revealed viral skin lesions and viral antigen in ganciclovir-treated mice. Given that neither DRG lesions nor viral antigen were found in mice treated with effective doses of either ganciclovir or cidofovir, both drugs were capable of limiting BV infection. Despite developing signs of neurologic involvement, ganciclovir-treated mice did survive the infection. In contrast, if neurologic signs developed in cidofovir-treated mice, with only rare exceptions, the infection inexorably progressed to death. This outcome suggests that ganciclovir is more effective than cidofovir in controlling the infection once it is established in the nervous system. This difference in efficacy against established BV infections was substantiated by the increased likelihood of survival of ganciclovir-treated mice in which topical administration was delayed until 24 h after inoculation with BV.
Regarding the comparison of the dose-response survival curves for ganciclovir and cidofovir, the major difference between these 2 drugs was in potency, with cidofovir being several orders of magnitude more potent than ganciclovir. In addition, there was more variability in the dose-response curves between experiments performed for ganciclovir than for cidofovir, although the mean curves of both followed similar shapes, as shown by the similar values for the Hill slope. The variability in ganciclovir experiments may have reflected the higher drug concentrations that needed to be dissolved in the PLO vehicle, resulting in more variable dispersion of ganciclovir throughout the vehicle.
The poor efficacy of topical acyclovir in our current study is consistent both with a previous study using parenteral drug delivery in mice, where it was completely ineffective,8 and in studies in rabbits where acyclovir efficacy was poor at best, despite the use of high drug concentrations and longer treatment regimens.4,51 There are several published cases of zoonotic BV infection where treatment with acyclovir failed to prevent advancement of the infection (that is, patients treated with acyclovir developed an overt or lethal infection).12,22 Together, these results raise concerns regarding the use of acyclovir for prophylactic treatment in humans after BV exposure incidents. If acyclovir has comparable in vivo efficacy against BV in humans as in mice and rabbits, its poor efficacy will allow BV to replicate sufficiently to invade and replicate within the CNS resulting in infections that are much more difficult to treat effectively. Similar concerns apply to penciclovir as well, given its poor efficacy in our current study and a previous in vivo study.8
Results with the ganciclovir-resistant mutant were somewhat unexpected, given that TK mutants of HSV can enter and replicate within the CNS.21 The ganciclovir-resistant BV mutant readily replicated in the epidermal and dermal layers of the skin, as evidenced through lesion development. However, only very mild, transient symptoms indicative of neurologic involvement were evident, even at a virus dose equivalent to 100 LD50 of the parental virus. Therefore, the ganciclovir-resistant BV TK mutant appears to lack the ability to invade and replicate within the CNS in mice. The viral TK phosphorylates thymidine (and to a lesser extent other nucleosides) to the monophosphate nucleotide form, which is an essential precursor for DNA replication. Because dNTPs are not present at high levels in quiescent cells, TK activity is particularly important if the virus is to replicate efficiently in nonreplicating cells, such as neurons. The inability of the ganciclovir-resistant TK mutant to replicate within the CNS raises the possibility that viral manipulation of cellular pools of DNA replication precursors could be a critical factor when infecting neurons and therefore could be an essential determinant of the neurovirulence of BV in nonmacaques.
All evidence to date indicates that once BV invades the CNS, even systemically administered drugs are not highly efficacious; consequently, prevention of BV movement into the CNS should be a primary goal of therapy. Although current recommendations mandate the use of acyclovir (or valacyclovir), penciclovir, or ganciclovir after a BV exposure incident, all experimental studies in animals and a few zoonotic case histories indicate that these drugs are not highly effective against BV. The high efficacy of topically applied cidofovir after superficial injury of the skin, the ease and brevity of treatment necessary for cidofovir efficacy, the lack of significant toxicity when cidofovir is applied topically, the efficacy of cidofovir in treating HSV infections in humans,42,43,50 and the availability of licensed cidofovir formulations all support the use of topical cidofovir as a routine prophylactic treatment immediately after a potential BV exposure incident, particularly in situations where the probability of BV transmission is increased.
Acknowledgments
Research reported in this publication was supported by the Office of the Director of the National Institutes of Health under award number R24OD022013 and by the American College of Laboratory Animal Medicine Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- 1.Almeida H, Amaral MH, Lobão P, Lobo JM. 2012. Pluronic F-127 and pluronic lecithin organogel (PLO): main features and their applications in topical and transdermal administration of drugs. J Pharm Pharm Sci 15:592–605. 10.18433/J3HW2B. [DOI] [PubMed] [Google Scholar]
- 2.Artenstein AW, Hicks CB, Goodwin BS, Jr, Hilliard JK. 1991. Human infection with B virus following a needlestick injury. Rev Infect Dis 13:288–291. 10.1093/clinids/13.2.288. [DOI] [PubMed] [Google Scholar]
- 3.Black DH, Maxwell LK, Eberle R. 2009. Characterization of a spontaneous drug-resistant mutant of monkey B virus (Macacine herpesvirus 1). Arch Virol 154:1495–1497. 10.1007/s00705-009-0452-3. [DOI] [PubMed] [Google Scholar]
- 4.Boulter EA, Thornton B, Bauer DJ, Bye A. 1980. Successful treatment of experimental B virus (Herpesvirus simiae) infection with acyclovir. Br Med J 280:681–683. 10.1136/bmj.280.6215.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boulter EA, Zwartouw HT, Thornton B. 1981. Postexposure immunoprophylaxis against B virus (Herpesvirus simiae) infection. Br Med J (Clin Res Ed) 283:1495–1497. 10.1136/bmj.283.6305.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breshears MA, Eberle R, Ritchey JW. 2001. Characterization of gross and histological lesions in Balb/c mice experimentally infected with herpesvirus saimiri 1 (HVS1). J Comp Pathol 125:25–33. 10.1053/jcpa.2001.0473. [DOI] [PubMed] [Google Scholar]
- 7.Breshears MA, Eberle R, Ritchey JW. 2005. Temporal progression of viral replication and gross and histological lesions in Balb/c mice inoculated epidermally with Saimiriine herpesvirus 1 (SaHV-1). J Comp Pathol 133:103–113. 10.1016/j.jcpa.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Brush LA, Black DH, McCormack KA, Maxwell LK, Wright G, Ritchey JW, Payton ME, Eberle R. 2014. Papiine herpesvirus 2 (HVP2) as a predictive model for drug sensitivity of Macacine herpesvirus 1 (BV). Comp Med 64:386–393. [PMC free article] [PubMed] [Google Scholar]
- 9.Bryan BL, Espana CD, Emmons RW, Vijayan N, Hoeprich PD. 1975. Recovery from encephalomyelitis caused by Herpesvirus simiae. Report of a case. Arch Intern Med 135:868–870. 10.1001/archinte.1975.00330060112017. [DOI] [PubMed] [Google Scholar]
- 10.Centers for Disease Control and Prevention (CDC). 1998. Fatal cercopithecine herpesvirus 1 (B virus) infection following a mucocutaneous exposure and interim recommendations for worker protection. MMWR Morb Mortal Wkly Rep 47:1073–1076, 1083. [PubMed] [Google Scholar]
- 11.Cohen JI, Davenport DS, Stewart JA, Deitchman S, Hilliard JK, Chapman LE, B Virus Working Group. 2002. Recommendations for prevention of and therapy for exposure to B virus (cercopithecine herpesvirus 1). Clin Infect Dis 35:1191–1203. 10.1086/344754. [DOI] [PubMed] [Google Scholar]
- 12.Davenport DS, Johnson DR, Holmes GP, Jewett DA, Ross SC, Hilliard JK. 1994. Diagnosis and management of human B virus (Herpesvims simiae) infections in Michigan. Clin Infect Dis 19:33–41. 10.1093/clinids/19.1.33. [DOI] [PubMed] [Google Scholar]
- 13.Davidson WL, Hummeler K. 1960. B virus infection in man. Ann N Y Acad Sci 85:970–979. [DOI] [PubMed] [Google Scholar]
- 14.De Clercq E. 2011. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates: the magic of the phosphonate bond. Biochem Pharmacol 82:99–109. 10.1016/j.bcp.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 15.Eberle R, Maxwell LK, Nicholson S, Black D, Jones-Engel L. 2017. Genome sequence variation among isolates of monkey B virus (Macacine alphaherpesvirus 1) from captive macaques. Virology 508:26–35. 10.1016/j.virol.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elmore D, Eberle R. 2008. Monkey B virus (Cercopithecine herpesvirus 1). Comp Med 58:11–21. [PMC free article] [PubMed] [Google Scholar]
- 17.Engel GA, Jones-Engel L, Schillaci MA, Suaryana KG, Putra A, Fuentes A, Henkel R. 2002. Human exposure to herpesvirus B-seropositive macaques, Bali, Indonesia. Emerg Infect Dis 8:789–795. 10.3201/eid0808.010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fierer J, Bazely P, Braude AI. 1973. Herpes B virus encephalomyelitis presenting as ophthalmic zoster. A possible latent infection reactivated. Ann Intern Med 79:225–228. 10.7326/0003-4819-79-2-225. [DOI] [PubMed] [Google Scholar]
- 19.Focher F, Lossani A, Verri A, Spadari S, Maioli A, Gambino JJ, Wright GE, Eberle R, Black DH, Medveczky P, Medveczky M, Shugar D. 2007. Sensitivity of monkey B virus (Cercopithecine herpesvirus 1) to antiviral drugs: role of thymidine kinase in antiviral activities of substrate analogs and acyclonucleosides. Antimicrob Agents Chemother 51:2028–2034. 10.1128/AAC.01284-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamuy R, Berman B. 1998. Treatment of herpes simplex virus infections with topical antiviral agents. Eur J Dermatol 8:310–319. [PubMed] [Google Scholar]
- 21.Harris W, Collins P, Fenton RJ, Snowden W, Sowa M, Darby G. 2003. Phenotypic and genotypic characterization of clinical isolates of herpes simplex virus resistant to aciclovir. J Gen Virol 84:1393–1401. 10.1099/vir.0.18880-0. [DOI] [PubMed] [Google Scholar]
- 22.Holmes GP, Hilliard JK, Klontz KC, Rupert AH, Schindler CM, Parrish E, Griffin DG, Ward GS, Bernstein ND, Bean TW, Ball MR, Sr, Brady JA, Wilder MH, Kaplan JE. 1990. B virus (Herpesvirus simiae) infection in humans: epidemiologic investigation of a cluster. Ann Intern Med 112:833–839. 10.7326/0003-4819-112-11-833. [DOI] [PubMed] [Google Scholar]
- 23.Huff JL, Eberle R, Capitano J, Zhou SS, Barry PA. 2003. Differential detection of B virus and rhesus cytomegalovirus in rhesus macaques. J Gen Virol 84:83–92. 10.1099/vir.0.18808-0. [DOI] [PubMed] [Google Scholar]
- 24.Huff JL, Barry PA. 2003. B-virus (Cercopithecine herpesvirus 1) infection in humans and macaques: potential for zoonotic disease. Emerg Infect Dis 9:246–250. 10.3201/eid0902.020272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones-Engel L, Engel GA, Heidrich J, Chalise M, Poudel N, Viscidi R, Barry PA, Allan JS, Grant R, Kyes R. 2006. Temple monkeys and health implications of commensalism, Kathmandu, Nepal. Emerg Infect Dis 12:900–906. 10.3201/eid1206.060030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramer T, Enquist LW. 2013. Directional spread of alphaherpesviruses in the nervous system. Viruses 5:678–707. 10.3390/v5020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krug PW, Schinazi RF, Hilliard JK. 2010. Inhibition of B virus (Macacine herpesvirus 1) by conventional and experimental antiviral compounds. Antimicrob Agents Chemother 54:452–459. 10.1128/AAC.01435-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobe DC, Spector T, Ellis MN. 1991. Synergistic topical therapy by acyclovir and A1110U for herpes simplex virus induced zosteriform rash in mice. Antiviral Res 15:87–100. 10.1016/0166-3542(91)90027-O. [DOI] [PubMed] [Google Scholar]
- 29.Nanda M, Curtin VT, Hilliard JK, Bernstein ND, Dix RD. 1990. Ocular histopathologic findings in a case of human herpes B virus infection. Arch Ophthalmol 108:713–716. 10.1001/archopht.1990.01070070099044. [DOI] [PubMed] [Google Scholar]
- 30.Ohsawa K, Black DH, Sato H, Rogers K, Eberle R. 2003. Sequence and genetic arrangement of the UL region of the monkey B virus (Cercopithecine herpesvirus 1) genome and comparison with the UL region of other primate herpesviruses. Arch Virol 148:989–997. 10.1007/s00705-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 31.Ohsawa K, Lehenbauer TW, Eberle R. 1999. Herpesvirus papio 2: a safer and sensitive alternative for serodiagnosis of B virus infection in macaque monkeys. Lab Anim Sci 49:605–616. [PubMed] [Google Scholar]
- 32.Palmer AE. 1987. Herpesvirus simiae: historical perspective. J Med Primatol 16:99–130. [PubMed] [Google Scholar]
- 33.Perelygina L, Zhu L, Zurkuhlen H, Mills R, Borodovsky M, Hilliard JK. 2003. Complete sequence and comparative analysis of the genome of herpes B virus (Cercopithecine herpesvirus 1) from a rhesus monkey. J Virol 77:6167–6177. 10.1128/JVI.77.11.6167-6177.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perrottet N, Decosterd LA, Meylan P, Pascual M, Biollaz J, Buclin T. 2009. Valganciclovir in adult solid organ transplant recipients: pharmacokinetic and pharmacodynamic characteristics and clinical interpretation of plasma concentration measurements. Clin Pharmacokinet 48:399–418. 10.2165/00003088-200948060-00006. [DOI] [PubMed] [Google Scholar]
- 35.Piret J, Boivin G. 2010. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother 55:459–472. 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rehman K, Zulfakar MH. 2014. Recent advances in gel technologies for topical and transdermal drug delivery. Drug Dev Ind Pharm 40:433–440. 10.3109/03639045.2013.828219. [DOI] [PubMed] [Google Scholar]
- 37.Remé T, Jentsch KD, Steinmann J, Kenner S, Straile U, Buse E, Sauerbrei A, Kaup FJ. 2009. Recommendation for post-exposure prophylaxis after potential exposure to herpes b virus in Germany. J Occup Med Toxicol 4:1–5. 10.1186/1745-6673-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ritchey JW, Payton ME, Eberle R. 2005. Clinicopathological characterization of monkey B virus (Cercopithecine herpesvirus 1) infection in mice. J Comp Pathol 132:202–217. 10.1016/j.jcpa.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Ritz N, Curtis N, Buttery J, Babl FE. 2009. Monkey bites in travelers: should we think of herpes B virus? Pediatr Emerg Care 25:529–531. 10.1097/PEC.0b013e3181b0a45c. [DOI] [PubMed] [Google Scholar]
- 40.Rogers KM, Deatheridge M, Breshears MA, Chapman S, Black D, Ritchey JW, Payton M, Eberle R. 2009. Type I IFN response to Papiine herpesvirus 2 (Herpesvirus papio 2; HVP2) determines neuropathogenicity in mice. Virology 386:280–289. 10.1016/j.virol.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogers KM, Ritchey JW, Payton M, Black DH, Eberle R. 2006. Neuropathogenesis of Herpesvirus papio 2 in mice parallels Cercopithecine herpesvirus 1 (B virus) infections in humans. J Gen Virol 87:267–276. 10.1099/vir.0.81476-0. [DOI] [PubMed] [Google Scholar]
- 42.Rosen T. 2017. Recurrent herpes labialis in adults: new tricks for an old dog. J Drugs Dermatol 16:s49–s53. [PubMed] [Google Scholar]
- 43.Sacks SL, Shafran SD, Diaz-Mitoma F, Trottier S, Sibbald RG, Hughes A, Safrin S, Rudy J, McGuire B, Jaffe HS. 1998. A multicenter phase I/II dose escalation study of single-dose cidofovir gel for treatment of recurrent genital herpes. Antimicrob Agents Chemother 42:2996–2999. 10.1128/AAC.42.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmid-Wendtner MH, Korting HC. 2004. Penciclovir cream—improved topical treatment for herpes simplex infections. Skin Pharmacol Physiol 17:214–218. 10.1159/000080214. [DOI] [PubMed] [Google Scholar]
- 45.Simon MA, Daniel MD, Lee-Parritz D, King NW, Ringler DJ. 1993. Disseminated B virus infection in a cynomolgus monkey. Lab Anim Sci 43:545–550. [PubMed] [Google Scholar]
- 46.Spector T, Averett DR, Nelson DJ, Lambe CU, Morrison RW, Jr, St Clair MH, Furman PA. 1985. Potentiation of antiherpetic activity of acyclovir by ribonucleotide reductase inhibition. Proc Natl Acad Sci USA 82:4254–4257. 10.1073/pnas.82.12.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spector T, Harrington JA, Morrison RW, Jr, Lambe CU, Nelson DJ, Averett DR, Biron K, Furman PA. 1989. 2-Acetylpyridine 5-[(dimethylamino)thiocarbonyl]-thiocarbonohydrazone (A1110U), a potent inactivator of ribonucleotide reductases of herpes simplex and varicella-zoster viruses and a potentiator of acyclovir. Proc Natl Acad Sci USA 86:1051–1055. 10.1073/pnas.86.3.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spector T, Lobe DC, Ellis MN, Blumenkopf TA, Szczech GM. 1992. Inactivators of herpes simplex virus ribonucleotide reductase: hematological profiles and in vivo potentiation of the antiviral activity of acyclovir. Antimicrob Agents Chemother 36:934–937. 10.1128/AAC.36.5.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weigler BJ. 1992. Biology of B virus in macaque and human hosts: a review. Clin Infect Dis 14:555–567. 10.1093/clinids/14.2.555. [DOI] [PubMed] [Google Scholar]
- 50.Zabawski EJ, Jr, Cockerell CJ. 1998. Topical and intralesional cidofovir: a review of pharmacology and therapeutic effects. J Am Acad Dermatol 39:741–745. 10.1016/S0190-9622(98)70046-5. [DOI] [PubMed] [Google Scholar]
- 51.Zwartouw HT, Humphreys CR, Collins P. 1989. Oral chemotherapy of fatal B virus (Herpesvirus simiae) infection. Antiviral Res 11:275–283. 10.1016/0166-3542(89)90037-5. [DOI] [PubMed] [Google Scholar]