

Abstract
SUMMARY
Background:
Based on our preclinical work, combination of PARPis with agents that inhibit HRR such as PI3K inhibitors (PI3Kis) may sensitize HRR proficient EOCs to PARPis. We aimed to assess the safety, identify the recommended phase 2 dose (RP2D) and explore preliminary sign of activity of the PARPi olaparib and the PI3Ki alpelisib in ovarian cancer.
Methods:
In this multicenter, open-label, phase 1b, classic 3+3 dose-escalation study of alpelisib and olaparib (ClinicalTrials.gov, ), patients aged ≥18 years with recurrent EOC of high-grade serous histology or any histology but with known gBRCAmut or recurrent triple negative breast cancer (BC) or any histology with known gBRCAmut were enrolled; additional EOC patients were enrolled in a dose expansion cohort. Patients received alpelisib 200–300mg orally once daily and olaparib 100–200mg (tablet formulation) orally twice daily. Primary endpoint was determination of the RPD2 and the maximum tolerated dose (MTD). Analyses included all patients who received at least one dose of the study drugs. The trial is active but closed to enrolment; follow-up for patients who completed treatment is ongoing.
Findings:
Thirty-four patients were enrolled into dose-escalation (n=28) and EOC expansion (n=6) cohorts; 30 patients had EOC and 4 had BC (all in dose-escalation). Two EOC patients never received study treatment because of ineligibility. MTD and RP2D were defined as alpelisib 200mg orally once daily and olaparib 200mg orally twice daily. Considering all dose levels, the most common treatment-related grade 3–4 adverse events were hyperglycemia (5 [15.6%) of 32 patients), nausea (3 [9.4%]), and alanine aminotransferase concentration increase (3 [9.4%]). No treatment-related deaths occurred. Dose-limiting toxicities included hyperglycemia and fever with decreased neutrophil count. Of the 28 EOC patients, 10 (35.7%) achieved a partial response (PR) and 14 (50%) had stable disease (SD) according to Response Evaluation Criteria in solid tumors 1.1.
Interpretation:
Combining alpelisib and olaparib is feasible with no unexpected toxicities. The observed activity of the olaparib/alpelisib in BRCAwt platinum resistant EOC was higher than expected compared to either olaparib or alpelisib monotherapies and warrants further investigation.
RESEARCH IN CONTEXT
Evidence before this study
We searched PubMed on September 29, 2018, for clinical studies exploring combinations between poly(ADP-Ribose) Polymerase (PARP) Inhibitors (PARPis) and other agents in patients with advanced and/or metastatic cancers using the following search terms: “combination” AND “PARP inhibitor OR olaparib OR niraparib OR rucaparib OR talazoparib OR veliparib” and refining research for clinical trials. This search revealed several trials exploring combinations of PARPis with chemotherapy and a few trials exploring combinations of PARPis with targeted agents. In general, none of the trials of chemotherapy combinations indicated evidence of clinical synergism between the PARPi and the cytotoxic agent, at least in part because these combinations require attenuation of PARPi or cytotoxic agent dosing. Similarly, to date, no evidence of clinical synergism between PARPis and targeted agents has been reported, at least in tumors which are homologous recombination repair (HRR) proficient (the previously reported olaparib/cediranib study included only patients with recurrent platinum sensitive ovarian cancer, a setting enriched for HRR deficiency). Based on preclinical work supporting synergism of PI3Ki and PARPis we initially performed a phase 1b dose escalation study of the pan-PI3Ki buparlisib (BKM120) and the PARPi olaparib for the treatment of recurrent ovarian and breast cancer but CNS toxicity precluded meaningful escalation of buparlisib. To overcome this problem, we evaluated the alpha isoform-specific PI3-Kinase inhibitor alpelisib (BYL719), which has not demonstrated CNS toxicity, in combination with olaparib in a phase 1b study with dose expansion in patients with ovarian cancer.
Added value of the study
This hypothesis-driven, investigator-initiated phase 1b dose-escalation clinical trial of alpelisib and olaparib with dose expansion in patients with ovarian cancer showed that the olaparib/alpelisib combination is feasible and exhibits clinical evidence of synergism in BRCAwt (somatic and germline), platinum-resistant ovarian carcinomas, i.e. in tumors enriched for HRR proficiency. To our knowledge, this is the first time that clinical evidence of synergism between a PARPi and a targeted agent or chemotherapy is reported in BRCAwt, platinum-resistant ovarian cancer. The RECIST 1.1 objective response rate of olaparib/alpelisib of 33.3% in these cancers is significantly higher than that expected from olaparib monotherapy (~4–5%) or alpelisib monotherapy (< 5%) in this setting. Furthermore, this study highlights a novel mechanism of action, i.e. use of a PI3K inhibitor to sensitize HRR-proficient ovarian cancers to PARPis. Of note, targeted next generation sequencing revealed objective responses of olaparib/alpelisib even in patients with tumors without any HRR and PI3K pathway alterations, who had undergone multiple prior lines of cytotoxic therapy. Additionally, the median duration of response and median progression-free and overall survival in this heavily pretreated study population were very promising.
Implications of all the available evidence
Our study has demonstrated that the combination of alpelisib and olaparib exhibits synergistic activity in BRCAwt, platinum-resistant ovarian cancers, i.e. in tumors enriched for HRR proficiency, thereby expanding potential use of PARPis beyond the setting of HRR deficiency where they are currently EMA and FDA approved. These results, as well as the mechanistic rationale behind PARPi/PI3Ki combinations, may be applicable not only to BRCAwt, platinum-resistant ovarian cancers, but also to other solid tumors with or without PI3K pathway alterations, including BRCAwt breast cancer, prostate, colorectal and endometrial cancers. Additional work in breast cancer may be particularly compelling, given the recent promising results of the phase 3, global, double-blind, SOLAR-1 study of alpelisib in combination with fulvestrant in hormone receptor-positive HER2 negative breast cancer whereby hyperactivation of the PI3K pathway can occur due to PIK3CA mutations in ~40% of patients.
INTRODUCTION
Approximately 50% of high grade serous ovarian carcinoma (HGSOC) harbor genetic or epigenetic alterations of genes in the homologous recombination repair (HRR) pathway(1). HRR deficient cancers are sensitive to PARP inhibitors (PARPis) and this synthetic lethal interaction is being exploited therapeutically in epithelial ovarian cancer (EOC) whereby three PARPi, i.e. olaparib, rucaparib and niraparib, have received EMA and FDA approval as monotherapy either in patients with germline or somatic BRCA1/2 mutations or as maintenance therapy after platinum chemotherapy in platinum sensitive recurrent EOC (2–5).
The promise of PARPis in the management of EOC is tempered, however, by the fact that EOCs with intrinsic, de novo, HRR proficiency respond less than HRR-deficient carcinomas (5, 6). Furthermore, the most prevalent mechanism of acquired PARPi resistance in HRR deficient cancers is acquisition of HRR proficiency as a consequence of secondary genetic or epigenetic events (such as secondary mutations in BRCA1/2 or RAD51C/D or reversal of BRCA1 promoter methylation) that restore HRR proficiency and confer PARPi resistance(2, 7–9). Taken together, HRR proficiency, either de novo (which is present in as high as 50% of HGSOC) or acquired (which represents the most important mechanism of PARPi resistance in HRR deficient carcinomas) poses a significant challenge for the successful use of PARPis in EOC.
Combinations of PARPis with agents that inhibit HRR may represent an effective strategy to sensitize EOCs with de novo or acquired HRR proficiency to PARPis and potentially expand use of these agents beyond HRR deficient EOCs. In this regard, previous work by us and others has demonstrated that PI3K inhibition (PI3Ki) leads to downregulation of BRCA1 and BRCA2 (BRCA1/2) and abrogation of HRR, increased DNA damage, gain in poly(ADP-ribosyl)ation, and subsequent sensitization to PARPis. Importantly, synergism between PI3Ki and PARPi is observed both in vitro and in vivo and in HRR proficient as well as HRR deficient models(10, 11). Mechanistically, downregulation of BRCA1/2 appears to be mediated by ERK-dependent activation of the ETS transcription factor, which suppresses BRCA1/2 gene transcription, thereby causing a deficiency in HRR and concomitant PARPi sensitivity (10, 12). The enhanced DNA damage induced by PI3Ki may also be a consequence of impaired production of nucleotides needed for DNA synthesis and repair(13). Specifically, PI3Ki disproportionately affects the nonoxidative pentose phosphate pathway that delivers ribose-5-phosphate required for synthesis of ribonucleotides, ultimately leading to a decrease in all four nucleotide triphosphates(13).
To evaluate the synergism between PI3Ki and PARPi in the clinic, we initially performed a phase 1b dose escalation study of the pan-PI3Ki buparlisib (BKM120) and the PARPi olaparib for the treatment of recurrent ovarian and breast cancer (BC)(14). However, CNS toxicity (depression and anxiety observed in 36% and 28% of patients, respectively) and grade 3 transaminase elevation prevented meaningful dose escalation of BMK120. Not unexpectedly, in that study, the anticancer activity of olaparib/buparlisib in EOC patients [70% of whom harbored germline BRCA mutations (gBRCAmut)] was similar to the historical activity of olaparib monotherapy in that population, i.e., there was no evidence that addition of buparlisib at the doses administered contributed any additional activity to olaparib monotherapy. To overcome the problem of CNS toxicity and transaminase abnormalities and search for preliminary clinical evidence of synergism between PI3Ki and PARPi, we have now evaluated the alpha specific PI3-Kinase inhibitor alpelisib (BYL719), which has not demonstrated CNS toxicity, in combination with olaparib in a phase 1b study. Of note, alpelisib, in combination with endocrine therapy (fulvestrant) showed promising results in hormone receptor-positive HER2 negative breast cancer with PIK3CA mutations in the recently reported phase 3, global, double-blind SOLAR-1 study (15).
METHODS
Study Design and Participants
This was a multicenter, open-label, phase 1b, classic 3+3 dose-escalation clinical trial (Figure 1). Key eligibility criteria included confirmed diagnosis of either recurrent ovarian, fallopian tube, or primary peritoneal cancer (collectively referred to as ‘EOC’) of high-grade serous histology or any histology but with known gBRCAmut or confirmed diagnosis of recurrent BC of TNBC histology or any histology with known gBRCAmut. Other eligibility criteria included age ≥18 years, ability to provide informed consent, Eastern Cooperative Oncology Group performance status of 1 or less, estimated life expectancy of at least 4 months, adequate bone marrow (haemoglobin >9.0 g/dL, absolute neutrophil count >1500 per μL, and platelet count>100 000 per μL), liver function (total bilirubin concentration<upper limit of normal [ULN], alanine and aspartate aminotransferase concentration <2.5 × ULN [<5 × ULN for patients with liver metastases], prothrombin time international normalised ratio<1.5, kidney function (serum creatinine concentration < ULN or calculated creatinine clearance ≥ 35 mL/min using Cockcroft-Gault formula), fasting serum amylase ≤ 2 x institutional ULN, fasting serum lipase ≤ institutional ULN, fasting plasma glucose (FPG) ≤ 140 mg/dL (7.7 mmol/L) and glycosylated Hemoglobin (HbA1c) ≤ 6.4% (both criteria have to be met). Participants must have measurable disease by RECIST 1.1 criteria or evaluable cancer via CA125 GCIG criteria. There must be absence of major comorbidities including but not limited to myocardial infarction within the last 6 months, impairment of gastrointestinal (GI) function or GI disease that may significantly alter the absorption of oral drugs, ongoing or active infection or acute or chronic liver, renal, lung disease or pancreatitis or psychiatric illness/social situations that would limit compliance with study requirements. There was no line limit on prior therapies; patients with recurrent, metastatic TNBC must have had at least 1 chemotherapy regimen for metastatic breast cancer or have developed metastatic breast cancer within 1 year of completion of adjuvant chemotherapy. Prior therapy for platinum-sensitive OC patients must have included 2 prior platinum-based chemotherapy regimens. Platinum sensitive, resistant or refractory disease was allowed. Prior PARPi and PI3K use was allowed for patients in dose escalation but not in the dose expansion cohort. Tumor assessment by RECIST 1.1 occurred every two cycles.
Figure 1.
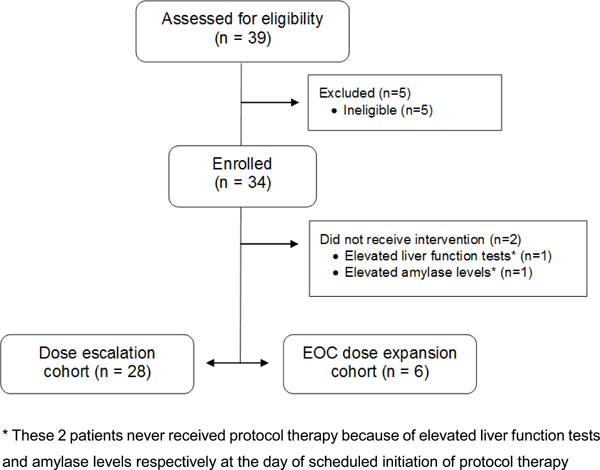
Trial Profile.
The clinical trial was approved by the institutional review boards of all participating institutions, and the US Food and Drug Administration (ClinicalTrial:NCT01623349). All procedures involving human participants were carried out in accordance with the Declaration of Helsinki. Written informed consent was obtained from patients or guardians before enrollment in the study.
Procedures
Olaparib was administered twice daily (tablet formulation) and alpelisib once daily on a 28-day cycle; both were administered continuously. Four dose levels were planned: the starting dose level (DL0) of alpelisib 250 mg q.d. plus olaparib 100 mg b.i.d., DL1 of alpelisib 250mg q.d. with olaparib 200 mg b.i.d., DL2 of alpelisib 300 mg q.d. with olaparib 200 mg b.i.d. and DL3 of alpelisib 200 mg q.d. with olaparib 200 mg b.i.d. Treatment continued indefinitely until progression, unacceptable toxicity, or patient refusal, or intercurrent illness that prevents further administration of treatment, or general or specific changes in the participant’s condition rendering the participant unacceptable for further treatment in the opinion of the treating investigator. Dose modifications and reductions followed prespecified rules (described in detail in the protocol included in the appendix pp 94–129). Generally, adverse events (AEs) worse than grade 2 caused study drugs’ delay (both study drugs in Cycle 1, and offending drug in Cycle 2 and beyond) until toxicity resolved to grade ≤1. If AEs resolved to grade ≤1 within 7 days, drugs were resumed at the same dose level; if AEs resolved to grade ≤1 within 8–28 days, protocol treatment was restarted at the next lowest dose of drug(s) causing the toxicity. If the toxicity did not resolve within 28 days, the participants were removed from the study. Similar criteria were followed if the patients developed a DLT. Intrapatient dose escalation was allowed.
Tumor assessment by RECIST 1.1 occurred every two cycles (8 weeks) and included assessment of chest, abdomen and pelvis via CT or MRI scan. Tumor assessment was done every 12 weeks after completion of cycle 16. PK blood samples were collected for alpelisib and olaparib on Day 1, time 0 and then days 8 and 15 of cycle 1 at 0, 1, 2, 4, and 8 hours after dosing both agents. Toxic effects were monitored weekly during cycles 1 and 2 and then every Day 1 of each cycle by means of both blood tests and clinical examination. Toxicities were assessed by CTCAE v 4.03. A dose limiting toxicity (DLT) was defined as study treatment-related toxicity during the first 4 weeks of treatment, and included 1) any grade 3 or 4 non-hematologic event excluding fatigue, nausea and/or vomiting, constipation or diarrhea, electrolyte imbalances, rash that resolved to ≤ grade 2 within ≤ 5 days, and/or grade 3 hypertension controlled with anti-hypertensive therapy, 2) any of the following hematologic events: a) Grade 4 neutropenia of > 7 days duration, b) febrile neutropenia, c) grade 4 thrombocytopenia or bleeding with grade 3 thrombocytopenia, d) requirement for repeated blood transfusion within 4–6 weeks, and/or e) all other Grade 4 hematologic toxicities, 3) inability to take 75% or more of the planned dose, 4) any grade 5 event related to study treatment or 5) any grade 3 or 4 event considered dose-limiting.
Tumor DNA from archival, formalin-fixed tissues was analyzed at the Dana-Farber Cancer Institute (DFCI) using targeted panel next-generation sequencing (Oncopanel assay). The Oncopanel targeted sequencing test at Dana-Farber Cancer Institute is a CLIA approved test that consists of next-generation sequencing (NGS) of formalin-fixed tumor samples covering exons of over 300 cancer-associated genes, plus intronic regions of genes involved in somatic rearrangements. Oncopanel tests are reviewed by molecular pathologists and report mutations, insertions/deletions, copy number variations, and structural variants in the targeted genes (16–18).
Outcomes
The primary objectives of this study were to determine the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) of the combination of alpelisib and olaparib. Secondary objectives included determination of safety, observed toxicities, PK and preliminary activity of olaparib/alpelisib as measured by objective response rate by RECIST 1.1. Duration of response and progression-free survival were exploratory endpoints that were included and defined in the protocol section 10 (Appendix p 144–150). Furthermore, all patients were followed for overall survival per protocol Section 5.6 (Appendix p 93) and protocol Section 9.0 (Appendix p140–141). Exploratory correlative endpoints included preliminary evidence of activity of olaparib/alpelisib in patients with and without germline and/or somatic BRCA mutations, in platinum resistant/refractory disease, and in molecularly defined subgroups of patients with or without HRR and PI3K pathway alterations as identified by the Oncopanel assay.
Statistical Analysis
We designed this clinical trial as a 3+3 dose escalation study, with dose escalation if 0/3 or 1/6 participants experienced a dose limiting toxicity (DLT) during the first cycle of therapy (first 28 days) which has he conventional probability of dose escalation of 0.49 if the true rate of DLT is 30% in this disease setting. There is a well-established monotonic dose-efficacy relationship for olaparib (19) as well as similar monotonic dose-efficacy relationships for alpelisib (20) and other PI3K inhibitors (21). Additionally, given our hypothesis (based on our preclinical work) that the synergism between PARPis and PI3Kis depends on inhibition of the PI3K pathway which is dose dependent, we hypothesized a monotonic efficacy for the combination of olaparib and alpelisib. Once the MTD was determined, 6 EOC patients were enrolled into an expansion cohort to further determine the safety and tolerability of the RP2D and for secondary objectives including preliminary activity of the combination and translational endpoints. We assessed safety and efficacy in all patients who received at least one dose of either of the study drugs.
Platinum refractory disease was defined as either relapse less than 2 months after the last platinum-based therapy or relapse during platinum therapy. Platinum-resistance was defined as relapse within 2 to 6 months after last dose of platinum-based chemotherapy. Platinum sensitivity was defined as a relapse greater than 6 months after last dose of platinum-based chemotherapy. At each radiographic assessment, Stable Disease (SD) was defined by RECIST 1.1 as neither sufficient shrinkage to qualify for partial response (PR) nor sufficient increase to qualify for progressive disease (PD), taking as reference the smallest sum diameters while on study. Given that RECIST v1.1 criteria specify only requiring confirmation when ORR is the primary endpoint and given that this was a phase 1b study, the schedule of assessments by protocol did not require a confirmatory scan. There was no independent radiology review. Duration of response (DOR) was defined as from the first date of the overall response to the date of documented disease progression or the date off treatment, whichever occurred first. Best overall response was defined to be stable disease if a PR is not observed on treatment, and if criteria for progression are not met at the first restaging, and duration of stable disease was dichotomized at 6 months. Progression free survival (PFS) was defined as the time in months from registration to documented disease progression (per RECIST 1.1) or clinical progression or death from any cause, whichever occurred first. Patient demographics and adverse event frequencies were summarized using descriptive statistics. PFS, DOR and overall survival (OS) analyses were summarized using the Kaplan-Meier product limit estimator. 95% confidence intervals (CI) are reported for events rates at landmark times and for median survival using Greenwood’s formula. The association of patient and disease characteristics to objective response was explored using Fisher exact tests. All statistical analyses were performed using R v3.4.4. This study is registered with ClinicalTrials.gov, number ClinicalTrial:NCT01623349.
Role of the funding source
The study funders had no role in study design, data collection, data analysis, data interpretation or writing of the report. All authors had full access to all the data in the study and the corresponding author had final responsibility to submit the report for publication.
RESULTS
Patient accrual
The preclinical evaluation of the olaparib/alpelisib trial, including tolerability and efficacy experiments in ovarian cancer patient derived xenograft models, proof of mechanism and target engagement studies are presented in detail in the Appendix pp 9–15. Between 2014/10/03 and 2016/12/21 (Figure 1), 34 patients were enrolled into the dose escalation (n=28) and ovarian cancer expansion (n=6) cohorts of this study; 30 patients had EOC (24 in dose escalation cohort and 6 in the EOC expansion cohort). Four patients had BC, all in the dose escalation cohort. Two EOC patients never received study treatment because they were deemed ineligible for the study and therefore are excluded from all analyses. At the time of cut-off (2/15/2018), only 2 EOC patients remained on protocol treatment, with a median follow-up of 12 months (IQR 8–17 months). The characteristics of the EOC patients are summarized in Table 1; 10 (36%) were gBRCAmut and 26 (93%) were platinum resistant/refractory. The median number of prior lines of anticancer therapies (not including hormonal therapies or radiation therapy) was 3, with a range of 1 to 8 prior lines (Table 1).
Table 1.
Clinicopathologic characteristics of the 28 EOC patients.
| Overall (n=28) | ||
|---|---|---|
| N | % | |
| Number of patients registered | 28 | 100.0 |
| Gender | 28 | 100.0 |
| Female | ||
| Age (median, IQR) | 60 | (55, 67) |
| Ethnicity | 23 | 82.1 |
| Non-Hispanic | ||
| Ethnicity Not Known | 5 | 17.9 |
| Race | 25 | 89.3 |
| White | ||
| Other | 3 | 10.7 |
| Germline BRCA mutation status | 1 | 3.6 |
| Unknown | ||
| Pathogenic Mutation | 10 | 35.7 |
| Wildtype | 17 | 60.7 |
| Platinum status | 23 | 82.1 |
| Platinum Resistant | ||
| Platinum Sensitive | 2 | 7.1 |
| Platinum Refractory | 3 | 10.7 |
| Carcinoma Type | 26 | 92.9 |
| Ovarian | ||
| Primary Peritoneal | 2 | 7.1 |
| Stage at diagnosis | 2 | 7.1 |
| IIB | ||
| IIC | 2 | 7.1 |
| IIIC | 16 | 57.1 |
| IV | 8 | 28.6 |
| Histology | 21 | 75 |
| High grade papillary serous | ||
| Poorly differentiated adenocarcinoma not otherwise specified | 5 | 17.9 |
| Other, Specify* | 2 | 7.1 |
| Lines of prior therapy (median, range) | 3 | (1,8) |
1 patient with carcinosarcoma and 1 patient with mixed high grade serous and transitional cell carcinoma
Dose escalation, PK studies and toxicities
Four dose levels were evaluated (Table 2). The starting dose level (DL0) of alpelisib 250 mg q.d. plus olaparib 100 mg b.i.d. was well tolerated without DLTs. When the olaparib dose was escalated to 200 mg b.i.d. with alpelisib 250 mg q.d. (DL1), there were no DLTs, but most patients had to dose reduce alpelisib to 200 mg q.d. after the DLT period. Subsequent escalation of alpelisib to 300 mg q.d. with olaparib 200 mg b.i.d. (DL2) was associated with 2 DLTs (G4 hyperglycemia and G4 neutropenia and fever) in 6 patients. Alpelisib was de-escalated back to 250 mg q.d. with olaparib 200 mg b.i.d. (DL1), but this dose level was also associated with 2 DLTs (G3 hyperglycemia and inability to take >75% of study drugs). This prompted de-escalation of alpelisib to 200 mg q.d. with olaparib 200 mg b.i.d. (DL3), which was associated with 1 DLT (G3 hyperglycemia) in 6 patients. Thus, DL3 was deemed safe and selected as the MTD; the 6 EOC patients on the expansion cohort were treated at this dose level. The maximum grade hyperglycemia by dose level is presented in the Appendix pp 1.
Table 2:
Dose levelsa explored, number of patients on each dose level, and whether DLTs were observed (n=34b).
| Alpelisib dose (once daily) | Olaparib dose (twice daily) | No of Ovarian patients | No. of Breast cancer patients | TOTAL | Number of DLTs | |
|---|---|---|---|---|---|---|
| Dose Level 0 (starting dose)a | 250 mg | 100 mg | 5 | 0 | 5b | None |
| Dose Level 1 | 250 mg | 200 mg | 3 | 1 | 4 | None |
| Dose Level 2 | 300 mg | 200 mg | 4 | 2 | 6b | 2 DLTs* |
| Dose Level 1 | 250 mg | 200 mg | 7 | 0 | 7 | 2 DLTs** |
| Dose Level 3 | 200 mg | 200 mg | 5 | 1 | 6 | 1 DLT*** |
| Ovarian Cancer Expansion at DL3 | 200 mg | 200 mg | 6 | 0 | 6 | |
| TOTAL | 30b | 4 | 34 |
Doses levels are presented in the order they were tested, i.e. initially dose level DL0 was tested, then DL1, then DL2, then DL1, then DL3 and then ovarian cancer expansion at DL3.
One EOC patient in DL0 and one EOC patient in DL2 never started protocol therapy because of elevated liver function tests elevated amylase levels at the day of scheduled initiation of protocol therapy
One patient had grade 4 hyperglycemia, and one patient had grade 4 neutropenia and fever
One patient had grade 3 hyperglycemia, and one patient was unable to receive >75% of study drugs
One patient had grade 3 hyperglycemia
Treatment related non-hematologic and hematologic toxicities that were G3+ or occurring in ≥10% of all treated EOC patients are listed in Table 3. There were no unexpected toxicities observed based on the known toxicities of olaparib and alpelisib and no irreversible toxicities. Unlike buparlisib, CNS toxicities such as depression and anxiety are not a known safety issue with alpelisib, and no CNS events higher than grade 1 were recorded in the study. Nausea, hyperglycemia (an expected toxicity of PI3Kis) and fatigue were the most common toxicities, mostly grades 1 and 2. Transaminase elevations, also a known toxicity of PI3Kis, were seen in 4 (12.5)% of patients who had increased alanine aminotransferase (3 with G3) and 2 (6.3%) of patients with increased aspartate aminotransferase (1 with G3); 4 (9.4%) patients exhibited lipase elevations (G3 in 2 patients), which were reversible. Anemia and thrombocytopenia, expected toxicities of olaparib, were observed in 7 (21.9%) and 5 (15.6%) of patients, respectively, all grade 1–2. Details on patients needing dose reduction per dose level are included in the Appendix pp 4. Three patients discontinued therapy for drug-related toxicity, two for hyperglycemia (G3 and G4 respectively) and one for nausea/vomiting (G2). Serious adverse events, as defined in the protocol (Appendix pp. 151) included hyperglycemia in 2 patients (both G4), febrile neutropenia G4 in 1 patient, G2 hypothyroidism in 1 patient, G2 small bowel fistula in 1 patient. There were no treatment-related deaths; furthermore, no deaths while on protocol treatment were observed.
Table 3.
Treatment-related toxicities that are grade 3+ or occurring in ≥10% of patients (n=32).
| Toxicity | Maximum Grade | ||
|---|---|---|---|
| 1 – 2 | 3 | 4 | |
| Nausea | 23 (71.9%) | 3 (9.4%) | - |
| Hyperglycemia | 17 (53.1%) | 3 (9.4%) | 2 (6.3%) |
| Fatigue | 18 (56.3%) | 2 (6.3%) | - |
| Diarrhea | 16 (50%) | - | - |
| Vomiting | 14 (43.8%) | 2 (6.3%) | - |
| Anorexia | 10 (31.3%) | - | - |
| Headache | 8 (25%) | - | - |
| Anemia | 7 (21.9%) | - | - |
| Constipation | 5 (15.6%) | - | - |
| Creatinine increased | 5 (15.6%) | - | - |
| Platelet count decreased | 5 (15.6%) | - | - |
| Rash acneiform | 4 (12.5%) | 1 (3.1%) | - |
| Rash maculo-papular | 4 (12.5%) | 1 (3.1%) | - |
| Serum amylase increased | 4 (12.5%) | 1 (3.1%) | - |
| Abdominal pain | 4 (12.5%) | - | - |
| Alanine aminotransferase increased | 1 (3.1%) | 3 (9.4%) | - |
| Dry skin | 4 (12.5%) | - | - |
| Dysgeusia | 4 (12.5%) | - | - |
| Dyspnea | 4 (12.5%) | - | - |
| Lipase increased | 2 (6.3%) | 2 (6.3%) | - |
| Mucositis oral | 4 (12.5%) | - | - |
| Investigations - Other, specify | 2 (6.3%) | - | 1 (3.1%) |
| Aspartate aminotransferase increased | 1 (3.1%) | 1 (3.1%) | - |
| Febrile neutropenia | - | - | 1 (3.1%) |
| GGT increased | - | 1 (3.1%) | - |
| Pain | - | 1 (3.1%) | - |
| Rectal pain | - | 1 (3.1%) | - |
| Renal Insufficiency | - | - | 1 (3.1%) |
Pharmacokinetic (PK) results for alpelisib and olaparib are presented in the Appendix pp 2–3. Steady state Cmax values determined on day 8, 2 hours post dosing for both olaparib and alpelisib, appear comparable to values when testing these agents as monotherapy in the phase 1 setting, with drug exposures increasing proportionally with increasing dose. Alpelisib Cmax results appeared unaffected by olaparib dosing, and olaparib Cmax results appeared unaffected by alpelisib dosing.
Objective Responses
Of the 28 EOC patients included in analysis, 10 (36%) had a partial response (PR), 14 (50%) had stable disease (SD), 3 (11%) had progressive disease (PD) and 1 (4%) was unevaluable for best overall response using RECIST 1.1, Figure 2. Of the 14 patients with SD, 8 (29% of all EOC patients) patients had SD for ≥6 months. Of the 4 BC patients in dose escalation, 3 patients had stable disease for 12, 3 and 10 cycles respectively while one patient was removed from protocol treatment before cycle 1 (this was the patient with the DLT of grade 3 hyperglycemia in DL3).
Figure 2. Response to study treatment.
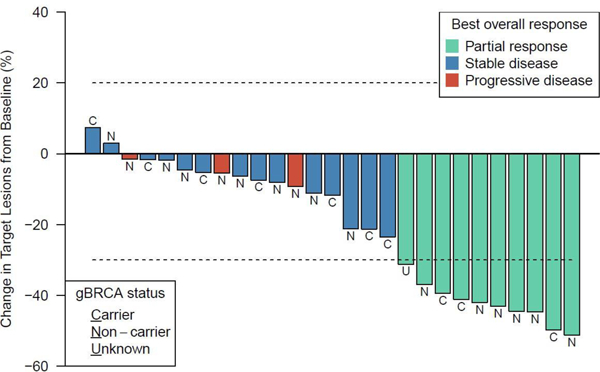
Summary of the maximum reduction in the sum of target lesion sizes in n=27 EOC patients with measurable disease and restaging scans; three patients had unequivocal progression of non-target lesions and/or new lesions at the first restaging and are indicated as progressive disease (PD). gBRCAmut carrier status is presented.
In EOC, the objective response rate (ORR) [complete (CR)+PR] was similar for gBRCAmut and gBRCAwt patients, 30% and 35% respectively, Table 4, (Fisher’s exact test p=0.42). Nine of 10 gBRCAmut and 16 of 17 gBRCAwt patients had platinum resistant/refractory disease; ORR was 33% (3 of 9 patients) and 31% (5 of 16) in gBRCAmut and gBRCAwt platinum resistant/refractory populations respectively. Among the 10 patients with PRs, 8 progressed while on treatment and 2 discontinued treatment because of intolerability. Best overall response by initial dose level is presented in the Appendix pp 4. Duration of response (Figure 3) ranged from 0.5 to 13.1 months, with a median of 5.5 months. A total of 23 PFS events are reported with a median PFS of 7.2 months (95% CI 4.9 – 9.0 months), Appendix pp 7. Eleven patients have died with a median OS of 21.3 months (95% CI 11.4–23.7 months), Appendix pp 7. Kaplan-Meier curves of PFS and OS by gBRCAmut status are presented in Appendix pp 8.
Table 4.
Best overall response by gBRCA mutation status (carrier, non-carrier, unknown) and overall.
| gBRCA mutation status | Overall | |||||||
|---|---|---|---|---|---|---|---|---|
| gBRCA mutation carrier | gBRCA mutation non-carrier | gBRCA mutation unknown | ||||||
| N | % | N | % | N | % | N | % | |
| Best Response | 3 | 30.0 | 6 | 35.3 | 1 | 100 | 10 | 35.7 |
| PR | ||||||||
| SD | 7 | 70.0 | 7 | 41.2 | - | - | 14 | 50.0 |
| PD | - | - | 3 | 17.6 | - | - | 3 | 10.7 |
| Unevaluable | - | - | 1 | 5.9 | - | - | 1 | 3.6 |
| Total | 10 | 100 | 17 | 100 | 1 | 100 | 28 | 100 |
Figure 3. Duration of response and time-to-progression.
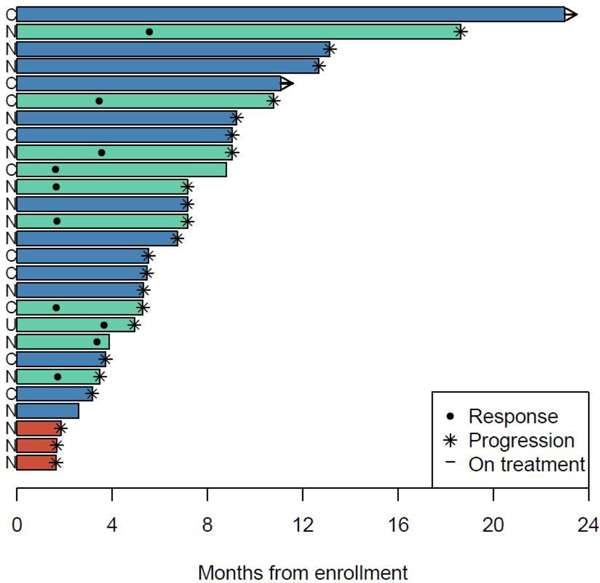
Summary of the duration of response for the 10 patients who achieved a partial response PR and time-to-progression (TTP) in the 18 patients who failed to achieve a PR. labeled by gBRCAmut carrier status and response. Duration and TTP are defined as the interval from date of enrollment to date of either progression or date of last disease assessment. Median duration among patients with a partial response was 5.5 months (maximum 13.1 months).
Targeted next generation sequencing and correlation with response
Tumor DNA was analyzed from archival, formalin-fixed tissues, using targeted next generation sequencing (Oncopanel) in 25 of the 28 EOC patients (Figure 4 and Appendix pp 5). Ten patients were gBRCAmut; Oncopanel data were available for 15 of the remaining 18 EOC patients. Of these 15 patients, 3 patients harbored tumors with somatic BRCA mutations (all BRCA2 mutations); one of these 3 patients exhibited an objective response (PR), Figure 4. Overall, the objective response rate in patients which were BRCAwt (germline and somatic) was 33% (4 out of 12 patients); all these patients had platinum resistant/refractory tumors.
Figure 4.
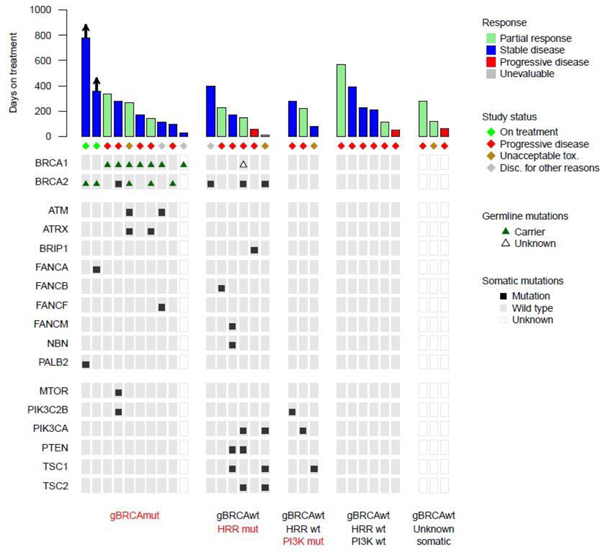
Genomic aberrations in the HRR and PI3K pathways in EOC patients on this study. Mutations were identified through targeted next-generation sequencing (Oncopanel).
Based on the Oncopanel analysis, a total of 6 patients harbored somatic mutations involving HRR genes (3 patients had BRCA2 somatic mutations as stated above), 3 patients had somatic mutations involving only the PIK3CA pathway, and 6 patients did not harbor any somatic mutations in the HRR and PIK3CA pathways (Figure 4). Objective responses were seen across all these patient cohorts; specifically, 2 of 6 patients with somatic HRR mutations, 1 of 3 patients with only PIK3CA mutations and 2 of 6 patients without HRR and PIK3CA mutations had a PR (Figure 4).
DISCUSSION
This phase 1b dose escalation study showed that combining alpelisib and olaparib produced no unexpected toxicities or safety signals and determined the maximum tolerated dose to be alpelisib to 200 mg q.d. with olaparib 200 mg b.i.d. which we consider as the RP2D. However, this recommended dose is merely one candidate dose along a contour of dose combinations, and the efficacy data across contrasting doses with similar DLT outcomes (i.e. between DL1 and DL3) appear to be consistent (Appendix pp 4). Adverse events that defined DLTs included hyperglycemia and fever with decreased neutrophil count. Regarding hyperglycemia, our study allowed patients with diabetes mellitus to participate provided their fasting glucose and Hgb A1C levels were less than 140 mg/dl (7.7 mmol/L) and 6.4%, respectively. However, more stringent criteria are currently used in trials of PI3Kis to decrease the frequency of hyperglycemia which is an otherwise expected toxicity of PI3Kis. Unlike the olaparib/buparlisib study(14), CNS toxicities (depression and anxiety) were not an issue with olaparib/alpelisib. Furthermore, transaminase elevations (another known toxicity of PI3Kis) were seen less frequently than olaparib/buparlisib (12.5% vs 20%, respectively) and, unlike olaparib/buparlisib, were not dose limiting. Finally, although nausea, fatigue and vomiting were observed in 50% or more of patients, these toxicities were mostly grade 1 and 2, easily manageable, and comparable to those observed with olaparib monotherapy (77% nausea, 63% fatigue and 40% vomiting in the recently reported SOLO1 study(22)).
The rationale for the combination of a PI3Ki with a PARPi was based on preclinical work performed initially in breast cancer models showing in vivo synergy of this combination(10, 11). The combination of alpelisib and olaparib was further supported by preclinical work in ovarian cancer PDX models (presented in the Appendix pp 9–15), which showed that alpelisib inhibits HRR and sensitizes ovarian cancer models with de novo or acquired HRR proficiency to olaparib.
In the previously published olaparib/buparlisib trial(14), the objective response rate of olaparib/buparlisib was 29% in patients with gBRCAmut and 12% in patients with gBRCAwt. This response rate was similar to the expected response rate of olaparib monotherapy in gBRCAmut EOC (i.e. ≥60% in platinum sensitive(3, 6) and 28% in platinum resistant gBRCAmut EOC(3, 6)) and gBRCAwt EOC (i.e. 50% in platinum sensitive(3, 6) and ~5% in platinum resistant gBRCAwt EOC(3, 6)), suggesting that addition of buparlisib did not enhance the activity of olaparib monotherapy. This apparent lack of clinical synergism in the olaparib/buparlisib study may have been, at least partly, related to the fact that CNS toxicities precluded dose escalation of buparlisib.
Unlike the olaparib/buparlisib trial where 70% of EOC patients were gBRCAmut, in the current olaparib/alpelisib study, 64% of EOC patients were gBRCAwt and 93% of EOC patients were platinum resistant/refractory, i.e. reflecting a population of patients with EOC tumors that were highly enriched for de novo and acquired HRR proficiency. This population of EOC patients afforded us the unique opportunity to obtain some preliminary clinical evidence that alpelisib may sensitize HRR proficient EOC to olaparib, which was consistent with the mechanistic rationale behind this study. Notably, we observed an ORR of 31.3% in gBRCAwt platinum resistant patients, and 33.3% in BRCAwt (germline and somatic) platinum resistant patients, a setting where the ORR of olaparib and other PARPis as monotherapies is only ~5%(3, 5, 6, 23–25). In this regard, the olaparib alone arm of NRG-GY005 (ClinicalTrial:NCT02502266), a large randomized trial in platinum resistant EOC was recently discontinued, due to limited activity of olaparib as monotherapy in this population. Even in EOCs that are gBRCAwt but HRD positive and platinum resistant, the response to PARPi niraparib monotherapy in the recently reported QUADRA study was only 10%(26). Taken together, these prior studies highlight that olaparib (and PARPis in general) have minimal single agent activity in the gBRCAwt platinum resistant setting, in stark contrast to the preliminary activity of olaparib/alpelisib in this population observed in the present small Phase 1b study.
The median duration of response and median PFS and OS observed in this population were also promising and in line with the results of AURELIA study in platinum resistant ovarian cancer (27) despite the fact that AURELIA involved a significantly less heavily pretreated patient population as it allowed patients with only up to 2 prior lines of anticancer therapy and more than 55% of patients had received only one prior line of anticancer therapy. In contrast, the current study included a patient population with a median of 3 prior lines of cytotoxic therapy (without counting prior hormonal therapy and radiation therapy as separate lines) and included patients with as many as 8 prior lines of therapy.
Furthermore, when we analyzed BRCAwt tumors for HRR and PI3K pathway alterations, we noted consistent objective responses across all different populations including 2 (33.3%) of 6 patients without any HRR and PI3K pathway alterations (Figure 4), one of which had platinum refractory disease and 5 prior lines of therapy. It is also important to underscore that PI3Kis, as monotherapy, have very modest activity in EOC(28, 29). Even in EOC that harbor PIK3CA mutations, responses are infrequently seen with PI3Ki monotherapy; monotherapy ORR in PIK3CA mutated cancers was 0% in one trial(30) with responses observed only with combinations of PI3Kis with other, mainly cytotoxic, agents(30).
The response rate of olaparib/alpelisib in gBRCAmut patients was 30% in all patients and 33.3% in gBRCAmut patients with platinum resistant disease. This activity is not significantly different than the activity of olaparib alone in this ovarian cancer population suggesting that alpelisib does not augment the efficacy of olaparib in the gBRCAmut ovarian cancer population. This is consistent with the mechanistic rationale behind this study, i.e., that the synergism between these agents reflects inhibition of HRR and consequent sensitization to PARPis specifically in HRR proficient tumors, with less synergism expected in tumors enriched for HRR deficiency.
We acknowledge certain limitations of this study. This was a Phase 1b clinical trial, with a small number of patients and with the known limitations of 3+3 designs in selecting optimal doses under both toxicity and biological activity (31–33). Response rate by RECIST 1.1 was a secondary endpoint with the goal of determining the preliminary activity of the olaparib/alpelisib combination. We analyzed the clinical response in different and distinct subsets of patients to assess whether responses are consistently observed across these subsets, and to evaluate whether these clinical observations support our hypothesis (formulated by our preclinical work included in the Appendix pp 9–15) that alpelisib can sensitize HRR-proficient ovarian cancers to olaparib. The analyses we presented are largely descriptive of the treatment effects and summarized without conducting a hypothesis test against a null. We fully acknowledge that such inferences of patient subsets would require a larger confirmatory study to be adequately powered and will be a critical component of further examination of olaparib/alpelisib in this disease. These limitations notwithstanding, our study indicated that the combination of alpelisib and olaparib produced no unexpected toxicities or safety signals and exhibited preliminary clinical evidence of synergism in gBRCAwt and BRCAwt (somatic and germline) platinum resistant ovarian cancers, i.e. in tumors enriched for HRR proficiency. To our knowledge, this is the first time that clinical evidence of synergism between a PI3Ki and a PARPi is reported in EOC. The activity of the olaparib/alpelisib combination in this setting appears higher than expected from either olaparib or alpelisib monotherapies and warrants further investigation.
Supplementary Material
ACKNOWLEDGMENTS
We thank all the patients and their families for participation in the present trial. This investigator-initiated trial was supported by the Stand Up To Cancer-Ovarian Cancer Research Fund Alliance-National Ovarian Cancer Coalition Ovarian Cancer Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT16–15). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. This work was also supported by the Breast Cancer Research Foundation. We thank Novartis for providing funding and BYL719 and AstraZeneca for providing olaparib.
Funding: Stand-Up-To-Cancer Ovarian Cancer Dream Team, Breast Cancer Research Foundation
Footnotes
DECLARATION OF INTERESTS
Dr. Konstantinopoulos reports other from AstraZeneca, other from Novartis, during the conduct of the study; other from AstraZeneca, other from Pfizer, other from Merck, outside the submitted work. Dr. Aghajanian reports personal fees from Tesaro, personal fees from Immunogen, personal fees from Clovis, personal fees from Mateon Therapeutics, personal fees from Cerulean Pharma, outside the submitted work. Dr. Westin reports grants and personal fees from AstraZeneca, grants and personal fees from Clovis, grants and personal fees from Tesaro, grants from Bayer, personal fees from Pfizer, grants and personal fees from Merck, grants from Cotinga Pharmaceuticals, grants from Novartis, personal fees from MediVation, grants and personal fees from Roche/Genentech, outside the submitted work. Dr. Coleman reports personal fees from Tesaro, personal fees from Agenus, grants from Merck, grants from Abbvie, grants and personal fees from Roche/genentech, grants and personal fees from Clovis, grants and personal fees from AstraZeneca, grants and personal fees from Janssen, grants from Esperance, grants and personal fees from Oncomed, grants and personal fees from Novartis, grants and personal fees from Genmab, personal fees from Eisai, personal fees from Gamamab, personal fees from incyte, outside the submitted work. Dr. Matulonis reports personal fees from Astrazeneca, personal fees from Myriad Genetics, personal fees from Clovis, personal fees from Merck, personal fees from Eli Lilly, personal fees from Mersana, personal fees from Geneos, personal fees from Fuji Film, from 2X Oncology, personal fees from Cerulean, personal fees from Immunogen, outside the submitted work. Dr. Barry reports other research support (institution) from Pfizer, outside the submitted work. Dr. Liu reports personal fees from AstraZeneca, personal fees from Tesaro, Inc, personal fees from Mersana Therapeutics, personal fees from Clovis Oncology, outside the submitted work. Dr. Winer reports personal fees from Genentech, personal fees from Infinite MD, personal fees from Lilly, personal fees from Leap Therapeutics, personal fees from Carrick Therapeutics, personal fees from GSK, other from Verastem, outside the submitted work. Dr. O’Cearbhaill reports personal fees from Clovis, personal fees from Tesaro, outside the submitted work. Dr. Mayer reports other from Pfizer, other from Myriad, from null, from null, rom null, during the conduct of the study; other from Eisai, other from Pfizer, other from Lilly, other from Context Therapeutics, outside the submitted work. Dr. Cadoo reports other from Astra Zeneca, other from Astra Zeneca, other from Syndax Pharmaceuticals, outside the submitted work. Dr. Kaufmann reports grants from Stand Up to Cancer, during the conduct of the study. Dr. Wulf reports grants from SU2C-AACR-DT0209, grants from Mary Kay Ash Foundation, grants from Ovarian Cancer Research Foundation, grants from Breast Cancer Alliance, grants from Breast Cancer Research Foundation, grants from NIH RO1 1R01CA226776–01, during the conduct of the study; In addition, Dr. Wulf has a patent Application 14/348810 Compositions and Methods for the Treatment of proliferative diseases pending, and a patent US 20090258352 A1 Pin as a marker for abnormal cell growth licensed to Cell Signaling; R&D Systems. Dr. Shapiro reports grants and personal fees from Lilly, grants and personal fees from Pfizer, grants and personal fees from Merck/EMD Serono, grants and personal fees from Sierra Oncology, personal fees from Roche, personal fees from Bicycle Therapeutics, personal fees from Fusion Pharmaceuticals, personal fees from G1 Therapeutics, personal fees from Cybrexa Therapeutics, personal fees from Bayer, personal fees from Ipsen, personal fees from Astex, personal fees from Almac, outside the submitted work. Dr. Palakurthi reports other from Elstar Therapeutics, outside the submitted work. Dr. Cantley is a founder and member of the SAB of Agios Pharmaceuticals and of Petra Pharmaceuticals. These companies are developing novel therapies for cancer. His laboratory also receives some financial support from Petra Pharmaceuticals. No drugs from these companies are discussed in this manuscript. Dr. Mills reports grants and personal fees from AstraZeneca, other from Catena Pharmaceuticals, grants and personal fees from Critical Outcome Technologies, grants and personal fees from ImmunoMET, other from Ionis, personal fees from PDX Pharmaceuticals, personal fees from Signalchem Lifesciences, personal fees from Symphogen, grants and personal fees from Takeda/Millennium Pharmaceuticals, personal fees from Tarveda, personal fees from Spindle Top Ventures, grants from Adelson Medical Research Foundation, grants from Breast Cancer Research Foundation, grants from Komen Research Foundation, grants from Nanostring, grants from Ovarian Cancer Research Foundation, grants from Prospect Creek Foundation, grants and personal fees from Pfizer, during the conduct of the study; In addition, Dr. Mills has a patent Nanostring licensed to Nanostring, and a patent Myriad Genetics licensed to Myriad. Dr. Swisher reports grants from Stand up to Cancer, during the conduct of the study; personal fees from johnson and johnson, outside the submitted work. Drs. Luo, Birrer, Kirschmeier, Buss, D’Andrea, Kochupurakkal and Eismann have nothing to disclose. Ms. Farooq, Mrs. Whalen and Mrs. Curtis have nothing to disclose.
DATA SHARING STATEMENT
Upon email request, we would be happy to provide fully de-dentified analysis datasets including: i) Dose-level information for the subjects (one record per subject), ii)Toxicity listings (multiple records per subject) and iii) baseline characteristics and efficacy data for the EOC subgroup (one record per subject).
REFERENCES
- 1.TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011. June 30;474(7353):609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015. November;5(11):1137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol. 2016. June;27(6):1013–9. [DOI] [PubMed] [Google Scholar]
- 4.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. 2016. December 01;375(22):2154–64. [DOI] [PubMed] [Google Scholar]
- 5.Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016. January;18(1):75–87. [DOI] [PubMed] [Google Scholar]
- 6.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011. September;12(9):852–61. [DOI] [PubMed] [Google Scholar]
- 7.Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017. September;7(9):984–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008. February 28;451(7182):1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008. April 15;68(8):2581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012. November;2(11):1036–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012. November;2(11):1048–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehman FL, Lord CJ, Ashworth A. The promise of combining inhibition of PI3K and PARP as cancer therapy. Cancer Discov. 2012. November;2(11):982–4. [DOI] [PubMed] [Google Scholar]
- 13.Juvekar A, Hu H, Yadegarynia S, Lyssiotis CA, Ullas S, Lien EC, et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc Natl Acad Sci U S A. 2016. July 26;113(30):E4338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matulonis UA, Wulf GM, Barry WT, Birrer M, Westin SN, Farooq S, et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann Oncol. 2017. March 1;28(3):512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.André F, Ciruelos EM, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. , editors. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the Phase 3 SOLAR-1 trial. European Society of Medical Oncology (ESMO); 2018; Munich, Germany. [Google Scholar]
- 16.Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI insight. 2016. November 17;1(19):e87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer discovery. 2012;2(1):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of OncoPanel: A Targeted Next-Generation Sequencing Assay for the Detection of Somatic Variants in Cancer. Arch Pathol Lab Med. 2017. June;141(6):751–8. [DOI] [PubMed] [Google Scholar]
- 19.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010. July 24;376(9737):245–51. [DOI] [PubMed] [Google Scholar]
- 20.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014. May;13(5):1117–29. [DOI] [PubMed] [Google Scholar]
- 21.Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez SM, et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017. July;7(7):704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2018. October 21. [DOI] [PubMed] [Google Scholar]
- 23.Domchek SM, Aghajanian C, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016. February;140(2):199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010. May 20;28(15):2512–9. [DOI] [PubMed] [Google Scholar]
- 25.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013. August;14(9):882–92. [DOI] [PubMed] [Google Scholar]
- 26.Moore K, Secord A, Geller M, Miller D, Cloven N, Fleming G, et al. QUADRA: A phase 2, open-label, single-arm study to evaluate niraparib in patients (pts) with relapsed ovarian cancer (ROC) who have received ≥3 prior chemotherapy regimens. ASCO Meeting 2018; 2018; Chicago, IL. J Clin Oncol 36, 2018. (suppl; abstr 5514); 2018. [Google Scholar]
- 27.Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol. 2014. May 1;32(13):1302–8. [DOI] [PubMed] [Google Scholar]
- 28.Matulonis U, Vergote I, Backes F, Martin LP, McMeekin S, Birrer M, et al. Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol Oncol. 2015. February;136(2):246–53. [DOI] [PubMed] [Google Scholar]
- 29.Rodon J, Brana I, Siu LL, De Jonge MJ, Homji N, Mills D, et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014. August;32(4):670–81. [DOI] [PubMed] [Google Scholar]
- 30.Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012. Mar 10;30(8):777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandrekar SJ, Cui Y, Sargent DJ. An adaptive phase I design for identifying a biologically optimal dose for dual agent drug combinations. Stat Med. 2007. May 20;26(11):2317–30. [DOI] [PubMed] [Google Scholar]
- 32.Paoletti X, Ezzalfani M, Le Tourneau C . Statistical controversies in clinical research: requiem for the 3 + 3 design for phase I trials. Ann Oncol. 2015. September;26(9):1808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin G, Li Y, Ji Y. Bayesian dose-finding in phase I/II clinical trials using toxicity and efficacy odds ratios. Biometrics. 2006. September;62(3):777–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.