

Abstract
The C-terminal domain (CTD) of the largest subunit in eukaryotic RNA polymerase II has a repetitive heptad sequence of Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7 which is responsible for recruiting transcriptional regulatory factors. The seventh heptad residues in mammals are less conserved and subject to various post-translational modifications, but the consequences of such variations are not well understood. In this study, we use ultraviolet photodissociation mass spectrometry, kinetic assays, and structural analyses to dissect how different residues or modifications at the seventh heptad position alter Tyr1 phosphorylation. We found that negatively charged residues in this position promote phosphorylation of adjacent Tyr1 sites, whereas positively charged residues discriminate against it. Modifications that alter the charges on seventh heptad residues such as arginine citrullination negate such distinctions. Such specificity can be explained by conserved, positively charged pockets near the active sites of ABL1 and its homologues. Our results reveal a novel mechanism for variations or modifications in the seventh heptad position directing subsequent phosphorylation of other CTD sites, which can contribute to the formation of various modification combinations that likely impact transcriptional regulation.
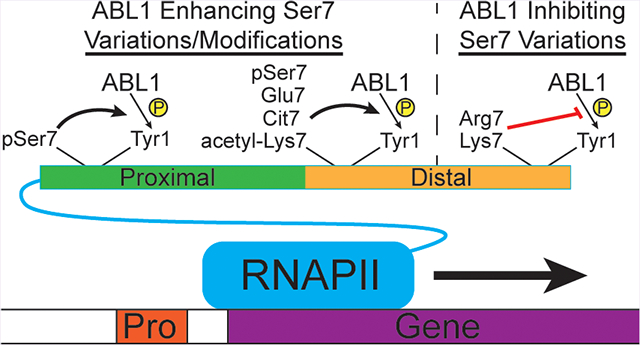
Grphical Abstract
The C-terminal domain (CTD) of RPB1, the largest subunit of eukaryotic RNA polymerase II (RNAPII), is a major hub for post-translational modifications (PTMs) that regulates protein recruitment throughout the transcriptional cycle.1−4 The consensus CTD heptad sequence of Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7 is conserved in eukaryotes2,3 and is essential for cellular survival.5−7 A wide variety of modifications can occur on the CTD, including phosphorylation, glycosylation, and even methylation and acetylation on nonconsensus residues.3,8 Of all the possible CTD PTMs, phosphorylation of Ser2 and Ser5 is critical for temporally recruiting certain regulatory factors during the transcription cycle.3,4 The other three nonproline residues in the CTD consensus sequence (namely, Tyr1, Thr4, and Ser7) have also been found to be phosphorylated in vivo,3 although the biological context and transcriptional implications of phosphorylation at these sites are not yet fully understood.
Of the seven residues in CTD heptad repeats, the seventh (typically Ser7) is the least conserved among species containing CTDs.3 The CTDs from unicellular eukaryotes like yeast predominantly contain consensus repeats, whereas mutations at the seventh position are frequently found in CTDs from multicellular eukaryotes.3 The biological significance of the divergence of identities of the seventh residue is yet to be understood but has been speculated to allow for functional diversification during transcription.2,3 In human RNAPII, the first half of the 52 heptad repeats in human CTD proximal to the core of RPB1 adheres closely to the consensus sequence. In contrast, the distal 26 repeats deviate greatly from the consensus sequence predominantly at the seventh position, varying from positively charged lysine and arginine residues to a negatively charged glutamate and several small polar residues (Figure 1A). In addition to the divergence of residue identity in the seventh CTD position, various chemical modifications have been detected at this site during eukaryotic transcription. Phosphorylation of Ser7 during transcription is an evolutionarily conserved mechanism in eukaryotes and appears to peak during the early stages of transcription in vivo.9,10 A Ser7 substitution to arginine in the human distal CTD (Arg1810) can be citrullinated, which helps recruit the positive transcription elongation factor (P-TEFb) to RNAPII thereby promoting the transition from transcriptional pausing to elongation.11 Additionally, acetylation and methylation of Lys7, as well as methylation of Arg7 residues, have been reported in human CTD.12−14 The diverse amino acid and modification possibilities at the seventh position of the RNAPII CTD heptad repeats raise questions as to whether these alterations can affect other CTD modifications and in turn regulate CTD protein recruitment and transcription.
Figure 1.

Mapping of ABL1 phosphorylation on the human distal CTD. (A) Overall map of 6xHis-GST linked human distal CTD phosphorylated by ABL1. Trypsin cleavage sites are indicated by horizontal bars. Major and minor ABL1 phosphorylation sites within each peptide are shown in dark and light blue boxes, respectively. Ser7 variations are colored based on their side chain character (red, negative charge; purple, positive charge; green, polar; and orange, nonpolar). CTD heptads that could not be mapped are enclosed in colored boxes. (B−G) Results from LC-MS/MS of singly phosphorylated species in distal CTD treated with ABL1. Extracted chromatograms and pie charts indicating relative abundances of singly phosphorylated species derived from UVPD-MS are shown. The numbers above the peptide sequences and next to the LC-MS peaks designate the corresponding distal CTD repeat. Vertical bars in the sequences indicate trypsin cleavage sites.
Unlike the poorly conserved Ser7 position, the Tyr1 residue found next to it in CTD heptads is one of the most conserved in the CTD.3 The chemical properties of the Tyr1 side-chain appear to be essential for proper CTD function during transcription. Mutation of Tyr1 to alanine abolishes phosphorylation of Ser2 and Ser5 by CTD kinases such as TFIIH and Erk2 in vitro.15 Even making conservative Tyr1 mutations to phenylalanine in human cells leads to a loss of viability pointing to an essential role for Tyr1 phosphorylation.16 Phosphorylation of Tyr1 residues has been detected in both unicellular and multicellular eukaryotes17−20 and has been implicated in RNAPII stability,21 antisense transcription,16 and accurate termination.18,22 The Abelson murine leukemia viral oncogene homologue 1 (ABL1) tyrosine kinase was the first RNAPII Tyr1 kinase identified in human cells, but other tyrosine kinases likely complement its function.23,24 ABL1 phosphorylation of Tyr1 has been linked to DNA damage response24 and more recently to RNAPII-dependent production of damage responsive antisense transcripts that mediate DNA repair.25 During transcription in vivo, Tyr1 phosphorylation predominantly occurs after Ser7 phosphorylation near the transcription start site.16 The positional and temporal closeness of Ser7 and Tyr1 led us to investigate whether the identities and PTM states of the seventh residue in CTD heptads can affect the neighboring Tyr1 phosphorylation status.
To examine the placement of Tyr1 phosphorylation on the CTD by ABL1, we used ultraviolet photodissociation (UVPD) coupled to liquid chromatography-tandem mass spectrometry (LC-MS/MS) to accurately localize Tyr1 sites of ABL1 kinase26 phosphorylation on the human distal CTD. Surprisingly, the charge of the residue in the seventh position of heptad repeats strongly correlated with the phosphorylation level of the adjacent Tyr1 residue. Using a combination of kinetic kinase assays and mass spectrometry on different CTD variants, we found that ABL1 preferentially phosphorylates Tyr1 when directly adjacent to a negatively charged residue in the seventh CTD position. This preference can be explained structurally by the presence of a positively charged pocket in ABL1 next to the tyrosine binding site that appears to recognize residues at the seventh position in heptad repeats. In contrast, positively charged residues in the seventh heptad position, such as arginine and lysine, appear to inhibit phosphorylation of Tyr1 sites adjacent to them. Tyr1 site preference can be altered when the seventh residue is modified, such as when Arg1810 is citrullinated. The positively charged pocket dictating the selection of Tyr1 found in ABL1 is conserved in other nuclear tyrosine kinases that might have overlapping function with ABL1. Our biochemical analysis demonstrates that the identity and modification state of the seventh CTD residue can significantly alter ABL1 recognition of Tyr1, which may regulate CTD function during transcription or other CTD-dependent processes.
RESULTS AND DISCUSSION
Mapping ABL1 Phosphorylation on the Human Distal CTD
To determine how the sequence variations in the distal CTD impact Tyr1 phosphorylation, we utilized tandem mass spectrometry to characterize the site and relative amount of phosphorylation of ABL1 on purified human distal CTD. However, CTD phosphorylation patterns can be difficult to determine using mass spectrometry owing to several technical challenges. First, it is difficult to uniformly proteolyze long, mostly consensus CTD sequences into suitable peptide segments for mass spectrometric mapping due to the lack of protease cutting sites.19,20 The human CTD sequence can be characterized as two distinct halves: the proximal CTD containing the first 26 repeats that are mostly consensus sequence and the distal CTD containing the last 26 repeats that have many more sequence variations. In particular, the distal CTD contains several Arg7 and Lys7 mutations that can be proteolyzed with trypsin into peptides mostly under 30 amino acids long, which simplifies mapping of phosphosites using LC-MS/MS. Second, the repetitive sequence of the CTD can prevent accurate localization of phosphorylation sites along the full-length CTD. Trypsin cleavage of the human distal CTD, however, generates mostly unique fragments that can be differentiated by mass due to the many residue changes from the consensus sequence. Finally, using traditional ion activation methods, such as low-energy, collision-induced dissociation (CID), to characterize peptide sequences is often inadequate for confidently localizing phosphorylation sites owing to a prevalent loss of labile modifications during the activation step, a process that is particularly problematic for CTD sequences that contain several potential phosphosites. To overcome this challenge, we employed ultraviolet photodissociation mass spectrometry (UVPD-MS), in which ions are energized via absorption of high-energy photons resulting in more diagnostic fragmentation patterns.27 Moreover, the fast activation process of UVPD does not promote cleavage of PTMs. The resulting rich UVPD mass spectra afford enhanced sequence coverage and characterization of phosphorylated CTD.15 Thus, we were able to interrogate the effects of distal CTD variations on Tyr1 phosphorylation with high confidence using UVPD-MS.
A construct containing the 26 human distal CTD heptad repeats from residues 1769−1970 was expressed as a 6xHis-GST linked recombinant protein (Table S1) and treated with the ABL1 kinase domain (residues 229−511; Figure 1A). After phosphorylation, the GST linker was cleaved, and the CTD protein was digested by trypsin protease to cut after the single Arg7 and eight Lys7 sites in the distal sequence (Figure 1A). The resulting CTD heptads were separated using liquid chromatography and activated using UVPD, providing means by which to map ABL1 phosphorylation on approximately 70% of the distal CTD sequence (Figure 1A). The mapped regions included every heptad in the distal CTD except for those in the long stretch from repeats 26−31 and the two repeats 39−40 which would have been proteolyzed into single heptads owing to repeating Lys7 variations (Figure 1A). We examined ABL1 phosphorylation on recombinant CTD or synthetic peptides containing similar sequences found in these missing regions in later experiments. In all of the proteolyzed peptides that were analyzed by UVPD-MS, we confirmed that Tyr1 is the only target site in the CTD for ABL1 phosphorylation (Figure 1B–G and Table S2). For each segment of the mapped human distal CTD, we could discern relative ABL1 target site preferences based on the intensity of the peak for each species. We found that certain phosphorylation species were much more abundant than others, suggesting that the sequence composition of the CTD heptads affects ABL1 specificity (Figure 1B–G). For example, in heptads 36−38, which contain the sole Glu7 variation in the distal CTD, we found that roughly 86% of the singly phosphorylated species exhibit phosphorylation of the Tyr1 residue directly adjacent to the Glu7 (Figure 1C). Phosphorylation of the other two Tyr1 sites in these repeats represented approximately 14% of the observed species, indicating that the Tyr1 residue adjacent to Glu7 was a highly preferred substrate (Figure 1C). This result is striking because glutamate can act as a phosphoserine mimic which expands the potential for ABL1 phosphosite regulation. In contrast, the peptides with Tyr1 phosphorylation adjacent to Arg/Lys7 mutations were the least abundant species in each case (Y32 in Figure 1B, Y36 in 1C, Y41 in 1D, Y43 in 1E, and Y46/48 in 1F). In particular, Tyr1 sites next to Lys7 were very rarely phosphorylated in the peptides containing heptads 41−42 and 46−49 (Figure 1D and F). Altogether, our UVPD-MS analysis of ABL1-treated human distal CTD revealed a novel phosphorylation pattern that suggests variations in the seventh CTD position dictate placement of Tyr1 phosphorylation.
Glutamate in the Seventh CTD Position Promotes ABL1 Phosphorylation of Adjacent Tyr1 Sites
Our mapping of ABL1 phosphorylation of the distal CTD identified the Tyr1 residue directly adjacent to the lone Glu7 mutation (Glu1852) as a highly preferable site for phosphorylation. Since there are other variations to the consensus sequence in heptads 36−38, we wanted to decipher whether Glu1852 was specifically promoting ABL1 phosphorylation of the adjacent Tyr1 sites. To test if glutamate in the seventh CTD position alone is sufficient to specify the phosphorylation of Tyr1, we used UVPD-MS to map Tyr1 phosphorylation sites on CTD substrates with the only difference as Ser7Glu mutations in certain heptads. We first generated a recombinant 6xHis-GST linked CTD containing three heptad repeats with some of the substrates containing site-specific Ser7 variations in order to map Tyr1 phosphorylation sites (Table S1). This design has several advantages: after 3C proteolysis to remove the purification tags, the three-repeat sequence is short enough to be amenable to a bottom-up LC-MS/MS workflow using UVPD without the requirement of further CTD proteolysis. This three-repeat CTD design is also long enough for ABL1 recognition and phosphorylation as shown by the appearance of Tyr1 phosphorylation peaks in the spectra for the variant containing consensus sequence in all three-repeats (3xWT; Figure 2A and Table S3). The LC-MS trace of singly phosphorylated species in this sample revealed two peaks of comparable abundance and a third peak with very low abundance (Figure 2A). UVPD-MS analysis identified the Tyr1 residues of the first and second repeats as the sites of major ABL1 phosphorylation (Figure 2A). We observed only minor amounts of phosphorylation of the last repeat (Figure 2A), possibly owing to the higher conformational entropy of this repeat being at the end of the sequence which could reduce ABL1 recognition. However, the roughly similar amounts of phosphorylation of the first and second repeats show that our recombinant CTD substrate design does not elicit any major pre-existing bias for ABL1.
Figure 2.

ABL1 preferentially phosphorylates Tyr1 directly adjacent to negatively charged residues specifically in the seventh CTD position. (A) LC-MS trace of singly phosphorylated species in three-repeat wild-type CTD substrate (3xWT) treated with ABL1. Phosphorylation sites were localized using UVPD-MS and are indicated on the chromatogram by repeat number. The GPGSGM amino acid sequences at the N-termini are retained after 3C proteolysis of the 6xHis-GST tags prior to LC-MS/MS analysis. The sites of major and minor phosphorylation are indicated by dark or light blue boxes, respectively. (B,C) Extracted chromatograms of singly phosphorylated species in three-repeat CTD substrates with Ser7Glu mutations (3xS7E Mid and Sp) treated with ABL1. The glutamate mutations in each sequence are shown in red. (D) Extracted chromatograms of singly phosphorylated species in three-repeat CTD substrate with Ser2Glu mutation (3xS2E Mid) treated with ABL1.
To determine if glutamate in the seventh CTD position can dictate ABL1 phosphorylation site preference, we designed three-repeat CTD constructs with Glu7 mutations in alternating heptads. To avoid positional bias, we made two three-repeat constructs with Glu7 in either the middle heptad (3xS7E Mid) or the first and third heptads (3xS7E Sp; Table S1). In the 3xS7E Mid construct, we observed one major phosphorylation species when treated with ABL1 corresponding to phosphorylation of the Tyr1 site directly adjacent to the middle Glu7 mutation (Figure 2B and Table S3). In the 3xS7E Sp sample, we observed a majority of ABL1 phosphorylation of the first and third Tyr1 residues that are adjacent to Glu7 variations with a very small amount of phosphorylation at the second Tyr1 (Figure 2C). These results support our distal CTD mapping that Glu1852 is a major factor in promoting ABL1 phosphorylation of the adjacent Tyr1 residue. Thus, there is a strong preference of Tyr1 phosphorylation by ABL1 when negatively charged Glu7 is the preceding residue.
Since the replacement of Ser7 with glutamate promotes ABL1 phosphorylation of adjacent Tyr1 residues, we were curious as to whether the mutation of Ser2 to glutamate could elicit the same effect due to its proximity. To test this, we designed a three-repeat substrate with the middle Ser2 mutated to glutamate (3xS2E Mid; Table S1). ABL1 phosphorylation of this substrate resulted in near-equivalent amounts of Tyr1 phosphorylation of all Tyr1 sites (Figure 2D and Table S3). ABL1 phosphorylation of 3xS2E Mid stands in stark contrast to the preferential phosphorylation observed with the 3xS7E substrates but is almost identical to the 3xWT phosphorylation pattern (Figure 2A and D). Therefore, the negatively charged residue specifically in the seventh CTD position primes phosphorylation of adjacent Tyr1 sites by ABL1, supporting our finding that the Ser7 variation Glu1852 in the human distal CTD directs ABL1 phosphorylation of the adjacent Tyr1.
Although Glu7 replacement of Ser7 in human CTD is infrequent, the chemical structure and properties of glutamate closely mimic phosphoserine and phosphothreonine. Phosphorylation of Ser7 is conserved from yeast to human9,10,28 and has been associated with snRNA transcription.29 Thus, we speculated that Ser/Thr7 phosphorylation could direct Tyr1 phosphorylation. To test whether ABL1 preferentially phosphorylates Ser7 phosphorylated substrates, we measured the kinetic activity of ABL1 toward a 26 repeat wild-type yeast CTD (26xWT) in comparison to a similar construct with every Ser7 replaced by glutamate residues (26xS7E; Table S1). We observed an increase in ABL1 reactivity toward the 26xS7E substrate with a kcat/Km of 0.4 mM−1 s−1 compared to 0.3 mM−1 s−1 for the 26xWT substrate (Figure 3A). While the 33% increase in ABL1 kinetic efficiency for our 26xS7E substrate is not particularly large, we suspect that this places advantages on Tyr1 preceded by Glu7 or phosphoryl-Ser7 residues for ABL1 phosphorylation.
Figure 3.

ABL1 preferentially phosphorylates Tyr1 directly adjacent to phosphoserine or phosphothreonine mimics in the seventh CTD position. (A) Radiolabeled kinetics of ABL1 against 26 repeat wild-type (26xWT, blue) and Ser7Glu (26xS7E, green) CTD substrates. (B) Extracted chromatogram of singly phosphorylated, consensus diheptad species (YSPTSPSYSPTSPE) from the 26 repeat S7E spaced CTD substrate (26xS7E Sp) treated with ABL1 and GluC protease. GluC protease sites are designated by vertical bars, and glutamates are shown in red in the sequence. (C,D) UVPD-MS spectra of singly phosphorylated, consensus diheptad species (YSPTSPSYSPTSPE) from the 26xS7E Sp substrate treated with ABL1 and GluC protease. Labels correspond to backbone cleavage sites as shown in the peptide diagram in C. Asterisks indicate retention of phosphorylation allowing for localization. (E,F) Extracted chromatograms of singly phosphorylated, nonconsensus diheptad species (YSPTSPAYSPTSPE and YSPTSPNYSPTSPE) from the 26xS7E Sp substrate treated with ABL1 and GluC protease. Mutations in the seventh CTD position are denoted by color based on the character of the side chain (orange for hydrophobic alanine and green for polar asparagine).
To test this possibility, we placed glutamate phosphomimics strategically in full-length CTD to see if the positions of these residues dictate ABL1 patterning of the CTD. This strategy was chosen in lieu of substrates with Ser7 phosphorylated in vitro by the Ser7 kinase TFIIH since this kinase predominantly phosphorylates Ser5.10,28 To isolate the effect of Ser/Thr7 phosphorylation on ABL1 recognition, we designed a 26-repeat yeast CTD with every other heptad containing Glu7 mutations to mimic Ser7 phosphorylation (26xS7E Sp; Table S1). The construct design is advantageous for LC-MS/MS analysis of a long CTD substrate, as the Glu7 residues serve as GluC protease cleavage sites and the spaced arrangement of these mutations allows dissection of which Tyr1 sites are preferred by ABL1. After treating 26xS7E Sp with ABL1 followed by GluC proteolysis, we observed two clearly separated peaks for the main singly phosphorylated diheptad fragments (YSPTSPSYSPTSPE) with one of these species being highly abundant (>90%; Figure 3B). UVPD-MS analysis of these peaks showed that the major species contained phosphorylation of Tyr1 sites that were adjacent to the Glu7 residues (Figure 3C), and the minor species contained phosphorylation of Tyr1 sites next to consensus Ser7 residues (Figure 3D and Table S4). Furthermore, we were able to identify Tyr1 phosphorylation sites on a few heptads in our construct that have different Ser7 variations which exist both in yeast and human CTD (Figure 3D and E). In both of the mapped diheptad peptide sets with either Ala7 or Asn7 variations (YSPTSPAYSPTSPE and YSPTSPNYSPTSPE), the Tyr1 sites adjacent to Glu7 were preferentially phosphorylated by ABL1 (Figure 3D and E). Overall, our results with full-length CTD substrates suggest that negatively charged Glu7 accommodates preferential phosphorylation of adjacent Tyr1 sites. Since glutamate resembles phosphorylated serine, Tyr1 subsequent to phosphoryl-Ser7 residues is also likely a preferred site of phosphorylation by ABL1.
Structural Rationale for ABL1 Regulation by Mutations in the Seventh CTD Position
Our UVPD-MS analyses show that the negatively charged residue glutamate strongly promotes phosphorylation of adjacent Tyr1 residues by ABL1. To understand why this specificity exists, we analyzed the structure of ABL1 and how it recognizes a substrate. Kinase−substrate complexes are transient and hard to capture by X-ray crystallography. Thus, researchers have designed ATP-peptide bisubstrate analogs that can be used in crystallography to capture the substrate-binding mode of tyrosine kinases.30,31 These bisubstrate analogs contain ATP covalently linked to a nitrophenylalanine as a part of a short peptide.30 The tight binding of ATP to the kinase stabilizes the complex, thus allowing visualization of the phosphotyrosinemimicking nitrophenylalanine and peptide residues within the tyrosine kinase substrate-binding pocket.30 In the structure of ABL1 bound to an ATP-peptide bisubstrate analog,31 we see that the nitrophenylalanine is locked into place by Gln252 and Lys400 (Figure 4A). An electrostatically positive pocket is located next to the nitrophenylalanine mimicking phosphotyrosine which accommodates the residue that normally precedes the substrate tyrosine (Figure 4B). Although the structure of CTD bound by ABL1 is not available, Tyr1 would likely occupy a similar position as the nitrophenylalanine in the ATP-peptide bisubstrate analog bound structure. This would place the residue preceding bound Tyr1, the seventh residue from the previous heptad, into this positive pocket directly next to the active site. Indeed, when we modeled Glu7 into the bisubstrate analog structure it situated within ~3.6 Å of the positively charged side chain of Arg367 of ABL1 (Figure 4C). The close proximity of these residues would suggest the formation of a salt bridge between Glu7 of CTD and Arg367 of ABL1. Furthermore, salt bridge formation would seem to be stabilized by the indole group of Trp405 forming a cation−π interaction with the side chain of Arg367 (~3.4 Å; Figure 4C). This tightly coordinated system of interactions between Glu7 and Arg367/Trp405 provides a structural explanation for why negatively charged residues in the seventh CTD position are preferentially recognized by ABL1. Similarly, phosphoserine modeled into the same pocket would be situated even closer to the side chain of Arg367 than Glu7 with favorable interaction (~2.6 Å; Figure 4D). Likewise, phosphorylated Thr7 residues in the distal CTD would closely resemble phosphorylated Ser7 residues and likely form favorable electrostatic interaction with the ABL1 Arg/Trp pocket to promote Tyr1 phosphorylation. On the other hand, when we mutated Ser2 to glutamate we did not see a similar effect on ABL1 specificity (Figure 2D), suggesting that phosphorylated Ser2 would not be recognized by the Arg/Trp pocket. Assuming that the binding of CTD to ABL1 is similar to its canonical substrates in terms of N- to C-terminal orientation, the Glu2 results are consistent with our prediction that the placement of a negatively charged residue in the position preceding but not following the target tyrosine would affect ABL1 recognition. Altogether, the Arg/Trp pocket of ABL1 appears to form favorable hydrophilic interactions with negatively charged or phosphorylated residues specifically in the seventh CTD position explaining why ABL1 preferentially phosphorylates Tyr1 sites adjacent to Glu7 mutations.
Figure 4.

Structural rationale for negatively charged residues in the seventh CTD position directing ABL1 phosphorylation of Tyr1. (A) Structure of ABL1 (light blue) bound to an ATP-peptide bisubstrate analog (dark green) based on the SRC sequence (PDB: 2G1T). The ABL1 surface is represented as a gray transparent layer around the protein. (B) Electrostatic region of ABL1 preceding the nitrophenylalanine binding pocket (blue−positive, red−negative). (C,D) Modeling of a phosphoserine mimic or phosphoserine (Glu or pSer, respectively; white) into the Ile106 position of the ATP-peptide bisubstrate analog. The most likely rotamer for Glu and pSer was selected for modeling. Predicted salt bridges between the modeled Glu/pSer and Arg367 of ABL1 are shown in yellow.
Arg1810 and Its Citrullination Change the Preference of Tyr1 Phosphorylation by ABL1
Our structural analysis of ABL1 binding to substrate highlights a basic pocket close to the active site that likely affects substrate recognition. Because negatively charged residues in the seventh CTD position appear to enhance recognition of Tyr1 by ABL1, we next asked whether positively charged residues like arginine would instead hinder ABL1 recognition. The human distal CTD contains a single seventh heptad arginine variation, Arg1810, in the 31st heptad repeat (Figure 1A). In our distal CTD mapping, phosphorylation of the Tyr1 following Arg1810 was rarely detected, accounting for only 1% of the products for repeats 32−35 (Figure 1B). To test the effect of arginine substitution on ABL1 preference directly, we designed a 6xHis-GST linked three-repeat CTD substrate with Ser7 replaced with arginine in the first and last heptad (3xS7R Sp, Table S1). We then used UVPD-MS to map where ABL1 phosphorylation occurred on this substrate (Table S5). As predicted, we observed increased phosphorylation of the Tyr1 adjacent to Ser7 compared to the Arg7 residues in the 3xS7R Sp substrate accounting for approximately 81% of the phosphorylated products (Figure 5A). Examining the structure of ABL1 bound to an ATP-peptide bisubstrate with arginine modeled into the position preceding the substrate tyrosine, we see that this arginine would extend into the positively charged pocket and likely cause unfavorable charge repulsion with Arg367 (Figure 5B). This repulsive structural interaction explains why Tyr1 following Arg1810 in the human distal CTD is a very poor substrate for ABL1 phosphorylation.
Figure 5.

Arginine and its citrullination in the 7th CTD position alter ABL1 preference for Tyr1 sites. (A) Extracted chromatograms of singly phosphorylated species in three-repeat CTD substrate with Ser7Arg mutations (3xS7R Sp) treated with ABL1. Phosphorylation sites were localized using UVPD-MS and are indicated on the chromatogram by repeat number. The GPGSGM amino acid sequence at the N-terminus is left over from 3C proteolysis of the 6xHis-GST tags prior to mass spectrometry. The sites of major and minor phosphorylation were indicated by dark or light blue boxes, respectively, and the arginine mutations are shown in purple. (B) Structure of ABL1 (light blue) bound to the ATP-peptide bisubstrate analog of SRC (green) with Ile106 modeled as an arginine (white). (C,D) Extracted chromatograms of peptides derived from human distal CTD repeats 30−33 containing either wild-type Arg1810 or citrullinated Arg1810 (Cit1810, green) treated with ABL1. (E) UVPD-MS spectrum of the novel peak in ABL1 treated Cit1810 peptide corresponding to phosphorylation of the middle tyrosine (peak 2).
While Arg1810 is the only Arg7 mutation in the human CTD, it has been found to affect transcription in different ways through differential post-translational modifications.12,32 In particular, citrullination of Arg1810 appears to play an important role in enhancing the transcription of specific genes.11 Citrulline (Cit) is almost identical in structure to arginine except that the guanidinium group is replaced with a carbamide, resulting in a loss of positive charge (Figure 5C,D). Because arginine in the seventh CTD position appears to hinder ABL1 phosphorylation of adjacent Tyr1 residues owing to its positive charge, we questioned whether neutralization of the positive charge of arginine through citrullination would reverse this effect. To examine how arginine citrullination affects ABL1 phosphorylation, we designed synthetic peptides with human CTD residues 1801−1822 containing either Arg1810 or Cit1810 and treated them with ABL1 to compare their phosphorylation patterns using UVPD-MS. Analysis of ABL1 phosphorylation on the native Arg1810 peptide revealed only two singly phosphorylated species corresponding to the two Tyr1 sites away from Arg1810 (Figure 5C and Table S5). This result is consistent with our 3xS7R Sp construct where we observed predominantly ABL1 phosphorylation of the Tyr1 sites away from the Arg7 residues (Figure 5A). In contrast, a novel phosphorylation peak was detected when the Cit1810 peptide was treated with ABL1 (Figure 5D). The UVPD-MS analysis confirmed identification of this new product as a phosphorylated species on the Tyr1 residue neighboring Cit1810 (Figure 5E). Thus, modifications that neutralize the positive charge of the arginine residue at the seventh CTD position can enhance recognition of adjacent Tyr1 sites by ABL1 and may thereby affect transcriptional regulation.
Lysines and Acetyl-Lysine Mimics in the Seventh CTD Position Affect the Preference of Tyr1 Phosphorylation by ABL1
There are eight Lys7 variations in the human distal CTD, and we suspected that their positively charged side chains like that of arginine would reduce ABL1 phosphorylation of adjacent Tyr1 sites. To evaluate the effect of lysines in the seventh CTD position on Tyr1 phosphorylation, we designed a 6xHis-GST linked three-repeat CTD substrate with Ser7 replaced by lysines in the first and third heptads (3xS7K Sp; Table S1) and mapped ABL1 phosphorylation using UVPD-MS (Table S6). In this construct, we observed two phosphorylation products of which the major peak consisted of Tyr1 phosphorylation adjacent to Ser7 (approximately 76%) and the minor peak of Tyr1 phosphorylation adjacent to Lys7 (Figure 6A). This result is consistent with the distal CTD mapping results in which Tyr1 sites next to Lys7 variations were less favored (Figure 1C−F). Like Arg7, the lack of preference can be explained by the positive pocket of Arg/Trp close to the active site of ABL1, which would disfavor the positively charged side chains of Lys7 variations (Figure 6B). Yet, the long flexible side chain of lysine does not appear to abolish the recognition of tyrosine, as a significant portion of Tyr1 phosphorylation adjacent to Lys7 residues in the 3xS7K Sp substrate still occurred (Figure 6A).
Figure 6.

Lysine and an acetylated lysine mimic (glutamine) in the 7th CTD position alter ABL1 preference for Tyr1 sites. (A) Extracted chromatograms of singly phosphorylated species in three-repeat CTD substrates with Ser7Lys mutations (3xS7K Sp) treated with ABL1. Phosphorylation sites were localized using UVPD-MS and are indicated on the chromatogram by repeat number UVPD-MS. The GPGSGM amino acid sequence at the N-terminus is retained after 3C proteolysis of the 6xHis-GST tags prior to mass spectrometry. The sites of major and minor phosphorylation were indicated by dark or light blue boxes, respectively, and the lysine mutations are shown in purple. (B) Structure of ABL1 (light blue) bound to the ATP-peptide bisubstrate analog of SRC (green) with Ile106 modeled as a lysine (white). (C,D) Extracted chromatograms of singly phosphorylated species in three-repeat CTD substrates with acetyl-lysine mimicking Ser7Gln mutations (3xS7Q Mid/Sp) treated with ABL1. (E,F) Structure of ABL1 (light blue) bound to the ATP-peptide bisubstrate analog of SRC (green) with Ile106 modeled as either acetyl-lysine mimicking glutamine or acetyl-lysine (white).
Because the positively charged side chains of lysines in the seventh heptad position seem to reduce the recognition of the adjacent Tyr1 by ABL1, we wondered whether modifications that neutralize their charge similar to arginine citrullination could affect ABL1 specificity. Acetylation of Lys7 (which neutralizes its positive charge) is enriched downstream of transcription start sites and is thought to play a role in enhancing expression of specific growth factors.13 To test whether lysine acetylation alters ABL1 preference, we generated 6xHis-GST linked three-repeat CTD substrates with Ser7 replaced with glutamine mutations (3xS7Q Mid/Sp) that resemble the chemical structure of acetyl-lysine. Unlike unmodified lysine, we observed a clear preference for ABL1 phosphorylation of the Tyr1 next to Gln7 compared to Ser7 in both of these substrates (Figure 6C,D). In the 3xS7Q Mid substrate, a highly abundant peak appears corresponding to the phosphorylation product of the tyrosine next to Gln7 (Figure 6C). The Tyr1 residues adjacent to glutamines in the 3xS7Q Sp substrate were preferably phosphorylated by ABL1, but the tyrosine next to Ser7 also accounted for a portion of the overall products (Figure 6D). The reversal in Tyr1 preference is surprising but easily understood by the structure of ABL1 in complex with its substrate. Glutamine can be accommodated by the positively charged Arg/Trp pocket, potentially engaging in favorable hydrogen bonding with Arg367 of ABL1 (Figure 6E). Similar to glutamine, an acetylated Lys7 would carry no positive charge and would likely be in a similar position where a hydrogen bond can be formed between its secondary amine and Arg367 (Figure 6F). Overall, positively charged residues in the seventh heptad position can make ABL1 phosphorylation of adjacent Tyr1 sites less favorable, whereas post-translational modified cations that neutralize these positive charges remove this inhibitory effect on ABL1 preference.
The Positively Charged Binding Pocket of ABL1 Is Evolutionarily Conserved among Several Nuclear Tyrosine Kinases
Our structural analysis of the ABL1 pocket composed of Arg367 and Trp405 bound to modeled Ser7 mutated and/or modified substrates provides a structural explanation for the substrate specificity of ABL1 in phosphorylating human RNAPII CTD. Substantial evidence has implicated ABL1 as a major RNAPII Tyr1 CTD kinase, particularly in response to DNA damage response.24,25 However, knockout of ABL1 does not abolish phosphorylation of Tyr1 in cells.24 Other tyrosine kinases have been attributed to this complementation. In particular, ABL2 (which shares ~95% identity with ABL1) has been shown to phosphorylate Tyr1 of the CTD.33 Since many of these enzymes share conserved catalytic domains, we speculated whether this pocket that confers Ser7 variation/modification specificity in ABL1 is conserved among other tyrosine kinases. Several nonreceptor tyrosine kinases have been reported to shuttle to the nucleus,34−36 and when we aligned several of their sequences we found that the Arg367 and Trp405 residues from ABL1 are highly conserved (Figure 7A). Furthermore, we aligned all of these kinases that have available structures and found that their Arg/Trp residues almost completely overlap spatially with each other (Figure 7B). This is especially true for ABL2 (Figure 7B), which has near-identical kinase activity against RNAPII CTD as ABL1.33 The only exception in this group of kinases with the highly conserved Arg/Trp pocket was SRC kinase in which the Arg388 side chain adopts a different rotameric state due to variation in sequence resulting in a shorter Arg-containing loop (Figure 7B). Intriguingly, SRC is also the only tyrosine kinase that has been reported to not have observable kinase activity against the RNAPII CTD in cells.23 Broad conservation of the Arg/Trp pocket both in sequence and in structural conformation in nonreceptor tyrosine kinases could indicate that Tyr1 kinases other than ABL1 have similar recognition of Ser7 variation/modified RNAPII CTD.
Figure 7.

Evolutionary conservation of Arg/Trp pocket in tyrosine kinases. (A) Sequence alignment of catalytic and activation loops from human nonreceptor tyrosine kinases that have been reported to shuttle to the nucleus. The conserved arginine residues in the catalytic loop are highlighted in yellow, while the conserved tryptophan residues are highlighted in green. (B) Structural alignment of the predicted phospho-Ser7 binding pocket in nonreceptor tyrosine kinases. The colors of each kinase are shown, and the PDB codes for each structure used were: ABL1, 2G1T; ABL, 2XYN; ACK1, 1U46; FAK1, 1MP8; FES, 3CBL; JAK1, 3EYG; SYK, 1XBA; and SRC, 1FMK. The ATP-peptide bisubstrate analog from the ABL1 structure with the modeled phosphorylated serine (white) was shown to compare positioning of the arginine and tryptophan residues from the other kinase structures. (C) Model of Ser7 variations and modifications that enhance or inhibit Tyr1 phosphorylation. RNAPII (blue), gene (purple), promoter (red), proximal CTD (green), distal CTD (orange), and phosphates (yellow) are depicted as cartoons. Phosphorylated Ser7 (pSer7), citrullinated Arg7 (Cit7), acetylated Lys7 (acetyl-Lys7), and other Ser7 variations are shown next to bold arrows indicating enhanced Tyr1 phosphorylation or red lines indicating reduced Tyr1 phosphorylation. Lines for variations and modifications only indicate their general location in the CTD (i.e., in the proximal or distal CTD regions).
DISCUSSION
The consensus motif of Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7 is predominant in simple eukaryotic CTD sequences like those in yeast, but more complex organisms have higher numbers of variations from the consensus sequence, especially at the seventh position. CTD heptads with different residues in the seventh position enable greater possibilities of chemical modifications, such as Thr7 phosphorylation, Arg7 methylation and citrullination, and Lys7 methylation and acetylation.11,32,37 These different Ser7 variations and PTMs seem to have implications in eukaryotic transcription,9,11,12,32,37 but the molecular mechanisms for how they regulate these processes are still not completely understood.
In our study, we show how variations and PTMs at the seventh CTD position can affect the placement of phosphorylation by Tyr1 CTD kinases. Using UVPD-MS with single amino acid resolution and structural modeling of ABL1 bound to ATP-peptide bisubstrate analogs, we reveal how differently charged residues in the seventh CTD position either prime or disfavor ABL1 phosphorylation of adjacent Tyr1 residues (Figure 7C). Negatively charged Glu1852 of the human distal CTD promotes ABL1 phosphorylation of the adjacent Tyr1 residue (Figure 7C). Glu7 mutations also mimic phosphorylated Ser/Thr7 residues which have been identified as being important for snRNA transcription.9,38,39 A positively charged pocket close to the ABL1 active site containing arginine and tryptophan seems to be playing a critical role in recognizing negatively charged residues in the seventh heptad position through a stable electrostatic interaction (Figure 4C). In contrast, positively charged residues disfavor adjacent Tyr1 phosphorylation by ABL1 (Figure 7C) likely because of having a similar charge to that of the arginine in the conserved kinase pocket. PTMs that neutralize the positively charged residues in the seventh position of CTD heptads, like citrullination of Arg1810,11 negate this effect of specificity on ABL1 (Figure 7C). Furthermore, the eight Lys7 variations in the human distal CTD can be neutralized via acetylation,13 and glutamine mutations that mimic their acetylation reverse ABL1 distinction of Tyr1 residues (Figure 7C). All these variations and chemical modifications of the seventh residue in CTD heptads greatly expand the coding power of the CTD and allow implementation of additional layers of regulation. The cross-talk between the seventh and first residue of adjacent CTD heptads displays a wide variety of combinations, which could result in differential recruitment of regulating factors that eventually lead to alternative transcriptional outcomes.
SUMMARY
Using mass spectrometry, kinetic assays, and structural analysis, we report a novel aspect of seventh residue variations and modifications in CTD heptad repeats that alter phosphorylation preference of Tyr1 CTD kinases. Negatively charged residues in this position promote phosphorylation of neighboring Tyr1 residues due to a conserved positively charged pocket in Tyr1 CTD kinases. Positively charged residues disfavor phosphorylation of neighboring Tyr1 residues, but neutralizing PTMs of these residues eliminate this distinction. Our analysis provides key clues to the role that variations and modifications in the seventh heptad position play in CTD regulation and should help guide future studies into how they affect transcription or other CTD-dependent processes.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
CTD sequences used for mass spectrometry and kinetic assays were subcloned using ligation independent cloning (SLIC)40 into pET28a (Novagene) derivative vectors encoding a 6xHis-tag followed by a GST-tag and a 3C protease site. The distal CTD sequence encoding residues 1769−1970 was directly subcloned from pYFP-RPB1 αAmr.41 Cloning of the 26 repeat Saccharomyces cerevisiae CTDs from residues 1534−1733 has been described previously,15 whereas the 26 repeat S7E and S7E Sp CTD sequences were synthesized by Biomatik. The three-repeat CTD constructs were made by annealing oligos (IDT) with 5′ end overhangs and ligating them into SLIC digested vectors. The 6xHisGST-CTD proteins were expressed and purified, as previously described.15
BL21 (DE3) cells coexpressing the human ABL1 kinase domain (residues 229−511) and YopH phosphatase from Yersinia were a kind gift from the Kuriyan lab.26 One liter cultures of these BL21 cells were grown at 37 °C in Terrific Broth media (Thermo) containing 50 μg/mL kanamycin and 50 μg/mL streptomycin. Once these cultures reached an OD600 of 0.6−0.8, expression of ABL1 was induced with 0.25 mM isopropyl-β-D-thiogalactopyranoside (IPTG), and the cultures were allowed to grow an additional 16 h at 18 °C. Cells were pelleted, resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 5% glycerol, and 25 mM imidazole), and lysed via sonication at 90 A for 2.5 min of 1 s on/5 s off cycles on ice. The lysate was cleared by centrifugation at 15 000 rpm for 40 min at 4 °C. The supernatant was loaded over 2−3 mL of Ni-NTA beads (Qiagen), washed with lysis buffer, and finally eluted with elution buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 5% glycerol, and 250 mM imidazole). Protein fractions were pooled and dialyzed overnight at 4 °C in 3.5 kDa dialysis membranes (Thermo) against dialysis buffer (20 mM Tris-HCl at pH 8.0, 100 mM NaCl, 5% glycerol, and 1 mM DTT) supplemented with 0.5 mg of TEV protease to release the N-terminal 6xHis-tag. The protein was then loaded onto a DEAE anion exchange column (GE) equilibrated with Buffer A (20 mM Tris-HCl pH 8.0, 5% glycerol, and 1 mM DTT) and eluted with a linear gradient of Buffer A to Buffer B (20 mM Tris-HCl pH 8.0, 1 M NaCl, 5% glycerol, and 1 mM DTT). Peak fractions were analyzed by Coomassie Brilliant Blue (Thermo) staining of SDS-PAGE gels and the fractions containing ABL1 were pooled and buffer exchanged into the gel filtration buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 5% glycerol, and 1 mM DTT). The protein was loaded on an equilibrated Superdex 75 size exclusion column and eluted with gel filtration buffer. Fractions containing ABL1 were concentrated and flash frozen for storage at −80 °C.
Kinase Reactions and Sample Preparation for Mass Spectrometry
Kinase reactions for mass spectrometry were performed using 0.025 mg mL−1 of ABL1 (~0.7 μM) with 1 mg mL−1 of 6xHis-GST-CTD protein and 2 mM ATP in kinase reaction buffer (40 mM Tris-HCl at pH 7.5 and 20 mM MgCl2) for 16 h at 30 °C in an Eppendorf Mastercycler. The proteins were then digested with 3C protease 8−16 at RT (0.1 mg mL−1 6xHis-GST-CTD, 0.001 U/μL 3C, 50 mM Tris-HCl at pH 8, and 150 mM NaCl) to remove the 6xHis-GST purification tag. After 3C proteolysis, the 3xCTD samples were processed for mass spectrometry, whereas the 26 repeat distal and S7E spaced CTD proteins were further digested with trypsin or GluC protease, respectively. The distal CTD samples were exchanged into 100 mM ammonium bicarbonate (pH 7) using a 30k molecular weight cutoff filter. Trypsin was added to the filtrate at a ratio of approximately 1:20 to the distal CTD protein. The reaction was carried out overnight at 37 °C. Limited proteolysis of the 26xS7E Sp protein was conducted with GluC protease (1 mg mL−1 3C digested 26xS7E Sp, 0.1 M NH4HCO3, and 0.001 mg mL−1 GluC) at 37 °C for 2 h. Biological duplicates were prepared and analyzed independently to ensure replication of the experiments. All ABL1 phosphorylated peptides (digested 26x distal, 26xS7E Sp, 3xCTD and undigested distal 1801−1822 peptides) were cleaned up using Pierce C18 spin columns according to the manufacturer’s instructions and dried using a Savant DNA SpeedVac (Thermo Fisher Scientific). Lyophilized products were reconstituted in LC starting solvent conditions of 2% acetonitrile and 0.1% formic acid for separation prior to MS/MS analysis.
Liquid Chromatography−MS for Phosphate Localization
All peptides were separated using a Dionex Ultimate 3000 nano liquid chromatography (Thermo Scientific) plumbed for direct injection into a 75-μm ID Picofrit analytical column (New Objective, Woburn, MA). One microliter of each sample was injected (less than 700 ng each) for separations using 1.8 μm UChrom C18 analytical columns (NanoLCMS Solutions, Oroville, CA) that were packed in-house to 20 cm. Mobile phases A and B were composed of water and acetonitrile, respectively, each containing 0.1% formic acid. Separations were carried out using gradients that were optimized for various samples as follows: a linear gradient of 2% to 35% B in 40 min was used for 26-repeat yeast S7E Sp and three-repeat CTD (WT, S7E Sp, S7E Mid, S2E Mid, S7Q Sp, and S7Q Mid). A stepwise gradient of 2% to 25% B for 25 min then 25% to 40% B for another 30 min was used for the human distal CTD samples. A linear gradient of 2% to 35% B for 51 min was used for peptides containing residues 1801−1822 of human distal CTD and mutant three-repeat CTD (S7R Sp and S7K Sp). The flow rate was maintained at 0.300 μL/min for all samples. Eluted peptides were analyzed in positive polarity mode using an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using a NanoFlex electrospray source. The mass spectrometer was equipped with an excimer laser operated at 193 nm (Coherent, Santa Clara, CA) and modified to allow for ultraviolet photodissociation in the dual linear ion trap as described earlier.42 All spectra were acquired in the Orbitrap mass analyzer using resolution settings of 60K and 30K (at m/z 200) for MS1 and MS/MS events, respectively. Targeted peptides were activated using two laser pulses of 1.2 mJ for UVPD in the low-pressure ion trap.42
MS/MS spectra were deconvoluted in the XCalibur QualBrowser software using the Xtract algorithm with a signal-to-noise threshold of 3. Fragments were matched to the nine ion-types observed from UVPD of peptides (a, a+, b, c, x, x+, y, y−, z) using ProSight Lite.43 Phosphosites were localized by adding the mass of a phospho-group (+79.97 Da) at each of the possible tyrosine residues to identify fragment ions that contain the moiety and optimize peptide characterization. Relative abundance information on each phosphorylated species from human distal CTD was calculated from the peak area of the eluting peptide. Specifically, the ion current of the phosphopeptides precursor ion was summed across the elution profile and normalized to the total ion current for all phosphopeptide isomers. Because of the identical chemical makeup of the peptides, ionization efficiency was assumed to be the same for the compared species. The abundance of phosphorylated species should not be compared between peptides with different sequences.
Radiolabeled Kinase Assays
ATP [γ−32P] labeled, 3000 Ci/mmol was purchased from PerkinElmer. Kinase reactions with 0−100 μM of 6xHis-GST-yeast 26x WT or S7E CTD substrates (0.75 μM ABL1, 40 mM Tris-HCl pH 8, 20 mM MgCl2, 0.1 mg mL−1 BSA, and 2 mM ATP) were incubated at 30 °C for 30 min. Reactions were quenched with 0.5 mL of ice-cold 1 mM potassium phosphate buffer at pH 6.8 with 1 mM EDTA and stored on ice. In a vacuum filtration container, prewet 0.45 μm Protran BA85 nitrocellulose filters (Whatman) washed several times in the same 1 mM potassium phosphate buffer 6.8 were prepared for each kinase reaction. Each reaction was applied to a separate filter and washed three times each before the filters were removed and added to 1 mL of scintillation fluid (RPI) in glass vials. The radioactivity of each vial was then measured in a scintillation counter and plotted for kinetic fitting. Biological triplicates were prepared and analyzed independently to ensure replication of the experiments.
Structural Modeling of ABL1 Structures Bound to ATP-Peptide Bisubstrate Analogs
The structure of ABL1 bound to the Src ATP-peptide bisubstrate analog was retrieved from the PDB (2G1T). Chains B (ABL1) and F (ATP-peptide) were used in our analysis. We mutated Ile106 of the ATP-peptide into various residues using the built-in mutagenesis wizard in PyMOL (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC), which automatically selected the likeliest rotamer conformation. Electrostatic potential maps of ABL1’s surface were also generated in PyMOL. Structural alignment of nonreceptor tyrosine kinases in PyMOL was conducted with the A chains in each of the following PDB structures: ABL2, 2XYN; ACK1, 1U46; FAK1, 1MP8; FES, 3CBL; JAK1, 3EYG; SYK, 1XBA; and SRC, 1FMK.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by grants from the National Institutes of Health (R01 GM104896 to Y.Z. and 125882 to Y.Z. and J.S.B.) and Welch Foundation (F-1778 to Y.Z. and F-1155 to J.S.B.). The BL21 strain containing plasmids for expressing the ABL1 kinase domain and YopH protease was a kind gift from the Kuriyan lab. Funding from the UT System for support of the UT System Proteomics Core Facility Network is gratefully acknowledged.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00610.
All CTD sequences and identified ions in UVPD-MS analyses (PDF)
REFERENCES
- (1).Buratowski S (2003) The CTD code. Nat. Struct. Mol. Biol 10, 679–680. [DOI] [PubMed] [Google Scholar]
- (2).Corden JL (2013) RNA polymerase II C-terminal domain: Tethering transcription to transcript and template. Chem. Rev 113, 8423–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Eick D, and Geyer M (2013) The RNA polymerase II carboxy-terminal domain (CTD) code. Chem. Rev. 113, 8456–8490. [DOI] [PubMed] [Google Scholar]
- (4).Jeronimo C, Bataille AR, and Robert F (2013) The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chem. Rev 113, 8491–8522. [DOI] [PubMed] [Google Scholar]
- (5).Nonet M, Sweetser D, and Young RA (1987) Functional redundancy and structural polymorphism in the large subunit of RNA polymerase II. Cell 50, 909–915. [DOI] [PubMed] [Google Scholar]
- (6).West ML, and Corden JL (1995) Construction and analysis of yeast RNA polymerase II CTD deletion and substitution mutations. Genetics 140, 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schneider S, Pei Y, Shuman S, and Schwer B (2010) Separable functions of the fission yeast Spt5 carboxyl-terminal domain (CTD) in capping enzyme binding and transcription elongation overlap with those of the RNA polymerase II CTD. Mol. Cell. Biol 30, 2353–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Heidemann M, Hintermair C, Voss K, and Eick D (2013) Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta, Gene Regul. Mech 1829, 55–62. [DOI] [PubMed] [Google Scholar]
- (9).Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, and Eick D (2007) Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318, 1780–1782. [DOI] [PubMed] [Google Scholar]
- (10).Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, and Ansari AZ (2009) TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell 34, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sharma P, Lioutas A, Fernandez-Fuentes N, Quilez J, Carbonell-Caballero J, Wright RHG, Di Vona C, Le Dily F, Schuller R, Eick D, Oliva B, and Beato M (2019) Arginine Citrullination at the C-Terminal Domain Controls RNA Polymerase II Transcription. Mol. Cell 73, 84–96. e87 [DOI] [PubMed] [Google Scholar]
- (12).Sims RJ 3rd, Rojas LA, Beck DB, Bonasio R, Schuller R, Drury WJ 3rd, Eick D, and Reinberg D (2011) The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science 332, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schroder S, Herker E, Itzen F, He D, Thomas S, Gilchrist DA, Kaehlcke K, Cho S, Pollard KS, Capra JA, Schnolzer M, Cole PA, Geyer M, Bruneau BG, Adelman K, and Ott M (2013) Acetylation of RNA polymerase II regulates growth-factorinduced gene transcription in mammalian cells. Mol. Cell 52, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dias JD, Rito T, Torlai Triglia E, Kukalev A, Ferrai C, Chotalia M, Brookes E, Kimura H, and Pombo A (2015) Methylation of RNA polymerase II non-consensus Lysine residues marks early transcription in mammalian cells. eLife 4, No. e11215, DOI: 10.7554/eLife.11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mayfield JE, Robinson MR, Cotham VC, Irani S, Matthews WL, Ram A, Gilmour DS, Cannon JR, Zhang YJ, and Brodbelt JS (2017) Mapping the Phosphorylation Pattern of Drosophila melanogaster RNA Polymerase II Carboxyl-Terminal Domain Using Ultraviolet Photodissociation Mass Spectrometry. ACS Chem. Biol 12, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Descostes N, Heidemann M, Spinelli L, Schuller R, Maqbool MA, Fenouil R, Koch F, Innocenti C, Gut M, Gut I, Eick D, and Andrau JC (2014) Tyrosine phosphorylation of RNA polymerase II CTD is associated with antisense promoter transcription and active enhancers in mammalian cells. eLife 3, e02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yurko N, Liu X, Yamazaki T, Hoque M, Tian B, and Manley JL (2017) MPK1/SLT2 Links Multiple Stress Responses with Gene Expression in Budding Yeast by Phosphorylating Tyr1 of the RNAP II CTD. Mol. Cell 68, 913–925. e913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, and Cramer P (2012) CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science 336, 1723–1725. [DOI] [PubMed] [Google Scholar]
- (19).Schuller R, Forne I, Straub T, Schreieck A, Texier Y, Shah N, Decker TM, Cramer P, Imhof A, and Eick D (2016) HeptadSpecific Phosphorylation of RNA Polymerase II CTD. Mol. Cell 61, 305–314. [DOI] [PubMed] [Google Scholar]
- (20).Suh H, Ficarro SB, Kang UB, Chun Y, Marto JA, and Buratowski S (2016) Direct Analysis of Phosphorylation Sites on the Rpb1 C-Terminal Domain of RNA Polymerase II. Mol. Cell 61, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hsin JP, Li W, Hoque M, Tian B, and Manley JL (2014) RNAP II CTD tyrosine 1 performs diverse functions in vertebrate cells. eLife 3, e02112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shah N, Maqbool MA, Yahia Y, El Aabidine AZ, Esnault C, Forne I, Decker TM, Martin D, Schuller R, Krebs S, Blum H, Imhof A, Eick D, and Andrau JC (2018) Tyrosine-1 of RNA Polymerase II CTD Controls Global Termination of Gene Transcription in Mammals. Mol. Cell 69, 48–61. e46 [DOI] [PubMed] [Google Scholar]
- (23).Baskaran R, Dahmus ME, and Wang JY (1993) Tyrosine phosphorylation of mammalian RNA polymerase II carboxyl-terminal domain. Proc. Natl. Acad. Sci. U. S. A 90, 11167–11171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liu ZG, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, and Wang JY (1996) Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature 384, 273–276. [DOI] [PubMed] [Google Scholar]
- (25).Burger K, Schlackow M, and Gullerova M (2019) Tyrosine kinase c-Abl couples RNA polymerase II transcription to DNA double-strand breaks. Nucleic Acids Res. 47, 3467–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Seeliger MA, Young M, Henderson MN, Pellicena P, King DS, Falick AM, and Kuriyan J (2005) High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 14, 3135–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Brodbelt JS (2014) Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev 43, 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kim M, Suh H, Cho EJ, and Buratowski S (2009) Phosphorylation of the yeast Rpb1 C-terminal domain at serines 2, 5, and 7. J. Biol. Chem 284, 26421–26426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Egloff S, O’Reilly D, Chapman RD, Taylor A, Tanzhaus K, Pitts L, Eick D, and Murphy S (2007) Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science 318, 1777–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, and Cole PA (2001) Mechanism-based design of a protein kinase inhibitor. Nat. Struct. Biol 8, 37–41. [DOI] [PubMed] [Google Scholar]
- (31).Levinson NM, Kuchment O, Shen K, Young MA, Koldobskiy M, Karplus M, Cole PA, and Kuriyan J (2006) A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol. 4, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yanling Zhao D, Gish G, Braunschweig U, Li Y, Ni Z, Schmitges FW, Zhong G, Liu K, Li W, Moffat J, Vedadi M, Min J, Pawson TJ, Blencowe BJ, and Greenblatt JF (2016) SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature 529, 48–53. [DOI] [PubMed] [Google Scholar]
- (33).Baskaran R, Chiang GG, Mysliwiec T, Kruh GD, and Wang JY (1997) Tyrosine phosphorylation of RNA polymerase II carboxyl-terminal domain by the Abl-related gene product. J. Biol. Chem 272, 18905–18909. [DOI] [PubMed] [Google Scholar]
- (34).Cans C, Mangano R, Barila D, Neubauer G, and Superti-Furga G (2000) Nuclear tyrosine phosphorylation: the beginning of a map. Biochem. Pharmacol 60, 1203–1215. [DOI] [PubMed] [Google Scholar]
- (35).Wang L, Duke L, Zhang PS, Arlinghaus RB, Symmans WF, Sahin A, Mendez R, and Dai JL (2003) Alternative splicing disrupts a nuclear localization signal in spleen tyrosine kinase that is required for invasion suppression in breast cancer. Cancer Res. 63, 4724–4730. [PubMed] [Google Scholar]
- (36).Ahmed I, Calle Y, Sayed MA, Kamal JM, Rengaswamy P, Manser E, Meiners S, and Nur-E-Kamal A (2004) Cdc42dependent nuclear translocation of non-receptor tyrosine kinase, ACK. Biochem. Biophys. Res. Commun 314, 571–579. [DOI] [PubMed] [Google Scholar]
- (37).Voss K, Forne I, Descostes N, Hintermair C, Schuller R, Maqbool MA, Heidemann M, Flatley A, Imhof A, Gut M, Gut I, Kremmer E, Andrau JC, and Eick D (2015) Site-specific methylation and acetylation of lysine residues in the C-terminal domain (CTD) of RNA polymerase II. Transcription 6, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Egloff S, Szczepaniak SA, Dienstbier M, Taylor A, Knight S, and Murphy S (2010) The integrator complex recognizes a new double mark on the RNA polymerase II carboxyl-terminal domain. J. Biol. Chem 285, 20564–20569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ni Z, Xu C, Guo X, Hunter GO, Kuznetsova OV, Tempel W, Marcon E, Zhong G, Guo H, Kuo WW, Li J, Young P, Olsen JB, Wan C, Loppnau P, El Bakkouri M, Senisterra GA, He H, Huang H, Sidhu SS, Emili A, Murphy S, Mosley AL, Arrowsmith CH, Min J, and Greenblatt JF (2014) RPRD1A and RPRD1B are human RNA polymerase II C-terminal domain scaffolds for Ser5 dephosphorylation. Nat. Struct. Mol. Biol 21, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Li MZ, and Elledge SJ (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256. [DOI] [PubMed] [Google Scholar]
- (41).Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, and Singer RH (2007) In vivo dynamics of RNA polymerase II transcription. Nat. Struct. Mol. Biol 14, 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Klein DR, Holden DD, and Brodbelt JS (2016) Shotgun Analysis of Rough-Type Lipopolysaccharides Using Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem 88, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Fellers RT, Greer JB, Early BP, Yu X, LeDuc RD, Kelleher NL, and Thomas PM (2015) ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 15, 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.