

Abstract
Expansion of G4C2 repeats within the C9ORF72 gene is the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Such repeats lead to decreased expression of the autophagy regulator C9ORF72 protein. Furthermore, sense and antisense repeats are translated into toxic dipeptide repeat (DPR) proteins. It is unclear how these repeats are translated, and in which way their translation and the reduced expression of C9ORF72 modulate repeat toxicity. Here, we found that sense and antisense repeats are translated upon initiation at canonical AUG or near‐cognate start codons, resulting in polyGA‐, polyPG‐, and to a lesser degree polyGR‐DPR proteins. However, accumulation of these proteins is prevented by autophagy. Importantly, reduced C9ORF72 levels lead to suboptimal autophagy, thereby impairing clearance of DPR proteins and causing their toxic accumulation, ultimately resulting in neuronal cell death. Of clinical importance, pharmacological compounds activating autophagy can prevent neuronal cell death caused by DPR proteins accumulation. These results suggest the existence of a double‐hit pathogenic mechanism in ALS/FTD, whereby reduced expression of C9ORF72 synergizes with DPR protein accumulation and toxicity.
Keywords: amyotrophic lateral sclerosis, autophagy, C9ORF72, neurodegeneration, RAN translation
Subject Categories: Neuroscience, RNA Biology, Protein Biosynthesis & Quality Control
ALS‐linked sense and antisense repeats are translated into toxic peptides that require clearance by C9ORF72‐dependent autophagy.
Introduction
With one individual affected in ~50,000 people, amyotrophic lateral sclerosis (ALS) is the third most common neurodegenerative disease. ALS is characterized by degeneration of both upper and spinal motor neurons that leads to progressive skeletal muscle paralysis, ultimately resulting in death from respiratory failure generally 3–5 years after age of onset. ALS patients can present significant clinical, genetic, and histopathological overlaps with frontotemporal dementia (FTD), a presenile dementia affecting frontal and temporal brain regions (Lomen‐Hoerth et al, 2002; Ringholz et al, 2005; Neumann et al, 2006). Consequently, ALS and FTD are now considered as two extremes of a same disease continuum. This notion is emphasized by the identification of an expansion of GGGGCC (G4C2) repeats within the first intron of the C9ORF72 gene as the most common inherited cause for both ALS and FTD in Caucasian populations of northern Europe and North America (DeJesus‐Hernandez et al, 2011; Renton et al, 2011; Gijselinck et al, 2012; Majounie et al, 2012).
Expanded G4C2 repeats are pathogenic through three main non‐exclusive mechanisms. First, transcripts containing expanded repeats accumulate in RNA foci that recruit various RNA‐binding proteins, potentially altering their localization and functions (Almeida et al, 2013; Donnelly et al, 2013; Lagier‐Tourenne et al, 2013; Lee et al, 2013; Mizielinska et al, 2013; Cooper‐Knock et al, 2014; Haeusler et al, 2014; Conlon et al, 2016, 2018). Second, sense G4C2 and antisense C4G2 expanded repeats are repeat‐associated non‐AUG (RAN) translated into dipeptide repeat (DPR)‐containing proteins, which form inclusions throughout the brain of patients with C9‐ALS/FTD (Ash et al, 2013; Gendron et al, 2013; Mori et al, 2013; Zu et al, 2013), as well as in mice expressing expanded G4C2 repeats (Chew et al, 2015; O'Rourke et al, 2016; Peters et al, 2015; Jiang et al, 2016; Liu et al, 2016). Overexpression of DPR proteins using artificial ATG codons for their translation initiation leads to neurodegeneration in cell and animal models, notably through alteration of mitochondria (Dafinca et al, 2016; Lopez‐Gonzalez et al, 2016; Choi et al, 2019), DNA repair (Walker et al, 2017; Lopez‐Gonzalez et al, 2019), nuclear and nucleolar organization (White et al, 2019; Zhang et al, 2019), and/or nucleocytoplasmic transport (Kwon et al, 2014; May et al, 2014; Mizielinska et al, 2014; Wen et al, 2014; Zhang et al, 2015, 2016; Freibaum et al, 2015; Jovičić et al, 2015; Tao et al, 2015; Boeynaems et al, 2016; Khosravi et al, 2017). Third, expanded G4C2 repeats promote DNA epigenetic changes that lead to decreased expression of C9ORF72 mRNA and protein levels in C9‐ALS/FTD individuals (DeJesus‐Hernandez et al, 2011; Gijselinck et al, 2012; Almeida et al, 2013; Waite et al, 2014; van Blitterswijk et al, 2015; Xiao et al, 2015; Saberi et al, 2017; Frick et al, 2018; Viodé et al, 2018). Interestingly, knockdown of C9ORF72 expression in zebrafish or in human motor neuron differentiated from iPS cells leads to some neuronal cell dysfunctions and cell death (Ciura et al, 2013; Shi et al, 2018). These results suggest that haploinsufficiency of C9ORF72 may play a role in the pathogenesis of ALS/FTD. However, the absence of neurodegeneration in mice knockout for C9orf72 argues against a sole loss of function of C9ORF72 as a cause of ALS/FTD (Lagier‐Tourenne et al, 2013; Koppers et al, 2015; O'Rourke et al, 2016; Atanasio et al, 2016; Burberry et al, 2016; Jiang et al, 2016; Sudria‐Lopez et al, 2016). Hence, the pathological consequences of C9ORF72 reduced expression in ALS/FTD remain to be clarified. Similarly, by which molecular mechanisms sense G4C2 and antisense C4G2 repeats are translated, and how such translation modulates repeat toxicity are key points that remain to be investigated.
Using constructs in which expanded repeats were embedded within the natural C9ORF72 human sequence, we found that the G4C2 sense repeats are mostly translated into poly(glycine–alanine) (polyGA) and to a lesser extent into poly(glycine–arginine) (polyGR)‐containing proteins through initiation to near‐cognate start codons that are located upstream of the G4C2 repeats. Near‐cognate initiation codons are codons differing from the AUG sequence by one base (CUG, GUG, UUG, ACG, AAG, etc.), but that can still initiate translation provided they are embedded in a correct Kozak consensus sequence (Kozak, 1989; Peabody, 1989). Moreover, we found that the antisense C9ORF72 transcript containing expanded C4G2 repeats is mostly translated into a poly(proline–glycine) (polyPG)‐containing protein using a canonical AUG start codon embedded in a poor Kozak consensus sequence. As a result of their non‐optimal translation initiation, DPR proteins are expressed at low levels and consequently present limited toxicity in neuronal cells. Furthermore, accumulation of DPR proteins is prevented by their constant degradation through autophagy. As the C9ORF72 protein regulates autophagy (Amick et al, 2016; Sellier et al, 2016; Sullivan et al, 2016; Ugolino et al, 2016; Webster et al, 2016; Xiao et al, 2016; Yang et al, 2016; Jung et al, 2017; Chitiprolu et al, 2018; Ho et al, 2019), we tested whether C9ORF72 modulates DPR expression. Importantly, reduced expression of C9ORF72 increases the accumulation and toxicity of DPR proteins expressed under their natural sequences. Finally, we screened various pharmacological compounds known to activate autophagy and found that mTOR inhibitors and some phenothiazine derivatives, which are clinically approved antipsychotic drugs known to activate autophagy in neuronal cell culture (Tsvetkov et al, 2010), prevent the neuronal cell death caused by the synergic toxicity of DPR protein accumulation upon C9ORF72 reduced expression.
In conclusion, these results suggest an alternative molecular mechanism to RAN translation that may explain production of DPR proteins from C9ORF72 sense and antisense repeats. Furthermore, these data support a double‐hit mechanism in ALS/FTD, where the reduced expression of C9ORF72 synergizes the accumulation and toxicity of DPR proteins.
Results
Expanded sense G4C2 repeats are translated into polyGA and polyGR
To assess translation of the sense GGGGCC repeats, we cloned 80 G4C2 repeats embedded within the natural human C9ORF72 sequence and fused these repeats to the GFP sequence in all three possible frames (Fig EV1A). These frames were named according to the DPR protein potentially encoded by the expanded G4C2 repeats, namely glycine–alanine (GA), glycine–proline (GP), and glycine–arginine (GR). Cell transfection and immunoblotting against the GFP indicated that expanded G4C2 repeats embedded in the natural C9ORF72 sequence are predominantly translated into the GA frame and with a much lower efficiency in the GR frame (Fig 1A). In contrast, we found no or very limited translation of the G4C2 repeats into the GP frame. Direct observation of the GFP fluorescence confirmed marginal levels of polyPG (Fig EV1B), excluding a potential bias of aggregation and insolubility that would impair detection of this protein by immunoblotting. Furthermore, RT–qPCR quantification indicates similar levels of RNA expression for polyGA, polyGP, and polyGR constructs, indicating that the absence of polyGP at the protein level is not caused by a lack of its expression at the RNA level (Fig EV1C). As a further control, cell transfections of GFP‐tagged polyGA, polyGP, and polyGR expressed under artificial ATG start codons demonstrate expression of all three DPR proteins by fluorescence (Fig EV1D) and immunoblot analyses (Fig EV1E). Expression of polyGP into a stable protein when using an artificial ATG start codon suggests that its absence when using the natural C9ORF72 sequence is not due to a bias of insolubility and/or rapid degradation of this protein.
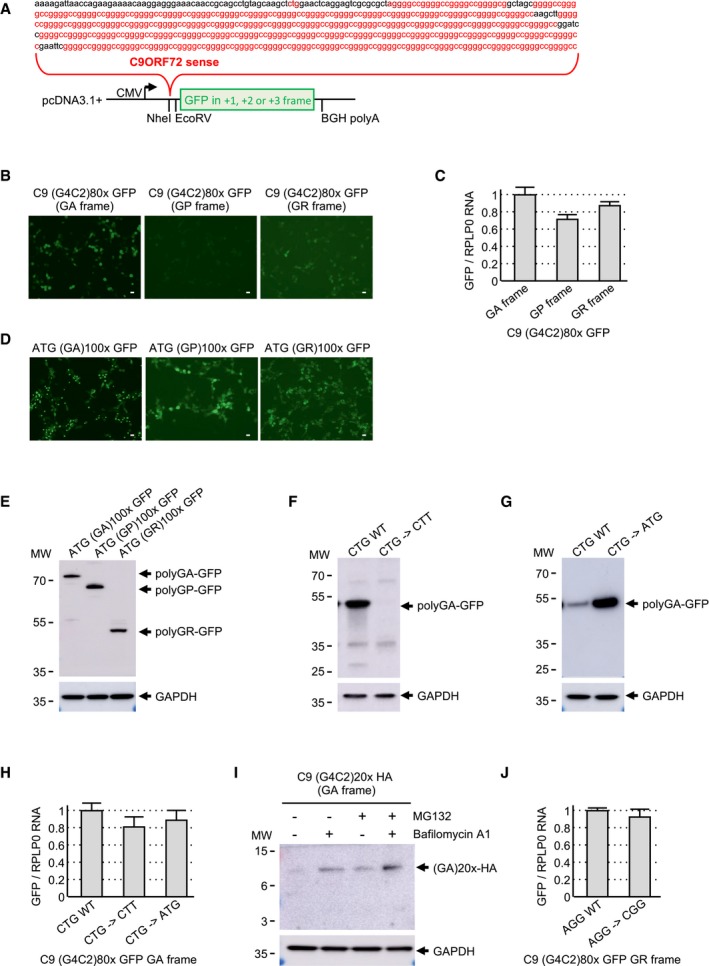
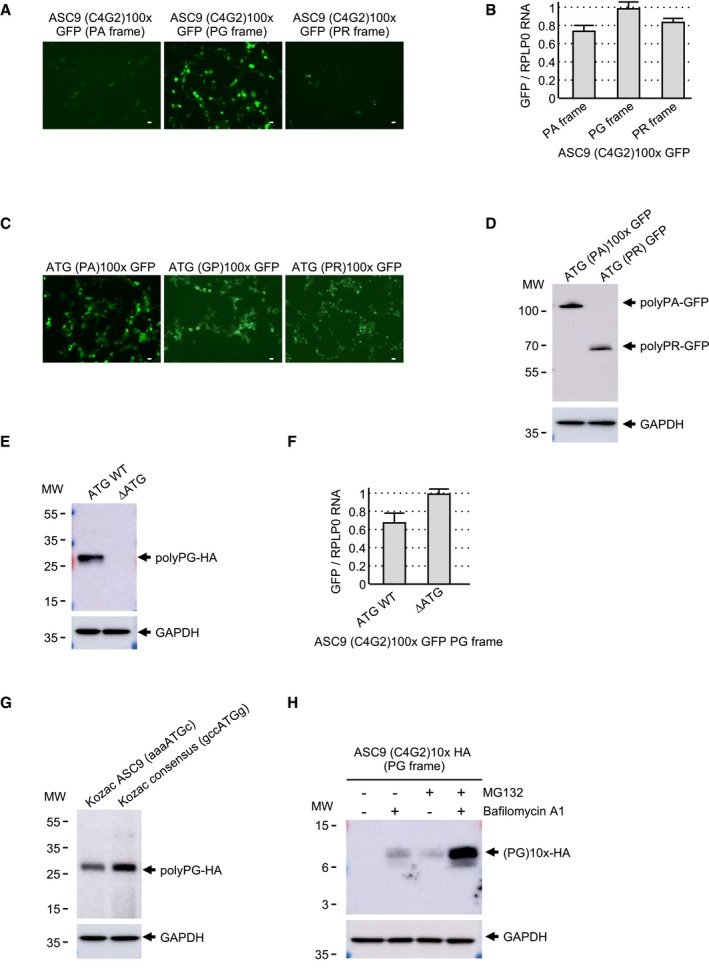
Figure EV1. Expanded G4C2 repeats are translated into polyGA .
-
AScheme and sequence of the human C9ORF72 sense transcript with 80 G4C2 repeats fused to the eGFP in the three possible frames and cloned into the pcDNA3.1 plasmid.
-
B, CFluorescence (B) and RT–qPCR (C) GFP analyses of HEK293 cells transfected for 24 h with 80 G4C2 repeats embedded in the human sense C9ORF72 sequence and fused in all three possible frames with the GFP deleted of its ATG.
-
D, EGFP expression (D) and immunoblotting (E) analysis of HEK293 cells transfected for 24 h with constructs expressing either polyGA, polyGP, or polyGR expressed under an artificial ATG start codon and fused to the GFP.
-
FImmunoblotting against the GFP or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with either a wild‐type or a mutant (CTG into CTT) construct containing 80 G4C2 repeats embedded in sense C9ORF72 fused to the GFP in the GA frame.
-
GImmunoblotting against the GFP or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with either a wild‐type or a mutant (CTG into ATG) construct containing 80 G4C2 repeats embedded in sense C9ORF72 fused to the GFP in the GA frame.
-
HRT–qPCR analysis of GFP expression of HEK293 cells transfected for 24 h with either wild‐type or mutant (CTG into CTT or ATG) constructs containing 80 G4C2 repeats embedded in sense C9ORF72 fused to the GFP in the GA frame.
-
IImmunoblotting against the HA‐tag or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with 20 G4C2 repeats embedded in the human sense C9ORF72 sequence fused to a HA‐tag in the GA frame and treated or not with MG132 and/or bafilomycin A1 for 15 h.
-
JRT–qPCR analysis of GFP expression of HEK293 cells transfected for 24 h with either wild‐type or mutant (AGG into CGG) constructs containing 80 G4C2 repeats embedded in sense C9ORF72 fused to the GFP in the GR frame.
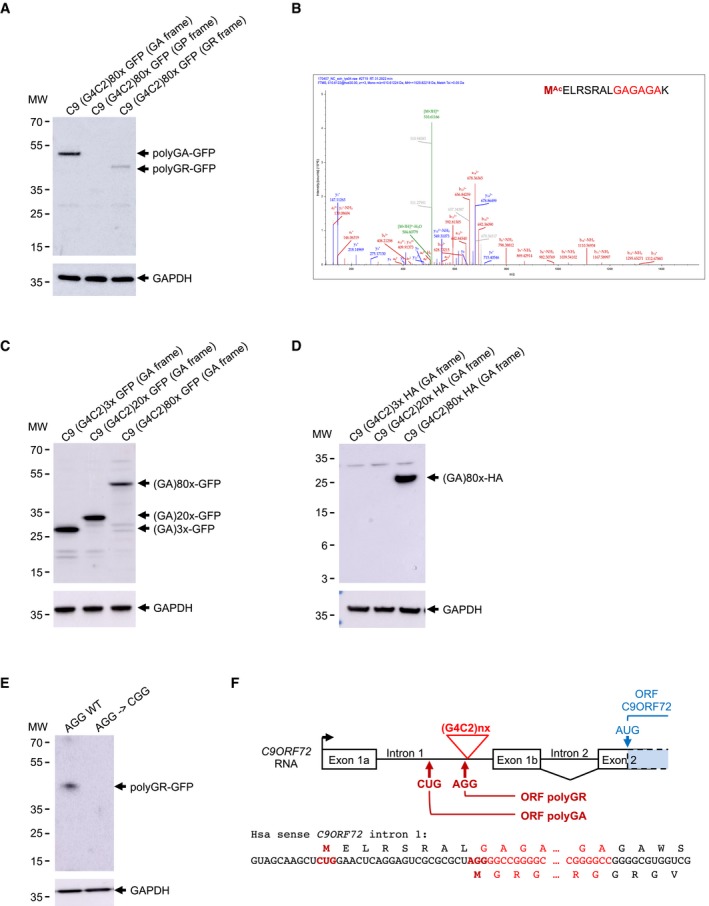
Figure 1. Expanded G4C2 repeats are translated into polyGA and polyGR .
-
AImmunoblotting against the GFP or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with 80 G4C2 repeats embedded in the human sense C9ORF72 and fused in all three possible frames with the GFP deleted of its ATG.
-
BLC‐MS/MS spectra of the N‐terminal part of the GFP‐immunoprecipitated and LysC‐digested polyGA protein expressed as in (A).
-
C, DImmunoblotting against the GFP‐ (C) or the HA‐tag (D) or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with 3, 20 or 80 G4C2 repeats embedded in the human sense C9ORF72 sequence fused to the GFP in the GA frame.
-
EImmunoblotting against the GFP or the GAPDH of proteins extracted from HEK293 cells transfected with either a wild‐type or a mutant (AGG into CGG) construct containing 80 G4C2 repeats embedded in sense C9ORF72 fused to the GFP in the GR frame.
-
FScheme of human C9ORF72 sense transcript with intron 1 retained. Expanded G4C2 repeats, CUG and AGG near‐cognate initiation codons, and polyGA and polyGR ORFs are indicated in red. C9ORF72 ORF is indicated in blue.
Next, we characterized the translation initiation sites of the polyGA and polyGR proteins. Expanded G4C2 repeats embedded in their natural C9ORF72 sequence were modified so that a lysine, target of the LysC enzyme, is present downstream of three G4C2 repeats in the GA frame. Immunoprecipitation of the polyGA DPR protein followed by LysC digestion and LC‐MS/MS analysis revealed a translation initiation to a CUG near‐cognate codon embedded in a correct Kozak consensus sequence and located 24 nucleotides upstream of the G4C2 repeats (Fig 1B). This CUG start site of polyGA is identical to the one found in three other independent studies (Green et al, 2017; Sonobe et al, 2018; Tabet et al, 2018). Mass spectrometry indicates that the initial amino acid of the polyGA DPR protein is a methionine, suggesting that the CUG codon is decoded by an initiator Met‐tRNA despite imperfect match. Mutation of this CTG codon into CTT abolished polyGA DPR protein expression (Fig EV1F), confirming the importance of this near‐cognate codon for translation of the G4C2 repeats in the GA frame. In contrast, mutation of the CTG codon into a canonical ATG start codon enhanced by fivefold to 10‐fold expression of polyGA (Fig EV1G), highlighting the weak translation initiation efficiency of the natural C9ORF72 sequence. As a control, RT–qPCR quantification shows that mutation of the CTG start codon does not modify polyGA expression at the RNA level (Fig EV1H), Next, cell transfection of various lengths of G4C2 repeats (3×, 20×, and 80×) fused to the GFP in the GA frame indicated that translation occurs with expanded G4C2 repeats of any lengths (Fig 1C). In contrast, fusion of G4C2 repeats to a smaller HA‐tag (9 amino acids, ~1 kDa) in the GA frame resulted in detection of polyGA with expanded (80×) G4C2 repeats, but only trace of proteins with 3 or 20 G4C2 repeats (Fig 1D). This is likely caused by the rapid degradation of these small proteins, as inhibition of the proteasome and autophagy degradation pathways allowed detection of the polyGA protein with 20 G4C2 repeats (Fig EV1I). These results suggest that translation initiation occurs at a CUG near‐cognate codon in the GA frame independently of the length of the G4C2 repeats. However, in absence of an expansion, the resulting protein is small and unstable, impairing its detection in normal conditions. In contrast, repeat expansion or fusion of the repeat to a large tag, such as the GFP protein, results in expression of a stable and detectable polyGA DPR protein.
Translation initiation of polyGA to a near‐cognate start codon questions whether a similar mechanism may occur for polyGP and polyGR. However, in the GP frame a stop codon is located just before the G4C2 repeats, preventing expression of a polyGP protein from any upstream putative near‐cognate initiation codons. Concerning the polyGR DPR protein, mass spectrometry failed to identify its N‐terminus, but its expression is likely to be dependent of a near‐cognate initiation mechanism as mutation of an AGG near‐cognate codon located just upstream of the G4C2 repeats and in the GR frame impairs expression of this protein (Fig 1E). As a control, mutation of this AGG codon does not affect RNA expression (Fig EV1J). This AGG codon is located just before the repeats, leading to a methionine adjacent to the GR repeats, which may explain why we failed to identify it by mass spectrometry. Of interest, translation initiation at this AGG near‐cognate codon is weak and results in limited expression of polyGR compared to polyGA (Fig 1A). To summarize this first part, translation initiation at either a CUG or an AGG near‐cognate codon is predicted to result in a polyGA or a polyGR DPR protein, which is composed of a short N‐terminus of either 8 or 1 amino acids, followed by a central repeated glycine–alanine or glycine–arginine stretch which length corresponds to the number of expanded G4C2 repeats and a C‐terminus ending in either C9ORF72 intron 2 or exon 2 according to splicing or retention of intron 2 (Fig 1F).
Expanded antisense C4G2 repeats are translated into polyPG
The C9ORF72 gene is transcribed in both sense and antisense directions. To assess the translation of the antisense CCCCGG repeats, we cloned one hundred C4G2 repeats, embedded within the natural human C9ORF72 antisense sequence, and fused to a HA‐tag in all three possible frames, namely proline–alanine (PA), proline–glycine (PG), and proline–arginine (PR). Cell transfection and immunoblotting against the HA‐tag indicated that expanded C4G2 repeats embedded in the antisense C9ORF72 sequence are predominantly translated into the PG frame (Fig 2A). Fusion of the C4G2 repeats in all three frames to the GFP followed by cell transfection and direct observation of the GFP fluorescence confirmed that translation occurs mostly in the PG frame (Fig EV2A). As a control, RT–qPCR analysis indicates similar RNA levels of polyPA, polyPG, and polyPR (Fig EV2B). Furthermore, polyPA and polyPR proteins are correctly expressed when cloned downstream of artificial ATG start codons (Fig EV2C and D), excluding that the non‐detection of these proteins is caused by a potential bias of protein insolubility and/or instability.
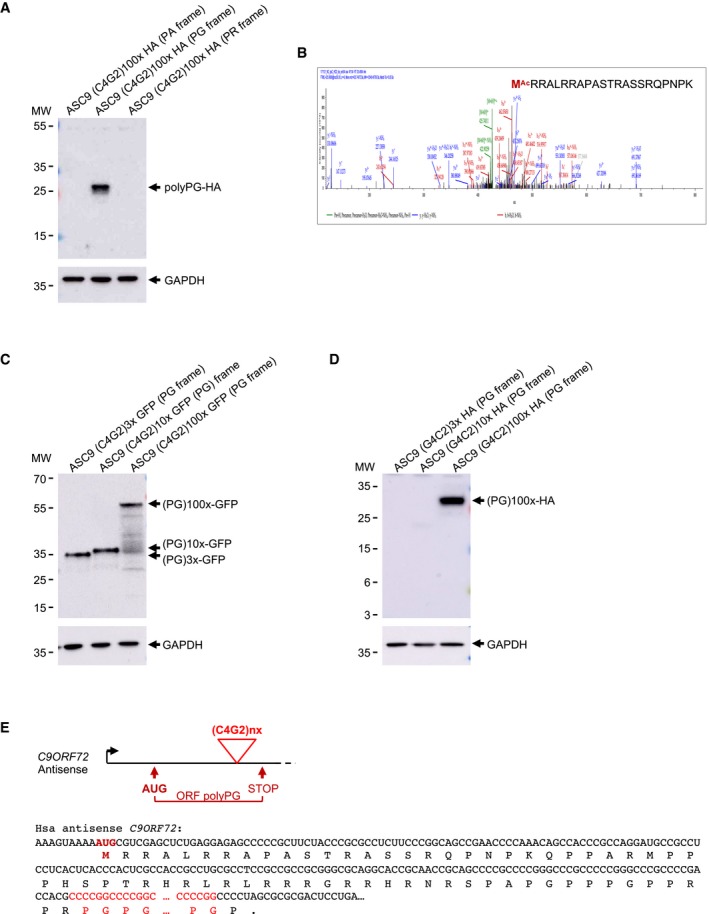
Figure 2. Expanded C4G2 repeats are translated into polyPG .
-
AImmunoblotting against the HA‐tag or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with 100 C4G2 repeats embedded in the human antisense C9ORF72 and fused in all three possible frames with a HA‐tag.
-
BLC‐MS/MS spectra of the N‐terminal part of the HA‐immunoprecipitated and LysC‐digested polyPG protein expressed as in (A).
-
C, DImmunoblotting against the GFP‐ (C) or the HA‐tag (D) or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with 3, 10, or 100 C4G2 repeats embedded in the human antisense C9ORF72 sequence fused to the GFP in the PG frame.
-
EScheme of human antisense C9ORF72 transcript. Expanded C4G2 repeats, AUG initiation codon, and polyPG ORF are indicated in red.
Figure EV2. Expanded C4G2 repeats are translated into polyPG .
-
A, BFluorescence (A) and RT–qPCR (B) GFP analyses of HEK293 cells transfected for 24 h with 100 C4G2 repeats embedded in the human antisense C9ORF72 sequence and fused in all three possible frames with the GFP deleted of its ATG.
-
C, DGFP expression (C) and immunoblotting (D) analysis of HEK293 cells transfected for 24 h with constructs expressing either polyPA or polyPR expressed under an artificial ATG start codon and fused to the GFP. Fluorescence of ATG‐polyGP from Fig EV1D is shown as control.
-
EImmunoblotting against HA or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with either a wild‐type or a mutant (∆ATG) construct containing 100 C4G2 repeats embedded in the antisense C9ORF72 sequence fused to a HA‐tag in the PG frame.
-
FRT–qPCR GFP expression analysis of HEK293 cells transfected for 24 h with either a wild‐type or a mutant (∆ATG) construct containing 100 C4G2 repeats embedded in the antisense C9ORF72 sequence fused to the GFP in the PG frame.
-
GImmunoblotting against the HA or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with either a wild‐type or a Kozak consensus mutant construct containing 100 C4G2 repeats embedded in antisense C9ORF72 fused to the HA‐tag in the PG frame.
-
HImmunoblotting against the HA‐tag or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with 10 C4G2 repeats embedded in the human antisense C9ORF72 sequence fused to a HA‐tag in the PG frame and treated or not with MG132 and/or bafilomycin A1 for 15 h.
As C4G2 repeats appear to be translated mostly into polyPG, we next investigated the translation initiation mechanism of this protein. HA immunoprecipitation followed by LysC digestion and LC‐MS/MS analysis revealed translation initiation to a canonical AUG start codon located 195 nucleotides upstream of the repeats (Fig 2B). Deletion of the C9ORF72 antisense sequence containing this initiation codon abolished polyPG expression (Fig EV2E), demonstrating the importance of this initiation sequence for polyPG translation. As a control, RT–qPCR indicated that deletion of the sequence containing this ATG does not alter RNA expression (Fig EV2F). Of interest, this AUG start codon is located in a poor Kozak sequence and its mutation into a consensus Kozak sequence enhanced expression of the polyPG protein by threefold to fivefold (Fig EV2G). Consistent with translation starting before the repeats, expression of polyPG fused to the GFP is independent of the C4G2 expansion size (Fig 2C). In contrast, fusion of expanded (100×) or non‐expanded (10×) C4G2 repeats to the small HA‐tag resulted in a detectable polyPG protein only with 100 G4C2 repeats (Fig 2D). The absence of detection of the small polyPG protein with 10 C4G2 repeats fused to the HA‐tag is likely due to its instability and rapid turnover as inhibition of the proteasome and autophagy degradation pathways allowed its accumulation and detection (Fig EV2H). These data indicate that the polyPG DPR protein is translated through initiation to a canonical AUG start codon, independently of the repeats. However, in the absence of a C4G2 expansion, the resulting polyPG protein is small and likely too unstable to be significantly observed in control conditions. In contrast, either expansion of the repeats or fusion to the GFP protein increases the size and the stability of this protein, enabling its detection. In summary, antisense C4G2 repeats are translated into a polyPG protein, which is predicted to be composed of a N‐terminus of 65 amino acids rich in proline and arginine, a central repeated proline–glycine stretch which length corresponds to the number of expanded C4G2 repeats and a stop codon located just after the repeats (Fig 2E).
Decreased expression of C9ORF72 synergizes DPR protein toxicity
Next, we tested the toxicity of the polyGA, polyPG, and polyGR DPR proteins expressed under their natural sense or antisense C9ORF72 human sequences. Consistent with their low expression, their transfection into neuronal cells resulted in low toxicity with only 10–20% of neuronal cell death (Fig 3A). This toxicity is likely due to translation of the repeats into DPR proteins and not caused by G4C2 or C2G4 RNA toxicity, as mutation or deletion of DPR protein initiation codons (CTG for polyGA, AGG for polyGR, and ATG for polyPG) resulted in negligible cell death (Fig 3A). As positive controls, transfection of polyGA, polyPG, and polyGR cloned downstream of canonical ATG start codons embedded in consensus Kozak sequences (gccATGg) resulted in higher DPR protein expression (Figs EV1C and EV2C) and consequently in higher neuronal cell toxicities, especially for polyGR (Fig 3A). These results are similar to the toxicity reported for DPR proteins driven by artificial ATG start codons and where polyGR are more toxic than polyGA or polyGP (May et al, 2014; Mizielinska et al, 2014; Yamakawa et al, 2015; Lopez‐Gonzalez et al, 2016, 2019; Schludi et al, 2017; Zhang et al, 2018; Choi et al, 2019). DPR proteins form protein aggregates that are degraded by autophagy (Cristofani et al, 2018). Indeed, bafilomycin A1‐mediated inhibition of autophagy enhances the accumulation of DPR proteins (Figs 3C and D, and EV3A and B). As the C9ORF72 protein regulates autophagy and as C9ORF72 protein expression is reduced in neuronal tissue from C9‐positive ALS/FTD individuals, we tested whether decreased expression of C9ORF72 may modulate the expression and toxicity of DPR proteins. Immunoblotting indicated that siRNA‐mediated depletion of C9ORF72 expression leads to an accumulation of polyGA, polyPG, and polyGR DPR proteins expressed under their natural start codons (Fig 3B–D). However, DPR accumulation upon partial decreased expression of C9ORF72 was milder compared to the complete block of autophagy induced by bafilomycin treatment (Fig 3C and D). These results are consistent with C9ORF72 decreased expression causing only a partial alteration of autophagy (Sellier et al, 2016). In that aspect, siRNA‐mediated depletion of C9ORF72 impaired the recruitment of the P62/SQSTM1 autophagy adaptor protein to polyGA aggregates (Fig EV3C). As a control, decreased expression of C9ORF72 does not modify co‐localization of ubiquitin with polyGA inclusions (Fig EV3D). These data suggest that C9ORF72 plays an early role in autophagy at the step of autophagosome formation. Furthermore, siRNA‐mediated depletion of C9ORF72 leads to increased lysosomal labeling (Fig EV3E). As a control, endosome labeling is not altered by decreased expression of C9ORF72 (Fig EV3F). These observations are consistent with recent reports showing a role of the C9ORF72/SMCR8 complex at lysosomes (Amick et al, 2016; Sullivan et al, 2016; Corrionero & Horvitz, 2018; Lan et al, 2019), and suggest a complex function of C9ORF72 both at early and at late steps of the autophagic process. As decreased expression of C9ORF72 promotes DPR protein accumulation, we then tested whether C9ORF72 may modulate DPR toxicity. Importantly, reduced expression of C9ORF72 substantially increased the neuronal cell death induced by polyGA, polyPG, and polyGR DPR proteins expressed under their natural start codons (Fig 3E). Of technical interest, toxicity induced by expression of polyGA, polyPG, and polyGR proteins expressed under artificial ATG start codons was less sensitive to C9ORF72 loss, probably due to the already high expression and toxicity of these ATG‐driven DPR proteins (Fig 3E). As negative controls, no toxicity was observed when the number of G4C2 repeats was reduced to control size or when DPR protein start codons (CUG for polyGA, AGG for polyGR, or AUG for polyPG) were deleted (Fig 3E). These controls indicate that G4C2 and C4G2 expanded repeats are likely not overly toxic at the RNA level. Furthermore, these results suggest a double‐hit mechanism where the decreased expression of C9ORF72 synergizes the toxicity of DPR proteins expressed under their natural start codons.
Figure 3. Decreased expression of C9ORF72 synergizes DPR protein toxicity.
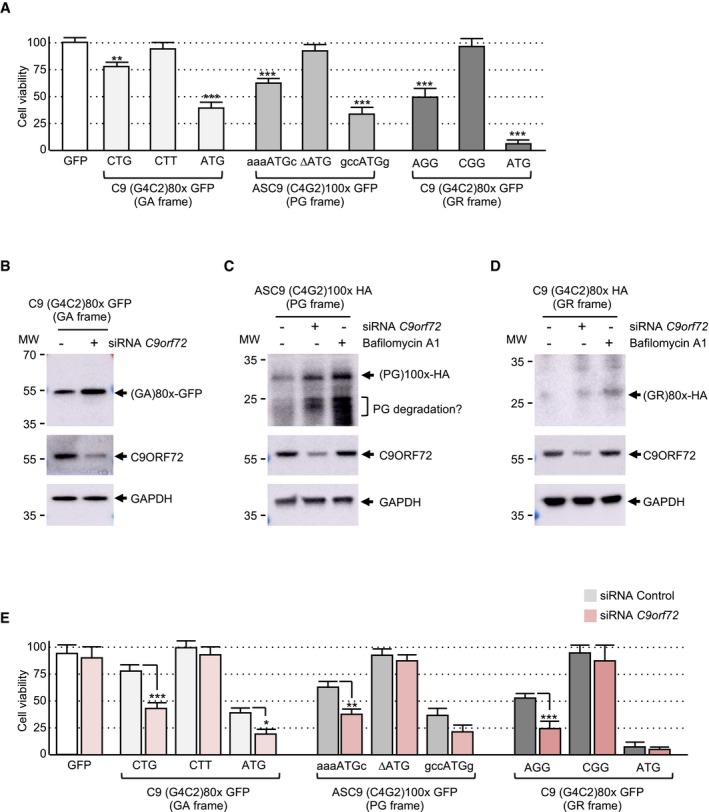
- Cell viability (TO‐PRO‐3 FACS staining) of GT1‐7 neuronal cells transfected for 24 h with either wild‐type or indicated mutant constructs containing either 80 G4C2 repeats or 100 C4G2 repeats embedded in sense or antisense C9ORF72 fused to the GFP in the GA, PG, or GR frame.
- Immunoblotting against the GFP, endogenous C9ORF72, or the GAPDH of proteins extracted from Neuro2A cells transfected for 24 h with 80 G4C2 repeats embedded in the human sense C9ORF72 sequence fused to the GFP in the GA frame and with either a control siRNA or a siRNA targeting C9orf72 mRNA.
- Immunoblotting against the HA‐tag, endogenous C9ORF72, or GAPDH of proteins extracted from Neuro2A cells transfected for 24 h with 100 C4G2 repeats embedded in the human antisense C9ORF72 sequence fused to a HA‐tag in the PG frame and with either a control siRNA or a siRNA targeting C9orf72 mRNA or treated with bafilomycin A1 for 15 h.
- Immunoblotting against the HA‐tag, endogenous C9ORF72, or GAPDH of proteins extracted from Neuro2A cells transfected for 24 h with 80 G4C2 repeats embedded in the human sense C9ORF72 sequence fused to a HA‐tag in the GR frame and with either a control siRNA or a siRNA targeting C9orf72 mRNA or treated with bafilomycin A1 for 15 h.
- Cell viability (TO‐PRO‐3 FACS staining) of GT1‐7 neuronal cells co‐transfected for 24 h with either a control siRNA or a siRNA targeting C9orf72 mRNA and wild‐type or mutant constructs containing either 80 G4C2 repeats or 100 C4G2 repeats embedded in sense or antisense C9ORF72 fused to the GFP in the GA, PG, or GR frame.
Figure EV3. Decreased expression of C9ORF72 synergizes DPR toxicity.
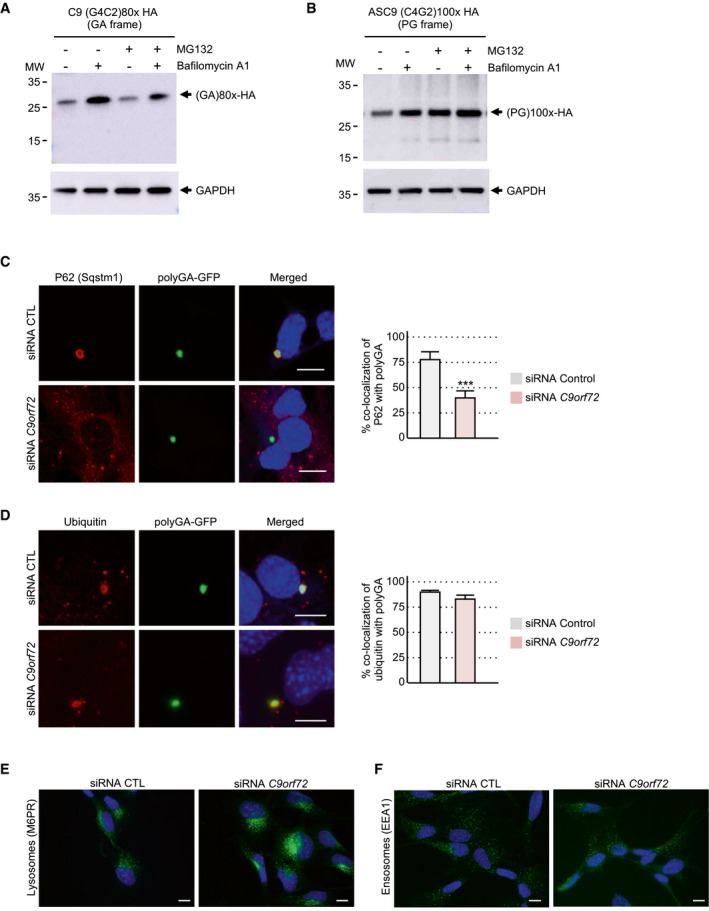
-
AImmunoblotting against the HA‐tag or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with 80 G4C2 repeats embedded in the human sense C9ORF72 sequence fused to a HA‐tag in the GA frame and treated or not with MG132 and/or bafilomycin A1.
-
BAs in (A) but with cells transfected with 100 C4G2 repeats embedded in the human antisense C9ORF72 sequence fused to a HA‐tag in the PG frame.
-
C, DLeft panel, representative images of immunofluorescence labeling of endogenous P62/SQSTM1 (C) or ubiquitin (D) and the GFP in GT1‐7 neuronal cells co‐transfected for 24 h with either a control siRNA or a siRNA targeting C9orf72 mRNA and a construct containing 80 G4C2 repeats embedded in sense C9ORF72 fused to the GFP in the GA frame. Right panel, quantification of the percent of co‐localization of P62 or ubiquitin with polyGA aggregates.
-
EImmunofluorescence labeling of M6PR in GT1‐7 neuronal cells transfected for 24 h with either a control siRNA or a siRNA targeting C9orf72 mRNA.
-
FImmunofluorescence labeling of EEA1 in GT1‐7 neuronal cells transfected for 24 h with either a control siRNA or a siRNA targeting C9orf72 mRNA.
Drugs activating autophagy reduce DPR protein accumulation and toxicity
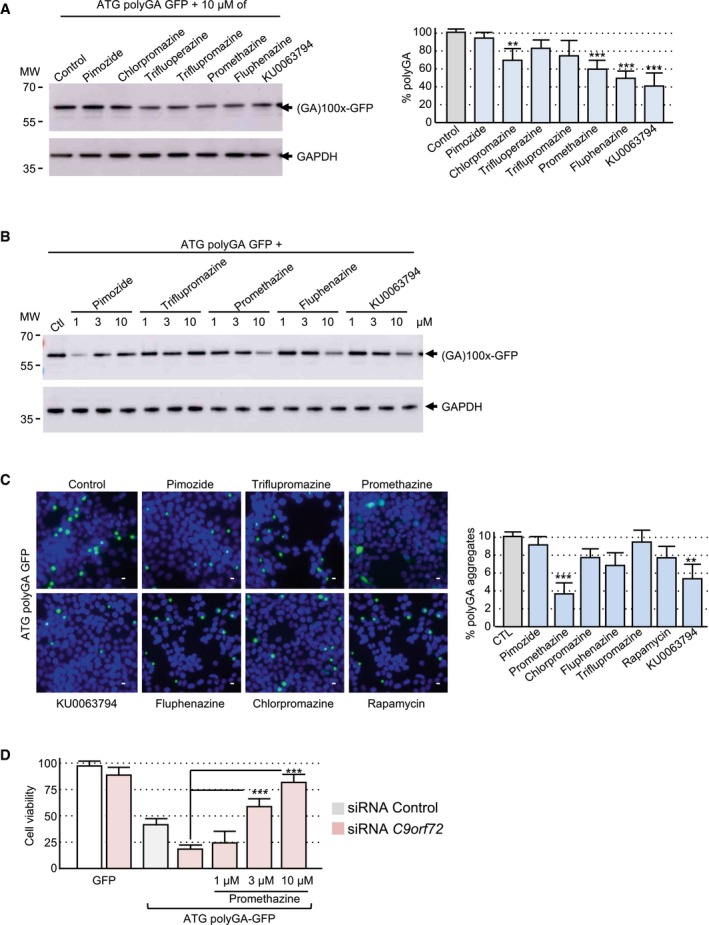
Autophagy is a highly regulated cellular process amendable to drug treatment (Rubinsztein et al, 2012; Galluzzi et al, 2017). Thus, we tested whether drugs known to activate autophagy may bypass C9ORF72 decreased expression and correct DPR protein aggregation and toxicity. The compounds tested included Torin1, KU0063794, and rapamycin, which are well‐characterized mTOR inhibitors and thus activators of autophagy, and various phenothiazine derivatives, which are clinically approved antipsychotic drugs known to activate autophagy in neuronal cell culture (Tsvetkov et al, 2010). Interestingly, addition of not only Torin1, but also to a lesser extent either promethazine, chlorpromazine, or fluphenazine, decreases the accumulation of polyGA and polyPG DPR proteins expressed under their natural start codons (Fig 4A and B). These compounds can also decrease accumulation of a polyGA‐GFP‐tagged protein expressed under an artificial ATG start codon (Fig EV4A and B). Direct observation of the GFP fluorescence confirmed that these compounds decrease the accumulation of polyGA protein aggregates (Fig EV4C). Next, we tested whether boosting autophagy may correct the toxicity of DPR protein expression. As inhibiting mTOR has many adverse consequences in animals (Zhang et al, 2011), we focused on phenothiazine derivatives, which are clinically approved drugs. Importantly, addition of promethazine alleviated neuronal cell death caused by the synergic toxicity caused by DPR protein accumulation upon C9ORF72 reduced expression (Fig 4C). Moreover, promethazine was also able to reduce cell death caused by expression of polyGA under an artificial ATG start codon (Fig EV4D). Overall, these results suggest that modulating autophagy could be of therapeutic interest in ALS/FTD to prevent the toxic accumulation of DPR proteins.
Figure 4. Promethazine reduces DPR protein accumulation and toxicity.
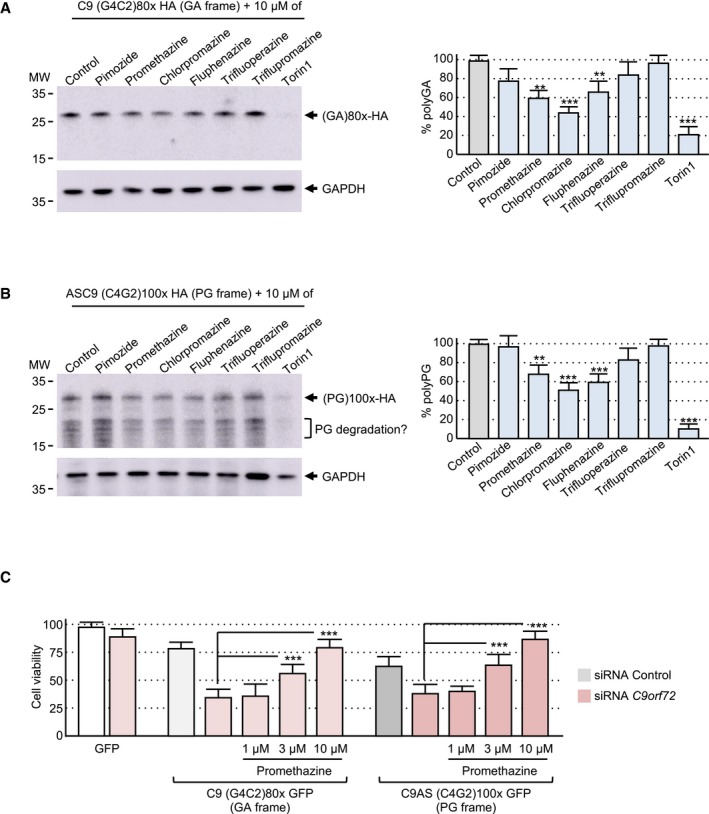
- Left panel, immunoblotting against the HA‐tag or the GAPDH of proteins extracted from Neuro2A cells transfected for 24 h with a construct expressing 80 G4C2 repeats embedded in the human sense C9ORF72 sequence fused to a HA in the GA frame and treated with 10 μM of the indicated compound for 15 h. Right panel, quantification of polyGA expression relative to the GAPDH.
- Left panel, immunoblotting against the HA‐tag or the GAPDH of proteins extracted from Neuro2A cells transfected for 24 h with a construct expressing 100 C4G2 repeats embedded in the human antisense C9ORF72 sequence fused to a HA‐tag in the PG frame and treated with 10 μM of the indicated compound for 15 h. Right panel, quantification of polyPG expression relative to the GAPDH.
- Cell viability (TO‐PRO‐3 FACS staining) of GT1‐7 neuronal cells treated with 1, 3, or 10 μM of promethazine and co‐transfected for 24 h with either a control siRNA or a siRNA targeting C9orf72 mRNA and a construct expressing either 80 G4C2 repeats or 100 C4G2 repeats embedded in sense or antisense C9ORF72 fused to the GFP in the GA or PG frame.
Figure EV4. Promethazine reduces DPR protein accumulation and toxicity.
- Left panel, immunoblotting against the GFP or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with a construct expressing under an artificial ATG start codon 100 GA repeats fused to the GFP (ATG (GA)100× GFP) and treated 15 h with 10 μM of the indicated drug. Right panel, quantification of polyGA expression relative to the GAPDH.
- Immunoblotting against the GFP or the GAPDH of proteins extracted from HEK293 cells transfected for 24 h with ATG (GA)100× GFP and treated 15 h with 1, 3, or 10 μM of the indicated drug.
- Left panel, GFP fluorescence of HEK293 cells transfected for 24 h with ATG (GA)100× GFP and treated with 10 μM of the indicated compound. Scale bars, 10 μm. Nuclei were counterstained with DAPI. Right panel, quantification of polyGA aggregates.
- Cell viability (TO‐PRO‐3 FACS staining) of GT1‐7 neuronal cells treated with 1, 3, or 10 μM of promethazine and co‐transfected for 24 h with ATG (GA)100× GFP and either a control siRNA or a siRNA targeting C9orf72 mRNA.
Discussion
An expansion of G4C2 repeats within the first intron of the C9ORF72 gene is the main genetic cause of ALS/FTD (DeJesus‐Hernandez et al, 2011; Renton et al, 2011). Sense and antisense transcripts containing these repeats are RAN‐translated into toxic DPR proteins (Ash et al, 2013; Gendron et al, 2013; Mori et al, 2013; Zu et al, 2013), which is a major pathogenic event in cell and animal models of ALS/FTD (Tran et al, 2015; Jiang et al, 2016; Liu et al, 2016; Moens et al, 2018). However, the molecular mechanisms by which expanded sense G4C2 and antisense C4G2 repeats are translated remain to be fully characterized.
Using repeats embedded in their natural human sequences, we found that expanded antisense C4G2 repeats are mostly translated into polyPG through initiation to a canonical AUG start codon located upstream of the repeats. In contrast, sense G4C2 repeats are translated mostly into polyGA and to a lesser extent into polyGR, through initiation to a CUG and an AGG near‐cognate codon, respectively. In support of these results, translation initiation of polyGA from the exact same CUG near‐cognate codon was independently identified in three other studies (Green et al, 2017; Sonobe et al, 2018; Tabet et al, 2018), and deletion of the sequence containing this CUG codon abolishes polyGA expression in neurons differentiated from C9‐iPS cells with ~1,000 G4C2 repeats (Almeida et al, 2019). Of interest, expanded CGG repeats in the neurodegenerative disease fragile X‐associated tremor/ataxia syndrome (FXTAS) are also translated into a toxic protein (FMRpolyGlycine) through initiation to a near‐cognate start codon located upstream of the repeats (Todd et al, 2013; Kearse et al, 2016; Sellier et al, 2017). Near‐cognate initiation codons are codons differing from the AUG sequence by one base, but that can still initiate translation provided they are embedded in a correct Kozak sequence (Kozak, 1989; Peabody, 1989). Ribosome profiling demonstrated that initiation to near‐cognate codons is more common than previously thought and may occur in up to ~30% of mammalian genes (Ingolia et al, 2011; Fritsch et al, 2012). These data suggest that translation initiation at near‐cognate codons located upstream of the repeats could be a common pathogenic mechanism in microsatellite expansion diseases, a hypothesis that may warrant further investigations (Gao et al, 2017). However, translation initiation at near‐cognate codons should occur independently of the presence of downstream repeats, but DPR proteins are observed only in carriers of G4C2 repeat expansions and not in control individuals. Of interest, we noted that in the absence of an expansion, translation does indeed occur, but the resulting proteins are small and unstable and consequently hardly detectable. In contrast, repeat expansion increases the size and the stability of these DPR proteins, enabling their detection. As a note of caution, our experiments are based on cell transfection with constructs containing a limited number of repeats. Thus, it remains to test whether larger expansions would employ a different mechanism of translation, notably RAN translation initiating directly within the repeats and/or translation frameshifting. In that aspect, G4C2 repeat translation frameshift from the GA frame, initiated at the CUG near‐cognate codon, to the GP and GR frames was recently reported in cell transfection assays (Tabet et al, 2018). However, deletion of this CUG codon abolishes polyGA translation, but does not affect polyGP and polyGR expression in C9‐iPS cells (Almeida et al, 2019). Similarly, we found only trace amounts of polyGP expressed from the sense G4C2 repeats, suggesting a very low rate of frameshifting from the GA frame to the GP frame. However, this could be due to the lower sensitivity of our assay (immunoblotting) compared to the more sensitive nanoluciferase and in vitro translation assays used in Tabet et al In summary, we found that DPR proteins are produced through a classic mechanism of translation initiation at canonical AUG or near‐cognate codons located upstream of the sense and antisense repeats, leading to expression of polyPG, polyGA, and, to a lesser degree, polyGR DPR proteins. Importantly, this expression pattern is similar to the one observed in patient tissues where inclusions of polyGA and polyGP/PG proteins are commonly observed, while fewer aggregates of polyGR are detected (Mackenzie et al, 2015; Davidson et al, 2016; Lee et al, 2017; Saberi et al, 2017).
A second important point of the present work is the synergic toxicity between C9ORF72 reduced expression and DPR protein expression (model presented in Fig 5). Indeed, we found that DPR proteins expressed under their natural sequences caused only limited toxicity, which is consistent with their limited expression. Mutation of their near‐cognate codons into AUG canonical start codons enhanced both DPR protein expression and toxicity, especially for the polyGR DPR protein, as consistently reported in various recent studies (Lopez‐Gonzalez et al, 2016, 2019; Zhang et al, 2018; Choi et al, 2019). Conversely, deletions of these DPR initiation codons abolished neuronal cell death, suggesting that expanded repeats cause little toxicity at the RNA level and that RAN translation initiating within the repeats is not sufficiently efficient to drive toxicity, at least with the limited number of repeats used in the present study. Of interest, accumulation and thus toxicity of DPR proteins expressed under their weak natural initiation codons are prevented by their constant degradation, notably by autophagy, a pathway regulated by the C9ORF72 protein. As reduced expression of C9ORF72 leads to suboptimal autophagy and as expression of C9ORF72 is decreased in brain tissues of C9‐positive ALS/FTD individuals, we tested whether reduced expression of C9ORF72 may modulate DPR protein accumulation and toxicity. Importantly, decreased expression of C9ORF72 increases the accumulation and thus the toxicity of polyGA, polyGR, and polyPG DPR proteins expressed under their native sequences. These data suggest that the sole reduction of C9ORF72 protein levels or the sole expression of DPR proteins under their native weak initiation codons may not be sufficient to drive pathogenicity in isolation. In contrast, expanded repeats in ALS/FTD may be pathogenic through a double‐hit mechanism where the suboptimal autophagy due to C9ORF72 reduced expression may enhance the accumulation and toxicity of DPR proteins expressed under their natural sequences. These results are reminiscent of stress conditions that increase expression and toxicity of DPR proteins (Green et al, 2017; Cheng et al, 2018; Sonobe et al, 2018; Westergard et al, 2019). Our results are also consistent with the decreased expression of C9ORF72 that synergizes the toxicity of expanded G4C2 repeats in C9‐BAC transgenic mice (Shao et al, 2019) or that enhances the toxicity of DPR proteins expressed under artificial ATG start codons in neurons differentiated from C9‐iPS cells (Shi et al, 2018). Interestingly, a model where both loss of C9ORF72 and expression of DPR proteins are required to observe neuronal cell death is consistent with the absence of neurodegeneration in BAC transgenic mice expressing DPR proteins from their natural sequences but with normal levels of C9orf72 (O'Rourke et al, 2016; Peters et al, 2015), as well with the absence of a neuronal phenotype in mice knockout for C9orf72 but without DPR protein expression (Lagier‐Tourenne et al, 2013; Koppers et al, 2015; O'Rourke et al, 2016; Atanasio et al, 2016; Burberry et al, 2016; Jiang et al, 2016; Sudria‐Lopez et al, 2016). Furthermore, this double‐hit mechanism is supported by the association of C9ORF72 mRNA decreased levels with patient survival (van Blitterswijk et al, 2015), and by the absence of clinical symptoms in individuals with a limited (30 to 70 repeats) number of G4C2 repeats, who show inclusions of DPR proteins but normal levels of the C9ORF72 protein (Gami et al, 2015; McGoldrick et al, 2018).
Figure 5. Model of C9ORF72 loss‐of‐function and DPR gain‐of‐function toxicity.
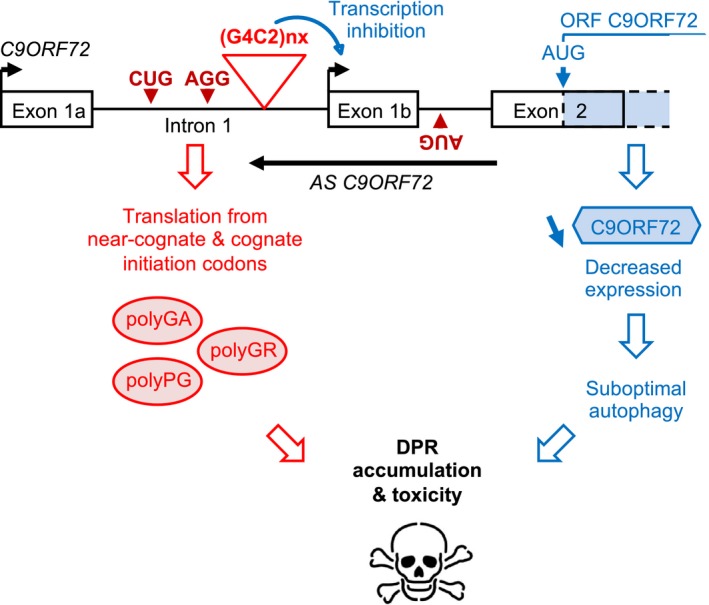
Expanded sense G4C2 and antisense C4G2 repeats are translated into polyGA, polyGR, and polyPG DPR proteins through initiation to near‐cognate codons or a cognate ATG codon embedded in a poor Kozak sequence. Concomitantly, expanded G4C2 repeats promote epigenetic DNA changes that inhibit promoter 1b activity, ultimately resulting in decreased expression of the C9ORF72 protein. Reduced expression of C9ORF72 leads to suboptimal autophagy that promotes the toxic accumulation of polyGA, polyGR, and polyPG DPR proteins, ultimately resulting in neuronal cell death.
Finally, our study provides an additional proof of concept that modulating autophagy may be of therapeutic interest in ALS/FTD. Indeed, previous studies have shown that stimulating autophagy is beneficial in SOD1, FUS, and TDP‐43 cell or animal models of ALS (Hetz et al, 2009; Crippa et al, 2010; Wang et al, 2012; Castillo et al, 2013; Barmada et al, 2014; Perera et al, 2017; Marrone et al, 2018, 2019). In agreement with these works, we found that compounds known to induce autophagy, notably promethazine, are able to bypass the reduced expression of C9ORF72 and can promote the clearance of DPR proteins, thus reducing their toxicity. Promethazine is a sedative and antihistamine drug with antipsychotic effects that belongs to a class of phenothiazine derivatives known to activate autophagy in neuronal cell cultures (Tsvetkov et al, 2010). Interestingly, these compounds ameliorate neuronal cell dysfunctions and prevent neuronal cell death in SOD1, TDP‐43, and FUS cellular and animal models of ALS (Barmada et al, 2014; Patten et al, 2017; Marrone et al, 2018, 2019). Furthermore, pimozide, a phenothiazine derivative related to promethazine, displayed some promising effects in a short randomized clinical trial of sporadic ALS (Patten et al, 2017). These results support the need for further investigations of autophagy activators in ALS/FTD.
In conclusion, these results suggest a model where the sole loss of C9ORF72 or the sole expression of DPR proteins under their native sense or antisense sequences is not sufficient to be pathogenic in isolation. In contrast, neuronal cell loss in ALS/FTD may result from a double‐hit mechanism where the suboptimal autophagy due to the reduced expression of C9ORF72 may synergize toxicity of other stress, notably the accumulation of toxic DPR proteins translated from their weak natural initiation codons (Fig 5). Of clinical importance, this double‐hit mechanism can be prevented by pharmaceutical compounds enhancing autophagy.
Materials and Methods
Constructions
Human sense C9ORF72 sequence containing 80 G4C2 repeats interrupted by NheI, HindIII, BamHI, and EcoRI restrictions sites was cloned into pcDNA3.1 fused to a HA‐tag or the GFP deleted of its ATG in all three frames. Human antisense C9ORF72 sequence containing 360 nts upstream of 100 C4G2 repeats was cloned into pcDNA3.1 fused to a HA‐tag or the GFP deleted of its ATG in all three frames. Mutations of size of the repeats, of initiation codons, or of Kozak sequences were achieved by oligonucleotide ligations. To insure stability of the expanded repeats, all plasmids were transformed into STBL3 bacterial strain (Invitrogen) and grown at room temperature (22°C).
Immunofluorescence
Coverslip were incubated for 15 min in PBS with 4% paraformaldehyde, washed with PBS, and incubated in PBS plus 0.5% Triton X‐100 during 10 min. The cells were washed three times with PBS, and the coverslips were incubated during 1 h with primary antibody against ubiquitin (ab134953, Abcam), EEA1 (ab109110, Abcam), M6PR (ab124767, Abcam), and P62/Sqstm1 (ab56416, Abcam). After washing with PBS, the coverslips were incubated with a goat anti‐mouse or anti‐rabbit secondary antibody conjugated with Alexa 488 or CY3 (Interchim SA) for 1 h, washed twice with PBS, and incubated for 3 min in PBS/DAPI (1/10,000 dilution). Coverslips were rinsed twice before mounting in Pro‐Long media (Molecular Probes) and were examined using a Leica microscope.
Cell cultures, transfections, treatments, and viability assay
Neuronal GT1‐7 cells were grown in DMEM 4.5 g/l glucose with 10% fetal calf serum, gentamicin, and penicillin at 37°C in 5% CO2. HEK293 cells were growth in DMEM 1 g/l glucose with 10% FCS and gentamicin at 37°C in 5% CO2. For transfection, cells were plated in DMEM and 0.1% fetal bovine serum and transfected for 24 h using Lipofectamine 2000 (Fisher Scientific) and/or RNAiMAX (Fisher Scientific) with either control siRNA or siRNA against C9orf72 (ON‐TARGETplus, Dharmacon). After 1–3 days, neurons were analyzed by FACS analysis for viability or by immunofluorescence or Western blotting. Cells were treated with 100 nM of bafilomycin A1, 1 μM MG132, or the indicated concentration of drug (Sigma) during 15 h before analysis. For cell viability, cells were detached by scraping and resuspended in PBS. TO‐PRO‐3 iodide (Fisher Scientific, T‐3605) was added at 20 nM to each sample and gently mixed just prior to analysis, and 30,000 cells were FACS‐analyzed.
Western blotting
Proteins were denatured 3 min at 95°C, separated on 4–12% Bis‐Tris Gel (NuPAGE), transferred on nitrocellulose membranes (Whatman Protran), blocked with 5% non‐fat dry milk in Tris‐buffered saline (TBS) buffer, incubated with anti‐GFP (ab290, Abcam), HA (ab130275, Abcam), C9ORF72 (3H10, homemade), and GAPDH (ab125247, Abcam) in TBS plus 5% non‐fat dry milk, washed three times, and incubated with anti‐rabbit or mouse peroxidase antibody (1:10,000, Cell Signaling) 1 h in TBS, followed by washing and ECL chemiluminescence revelation (Amersham ECL Prime).
Mass spectrometry analysis
HEK293 cells were transfected with HA‐ or GFP‐tagged plasmid using Lipofectamine 2000 (Fisher Scientific) for 24 h. Proteins were purified by immunoprecipitation against the HA or GFP, separated on 4–12% Bis‐Tris Gel (NuPAGE), and visualized by Coomassie blue staining. Gel bands were excised and subjected to in‐gel reduction in 10 mM DTT in 100 mM NH4HCO3 (Sigma‐Aldrich) for 1 h at 57°C, alkylated for 45 min in the dark with 55 mM iodoacetamide in 100 mM NH4HCO3 (Sigma‐Aldrich), washed in 25 mM NH4HCO3, dehydrated with acetonitrile, and dried in SpeedVac 5301 Concentrator (Eppendorf). Then, the gel pieces were rehydrated with 12.5 ng/μl LysC solution (Promega) in 50 mM NH4HCO3 and incubated overnight at 37°C. The peptides were extracted twice with acetonitrile/water/formic acid—45/45/10, v/v/v followed by a final extraction with acetonitrile/formic acid (FA)—95/05, v/v. Extracted peptides were then analyzed using an Ultimate 3000 nano‐RSLC (Thermo Scientific) coupled in line with an Orbitrap ELITE (Thermo Scientific). Peptides were separated on a C18 nanocolumn with a linear gradient of acetonitrile and analyzed within a Top 20 collision‐induced dissociation data‐dependent mass spectrometry with an inclusion list. Data were processed by database searching using SequestHT (Thermo Fisher Scientific) with Proteome Discoverer 1.4 software (Thermo Fisher Scientific) against a homemade database of all potential three frames translated proteins or peptides from the human C9ORF72 sense and antisense sequences. Precursor and fragment mass tolerance were set at 7 ppm and 0.5 Da, respectively. Oxidation (M) and N‐terminal acetylation were set as variable modification, and carbamidomethylation (C), as fixed modification. Peptides were filtered with the fixed value node of Proteome Discoverer 1.4.
RT–qPCR
RT–qPCR analyses were performed using the Transcriptor Reverse Transcriptase Kit (Roche) and the QuantiTect SYBR Green PCR Kit (Qiagen) in a LightCycler 480 (Roche) with 15 min at 94°C followed by 50 cycles of 15 s at 94°C, 20 s at 58°C, and 20 s at 72°C using aagttcatctgcaccaccg (fwd) and ttctgctggtagtggtcggcg (rev) primers to quantify the GFP expression, with RPLP0 mRNA as standard (fwd: gaagtcactgtgccagccca, rev: gaaggtgtaatccgtctcca). Data were analyzed using LightCycler 480 analysis software and the 2ΔC t method.
Statistical analysis
All cell experiments are represented as average ± standard error of mean (s.e.m.) with significance determined using Student's t‐test.
Author contributions
Experiments were performed by MB, VP, AG, FR, and CS. Data were collected and analyzed by LN, NC‐B, and CS. The study was designed, coordinated, and written by CS and NC‐B.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgments
We thank Prof. Mellon (UCSD, USA) for the gift of the GT1‐7 cells and Prof. Suzuki for the gift of the ATG DPR plasmids (Yamakawa et al, 2015). This work was supported by Fondation de France Thierry Latran #57486 “Model‐ALS” and #88187 “DRUG‐C9DPR” (NCB), AFM #18605 “Role of C9ORF72 in ALS” (NCB); ARSLA “Therapeutic approach for Amyotrophic Lateral Sclerosis” (NCB); ERC‐2012‐StG #310659 “RNA DISEASES” (NCB); ANR‐17‐CE16‐0001‐01 “C9ORF72 ALS” (NCB); ANR‐18‐CE16‐0019 “NewMutALS” (NCB); ANR‐10‐LABX‐0030‐INRT (IGBMC) and ANR‐10‐IDEX‐0002‐02 (IGBMC).
The EMBO Journal (2020) 39: e100574
Contributor Information
Chantal Sellier, Email: sellier@igbmc.fr.
Nicolas Charlet‐Berguerand, Email: ncharlet@igbmc.fr.
Data availability
The data that support the findings of this study are available within the article and its supplementary information files or from the corresponding authors on reasonable request.
References
- Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet‐Berguerand N, Karydas A et al (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC‐derived human neurons. Acta Neuropathol 126: 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida S, Krishnan G, Rushe M, Gu Y, Kankel MW, Gao FB (2019) Production of poly(GA) in C9ORF72 patient motor neurons derived from induced pluripotent stem cells. Acta Neuropathol 138: 1099–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amick J, Roczniak‐Ferguson A, Ferguson SM (2016) C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell 27: 3040–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, van Blitterswijk MM, Jansen‐West K, Paul JW III, Rademakers R, Boylan KB et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y et al (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 16: 23204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D et al (2014) Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 10: 677–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Gendron TF, Baker MC, DeJesus‐Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J et al (2015) Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol 130: 863–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I et al (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 12: 20877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki‐Uematsu S, Ghosh S, Singh A, Merkle FT et al (2016) Loss‐of‐function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8: 347ra93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, Hetz C (2013) Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9: 1308–1320 [DOI] [PubMed] [Google Scholar]
- Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F, Hayes LR, Lopez‐Gonzalez R, Drenner K, Jiang J et al (2018) C9ORF72 GGGGCC repeat‐associated non‐AUG translation is upregulated by stress through eIF2α phosphorylation. Nat Commun 9: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes‐Casey M, Lee CW, Jansen‐West K, Kurti A, Murray ME et al (2015) C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss, and behavioral deficits. Science 348: 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitiprolu M, Jagow C, Tremblay V, Bondy‐Chorney E, Paris G, Savard A, Palidwor G, Barry FA, Zinman L, Keith J et al (2018) A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun 9: 2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Lopez‐Gonzalez R, Krishnan G, Phillips HL, Li AN, Seeley WW, Yao WD, Almeida S, Gao FB (2019) C9ORF72‐ALS/FTD‐associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo . Nat Neurosci 22: 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E (2013) Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol 74: 180–187 [DOI] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL (2016) The C9ORF72 GGGGCC expansion forms RNA G‐quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 13: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Fagegaltier D, Agius P, Davis‐Porada J, Gregory J, Hubbard I, Kang K, Kim D, New York Genome Center ALS Consortium , Phatnani H et al (2018) Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. Elife 7: e37754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper‐Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J et al (2014) Sequestration of multiple RNA recognition motif‐containing proteins by C9orf72 repeat expansions. Brain 137(Pt 7): 2040–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Horvitz HR (2018) A C9orf72 ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Curr Biol 28: 1522–1535.e5 [DOI] [PubMed] [Google Scholar]
- Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E, Marino M, Carra S et al (2010) The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 19: 3440–3456 [DOI] [PubMed] [Google Scholar]
- Cristofani R, Crippa V, Vezzoli G, Rusmini P, Galbiati M, Cicardi ME, Meroni M, Ferrari V, Tedesco B, Piccolella M et al (2018) The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72‐related neurodegenerative diseases. Cell Stress Chaperones 23: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H, Vowles J, Douglas AG, Fletcher‐Jones A, Browne C et al (2016) C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell‐derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34: 2063–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson Y, Robinson AC, Liu X, Wu D, Troakes C, Rollinson S, Masuda‐Suzukake M, Suzuki G, Nonaka T, Shi J et al (2016) Neurodegeneration in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP‐43 pathology and not associated with aggregated forms of dipeptide repeat proteins. Neuropathol Appl Neurobiol 42: 242–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez‐Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525: 129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick P, Sellier C, Mackenzie IRA, Cheng CY, Tahraoui‐Bories J, Martinat C, Pasterkamp RJ, Prudlo J, Edbauer D, Oulad‐Abdelghani M et al (2018) Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun 6: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch C, Herrmann A, Nothnagel M, Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak M, Hampe J et al (2012) Genome‐wide search for novel human uORFs and N‐terminal protein extensions using ribosomal footprinting. Genome Res 22: 2208–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Bravo‐San Pedro JM, Levine B, Green DR, Kroemer G (2017) Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 16: 487–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gami P, Murray C, Schottlaender L, Bettencourt C, De Pablo Fernandez E, Mudanohwo E, Mizielinska S, Polke JM, Holton JL, Isaacs AM et al (2015) A 30‐unit hexanucleotide repeat expansion in C9orf72 induces pathological lesions with dipeptide‐repeat proteins and RNA foci, but not TDP‐43 inclusions and clinical disease. Acta Neuropathol 130: 599–601 [DOI] [PubMed] [Google Scholar]
- Gao FB, Richter JD, Cleveland DW (2017) Rethinking Unconventional Translation in Neurodegeneration. Cell 171: 994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang YJ, Jansen‐West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes‐Casey M, Chew J et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat‐associated non‐ATG translation in c9FTD/ALS. Acta Neuropathol 126: 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S et al (2012) A C9orf72 promoter repeat expansion in a Flanders‐Belgian cohort with disorders of the frontotemporal lobar degeneration‐amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11: 54–65 [DOI] [PubMed] [Google Scholar]
- Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ, Goldstrohm AC, Barmada SJ, Todd PK (2017) RAN translation at C9orf72‐associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun 8: 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH (2009) XBP‐1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 23: 2294–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WY, Tai YK, Chang JC, Liang J, Tyan SH, Chen S, Guan JL, Zhou H, Shen HM, Koo E et al (2019) The ALS‐FTD‐linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 15: 827–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis‐Downes M, Seelman A, Stauffer JE, Jafar‐Nejad P, Drenner K, Schulte D et al (2016) Gain of toxicity from ALS/FTD‐linked repeat expansions in C9ORF72 Is alleviated by antisense oligonucleotides targeting GGGGCC‐containing RNAs. Neuron 90: 535–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW III, Sun S, Herdy JR, Bieri G et al (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18: 1226–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Nayak A, Schaeffer V, Starzetz T, Kirsch AK, Müller S, Dikic I, Mittelbronn M, Behrends C (2017) Multiplex image‐based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. Elife 6: e23063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, Todd PK (2016) CGG repeat‐associated Non‐AUG translation utilizes a cap‐dependent scanning mechanism of initiation to produce toxic proteins. Mol Cell 62: 314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi B, Hartmann H, May S, Möhl C, Ederle H, Michaelsen M, Schludi MH, Dormann D, Edbauer D (2017) Cytoplasmic poly‐GA aggregates impair nuclear import of TDP‐43 in C9orf72 ALS/FTLD. Hum Mol Genet 26: 790–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78: 426–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1989) Context effects and inefficient initiation at non‐AUG codons in eucaryotic cell‐free translation systems. Mol Cell Biol 9: 5073–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL (2014) Poly‐dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345: 1139–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier‐Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA 110: E4530–E4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Sullivan PM, Hu F (2019) SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy 15: 871–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Chen HJ, Peres JN, Gomez‐Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C et al (2013) Hexanucleotide repeats in ALS/FTD form length‐dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5: 1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Baskaran P, Gomez‐Deza J, Chen HJ, Nishimura AL, Smith BN, Troakes C, Adachi Y, Stepto A, Petrucelli L et al (2017) C9orf72 poly GA RAN‐translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet 26: 4765–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP (2016) C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90: 521–534 [DOI] [PubMed] [Google Scholar]
- Lomen‐Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59: 1077–1079 [DOI] [PubMed] [Google Scholar]
- Lopez‐Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, Petrucelli L, Miller BL, Almeida S, Gao FB (2016) Poly(GR) in C9ORF72‐Related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC‐derived motor neurons. Neuron 92: 383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Gonzalez R, Yang D, Pribadi M, Kim TS, Krishnan G, Choi SY, Lee S, Coppola G, Gao FB (2019) Partial inhibition of the overactivated Ku80‐dependent DNA repair pathway rescues neurodegeneration in C9ORF72‐ALS/FTD. Proc Natl Acad Sci USA 116: 9628–9633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon‐Sanchez J et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol 11: 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone L, Poser I, Casci I, Japtok J, Reinhardt P, Janosch A, Andree C, Lee HO, Moebius C, Koerner E et al (2018) Isogenic FUS‐eGFP iPSC reporter lines enable quantification of FUS stress granule pathology that is rescued by drugs inducing autophagy. Stem Cell Reports 10: 375–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone L, Drexler HCA, Wang J, Tripathi P, Distler T, Heisterkamp P, Anderson EN, Kour S, Moraiti A, Maharana S et al (2019) FUS pathology in ALS is linked to alterations in multiple ALS‐associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol 138: 67–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf‐Strathmann J et al (2014) C9orf72 FTLD/ALS‐associated Gly‐Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128: 485–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D et al (2015) Quantitative analysis and clinico‐pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol 130: 845–861 [DOI] [PubMed] [Google Scholar]
- McGoldrick P, Zhang M, van Blitterswijk M, Sato C, Moreno D, Xiao S, Zhang AB, McKeever PM, Weichert A, Schneider R et al (2018) Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology 90: e323–e331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 126: 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345: 1192–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens TG, Mizielinska S, Niccoli T, Mitchell JS, Thoeng A, Ridler CE, Grönke S, Esser J, Heslegrave A, Zetterberg H et al (2018) Sense and antisense RNA are not toxic in Drosophila models of C9orf72‐associated ALS/FTD. Acta Neuropathol 135: 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science 339: 1335–1338 [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133 [DOI] [PubMed] [Google Scholar]
- O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J et al (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science 351: 1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten SA, Aggad D, Martinez J, Tremblay E, Petrillo J, Armstrong GA, La Fontaine A, Maios C, Liao M, Ciura S et al (2017) Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2: 97152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peabody DS (1989) Translation initiation at non‐AUG triplets in mammalian cells. J Biol Chem 264: 5031–5035 [PubMed] [Google Scholar]
- Perera ND, Sheean RK, Lau CL, Shin YS, Beart PM, Horne MK, Turner BJ (2017) Rilmenidine promotes MTOR‐independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 5: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J et al (2015) Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88: 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS/FTD. Neuron 72: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65: 586–590 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11: 709–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi S, Stauffer JE, Jiang J, Garcia SD, Taylor AE, Schulte D, Ohkubo T, Schloffman CL, Maldonado M, Baughn M et al (2017) Sense‐encoded poly‐GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co‐localize with TDP‐43 in dendrites of repeat‐expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol 135: 459–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schludi MH, Becker L, Garrett L, Gendron TF, Zhou Q, Schreiber F, Popper B, Dimou L, Strom TM, Winkelmann J et al (2017) Spinal poly‐GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol 134: 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Campanari ML, Corbier C, Gaucherot A, Kolb‐Cheynel I, Oulad‐Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E et al (2016) Loss of C9ORF72 partly impairs autophagy and synergizes the toxicity of Ataxin‐2 with intermediate size of polyglutamine expansion. EMBO J 35: 1276–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Buijsen RA, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A et al (2017) Translation of expanded CGG repeats into FMRpolyG Is pathogenic and may contribute to fragile X tremor ataxia syndrome. Neuron 93: 331–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q, Liang C, Chang Q, Zhang W, Yang M, Chen JF (2019) C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose‐dependent manner. Acta Neuropathol Commun 7: 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY et al (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 24: 313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonobe Y, Ghadge G, Masaki K, Sendoel A, Fuchs E, Roos RP (2018) Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol Dis 116: 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudria‐Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, Youssef SA, Harkema L, de Bruin A, Veldink JH et al (2016) Full ablation of C9orf72 in mice causes immune system‐related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol 132: 145–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F (2016) The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy‐lysosome pathway. Acta Neuropathol Commun 4: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabet R, Schaeffer L, Freyermuth F, Jambeau M, Workman M, Lee CZ, Lin CC, Jiang J, Jansen‐West K, Abou‐Hamdan H et al (2018) CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat Commun 9: 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, Xu G, Wang G, Ying Z (2015) Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation‐induced cytotoxicity. Hum Mol Genet 24: 2426–2441 [DOI] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V et al (2013) CGG repeat‐associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78: 440–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Almeida S, Moore J, Gendron TF, Chalasani U, Lu Y, Du X, Nickerson JA, Petrucelli L, Weng Z et al (2015) Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a Drosophila model of C9ORF72 FTD/ALS. Neuron 87: 1207–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S (2010) A small‐molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci USA 107: 16982–16987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolino J, Ji YJ, Conchina K, Chu J, Nirujogi RS, Pandey A, Brady NR, Hamacher‐Brady A, Wang J (2016) Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet 12: e1006443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viodé A, Fournier C, Camuzat A, Fenaille F, NeuroCEB Brain Bank , Latouche M, Elahi F, Le Ber I, Junot C, Lamari F et al (2018) New antibody‐free mass spectrometry‐based quantification reveals that C9ORF72 long protein isoform is reduced in the frontal cortex of hexanucleotide‐repeat expansion carriers. Front Neurosci 12: 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35: 1779.e5–1779.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C, Herranz‐Martin S, Karyka E, Liao C, Lewis K, Elsayed W, Lukashchuk V, Chiang SC, Ray S, Mulcahy PJ et al (2017) C9orf72 expansion disrupts ATM‐mediated chromosomal break repair. Nat Neurosci 20: 1225–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CK (2012) Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA‐binding protein 43. Proc Natl Acad Sci USA 109: 15024–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ et al (2016) The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 35: 1656–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB et al (2014) Antisense proline‐arginine RAN dipeptides linked to C9ORF72‐ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84: 1213–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergard T, McAvoy K, Russell K, Wen X, Pang Y, Morris B, Pasinelli P, Trotti D, Haeusler A (2019) Repeat‐associated non‐AUG translation in C9orf72‐ALS/FTD is driven by neuronal excitation and stress. EMBO Mol Med 11: e9423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MR, Mitrea DM, Zhang P, Stanley CB, Cassidy DE, Nourse A, Phillips AH, Tolbert M, Taylor JP, Kriwacki RW (2019) C9orf72 Poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol Cell 74: 713–728.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J (2015) Isoform‐specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 78: 568–583 [DOI] [PubMed] [Google Scholar]
- Xiao S, MacNair L, McLean J, McGoldrick P, McKeever P, Soleimani S, Keith J, Zinman L, Rogaeva E, Robertson J (2016) C9orf72 isoforms in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain Res 15: 43–49 [DOI] [PubMed] [Google Scholar]
- Yamakawa M, Ito D, Honda T, Kubo K, Noda M, Nakajima K, Suzuki N (2015) Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet 24: 1630–1645 [DOI] [PubMed] [Google Scholar]
- Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, Chen JF (2016) A C9ORF72/SMCR8‐containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2: e1601167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, Wang Z, Le W (2011) Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7: 412–425 [DOI] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525: 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen‐West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M et al (2016) C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 19: 668–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Gendron TF, Ebbert MTW, O'Raw AD, Yue M, Jansen‐West K, Zhang X, Prudencio M, Chew J, Cook CN et al (2018) Poly(GR) impairs protein translation and stress granule dynamics in C9orf72‐associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med 24: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen‐West K, O'Raw AD, Pickles SR, Prudencio M, Carlomagno Y et al (2019) Heterochromatin anomalies and double‐stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363: eaav2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Bañez‐Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA 110: E4968–E4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Data Availability Statement
The data that support the findings of this study are available within the article and its supplementary information files or from the corresponding authors on reasonable request.