

Abstract

We report herein the discovery of isoxazole amides as potent and selective SET and MYND Domain-Containing Protein 3 (SMYD3) inhibitors. Elucidation of the structure–activity relationship of the high-throughput screening (HTS) lead compound 1 provided potent and selective SMYD3 inhibitors. The SAR optimization, cocrystal structures of small molecules with SMYD3, and mode of inhibition (MOI) characterization of compounds are described. The synthesis and biological and pharmacokinetic profiles of compounds are also presented.
Keywords: SMYD3, MEKK2, inhibitors, isoxazole amides
Lysine methylation represents a fundamental regulatory mechanism that influences protein function.1,2 Lysine methylation often serves to recruit proteins with lysine specific reader domains thereby mediating transcriptional changes, as is the case for methylation of histones. However, lysine methylation can also alter the enzymatic activity of methylated proteins and/or promote/inhibit other post-translational modifications.1 SET and MYND Domain-Containing Protein 3 (SMYD3) is a protein lysine methyltransferase shown to catalyze methylation of histone and nonhistone proteins including methylation of Histone H4 at lysine 5 and Mitogen-activated protein kinase kinase 2 (MEKK2) at lysine 260.3,4
MEKK2 methylation by SMYD3 regulates the MEK/ERK kinase signaling pathway in RAS-driven tumors.4 In both lung and pancreatic models of cancer, abrogation of SMYD3 methylase activity inhibits tumorigenesis in response to RAS activation suggesting an opportunity to target the methyltransferase activity of SMYD3 in cancers with elevated RAS activity. Oncogenic RAS mutations are among the most frequent alterations in cancer; therefore, the development of compounds which inhibit signaling downstream of RAS represents a unique approach to target RAS-driven cancers.
Histone H4 is one of the five histone proteins; featuring a main globular domain and a long N-terminal tail. In addition to being critical to the structure of nucleosomes, histones are highly post-translationally modified, and these epigenetic modifications influence nucleosome positioning and the recruitment of transcriptional activators and/or inhibitors thereby regulating gene transcription. Epigenetic modifications play an important role in the regulation of many cellular processes including cell proliferation, differentiation, and cell survival. Global epigenetic dysregulation is common in cancer and includes global changes in DNA and/or histone methylation, dysregulation of noncoding RNAs, and nucleosome remodeling leading to aberrant activation or inactivation of oncogenes, tumor suppressors, and signaling pathways. However, unlike genetic mutations, these epigenetic changes can be reversed through selective inhibition of the enzymes involved. Thus, selective inhibitors of dysregulated methyltransferases could be useful in the treatment of proliferative diseases such as cancer.
SMYD3 amplification and/or increased expression has been observed in numerous solid tumors including those of colorectal, breast, prostate, ovarian, gastric, and hepatocellular origin and, in some instances, correlates with poor prognosis.6−9 Studies have reported that direct knockdown of SMYD3 via siRNA or shRNA decreases cancer cell line proliferation in a number of cancer cell types including breast, liver, colon, and prostate.3,6,10,11 Thus, there is evidence to suggest that inhibition of SMYD3 activity decreases cellular proliferation. Accordingly, compounds that inhibit SMYD3 activity hold promise as novel therapeutic agents for the treatment of cancer.
While small molecule SMYD3 inhibitors have been reported previously,12,13 this paper reports the SAR results of a novel class of compounds that exhibit good potency and excellent selectivity and pharmacokinetics profiles. Additionally, the synthesis, crystallography, mode of inhibition (MOI) characterization, and cellular mechanistic activity of these compounds are described.
Results and Discussion
To identify inhibitors of SMYD3, a high-throughput screen (HTS) was conducted using the GSK proprietary compound collection and a SMYD3 biochemical SPA format that measures the inhibition of MEKK2 methylation. Compound 1, an isoxazole amide analog which is closely related to another recently reported SMYD3 inhibitor, EPZ028862,13 was identified and presented as a good starting point for our chemistry effort (Table 1). The molecule has reasonable biochemical potency (IC50 = 5 μM), ligand efficiency (LE = 0.35), and physicochemical properties (Table 1). Compound 1 also possesses an excellent PK profile (vide infra). To facilitate chemistry efforts, we obtained the cocrystal structure of SMYD3 with compound 1 (Figure 1). Compound 1 is bound in the lysine tunnel of SMYD3 in close proximity to the SAM binding site. The cyclopropyl group of compound 1 points to SAM and the piperidine sulfonamide points toward the substrate peptide binding site. The other key observations are that a hydrogen bond interaction is made between the amide oxygen of compound 1 and the backbone amine of Thr184. This interaction mimics that made by the backbone carbonyl of the substrate lysine observed in the crystal structure of SMYD3 bound to a MEKK2 peptide substrate.14 The piperidine sulfonamide of compound 1 does not appear to make any significant interactions in the rather large and polar substrate pocket; therefore, focus was placed on optimization of this portion of the compound as will be described in more detail below.
Table 1. SAR.


Scintillation proximity assay.
Methylation of MEKK2 cellular mechanistic assay.
Not determined.
CLND: chemiluminescent nitrogen detection.
AMP: artificial membrane permeability.
Figure 1.
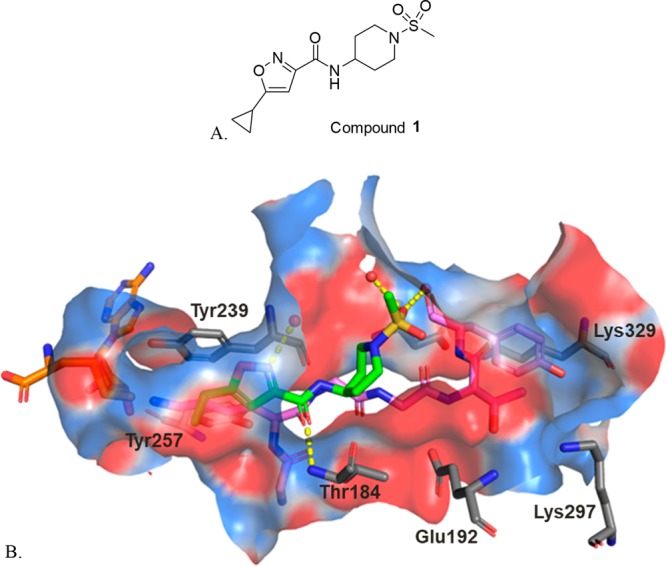
(A) Structure of compound 1; (B) Crystal structure of compound 1 (green carbons) bound to SMYD3 overlaid with a MEKK2 peptide (pink carbons) from PDB 5HQ8.14 The cofactor SAM is colored with orange carbons, and crystallographic waters are represented as red spheres. Key hydrogen bond interactions between compound 1 and SMYD3 are depicted with yellow dashes. The binding site surface is colored by electrostatic potential. An alternate conformation observed for the side chain of Glu192 has been omitted for clarity. All crystal structure figures were generated with PyMOL (The PyMOL Molecular Graphics System, Version 2.0.7 Schrodinger, LLC).
The initial SAR study around the left-hand side of the molecule, such as the replacement of cyclopropyl, isoxazole, and amide moieties, was largely not fruitful in terms of improving the potency. Although the small methyl and ethyl isoxazoles showed similar potency as 1, they provided no obvious advantage over cyclopropyl analogs. Modifications to the amide functionality that presumably disrupted the H-bond between the amide oxygen and the backbone NH of Thr184 were not tolerated for replacement and are in concert with the observation of the cocrystal structure. On the basis of cocrystal analysis, we shifted our focus to the right-hand side of the molecule. Gratifyingly, the potency of 1 can be improved 10-fold by introduction of a basic amine group, like compound 3. The crystal structure of this compound bound to SMYD3 was solved, and it was observed that the relatively small ethyl amine can bind in two conformations (Figure 2). To improve potency, larger groups and constraints were explored to more optimally direct the basic amine toward polar residues in the substrate pocket. Simply extending the amine side chain by one carbon (4) further improved the biochemical potency 10-fold and showed an EC50 potency of about 0.5 μM in a cellular mechanistic assay developed to quantify inhibition of MEKK2 methylation. Conversion of the primary amine to pyrrolidine (5 and 6) or piperidines (7–9) resulted in similar biochemical IC50 values and a slight improvement in cellular potency in some instances (presumably due to the enhancement of permeability). However, introduction of a ring constraint in the linker to form cyclohexane amines improved the potency up to 15-fold in the biochemical assay (10 and 11). The trans isomer 10 is more potent than the cis analog 11 in the biochemical and cellular assays. Further extending the amine by insertion of methylene on the right-hand side of compound 10 to form a cyclohexyl methyl amine (12) caused a loss of potency; however, inserting a methylene group in the left-hand side of molecule 10 to form methyl cyclohexyl amine (13) retained potency. Introduction of an aromatic phenyl ring as a linker between the sulfonamide and amine provided compounds 14–18. A crystal structure of compound 14 with SMYD3 reveals that the sulfonamide oxygens make water mediated hydrogen bond interactions with SMYD3 residues Ile214 and Met242 (Figure 3). The phenyl ring projects the aminomethyl group toward a highly charged, solvent rich region of the SMYD3 substrate binding site. The basic amine of compound 14 makes water mediated hydrogen bond interactions with Lys297 and Glu294. The overall loss of potency (14–18) compared to compound 13 may be due to the relative hydrophobicity and rigidity imposed by the aromatic phenyl.
Figure 2.
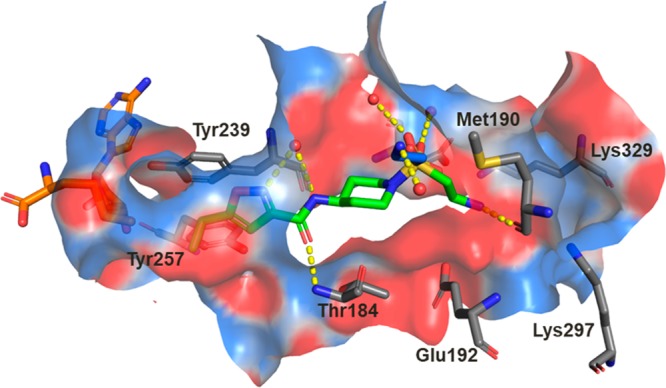
Crystal structure of compound 3 (green carbons) bound to SMYD3. A second conformation of the ethylamine of compound 3 observed in the crystal structure is colored with blue carbons, the cofactor SAM is colored with orange carbons, and crystallographic waters are represented as red spheres. Key hydrogen bond interactions are depicted with yellow dashes, and the binding site surface is colored by electrostatic potential. An alternate conformation observed for the side chain of Glu192 has been omitted for clarity.
Figure 3.
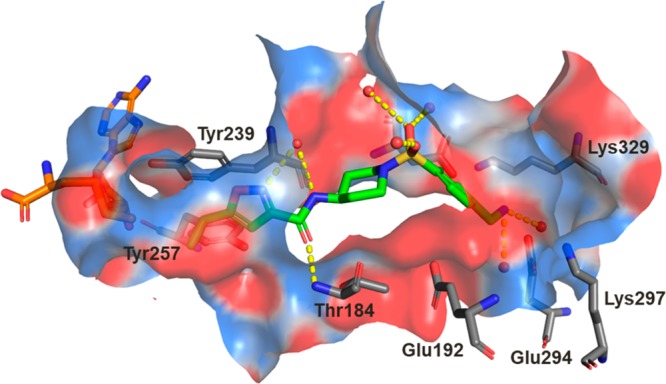
Crystal structure of compound 14 (green carbons) bound to SMYD3. The cofactor SAM is colored with orange carbons, and crystallographic waters are represented as red spheres. Key hydrogen bond interactions are depicted with yellow dashes, and the binding site surface is colored by electrostatic potential. A glycerol molecule that has partial occupancy with the water which is in hydrogen bonding distance to the isoxazole and amide nitrogens of compound 14 and alternate conformations observed for the side chains of Glu192 and Glu294 have been omitted for clarity.
Based on the excellent cellular potency, we focused on compound 13. While the basic amine group may be key to improving the potency in compound 13, a primary amine has been known to have some undesirable liability and associates with a very poor permeability (AMP < 10 nm/s, compound 13). Aiming to modulate the physiochemical properties, the amine group was substituted to form simple secondary amines (19–22) and a tertiary amine (23) that proved to be less effective both in terms of potency and permeability (Table 2). Introduction of branched alkyl groups on the amine (24–26) maintained good potency in cells and improved the permeability. Incorporating fluorinated alkyl groups (27–30) improved the biochemical and cellular potency further; in particular, compound 28 has an EC50 of 39 nM in the MEKK2me assay. A crystal structure of compound 28 with SMYD3 suggests that this compound makes many of the same interactions as compound 14 though the basic amine of compound 28 is stabilized by coming in close proximity to the negatively charged side chains of Glu192 and Asp241 rather than making water mediated interactions with the protein (Figure 4). Compound 28 further displaces several crytallographically observed water molecules with the trifluorobutane that projects the terminal CF3 group to bind in a hydrophobic pocket formed by the side chains of Leu290 and Cys333 and the aliphatic carbons of residues Glu294, Lys297, and Lys329.
Table 2. SAR (continued).


Scintillation proximity assay.
Methylation of MEKK2 cellular mechanistic assay.
Not determined.
CLND: chemiluminescent nitrogen detection.
AMP: artificial membrane permeability.
Figure 4.
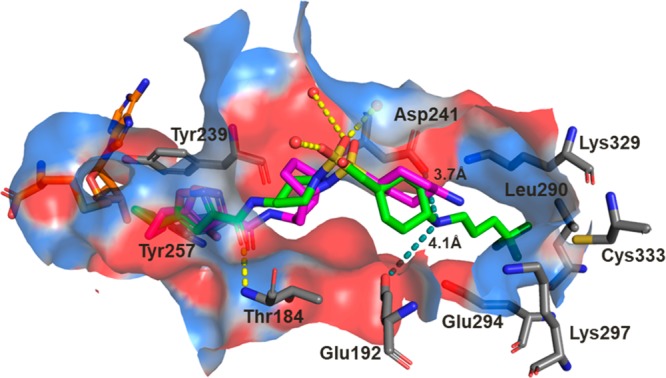
Crystal structure of compound 28 (green carbons) bound to SMYD3 overlaid with EPZ028862 (magenta carbons) from PDB 5V37. The cofactor SAH is colored with orange carbons, and crystallographic waters are represented as red spheres. Key hydrogen bond interactions between compound 28 and SMYD3 are depicted with yellow dashes, and the binding site surface is colored by electrostatic potential. The teal dashes depict measured distances between the amine of compound 28 and the side chains of Glu192 and Asp241. While there are two conformations observed in the crystal structure for the cyclopropyl group of compound 28, only one is depicted for clarity.
To balance both the potency and physicochemical properties, we substituted the amine with the benzyl group (31, Table 2). Although compound 31 suffered with poor solubility (CLND) and permeability, it showed a potency of 22 nM in the MEKK2me cellular assay. Substitution of the phenyl ring with fluorine retained excellent potency (compounds 32–34). The meta or ortho fluorine substitutions provided compounds (33 and 34) with balanced potency, solubility, and permeability. Replacement of fluorine with chlorine delivered compound 35 with excellent potency in the MEKK2me cellular assay (EC50 = 11 nM). The other chlorine analogs (36–37) were also very potent. Substitution of the phenyl ring with a methyl group (38–40) was less effective compared to fluorine or chlorine. Replacement of the phenyl ring with pyridine (41–43) caused a loss of potency; however, substitution of the pyridine ring with chlorine (44), methyl (45), and fluorine (46–48) regained the potency.
Applying the SAR learned, we revisited the cyclohexyl amine of 10 since it showed similar cellular potencies vs 13 (Table 1). Substituting the amine group with trifluorobutane provided compound 49 (Figure 5). Compound 49 exhibits IC50 potencies of 6.3 and 44 nM in biochemical and cellular MEKK2me assays, respectively. At higher doses (>400 nM), greater than 90% inhibition of MEKK2 methylation was achieved with compound 49 in cells.
Figure 5.

Structure of compound 49.
To evaluate the mode of inhibition of compounds in this series, 7, 28, and 49 were assessed by examining the effect of substrate concentration on the inhibition of SMYD3 utilizing the Cheng–Prusoff relationship for competitive, noncompetitive, and uncompetitive inhibition.15 IC50 values for these compounds showed no change with varying MEKK2 concentration indicative of MEKK2 noncompetitive inhibition (Figure 6A–C) while IC50 values decreased with increasing SAM concentration in a curve–linear fashion suggestive of uncompetitive behavior with respect to SAM binding yielding an αKi value of 17 ± 1 nM, 6 ± 0.5 nM, and 18 ± 2 nM for 7, 28, and 49, respectively (Figure 6D–F). Additionally, to confirm the mode of inhibition, initial velocity data was plotted as a function of varying substrate at different concentrations of compound 49 yielding a Ki of 18 ± 3 nM (Supporting Information Figure 1A and B). The double-reciprocal plot versus SAM resulted in a set of parallel lines consistent with an uncompetitive mode of inhibition (α = 0.13, Supporting Information Figure 1C) while the double-reciprocal plot versus MEKK2 yielded a set of lines that converged to the left of the y-axis consistent with a noncompetitive inhibitor (α = 0.93, Supporting Information Figure 1D). F-test analysis supported the mode of inhibition deduced by the double-reciprocal patterns.
Figure 6.

Mode of inhibition against SMYD3 by isoxazole series compounds. IC50 values were determined following a 60 min SMYD3 reaction and fitting the data to a 3-parameter dose–response equation. Top Panel: Data represents independent experiments showing IC50 values plotted as a function of [FL MEKK2]/Kmapp for compound 7 (A), 28 (B), and 49 (C, n = 1) where IC50 = Ki for noncompetitive inhibition. Bottom Panel: Data represents independent experiments showing IC50 values plotted as a function of [SAM]/Kmapp fit to an equation for uncompetitive inhibition αKi = IC50/(1 + (Km/[S])) for compound 7 (D), 28 (E), and 49 (F, n = 1).
Additionally, the ability of compounds 7 and 28 to inhibit SMYD3 methylation of a histone H4 substrate was investigated. Inhibition of SMYD3 methylation on recombinant histone H4 decreased roughly 10-fold in comparison to MEKK2 resulting in IC50 values of 167 nM and 67 nM for compounds 7 and 28, respectively (n = 3).
To assess whether inhibition of SMYD3 methyltransferase activity would affect MAPK signaling as previously suggested by knockout (KO) and knockdown (KD) studies,4,5 the inhibition of ERK1/2 phosphorylation was assessed in A549 cells following exposure to compounds 7, 10, and 49. No inhibition of ERK1/2 phosphorylation was observed (Figure 7). Thus, while compounds 7, 10, and 49 inhibit methylation of MEKK2 by SMYD3 in both biochemical and cellular assays, the inability of these inhibitors to affect ERK1/2 phosphorylation suggests that methylation of MEKK2 does not alter MEKK2 activity toward ERK1/2. These data suggest that the decrease in ERK1/2 phosphorylation observed upon SMYD3 knockout (KO) and knockdown (KD) studies is not due to alteration of MEKK2 methylation and thus SMYD3 may have other methyltransferase independent activities which contributed to the effects on MAPK signaling observed in KO/KD studies.
Figure 7.

Inhibition of SMYD3 methyltransferase activity does not affect ERK1/2 activation state. Following treatment of A549 cells for 72 h with compounds 7, 10, and 49, the cells were serum starved for 24 h and then treated with 25 ng/μL EGF for 15 min to stimulate MAPK signaling. Trametinib, a MEK1/2 kinase inhibitor, is included as a positive control. Lysates were immunoblotted with antibodies against the proteins indicated. Total ERK1/2, SMYD3, and tubulin are included as controls.
To evaluate the ability of compounds from this series to be used for in vivo studies, several compounds were examined in rat pharmacokinetic experiments (Table 3). In general, compounds from this series exhibited low blood clearance with high volume of distribution and long t1/2. Bioavailability was high with reasonable to good oral exposures. While some compounds have a relatively high volume of distribution, overall the PK profile is favorable.
Table 3. Pharmacokinetics of Selected Compoundsa.
| compounds | CL | Vd | t1/2 | DNAUCpo | F |
|---|---|---|---|---|---|
| 1b | 6 | 1.2 | 2.5 | 2142 | 79 |
| 7c | 37 | 4.5 | 2.7 | 123 | 27 |
| 14c | 311 | 2.3 | 0.2 | ||
| 17c | 68 | 6.7 | 1.7 | 295 | |
| 28c | 20 | 18.9 | 12.8 | 676 | 79 |
| 31c | 90 | 16.1 | 3.6 | 149 | 79 |
| 35d | 36 | 14.7 | 7.3 | 365 | 70 |
| 34d | 71 | 9.4 | 1.7 | 87 | 65 |
| 46d | 17 | 12.0 | 10.8 | 578 | 59 |
| 49d | 20 | 12.5 | 8.6 | 967 | 86 |
Sprague–Dawley rats. CL: mL/min/kg; Vd: L/kg; DNAUCpo: (ng·h/mL)/(mg/kg); t1/2: iv, hour; F: %.
iv: 1 mg/kg, po: 5 mg/kg, n = 3.
iv: 0.5 mg/kg, po: 2 mg/kg, n = 3.
iv: 1 mg/kg, po: 2 mg/kg, n = 3.
Compound 49 is selective against >350 kinases and 30 histone methyltransferases (<25% and <12% inhibition was observed at 10 μM versus kinase and HMT panels, respectively, Supporting Information Tables S2 and S3). The balanced and clean profile, excellent potency, and pharmacokinetic properties make compound 49 an excellent compound for use in evaluating the biological role of SMYD3 activity in both cells and animals.
Conclusion
While other small molecule SMYD3 inhibitors have been reported and a similar isoxazole analog of this series has been exemplified,12,13 the compounds disclosed here represent a detailed SAR study of this class. In particular, a close analog of this class, EPZ028862, showed excellent inhibition of SMYD3 and selectivity against SMYD2.13 Some of the analogs reported herein are more potent than previously reported inhibitors and have a better PK profile, including higher bioavailability, longer t1/2, and/or lower clearance. While all reported inhibitors bind in the same binding site of SMYD3 and the ends of each inhibitor reach the same binding elements, they do so through a different trajectory. EPZ028862 generally makes similar interactions as the compounds reported here, though in contrast the basic amine of compound 28 is observed to come in close proximity with the negatively charged Asp241 and Glu192 side chains while the basic amine of EPZ028862 is stabilized by solvent mediated interactions (Figure 4).13 Furthermore, the trifluorobutane group of compound 28 extends deeper into a hydrophobic region of the substrate binding site.
In summary, the compounds disclosed in this paper represent a novel structural class of SMYD3 inhibitors. Modification of the HTS hit, compound 1, with the structure guided SAR study led to compound 49, a potent, selective, orally bioavailable small molecule inhibitor of SMYD3 which can serve as a useful in vitro and in vivo tool molecule to investigate the role of SMYD3 methyltransferase activity in cancer and other disorders.
Acknowledgments
We thank Patrick Stoy and Guosen Ye for supporting the early chemistry hit discovery effort.
Glossary
Abbreviations
- SMYD3
SET and MYND Domain-Containing Protein 3
- MEKK2
mitogen-activated protein kinase kinase 2
- SAM
S-adenosyl methionine
- SAH
S-adenosyl-l-homocysteine
- H4K5
histone 4 lysine 5
- ERK
extracellular signal-regulated kinases
- siRNA
small interfering RNA
- shRNA
short hairpin RNA
- PK
pharmacokinetics
- IC50
half maximal inhibitory concentration
- CLND
chemiluminescent nitrogen detection
- AMP
artificial membrane permeability
- EGF
epidermal growth factor
- MOI
mode of inhibition
- HTS
high throughput screening
- SAR
structure activity relationship
- MAPK
mitogen-activated protein kinase
- ADME
absorption, distribution, metabolism, and excretion
- CL
clearance
- Vd
volume distribution
- DNAUC
dose normalized area under curve
- HMT
histone methyltransferase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00493.
ADME/PK methods, cellular assay conditions, biochemical assay conditions and MOI data, crystallography methods and data, kinase and HMT panels screening conditions and results, synthesis and data for compound 49 (PDF)
Accession Codes
Coordinates have been deposited in the Protein Data Bank with accession codes 6PAF, 6P6K, 6P7Z and 6P6G.
Author Present Address
⊥ RN: University of California Davis; WGB: Janssen Pharmaceuticals, Spring House, PA 19477; JL: Prelude Therapeutics; DAH: retired.
The authors declare no competing financial interest.
Supplementary Material
References
- Lanouette S.; Mongeon V.; Figeys D.; Couture J. F. The functional diversity of protein lysine methylation. Mol. Syst. Biol. 2014, 10, 724. 10.1002/msb.134974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Connolly J.; Biggar K. K. Beyond histones - the expanding roles of protein lysine methylation. FEBS J. 2017, 284 (17), 2732–2744. 10.1111/febs.14056. [DOI] [PubMed] [Google Scholar]
- Van Aller G. S.; Reynoird N.; Barbash O.; Huddleston M.; Liu S.; Zmoos A. F.; McDevitt P.; Sinnamon R.; Le B.; Mas G.; Annan R.; Sage J.; Garcia B. A.; Tummino P. J.; Gozani O.; Kruger R. G. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 2012, 7 (4), 340–3. 10.4161/epi.19506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur P. K.; Reynoird N.; Khatri P.; Jansen P. W.; Wilkinson A. W.; Liu S.; Barbash O.; Van Aller G. S.; Huddleston M.; Dhanak D.; Tummino P. J.; Kruger R. G.; Garcia B. A.; Butte A. J.; Vermeulen M.; Sage J.; Gozani O. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510 (7504), 283–7. 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y.; Grabocka E.; Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11 (11), 761–74. 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto R.; Furukawa Y.; Morita M.; Iimura Y.; Silva F. P.; Li M.; Yagyu R.; Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6 (8), 731–40. 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- Liu L.; Kimball S.; Liu H.; Holowatyj A.; Yang Z. Q. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 2015, 6 (4), 2466–82. 10.18632/oncotarget.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Liu Y.; Kong F.; Xin W.; Li X.; Liang H.; Jia Y. Elevated Levels of SET and MYND Domain-Containing Protein 3 Are Correlated with Overexpression of Transforming Growth Factor-beta1 in Gastric Cancer. J. Am. Coll Surg 2015, 221 (2), 579–90. 10.1016/j.jamcollsurg.2015.02.023. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Liu H.; Luo X.; Deng J.; Pan Y.; Liang H. Overexpression of SMYD3 and matrix metalloproteinase-9 are associated with poor prognosis of patients with gastric cancer. Tumor Biol. 2015, 36 (6), 4377–86. 10.1007/s13277-015-3077-z. [DOI] [PubMed] [Google Scholar]
- Kim J. M.; Kim K.; Schmidt T.; Punj V.; Tucker H.; Rice J. C.; Ulmer T. S.; An W. Cooperation between SMYD3 and PC4 drives a distinct transcriptional program in cancer cells. Nucleic Acids Res. 2015, 43 (18), 8868–83. 10.1093/nar/gkv874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira F. Q.; Costa-Pinheiro P.; Almeida-Rios D.; Graca I.; Monteiro-Reis S.; Simoes-Sousa S.; Carneiro I.; Sousa E. J.; Godinho M. I.; Baltazar F.; Henrique R.; Jeronimo C. SMYD3 contributes to a more aggressive phenotype of prostate cancer and targets Cyclin D2 through H4K20me3. Oncotarget 2015, 6 (15), 13644–57. 10.18632/oncotarget.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. H.; Boriack-Sjodin P. A.; Smith S.; Thomenius M.; Rioux N.; Munchhof M.; Mills J. E.; Klaus C.; Totman J.; Riera T. V.; Raimondi A.; Jacques S. L.; West K.; Foley M.; Waters N. J.; Kuntz K. W.; Wigle T. J.; Scott M. P.; Copeland R. A.; Smith J. J.; Chesworth R. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7 (2), 134–8. 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomenius M. J.; Totman J.; Harvey D.; Mitchell L. H.; Riera T. V.; Cosmopoulos K.; Grassian A. R.; Klaus C.; Foley M.; Admirand E. A.; Jahic H.; Majer C.; Wigle T.; Jacques S. L.; Gureasko J.; Brach D.; Lingaraj T.; West K.; Smith S.; Rioux N.; Waters N. J.; Tang C.; Raimondi A.; Munchhof M.; Mills J. E.; Ribich S.; Porter Scott M.; Kuntz K. W.; Janzen W. P.; Moyer M.; Smith J. J.; Chesworth R.; Copeland R. A.; Boriack-Sjodin P. A. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS One 2018, 13 (6), e0197372 10.1371/journal.pone.0197372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aller G. S.; Graves A. P.; Elkins P. A.; Bonnette W. G.; McDevitt P. J.; Zappacosta F.; Annan R. S.; Dean T. W.; Su D. S.; Carpenter C. L.; Mohammad H. P.; Kruger R. G. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure 2016, 24 (5), 774–781. 10.1016/j.str.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22 (23), 3099–108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.