

Abstract

Inhibition of neprilysin (NEP) is widely studied as a therapeutic target for the treatment of hypertension, heart failure, and kidney disease. Sacubitril/valsartan (LCZ696) is a drug approved to reduce the risk of cardiovascular death in heart failure patients with reduced ejection fraction. LBQ657 is the active metabolite of sacubitril and an inhibitor of NEP. Previously, we have reported the crystal structure of NEP bound with LBQ657, whereby we noted the presence of a subsite in S1′ that has not been explored before. We were also intrigued by the zinc coordination made by one of the carboxylic acids of LBQ657, leading us to explore alternative linkers to efficiently engage zinc for NEP inhibition. Structure-guided design culminated in the synthesis of selective, orally bioavailable, and subnanomolar inhibitors of NEP. A 17-fold boost in biochemical potency was observed upon addition of a chlorine atom that occupied the newly found subsite in S1′. We report herein the discovery and preclinical profiling of compound 13, which paved the path to our clinical candidate.
Keywords: Neprilysin, crystal structure, structure-based design, structure−activity relationship
NEP (EC 3.4.24.11; neprilysin, neutral endopeptidase, enkephalinase; atriopeptidase) is a zinc-dependent type II integral membrane endopeptidase belonging to the peptidase M13 family. It cleaves a variety of peptide substrates such as atrial natriuretic peptide (ANP, also known as ANF), brain natriuretic peptide (BNP), bradykinin, adrenomedullin, and endothelin-1.1 The increase in circulating ANP levels achieved by NEP inhibition has favorable implications for the treatment of congestive heart failure and chronic renal failure.2,3 Sacubitril/valsartan (LCZ696) is a drug approved to reduce the risk of cardiovascular death and hospitalization for patients with chronic heart failure (NYHA Class II–IV) and reduced ejection fraction (HFrEF).4 LCZ696 (Figure 1) is a sodium salt complex containing the anionic forms of the angiotensin II type 1 receptor antagonist valsartan and the NEP inhibitor prodrug sacubitril.5 Upon oral administration, LCZ696 provides rapid systemic exposure to sacubitril and valsartan. Sacubitril is converted by enzymatic cleavage of the ethyl ester to the active metabolite LBQ657 (compound 1, Figure 2b).6
Figure 1.

Schematic structural formula of LCZ696, showing sacubitril and valsartan, in their respective anionic forms.
Figure 2.

(a) Crystal structure of NEP (white carbon atoms) with LBQ657 (yellow) showcasing the S1′ subsite. The ligand binding pocket is shown in gray and zinc atom is in magenta. H-bonding interactions are shown as black dotted lines and crystallographic waters are shown as red spheres. The distance between the meta position of the aromatic ring and two S1′ residues (Arg717 and Thr721) is shown in green. (b) Schematic representation of the concepts studied in this paper.
We have reported the crystal structure of LBQ657 in complex with NEP, rationalizing its potency and selectivity.7 LBQ657 binds in the active site of NEP making an intricate network of H-bonding and van der Waals interactions that involves all functional groups of the compound giving rise to the high inhibitory potency of 5 nM8 (Figure 2a).
In the crystal structure of LBQ657 in complex with NEP we observed a small subsite in S1′ extending out from the meta-position of the phenyl ring toward the backbone carbonyl atom of Arg717 and side chain of Thr721 (Figure 2). To the best of our knowledge, interactions in this region were not explored at the time when we conducted the study, leading us to probe the structure–activity relationship (SAR) at this position. One other observation made from the cocrystal structure was the zinc coordination made by LBQ657. Unlike many previously reported NEP inhibitor complex structures,9−13 LBQ657 has a longer linker between the zinc coordinating group (carboxylate) and the chiral carbon atom that anchors the S1′ and S2′ binding moieties (Figure 2b). This led us to study alternative linkers to efficiently engage zinc for NEP inhibition. We investigated both of these findings by initially synthesizing a few rationally designed compounds. Encouraged by the results, we delved into a systematic exploration of the S1′ SAR. In this report we describe the medicinal chemistry optimization, SAR, validation of the binding mode by cocrystallization with NEP, and results from preclinical investigation of selected compounds.
As mentioned above, in four out of the seven previously reported NEP inhibitor cocrystal structures,10−13 the zinc coordinating moiety, which is either a thiol or phosphonic acid analog, is separated from the chiral anchor atom by a single linker atom which is either carbon or nitrogen. In LBQ657 there are two atoms including a chiral carbon atom that serves as the linker presenting the carboxylic acid for zinc coordination. We were curious to study the impact of modifying the linker group and hence made compounds 2–4, Table 1. A biochemical assay was developed to assess NEP inhibition in vitro using recombinant human NEP (Supporting Information). As shown in Table 1, removing the chiral methyl group in LBQ657 resulted in almost a 100-fold loss in NEP inhibitory activity (compound 2) while the dimethyl analog 3 was 3-fold less active. Based on these observations one may conclude that the chiral methyl group in LBQ657 rigidifies the linker and facilitates zinc coordination via the carboxylic acid. Shortening the linker length by one carbon atom (4) was equipotent to 3 indicating zinc coordination is plausible albeit less efficient. Considering the biochemical potency of 4 was within 5-fold of LBQ657, we characterized the in vitro absorption, distribution, metabolism, and elimination (ADME) parameters followed by in vivo rat pharmacokinetic (PK) profile. Compound 4 was stable in both rat and human liver microsomes (Clint < 3.42 μL/min/mg in rat and human) and hepatocytes (Clint = 2.1 and 0.68 μL/min per million cells in rat and human, respectively). When administered to Sprague–Dawley rats at 1 mg/kg IV, the plasma clearance of 4 was low (11 mL/min/kg) with a small volume of distribution (0.2 L/kg) and half-life of 2.5 h. Based on our experience with sacubitril the prodrug of LBQ657, the ethyl ester prodrug of 4 was administered orally at 3 mg/kg. The prodrug of 4 showed rapid absorption and complete hydrolysis to generate 4 with a bioavailability of ∼66%.
Table 1. Impact of Linker Length on NEP Inhibitory Activitya.
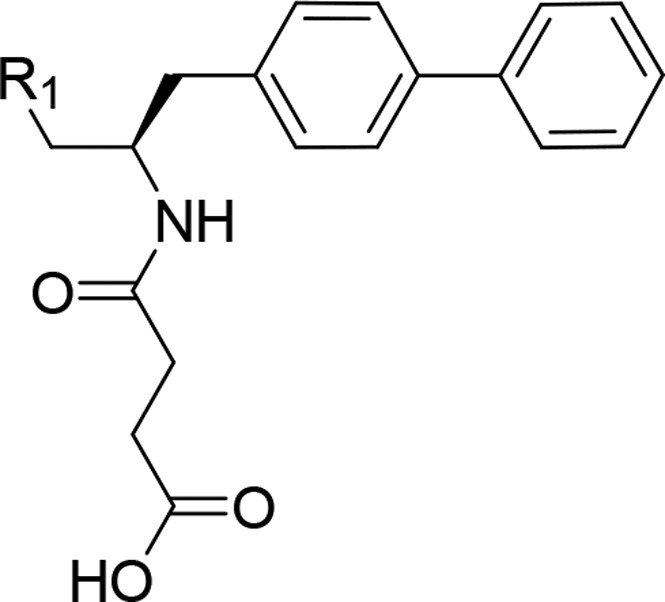
| Compound | R1 | hNEP IC50 (nM) |
|---|---|---|
| 1 (LBQ657) | CH(CH3)COOH (R) | 6 |
| 2 | CH2COOH | 550 |
| 3 | C(CH3)2COOH | 20 |
| 4 | COOH | 25 |
IC50 values are the mean of at least two independent assays.
Encouraged by the PK data, we decided to study the in vivo pharmacodynamics (PD) in an ANP potentiation model in conscious rats.6 In this animal model, conscious chronically cannulated adult male Sprague–Dawley rats were infused intravenously with rat ANP to saturate the natriuretic peptide clearance receptors and, thereby, shift the elimination of ANP to predominantly enzymatic degradation by NEP. Blood samples were withdrawn before dosing with compound or vehicle (baseline) and at various times up to 8 h postdosing to simultaneously assess PK/PD. PD readout was the potentiation (% baseline) of plasma ANP (PANP), which was used as an index of a surrogate for the extent and duration of NEP inhibition. When administered orally at 30 mg/kg, the ethyl ester prodrug of 4 rapidly (within 15 min) augmented PANP (Figure 3a). PANP levels remained elevated for almost the entire experiment, reflecting the compound’s long duration of action. Plasma levels of 4 remained >2 μM (Figure 3b). In this model, the prodrug of 4 was found to be more effective than sacubitril when administered at 30 mg/kg orally.
Figure 3.
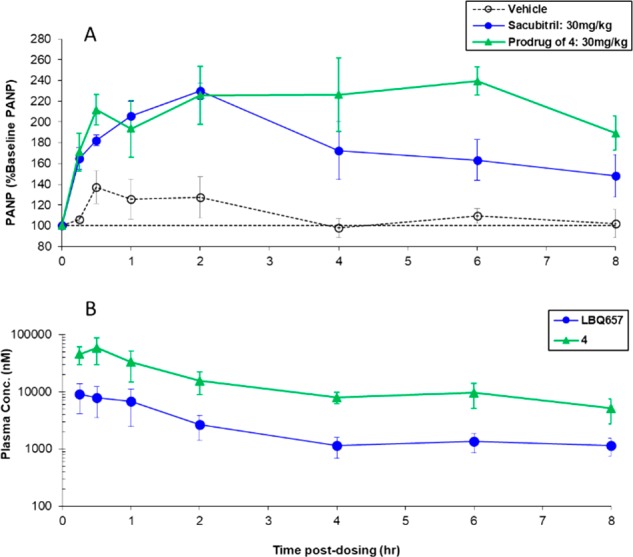
(a) Pharmacodynamic and (b) pharmacokinetic time courses of orally administered prodrug of 4 at 30 mg/kg in the ANP potentiation model in conscious rats.
Synthesis of 4 with one less chiral center was much simpler relative to LBQ657. The promising in vivo profile of 4 and its simplified synthesis resulted in the selection of 4 as a lead compound for SAR exploration of the S1′ binding moiety. In the LBQ657-bound NEP crystal structure, the distance between the meta position of the second phenyl ring (Figure 2) and the Arg717 backbone carbonyl was 5.3 Å. The side chain of Thr721 was at a distance of ∼5.6 Å. We started out by adding small substituents in this meta position (Table 2) to fill this space.
Table 2. SAR of S1′ Substitutiona.
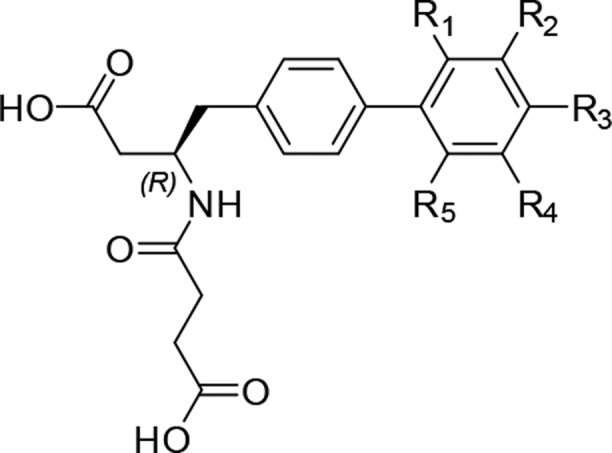
| Compound | R1 | R2 | R3 | R4 | R5 | hNEP IC50 (nM) |
|---|---|---|---|---|---|---|
| 4 | H | H | H | H | H | 25 |
| 5 | Cl | 8 | ||||
| 6 | F | 24 | ||||
| 7 | CH3 | 21.3 | ||||
| 8 | CN | 792 | ||||
| 9 | OCH3 | 4.5 | ||||
| 10 | OC2H5 | 413 | ||||
| 11 | CF3 | 1370 | ||||
| 12 | OCF3 | 212 | ||||
| 13 | Cl | 1.5 | ||||
| 14 | F | 6.7 | ||||
| 15 | NH2 | 44 | ||||
| 16 | CH3 | 1.6 | ||||
| 17 | C2H5 | 5.4 | ||||
| 18 | OCH3 | 60 | ||||
| 19 | CF3 | 111 | ||||
| 20 | OCF3 | 275 | ||||
| 21 | CN | 6.9 | ||||
| 22 | NO2 | 26 | ||||
| 23 | NHCOCH3 | 4862 | ||||
| 24 | Cl | 1950 | ||||
| 25 | F | 1500 | ||||
| 26 | Cl | OCH3 | 0.38 | |||
| 27 | OCH3 | Cl | 4182 | |||
| 28 | Cl | F | 54 |
IC50 values are the mean of at least two independent assays.
Compound 13 with chlorine at R2 was 17-times more active than its unsubstituted analog 4. Methyl analog 16 was equipotent to 13. The added substituents likely fill the S1′ subsite making van der Waals interaction with NEP residues thereby improving the binding affinity. The tolerance for bulk, H-bond donor/acceptor groups was also explored with various R2 substituents (13–23,Table 2) although no further improvement in potency was observed relative to 13 and 16. For additional confirmation that the improved potency seen in 13 was indeed a result of filling the space rather than adding a bulky group, chloro and fluoro substituents were added at ortho (5 and 6) and para position of the biphenyl ring (24 and 25), respectively. It was interesting to note that adding chlorine at the ortho position was 3-fold better compared to parent compound 4 while being ∼80-fold less active when substituted at the para position (24). This led us to further probe the ortho position although being conscious that bigger substituents would change the torsion between the two phenyl rings of the S1′ biphenyl group. Clearly, with the exception of compound 9 (vs 18), all substituents at the ortho position (5–12) and para position (24–25) were less active than the corresponding analogs at the meta position.
Considering the symmetrical nature of the phenyl ring, it was critical to understand the exact location of the methoxy group, so compounds 26 and 27 were synthesized. The significant loss of potency of 27 vs the synergistic improvement seen for 26 led us to conclude that the binding mode of 13 and 26 was comparable to LBQ657. Dimeta substitution was less explored as the initial data with 28 negated the beneficial effect of chloro substitution.
The profound impact on potency improvement upon chloro substitution was investigated on LBQ657. It was astounding to see a perfect translation of the SAR wherein a 20-fold improvement was seen for 30 (Table 3). The S-methyl isomer of 30 (compound 29) and the racemic analog 31 also had improved inhibitory potency relative to LBQ657.
Table 3. Testing SAR of S1′ Substitution in Pentanoic Acid Seriesa.
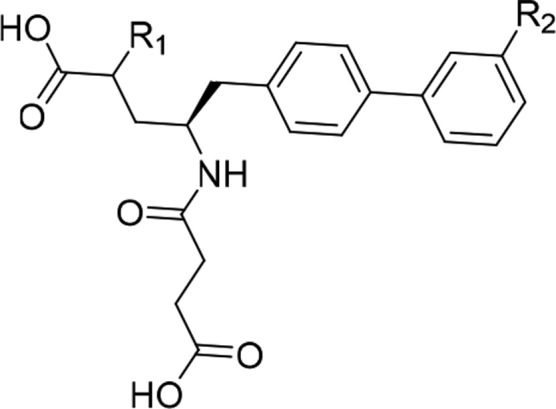
| Compound | R1 | R2 | hNEPIC50 (nM) |
|---|---|---|---|
| LBQ657 | (R)-CH3 | H | 6 |
| 29 | (S)-CH3 | Cl | 2 |
| 30 | (R)-CH3 | Cl | 0.3 |
| 31 | rac-CH3 | Cl | 0.8 |
IC50 values are the mean of at least two independent assays.
A typical synthesis of biphenylbutanoic acids is depicted in Scheme 1. Treatment of acid 32 with iodoethane in the presence of NaHCO3 afforded ethyl ester 33, which was then converted to 34 by performing Suzuki–Miyaura coupling. Deprotection of Boc of 34 by HCl/dioxane gave the amine hydrochloride 35. The amine was reacted with succinic anhydride to afford the amide 36 followed by hydrolysis of the ethyl ester under basic conditions to furnish diacid 13.
Scheme 1. Typical Synthetic Route Utilized for Biphenylbutanoic Acid Derivatives.

Reagents and conditions: (a) EtI, NaHCO3, DMF, rt; (b) 3-Cl-C6H4-B(OH)2, Pd(PPh3)4, aq. Na2CO3, DME, 95 °C; (c) 4 M HCl in 1,4-dioxane; (d) succinic anhydride, DIPEA, DCM; (e) 1 M aq. NaOH, THF, MeOH, rt; 1 M aq. HCl.
While most synthesized compounds were modeled in the crystal structure of NEP prior to synthesis, we sought to validate the binding mode experimentally. The crystal structure of NEP complexed with 13 was solved using experimental conditions described for LBQ657.7 The complex of 13 with NEP crystallized in the orthorhombic space group P212121 with two copies in the asymmetric unit diffracting to 2.54 Å resolution.
The structure was solved by molecular replacement. The overall binding mode of 13 is comparable to LBQ657 (Figure 4c) and validates our hypothesis of maintaining the zinc coordination with a shorter linker. The orientation of the carboxylic acid making zinc coordination is stabilized by interaction with His711 (Figure 4a and 4b). While we noted small movement in side chains of Phe106 and Asn542 relative to LBQ657 X-ray, this did not impact the H-bonding interaction of 13 with Asn542, Arg102, and Arg110. The succinic acid in S2′ has undergone a rotation of 75° relative to LBQ657. Owing to the flexible nature of the succinic acid, alternate orientations cannot be ruled out in solution. The amide carbonyl of 13 makes H-bonding interactions with the side chain of Arg717. In being consistent with our rationale, it was comforting to see the chloro group occupying the S1′ subsite pointing toward the backbone carbonyl of Arg717 and Thr721. It efficiently fills the available space surrounded by side chain from Met579, Asp650, Phe689, Arg717, and Ile718.
Figure 4.

Crystal structure of NEP complexed with 13 (yellow). Zinc atom is shown in magenta. H-bonding interactions are shown as black dotted lines. Chlorine is colored green. (a and b) Two different orientations of the binding site are shown to provide clarity of the interactions. (c) Overlay of crystal structure of LBQ657 (cyan) on 13.
We routinely profiled compounds in in vitro ADME assays, which gated compound selection for in vivo PK/PD studies in rat. The combination of in vitro potency and ADME profile resulted in selection of 13 and 26 for PK studies in rats. Oral administration of the ethyl ester prodrug of 13 and 26 showed rapid absorption and hydrolysis to 13 and 26 with bioavailability of 42% and 25%, respectively (Table 4). Both compounds showed low clearance (5 mL/min/kg) and similar apparent half-life after a single IV administration in rats. Considering 13 had ∼2-fold higher bioavailability than 26, we selected 13 for further in vivo profiling studies. Plasma clearance was low for 13 in monkey (3 mL/min/kg) and moderate in dog (29 mL/min/kg). The apparent half-life was 0.8 h for 13 in dog and ∼4 h in monkey.
Table 4. Pharmacokinetic Parameters of 13 and 26 in Sprague Dawley Rat Dosed at 1 mg/kg IV (Parent Compound) and 3 mg/kg PO (Ethyl Ester Prodrug)a.
| Administered Compound | Route | oral Cmax (nM) | AUC(0–7) (nM·Hours) | CL (mL/min/kg) | Vdss (L/kg) | Elimination t1/2 (IV) (Hours) | %F |
|---|---|---|---|---|---|---|---|
| 13 (active) | IV | 8730 | 5 | 0.1 | 0.8 | ||
| 36 (prodrug) | PO | 12600 | 10100 | 42 | |||
| 26 (active) | IV | 8100 | 5 | 0.1 | 1.0 | ||
| 39 (prodrug) | PO | 5380 | 5630 | 25 |
36 is the ethyl ester prodrug of 13; 39 is the ethyl ester prodrug of 26.
36, the prodrug of 13, was rapidly absorbed in all species after a single oral dose. However, the extent of absorbed prodrug converted to active drug was different among rat (99%), dog (5%), and monkey (69%).
The ethyl ester prodrug of 13 was tested in the rat ANP potentiation model. The prodrug (36) dose-dependently elevated PANP (Figure 5a). At the highest doses, PANP levels remained elevated for the entire experiment, reflecting the compound’s long duration of action. Plasma levels of 13 followed a similar profile (Figure 5b). Upon evaluation across an internal panel of ∼110 enzymes, receptors and ion channels 13 furnished IC50 values of >30 μM.
Figure 5.
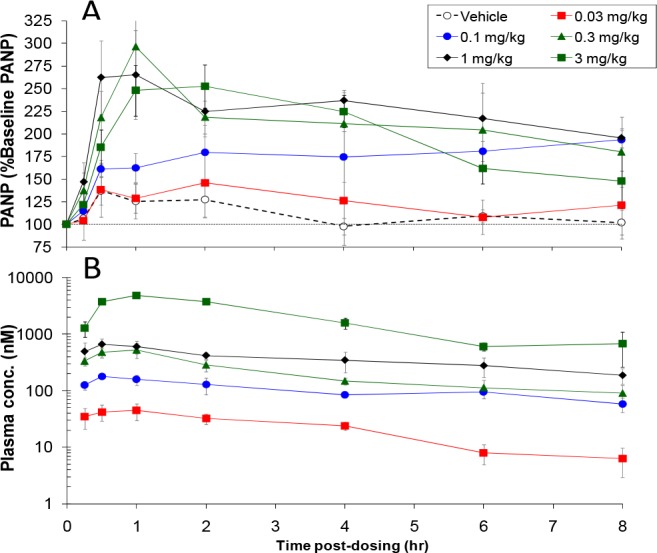
Dose-dependent potentiation of PANP (a) pharmacodynamics and (b) pharmacokinetic time courses of orally administered 36 (ethyl ester prodrug of 13) in the ANP potentiation model in conscious rats.
In conclusion, a structure-guided approach resulted in the identification of subnanomolar inhibitors of NEP. We took advantage of the simplified synthesis of the biphenyl butanoic acid series to quickly explore the SAR in the S1′ subsite. The potency drop with a less optimal linker (4) was compensated by addition of chlorine in the S1′ subsite. In our preclinical pharmacology model, 13 was found to be 100-fold more potent than sacubitril. While we profiled 13 as a potential clinical candidate, the early success encouraged us to explore other linkers and modifications of the S2′ binding moiety. Results from the new chemical series that evolved from the effort will be published in the future.
Acknowledgments
The authors would like to dedicate this paper in memory of Dr. Gary M. Ksander. The authors acknowledge the contribution of the Analytical Sciences group at the Novartis Institutes for BioMedical Research for support in generating the analytical details for the compounds described herein. The authors would like to thank Jaime Spear, Ivana Liric Rajlic, Danuta Lubicka, Ursula Bodendorf, Marion Kamke, Ingrid Bechtold, Susanne Worpenberg, Kathrin Dresen, Patrick Schweigler, Anna Bernardi, and Claude Logel for their excellent technical assistance.
Glossary
Abbreviations
- NEP
neprilysin
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- HFrEF
heart failure with reduced ejection fraction
- ARB
angiotensin II type-1 receptor antagonist
- SAR
structure activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00578.
Synthetic procedures, compound characterization, biochemical and pharmacokinetic study protocol, and crystallographic structure determination of NEP in complex with compound 13 (PDF)
Accession Codes
Atomic coordinates and structure factors for the crystal structure of NEP with compound 13 are deposited in the protein data bank with accession code 6THP.pdb. Authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
¥ Fourteen Emerson Street, Belmont, MA, 02478.
Author Present Address
¶ Janssen Pharmaceutical K. K., 3-5-2 Nishi-kanda, Chiyoda-ku, Tokyo 101-0065, Japan.
Author Present Address
○ Tesaro, 1000 Winter St, Waltham, MA 02451.
Author Present Address
€ Golda Och Academy, 1418 Pleasant Valley Way, West Orange, NJ 07052.
Author Present Address
# Rigel BioPharma Consulting LLC, 54 Cromwell Ct, Berkeley Heights, NJ 07922.
Author Present Address
□ Bristol-Myers Squibb, 7 Powderhorn Drive, Warren NJ, 07059.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author Contributions
∥ T.K. and R.G.K. contributed equally.
The authors declare no competing financial interest.
Author Status
† Deceased.
Supplementary Material
References
- Roques B. P.; Noble F.; Daugé V.; Fournié Zaluski M. C.; Beaumont A. Neutral Endopeptidase-24.11 - Structure, Inhibition, and Experimental and Clinical-Pharmacology. Pharmacol. Rev. 1993, 45, 87–146. [PubMed] [Google Scholar]
- Brunner-La Rocca H. P.; Kiowski W.; Ramsay D.; Sütsch G. Therapeutic benefits of increasing natriuretic peptide levels. Cardiovasc. Res. 2001, 51, 510–520. 10.1016/S0008-6363(01)00302-9. [DOI] [PubMed] [Google Scholar]
- Swainson C. P.; Craig K. C. Effects of Atrial-Natriuretic-Peptide (99–126) in Chronic Renal-Disease in Man. Nephrol., Dial., Transplant. 1991, 6, 336–341. 10.1093/ndt/6.5.336. [DOI] [PubMed] [Google Scholar]
- McMurray J. J. V.; Packer M.; Desai A. S.; Gong J. J.; Lefkowitz M. P.; Rizkala A. R.; Rouleau J. L.; Shi V. C.; Solomon S. D.; Swedberg K.; Zile M. R. Angiotensin-Neprilysin Inhibition versus Enalapril in Heart Failure. N. Engl. J. Med. 2014, 371, 993–1004. 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- Feng L.; Karpinski P. H.; Sutton P.; Liu Y.; Hook D. F.; Hu B.; Blacklock T. J.; Fanwick P. E.; Prashad M.; Godtfredsen S.; Ziltener C. LCZ696: a dual-acting sodium supramolecular complex. Tetrahedron Lett. 2012, 53, 275–276. 10.1016/j.tetlet.2011.11.029. [DOI] [Google Scholar]
- Gu J.; Noe A.; Chandra P.; Al-Fayoumi S.; Ligueros-Saylan M.; Sarangapani R.; Maahs S.; Ksander G.; Rigel D. F.; Jeng A. Y.; Lin T.-H.; Zheng W.; Dole W. P. Pharmacokinetics and Pharmacodynamics of LCZ696, a Novel Dual-Acting Angiotensin Receptor-Neprilysin Inhibitor (ARNi). J. Clin. Pharmacol. 2010, 50, 401–414. 10.1177/0091270009343932. [DOI] [PubMed] [Google Scholar]
- Schiering N.; D’Arcy A.; Villard F.; Ramage P.; Logel C.; Cumin F.; Ksander G. M.; Wiesmann C.; Karki R. G.; Mogi M. Structure of neprilysin in complex with the active metabolite of sacubitril. Sci. Rep. 2016, 6, 27909. 10.1038/srep27909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksander G. M.; Ghai R. D.; deJesus R.; Diefenbacher C. G.; Yuan A.; Berry C.; Sakane Y.; Trapani A. Dicarboxylic-Acid Dipeptide Neutral Endopeptidase Inhibitors. J. Med. Chem. 1995, 38, 1689–1700. 10.1021/jm00010a014. [DOI] [PubMed] [Google Scholar]
- Oefner C.; Roques B. P.; Fournie-Zaluski M. C.; Dale G. E. Structural analysis of neprilysin with various specific and potent inhibitors. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 392–396. 10.1107/S0907444903027410. [DOI] [PubMed] [Google Scholar]
- Oefner C.; Pierau S.; Schulz H.; Dale G. E. Structural studies of a bifunctional inhibitor of neprilysin and DPP-IV. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 975–981. 10.1107/S0907444907036281. [DOI] [PubMed] [Google Scholar]
- Oefner C.; D’Arcy A.; Hennig M.; Winkler F. K.; Dale G. E. Structure of human neutral endopeptidase (neprilysin) complexed with phosphoramidon. J. Mol. Biol. 2000, 296, 341–349. 10.1006/jmbi.1999.3492. [DOI] [PubMed] [Google Scholar]
- Sahli S.; Frank B.; Schweizer W. B.; Diederich F.; Blum-Kaelin D.; Aebi J. D.; Bohm H. J. Second-generation inhibitors for the metalloprotease neprilysin based on bicyclic heteroaromatic scaffolds: Synthesis, biological activity, and X-ray crystal-structure analysis. Helv. Chim. Acta 2005, 88, 731–750. 10.1002/hlca.200590051. [DOI] [Google Scholar]
- Glossop M. S.; Bazin R. J.; Dack K. N.; Fox D. N. A.; MacDonald G. A.; Mills M.; Owen D. R.; Phillips C.; Reeves K. A.; Ringer T. J.; Strang R. S.; Watson C. A. L. Synthesis and evaluation of heteroarylalanine diacids as potent and selective neutral endopeptidase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3404–3406. 10.1016/j.bmcl.2011.03.109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.