

Abstract

Hepatitis B virus (HBV) remains a major health concern with 260 million people having been infected globally, and approximately 680,000 deaths have occurred annually from cirrhosis and liver cancer. The modulation of HBV capsid assembly has emerged as a promising therapeutic approach for curing chronic HBV infection. Small-molecule capsid assembly modulators (CAMs) can broadly be classified as heteroaryldihydropyrimidines and sulfamoylbenzamides (SBAs). SBAs are capsid activators that inhibit viral replication by achieving capsid assembly before polymerase encapsulation. Herein, we report a novel series of HBV CAMs based on NVR 3-778, a potent CAM belonging to the SBA class. The lead compound (KR-26556) exhibited improved pharmacological activity and was examined through molecular docking studies.
Keywords: Hepatitis B, capsid assembly, small-molecules, anti-HBV, docking studies
Chronic hepatitis B (CHB), which is caused by hepatitis B virus (HBV), is a major health problem affecting approximately 260 million patients globally. CHB is the major cause of chronic liver diseases and hepatocellular carcinoma.1,2 Existing therapies for CHB include interferon alpha and several nucleoside/nucleotide analogs.3,4 However, existing treatment regimens cannot functionally cure HBV, and most patients require further treatment.5−7 Because of effective vaccination campaigns, the prevalence of HBV in children has been reduced to 1.3%. However, the initial birth dose vaccination is only 39%, indicating that a large population remains at risk for HBV infection.8
No FDA-approved therapeutics for CHB can completely cure the infection because of the lack of their ability to eradicate the covalently closed circular DNA (cccDNA) from the nuclei of infected hepatocytes.9 Improved suppression of viral replication and durable response rates require the development of new therapies for CHB infection. Studies are needed to identify and develop anti-HBV agents that operate by novel mechanisms distinct from current treatments.10,11
The core protein of HBV (HBc) plays multiple roles in the HBV life cycle. The key roles of HBc comprised capsid structure and assembly, regulation of transcription, interaction with host factors, and phosphorylation. HBc has become a privileged drug target for the development of innovative direct-acting antiviral agents.12 To improve the treatment of CHB, capsid assembly modulators (CAMs) have been developed and several have entered human clinical trials (Figure 1).9 At the onset of infection, the relaxed circular DNA is transformed into cccDNA in hepatocytes. Transcription of cccDNA results in the production of pregenomic RNA (pgRNA) and viral mRNAs, which in turn produce viral protein, precore, core, polymerase, HBV surface agents, and HBV X proteins.
Figure 1.

Representative CAMs.
The capsid formed from its building blocks (HBV core proteins) encloses pgRNA and viral polymerase during capsid assembly. The capsid serves as the site for reverse transcription, leading to the product on of viral DNA, and ultimately allows for the development of HBV DNA-containing infectious viral particles. Small molecules that bind to the core protein or capsid can interfere with efficient capsid assembly or disassembly and often hasten capsid assembly in the absence of HBV RNA or polymerase. These small molecules, referred to as capsid assembly modulators (CAMs), can interfere with pgRNA encapsidation and HBV DNA replication by accelerating or misdirecting the formation of capsid-like structures. Moreover, by blocking the formation of infectious HBV virions, CAMs also block the production of HBV RNA-containing particles in persistently infected cells and interfere with cccDNA formation when CAMs are present at the time of de novo infection.2
In our ongoing efforts to identify more effective CAMs, we discovered a more potent derivative of compound 3 (NVR 3-778) with significantly improved anti-HBV activity in vitro. The superimposed cocrystal structures of compound 2 (HepG2.2.15 EC50: 0.0064 μM), (PDB: 5WRE) and NVR 3-778 (HepG2.2.15 EC50: 0.26 μM) (PDB: 5T2P)13 with HBV capsid protein revealed that most binding interactions are similar. However, in compound 2, the thiazole moiety at position 2 of the 1,4-dihydropyrimidine core affords additional interactions with the HBV capsid protein (Figure 2a). The higher potency of compound 2 may originate from these additional interactions of the thiazole ring with HBV capsid protein. NVR 3-778 lacks such interactions, likely leading to its inferior potency compared to compound 2. Thus, we predicted that similar interactions could be introduced between NVR 3-778 and the HBV capsid protein by installing appropriate moieties on the benzamide core of NVR 3-778 (Figure 2b). Such modifications may improve potency.
Figure 2.

Research design. (a) Superimposition of compound 2 (in cyan) and NVR 3-778 (in magenta) bound to HBV capsid proteins. The key hydrophobic and hydrophilic interactions are shown. The thiazole moiety 1,4-dihydropyrimidine core of compound 2 with additional interactions can be seen. (b) Structures of compound 2 and NVR 3-778 highlighting the strategy of additional substitutions on the benzamide core of NVR 3-778.
Structure activity relationship (SAR) studies were performed to investigate the role of fluorine at position 4 of the benzamide core of NVR 3-778. Removal of fluorine at position 4 resulted in a several fold-loss in activity (compound 4, Table 1), indicating the essential role of fluorine at this position. We next placed a methoxy group at position 6 of compound 4 to form additional hydrogen bonding interactions (compound 5, Table 1). However, a complete loss of potency was observed for compound 5 (EC50 > 3.30 μM). In a similar manner, compound 6 with a hydroxyl group at position 6 of the benzamide core was found to be inactive (EC50 > 3.30 μM). Interestingly, methyl sulfide analogue (compound 7) considerably restored the potency with an EC50 of 0.72 μM. Compound 8 with an indole core which was designed to mimic the thiazole moiety of compound 2 was found to be inactive. Based on these findings, we next introduced a primary amine at position 6 of the benzamide core. A significant improvement in potency was observed (compound 9, EC50: 0.24 μM). However, the tertiary amine analogue (compound 10) was found to be inactive.
Table 1. SAR of Benzamide Core in the Absence of C-4 Fluoride.


Antiviral effect of compounds. After 65 h of treatment with compounds, the HepAD38 cells were harvested and intracellular HBV DNA was extracted and quantified (Real-time PCR Assay).
Cytotoxicity of compounds determined by cell viability assay using HepAD38 cells.
Reinstallation of a fluorine atom at position 4 of the benzamide core further improved the potency, and compound 11 (KR-26556) was found to be 9-fold more potent than NVR 3-778 (Table 2). Interestingly when the primary amine of compound 11 was replaced with a secondary methylamine moiety, the activity was completely diminished (compound 12). This demonstrated the crucial role of the NH2 group at position 6 of the benzamide core. Changing the position of the primary amine from 6 to 5 at the benzamide core resulted in a significant loss of potency (compound 13, Table 2). However, compound 13 was still slightly more potent than NVR 3-778. Moreover, hydrophobic groups were not tolerated at position 5 of the benzamide core. For instance, compound 14 with a bromo at position 5 was completely inactive (EC50 > 3.30 μM).
Table 2. SAR of Benzamide Core in the Presence of C-4 Fluoride.


Antiviral effect of compounds. After 65 h of treatment with compounds, the HepAD38 cells were harvested and intracellular HBV DNA was extracted and quantified (real-time PCR Assay).
Cytotoxicity of compounds deteremined by cell viability assay using HepAD38 cells.
To gain insight into the factor-enhanced viral inhibitory activity at the molecular level, molecular dynamics (MD) simulation was carried out to determine the binding mode of 11 against HBV core protein (Figure 3). The structure of 11 bound the HBV core based on the docking used as the starting structure in MD simulation. The proposed binding mode of 11 was very similar to that of NVR 3-778 in the same pocket at the BC interface of the core protein. The trifluoro-substituted phenyl group of 11 bound in a deep subpocket formed by Pro25, Leu30, Thr33, and Ser106 on the B chain and Val124 and Arg127 on the C chain. The benzamide oxygen of KR-26556 formed a key hydrogen bond with Trp102, where the benzamide nitrogen additionally hydrogen bonded with Thr128. The piperidyl group of 11 faced the solvent exposed area in a similar binding mode as NVR 3-778. Based on MD simulation, when 11 bound to the core protein, hydrogen bonding interactions were observed between the hydroxyl group of piperidyl and Thr142. Notably, the 2-amino substituent of benzamide of 11 interacted with Tyr118 through hydrogen bonding in a high-degree interactions fraction. This feature may be one reason for the high antiviral activity of 11 against HBV core protein, as this interaction was lacking in NVR 3-778 (see Supporting Information, Figure SI-6).
Figure 3.

Molecular dynamics simulation and interaction profile analysis of 11 for HBV core protein. (a) Predicted binding mode of 11 (yellow ball and stick model). The B and C chains are displayed in the ribbon model, colored in red and green, respectively. The hydrogen bonds are shown as green dashed lines and hydrophobic interactions are represented by pink dashed lines. (b) Contact histograms of HBV core protein with compound 8 are represented as a colored stacked bar plot between 50 and 150 ns of MD simulation. Only H-bonding (yellow-green colored bar) and representative hydrophobic interactions (lavender colored bar) are presented for clarity.
We subsequently focused on the safety assessment and in vitro pharmacokinetic profiling of 11 (Table 3). We first measured the inhibition of cytochrome P450 (CYP) by 11. Compound 11 was found to inhibit various CYP enzymes with IC50 values in the high micromolar range, showing a relatively safer profile. The cardiotoxicity of 11 was evaluated using hERG ligand binding assay and hERG K+ channel activity assay (IC50 of 8.78 and 10.9 μM, respectively). The high micromolar IC50 values of 11 in both the hERG assays were a preliminary validation that the compound might not be causing a considerable cardiotoxicity. We next evaluated the drug-like properties of this lead compound. Compound 11 showed promising liver microsomal and plasma stability in rat and human in vitro. The plasma protein binding levels were 98.67% and 99.27% in rat and human, respectively, in vitro presumably due to a high fraction of lipophilic fluorine atoms despite the fact that compound 11 possesses a lower CLogP of 1.64.
Table 3. Safety and in Vitro Pharmacokinetic Profile of Compound 11.
| CYP Inhibition Assaya (IC50, μM) | |
| 1A2 | 81.13 ± 13.26 |
| 2C9 | 17.02 ± 3.67 |
| 2C19 | 23.06 ± 1.96 |
| 2D6 | 32.58 ± 7.40 |
| 3A4 | 27.84 ± 1.92 |
| Cardiotoxicity (IC50, μM)b | |
| hERG ligand binding assay | 8.78 (n = 3) |
| hERG K+ channel activity assay | 10.9 (n = 3) |
| Liver Microsomal Phase I Stabilityc | |
| Rat (%) | 49.18 ± 2.8 |
| Human (%) | 50.24 ± 5.88 |
| Rat (t1/2, min) | 28.88 ± 0.72 |
| Plasma Stability (%)d | |
| Rat (%) | 90.10 ± 4.78 |
| Human (%) | 71.87 ± 2.20 |
| Plasma Protein Binding (%)e | |
| Rat (%) | 98.67 ± 0.09 |
| Human (%) | 99.27 ± 0.07 |
IC50 values (μM) in human liver microsomes using cocktail substrate assay (mean ± SD, n = 3).
See Supporting Information (p 8) for this part.
Liver microsomal phase I stability (% of remaining after 30 min), (mean ± SD, n = 3).
Experiment about percentage of remaining after 4 h incubation at 37 °C, (mean ± SD, n = 3).
Plasma protein binding rate (%) (mean ± SD, n = 3).
Lastly, compound 11 was evaluated for its in vivo pharmacokinetics. Table 4 shows the plasma concentration of 11 in rats after intravenous and oral administration. Compound 11 reaches the maximum plasma concentration (Cmax) after 1.58 h of oral administration indicating a moderate drug absorption. The half-lifes (T1/2) were found to be good after intravenous (6.67 h) and oral administration (3.44 h). 11 retains good exposure (AUC) after IV (6.3 μg.h/mL) and PO (5.05 μg.h/mL) administration. Encouraging clearance (CL) after IV administration (0.81 L/h/kg) along with a moderate volume of distribution (2.95 L/kg) was observed. Excellent oral bioavailability (80%) makes compound 11 suitable for IV as well as PO dosing.
Table 4. In Vivo Pharmacokinetic Parameters of Compound 11 in Male Rats.
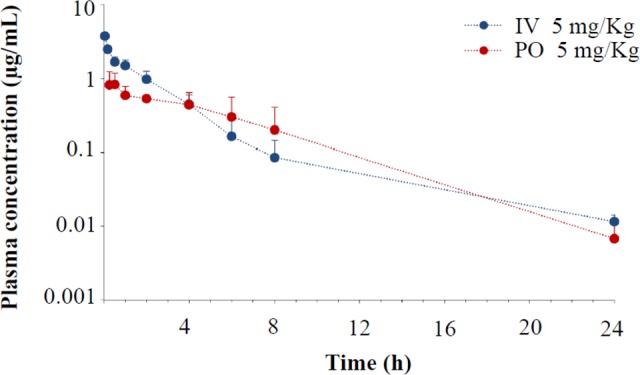
| Parametera | I.V.b (5 mg/kg) | P.O.c (5 mg/kg) |
|---|---|---|
| Tmax (h) | N/A | 1.58 ± 2.1 |
| Cmax (μg/mL) | N/A | 0.94 + 0.23 |
| T1/2 (h) | 6.67 ± 3.99 | 3.44 ± 0.37 |
| AUClast (μg·h/mL) | 6.3 ± 1.76 | 5.05 ± 2.37 |
| AUC, (μg·h/mL) | 6.42 ± 1.7 | 5.09 ± 2.39 |
| CL (L/h/kg) | 0.81 + 0.2 | N/A |
| Vss (L/kg) | 2.95 ± 1.01 | N/A |
| Ft (%) | N/A | 80.19 |
Each value is presented as mean ± standard deviation of at least three independent experiments.
Parameters from intravenous administration.
Parameters from oral administration.
Compounds 4–6 and 12 were prepared according to Scheme 1. Chlorosulfonylation was achieved by employing chlorosulfonic acid at 150 °C for 12 h. The resulting carboxylic acid with chlorosulfonyl groups at position C3 was converted to acid chlorides followed by a reaction with 3,4,5-trifluoroaniline to generate the desired amide bond at the C1 position. Finally, piperidin-4-ol was treated with the chlorosulfonyl intermediates to obtain compounds 4, 5, and 12. BBr3-mediated demethylation of compound 5 afforded compound 6.
Scheme 1. Synthetic Route via Acid Chloride.

Reagents and conditions: (a) ClSO3H, 0 °C, 150 °C, 12 h; (b) SOCl2, 80 °C, 12 h; (c) 3,4,5-trifluoroaniline, toluene, reflux; (d) piperidin-4-ol, MeCN, rt, 3 h; (e) 1.0 M BBr3 solution in dichloromethane, DCM, −10 °C to rt.
Syntheses of compounds 9–11 commenced with trifluoroacetamide protection of 2-aminobenzoic acids 9a and 11a. Chlorosulfonylation afforded the desired sulfonyl chlorides 9c and 11c, which were further reacted with piperidin-4-ol to furnish the desired intermediates 9d and 11d. Amidation using HATU in the presence of DIPEA afforded 9e and 11e. Trifluoroacetamide deprotection afforded compounds 9 and 11.
Dimethylation of compound 9 affords the dimethyl amino compound 10 (Scheme 2).
Scheme 2. Synthetic Route for Compounds 9–11.

Reagents and conditions: (a) Trifluoroacetic anhydride, DCM, rt, 4 h; (b) ClSO3H, 150 °C, 16 h; (c) 4-piperidinol, DIPEA, MeCN, rt, overnight; (d) 3,4,5-trifluoroaniline, HATU, DIPEA, DMF, rt–50 °C, overnight; (e) LiOH·H2O, THF:H2O, 50 °C, 48 h; (f) 1 N NaOH aqueous solution, MeOH, 30 °C, 2 h; (g) 35% formaldehyde aq solution, NaCNBH3, AcOH, MeCN, 0 °C, 1 h.
Scheme 3 illustrates the synthesis of compounds 13 and 14. Chlorosulfonylation of 3-bromo-4-fluorobenzoic acid afforded 3-bromo-5-(chlorosulfonyl)-4-fluorobenzoic acid, which was reacted with piperidin-4-ol to afford 3-bromo-4-fluoro-5-((4-hydroxypiperidin-1-yl)sulfonyl)benzoic acid. Amidation with 3,4,5-trifluoroaniline afforded compound 14, which was converted to compound 13 by Buchwald–Hartwig cross coupling with tert-butyl carbamate and deprotection (see the Supporting Information for the synthesis of other compounds).
Scheme 3. Synthesis of Compounds 13 and 14.

Reagents and conditions: (a) ClSO3H, 170 °C, 72 h; (b) 4-piperidinol, DIPEA, dioxane/water rt, 2 h; (c) 3,4,5-trifluoroaniline, HATU, DIPEA, DMF, rt, 24 h; (d) tert-butyl carbamate, Pd2(dba)3, Xphos, Cs2CO3, toluene, 100 °C, 2 h; (e) 4 N HCl in dioxane, EtOAc, rt, 8 h.
In conclusion, we successfully introduced chemical modifications on NVR 3-778 based on in silico studies to afford 11 with improved antiviral activity in vitro. The binding mode and importance of substitution at position 6 of the benzamide core were determined by MD studies. 11, with a strong potency, tolerable safety profile, and adequate drug-like properties, is a promising lead compound for anti-HBV drug discovery. Taken together, these findings may be helpful for completely eradicating CHB infection.
Acknowledgments
This study was supported by Korea Research Institute of Chemical Technology.
Glossary
Abbrebiations
- IV
intravenous
- PO
per os (by oral)
- Tmax (h)
time of maximum drug concentration
- Cmax (mg/mL)
maximum plasma concentration
- T1/2 (h)
terminal half-life
- AUClast (μg·h/mL)
areas under the plasma concentration–time curve from time zero to time of last measurable concentration
- AUC∞(μg·h/mL)
areas under the plasma concentration–time curve from time zero to time infinity
- CL (L/h/kg)
total clearance from plasma
- Vss (L/kg)
steady-state volume of distribution
- Ft (%)
bioavailability.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00550.
Detailed synthetic procedures and characterization data for key compounds, including copies of 1H and 13NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cole A. G. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr. Opin. Pharmacol. 2016, 30, 131–137. 10.1016/j.coph.2016.08.004. [DOI] [PubMed] [Google Scholar]
- Lam A. M.; Espiritu C.; Vogel R.; Ren S.; Lau V.; Kelly M.; Kuduk S. D.; Hartman G. D.; Flores O. A.; Klumpp K. Preclinical Characterization of NVR 3–778, a first-in-class capsid assembly modulator against hepatitis B virus. Antimicrob. Agents Chemother. 2019, 63, e01734–18. 10.1128/AAC.01734-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N.; Zhao F.; Jia H.; Rai D.; Zhan P.; Jiang X.; Liu X. Non-nucleoside anti-HBV agents: advances in structural optimization and mechanism of action investigations. MedChemComm 2015, 6, 521–535. 10.1039/C4MD00521J. [DOI] [Google Scholar]
- Wong V.; Chan H. Chronic hepatitis B: a treatment update. Semin. Liver Dis. 2013, 33, 122–129. 10.1055/s-0033-1345715. [DOI] [PubMed] [Google Scholar]
- Feng S.; Gao L.; Han X.; Hu T.; Hu Y.; Liu H.; Thomas A. W.; Yan Z.; Yang S.; Young J. A. T.; Yun H.; Zhu W.; Shen H. C. Discovery of small molecule therapeutics for treatment of chronic HBV infection. ACS Infect. Dis. 2018, 4, 257–277. 10.1021/acsinfecdis.7b00144. [DOI] [PubMed] [Google Scholar]
- Lampertico P.; Agarwal K.; Berg T.; Buti M.; Janssen H. L. A.; Papatheodoridis G.; Zoulim F.; Tacke F. EASL 2017 Clinical Practice Guidelines on the mananagment of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. 10.1016/j.jhep.2017.03.021. [DOI] [PubMed] [Google Scholar]
- Terrault N. A.; Bzowej N. H.; Chang K. M.; Hwang J. P.; Jonas M. M.; Murad M. H. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016, 63, 261–283. 10.1002/hep.28156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blach S.; Razavi H.; Razavi-Shearer D.; Edmunds J.; Jones E.; Apolloni A.; Hasso-Agopsowicz M.; De la Hoz-Restrepo F.; Vargas R.; Soares-Wieser K.; Ott J.. World Health Organization Global Hepatitis Report 2017. https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed November 24, 2019). [Google Scholar]
- Boucle S.; Lu X.; Bassit L.; Ozturk T.; Russell O. O.; Amblard F.; Coats S. J.; Schinazi R. F. Synthesis and antiviral evaluation of novel heteroarylpyrimidines analogs as HBV capsid effectors. Bioorg. Med. Chem. Lett. 2017, 27, 904–910. 10.1016/j.bmcl.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Wang G. A review of non-nucleoside anti-hepatitis B virus agents. Eur. J. Med. Chem. 2014, 75, 267–281. 10.1016/j.ejmech.2014.01.046. [DOI] [PubMed] [Google Scholar]
- Fanning G. C.; Zoulim F.; Hou J.; Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat. Rev. Drug Discovery 2019, 18, 827–844. 10.1038/s41573-019-0037-0. [DOI] [PubMed] [Google Scholar]
- Diab A.; Foca A.; Zoulim F.; Durantel D.; Andrisani O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antiviral Res. 2018, 149, 211–220. 10.1016/j.antiviral.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Hu T.; Zhou X.; Steffen W.; Fernando G.-A.; Zhiheng X.; Daitze W.; Mao Y.; Xiaojun T.; Zhou Y.; Shen F.; Zhisen Z.; Guozhi T.; Najera I.; Guang Y.; Shen H. C.; Young J. A. T.; Ning Q. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms. Sci. Rep. 2017, 7, 42374. 10.1038/srep42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.