

Abstract
Toll-like receptor 2 (TLR2) is suggested to initiate the activation of NLRP3 inflammasome, and considered to be involved in asthma. The findings that melatonin modulates TLRs-mediated immune responses, together with the suppressing effect of TLRs on endogenous melatonin synthesis, support the possibility that a feedback loop exists between TLRs system and endogenous melatonin synthesis. To determine whether TLR2-melatonin feedback loop exists in allergic airway disease and regulates NLRP3 inflammasome activity, wild-type (WT) and TLR2−/− mice were challenged with OVA to establish allergic airway disease model. Following OVA challenge, WT mice exhibited increased-expression of TLR2, activation of NLRP3 inflammasome and marked airway inflammation, which were all effectively inhibited in the TLR2−/− mice, indicating that TLR2-NLRP3 mediated airway inflammation. Meanwhile, melatonin biosynthesis was reduced in OVA-challenged WT mice, while such reduction was notably rescued by TLR2 deficiency, suggesting that TLR2-NLRP3-mediated allergic airway inflammation was associated with decreased endogenous melatonin biosynthesis. Furthermore, addition of melatonin to OVA-challenged WT mice pronouncedly ameliorated airway inflammation, decreased TLR2 expression and NLRP3 inflammasome activation, further implying that melatonin in turn inhibited airway inflammation via suppressing TLR2-NLRP3 signal. Most interestingly, although melatonin receptor antagonist luzindole significantly reduced the protein expressions of ASMT, AANAT and subsequent level of melatonin in OVA-challenged TLR2−/− mice, it exhibited null effect on leukocytes infiltration, Th2-cytokines production and NLRP3 activity. These results indicate that a TLR2-melatonin feedback loop regulates NLRP3 inflammasome activity in allergic airway inflammation, and melatonin may be a promising therapeutic medicine for airway inflammatory diseases such as asthma.
Keywords: allergic airway inflammation, melatonin synthesis, NLRP3 inflammasome, TLR2, feedback loop
Introduction
Asthma is one of the common respiratory diseases characterized by persistent or non-resolving allergen-driven inflammation. Toll-like receptors (TLRs), a member of pattern recognition receptors (PRRs), have been suggested to contribute to the initiation and perpetuation of airway inflammation, by virtue of their ability as the host's first line defense to recognize invading pathogens and aeroallergens (1). Within the TLRs family, TLR2 is regarded as the major one responsible for the sustaining airway inflammation and thus be most relevant to the onset of asthma (2–4). However, despite more and more advances in our understanding of the role of TLR2 in allergic asthma, the mechanism underlying TLR2 regulation in allergic airway inflammation is still elusive.
Recently, the other important type of intracellular PRRs, nucleotide-binding oligomerization domain-like receptors (NLRs) has been reported to be strongly associated with inflammatory diseases (5, 6). Particularly, as the best-characterized subtype shown to be expressed in airway, nucleotide-binding domain and leucine-rich repeat protein 3 (NLRP3) inflammasome is considered to be involved in the progress of asthma (7, 8). The NLRP3 inflammasome is a cytosolic protein complex composed of NLRP3, ASC, and pro-caspase-1. The activation of NLRP3 inflammasome proteolytically cleaves pro-caspase-1 into active caspase-1, which in turn promotes maturation of IL-1β and IL-18 (9). And these two NLRP3-associated cytokines are critical in the initiation and amplification of inflammatory process (5). Although NLRP3 inflammasome has been extensively investigated, its role in allergic airway inflammation is still controversial (10–14), and its regulatory networks remain elusive. TLRs and NLRs are two important kinds of PRRs, the formation and activation of NLRP3 inflammasome has been suggested to be initiated by TLRs (8, 15, 16). However, the crosstalk between TLR2 and NLRP3 inflammasome activity in the allergic airway diseases, as well as the underlying mechanism is not clear.
Melatonin (N-acetyl-5-methoxytrytamine), which is synthesized by the pineal gland or extra-pineal tissues (17), has been identified as a powerful immune-modulatory and anti-inflammatory molecule (18). Exogenous administration of melatonin has been reported to pronouncedly ameliorate airway inflammation (19, 20). Similarly, endogenous melatonin, which is produced from tryptophan (Trp) by converting 5-hydroxytryptamine (5-HT) successively by two key enzymes AANAT and ASMT, is also tightly associated with the pathogenesis of asthma (21, 22). It has been reported that endogenous melatonin synthesis is suppressed by activation of TLR9 (22), while other study shows that melatonin is able to inhibit TLRs-mediated inflammation (23), suggesting there may be a feedback loop between TLRs system and endogenous melatonin synthesis. More notably, it has been recently identified that a novel molecular target for melatonin is NLRP3 in murine model of septic response, liver injury and acute lung injury (24–26). However, it is uncertain whether TLR2-melatonin feedback loop exists in allergic airway disease, and regulates NLRP3 inflammasome activity.
To address these questions, we first examined whether TLR2 modulated melatonin biosynthesis and NLRP3 inflammasome activity by using TLR2−/− mice, and further revealed whether this effect of TLR2 was feedback regulated by melatonin by administrating exogenous melatonin or melatonin receptor antagonist luzindole in OVA-induced allergic airway inflammation.
Materials and Methods
Mice
Female C57BL/6 mice were obtained from Shanghai Laboratory Animal Center, TLR2 deficient (TLR2−/−) mice on C57BL/6 background were presented by Dr. ZG. Tian (Institute of Immunology, School of Life Sciences, University of Science and Technology of China). All experimental protocols were approved by the Animal Care and Use Committee of Anhui Medical University.
OVA-Induced Allergic Airway Inflammation and Interventions
The murine model of allergic airway inflammation was established according to previous study (27). Briefly, the mice (6–8 weeks old) were sensitized on day 0 with an intra-peritoneal injection of OVA (Sigma, St. Louis, MO, USA) emulsified with 1 mg potassium aluminum sulfate (Sangon Biotech, Shanghai, China) in 500 μl of saline. Airway inflammation was induced by inhalation of 1% aerosolized OVA 30 min per day, for consecutive 7 days. Control mice received saline-sensitization and inhalation of nebulized saline solution. The mice from WT-OVA-Melatonin group were administered with melatonin (10 mg/kg, Sigma), and the mice from TLR2−/−-OVA-Luzindole group were administered with luzindole (30 mg/kg, Sigma) 1 h before allergen-challenge through intra-peritoneal injection, respectively, the dose of melatonin and luzindole was used according to previous studies (19, 28). Experiments were performed with six mice per group. Mice were harvested 24 h following the last challenge.
Bronchoalveolar Lavage Fluid (BALF) Collection and Cellular Analysis
The left lung bronchoalveolar lavage was performed with 500 μl ice-cold phosphate-buffered saline (PBS) for three times, and the resultant BALF was centrifuged to separate the cellular components. The cells were re-suspended in 200 μl PBS. Total cell counts were performed using a hemocytometer, differential cell counts were evaluated morphologically by staining cytospin slides of BALF samples with Wright Stain solution (Sigma).
Lung Histopathological Examination
Immediately after collection of the BALF samples, lung tissues were fixed in 4% paraformaldehyde, and then embedded in paraffin before sagittally cutting into 5 μm sections. Subsequently, the sections were stained with hematoxylin/eosin (H&E) or periodic acid-Schiff (PAS) to analyze the inflammatory cells infiltration and global cell hyperplasia, respectively. Finally, a semi-quantitative method as described previously was used to evaluate the peribronchial inflammation and global cell hyperplasia (29, 30).
Immunohistochemistry
Lung tissue sections (5 μM) were incubated with TLR2 antibody at 4°C overnight. Second day, the sections were incubated with HRP-conjugated secondary antibody (Beijing Golden Bridge Biotechnology, Beijing, China) for 1 h at 37°C. After rinsed for three times, sections were stained by diaminobenzidine (DAB) and counterstained by hematoxylin, and dark-brown staining was considered positive. The image were obtained under an Carl Zeiss Axio Scope.A1 (Carl Zeiss, German).
Cytokines, 5-HT and Melatonin Determined by Enzyme-Linked Immunosorbent Assay (ELISA)
The level of OVA-specific IgE in serum was determined by ELISA using specific kits from Cusabio (Wuhan, China). The levels of IL-4, IL-13, IL-1β, IL-18, and 5-HT in BALF, the content of melatonin in lung homogenate were measured by ELISA kits from Cloud-Clone Crop (Wuhan, China) according to the protocols from the manufacturer.
Western Blotting Analysis
Lung tissues were homogenized in RIPA in the presence of a protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and Phosphatase Inhibitor Cocktail (Roche). The protein concentrations were determined by a BCA kit (Pierce, Rockford, IL, USA). Equal concentrations of protein (25 μg) were separated by SDS-PAGE on 12% acrylamide gels. The primary antibodies used were as follow: NLRP3 (Adipogen, San Diego, CA, USA), caspase 1 (p20) (Adipogen), IL-1β, pro-IL-1β (R&D Systems, Minneapolis, MN, USA), TLR2 (Millipore, Billerica, MA, USA), AANAT (Sigma), ASMT (Abcam, Boston, MA, USA) and GAPDH (KANGCHEN Biotech, Shanghai, China) for 1 h at 37°C, followed by at 4°C overnight. Blots were washed thrice with TBST and probed with appropriate secondary antibodies for 1 h at room temperature. After washing three times, the immune-reaction was analyzed using the ECL detection system (Pierce). The band density was determined by ImageJ software (NIH, Bethesda, MD, USA).
Statistics
Results are showed as mean ± SEM. A one-way analysis of variance followed by Bonferroni multiple comparison test was used to determine the differences among the groups, except for analysis of TLR2 expression, which was performed using independent-sample t-test (SPSS 19.0). P < 0.05 was accepted significant.
Results
TLR2 Is Required for OVA-Induced Murine Allergic Airway Inflammation
We first set out to confirm the role of TLR2 in murine allergic airway disease. WT and TLR2−/− mice were sensitized and challenged with OVA following the protocol showed in Figure 1A. Immunohistochemistry and western blot results showed that TLR2 protein expression was significantly increased in OVA-challenged WT mice in comparison with that of control mice (Figures 1B,C), and TLR2 was expressed on various types of cells, such as epithelial cells and leukocytes. Concomitantly, lung histology showed an increase in leukocyte recruitment to peribronchial and mucous cell metaplasia in OVA-challenged WT mice (Figures 1D–G). In sharp contrast, OVA-challenged TLR2−/− mice showed reductions in inflammatory cells recruitment (Figures 1D,F) and airway PAS+ cells (Figures 1E,G) in comparison with that of OVA-challenged WT mice.
Figure 1.

TLR2 is required for OVA-induced murine allergic airway inflammation. (A) Protocol of establishing allergic airway inflammation, and comparison of resolution of WT and TLR2−/− mice. (B) The expression of TLR2 in lung tissue from vehicle and OVA-challenged mice was analyzed by immunohistochemistry. (C) The protein expression of TLR2 in OVA-challenged WT mice analyzed by western blot and quantification of the protein expression of TLR2. (D) Histological evaluation of the airway inflammation by staining lung sections with H&E, arrows indicates infiltrated leukocytes. (E) Histological examination of mucus production in the lung sections stained with PAS, arrow heads indicates goblet cells. (F) Quantitative analysis of airway inflammation. (G) Quantitative of mucus production. Scale bar: 50 μm. **p < 0.01, ***p < 0.001.
TLR2 Is Required for OVA-Induced Inflammatory Cells Infiltration, IgE, and Th2 Cytokines Production
In addition to lung histological changes, airway challenged with OVA induced a significant increase in total BALF cellularity in comparison with that of control mice (Figure 2A). Further morphologic assessments of differentially stained BALF samples revealed that the increase in cellularity was resulted from a significant influx of neutrophils, lymphocytes, monocytes and eosinophils (Figures 2B–E). However, in comparison with WT mice, the total number or composition of the BALF cellularity in TLR2−/− mice post OVA challenge was significantly decreased except for monocytes, which trended to increase but did not reach statistical significance (Figures 2A–E). Meanwhile, the level of OVA-specific IgE in TLR2−/− mice was significantly lower than that of WT mice (Figure 2F). Furthermore, significant increase in the levels of Th2-associated cytokines including IL-4 and IL-13 was observed in OVA-challenge WT mice (Figures 2E,F). Similarly, significant differences between WT and TLR2−/− mice were observed that TLR2 deficiency significantly decreased the levels of these two Th2-associated cytokines post OVA challenge (Figures 2G,H). Together, these data supported the role of TLR2 in the development of allergic airway inflammation in this OVA model.
Figure 2.

TLR2 is required for OVA-induced inflammatory cells infiltration, IgE and Th2 cytokines production. (A) Total cell counts in the BALF of WT and TLR2−/− mice. (B–E) Differential cell counts in BALF of WT and TLR2−/− mice. (F) The level of OVA-specific IgE in serum. (G,H) Productions of IL-4 and IL-13 in BALF of WT and TLR2−/− mice were analyzed by ELISA. *p < 0.05, **p < 0.01, ***p < 0.001.
OVA-Induced Activation of NLRP3 Inflammasome and Decrease of Melatonin Biosynthesis Are TLR2 Dependent
We next questioned how TLR2 regulated allergic airway inflammation. It has been shown that NLRP3 inflammasome is associated with allergic airway disease in response to OVA (10). We next assessed the link between TLR2 and NLRP3 inflammasome activity. Our results showed that NLRP3, cleaved form of IL-1β and caspase 1(p20) were increased in OVA-challenged WT mice in comparison with those of control mice, while such increase was completely abrogated in TLR2−/− mice following OVA challenge (Figures 3A–D). Similarly, productions of NLRP3-associated IL-1β and IL-18 were markedly decreased in OVA-challenged TLR2−/− mice, comparable to those of control mice (Figures 3E,F). Taken together, in this OVA model, NLRP3 inflammasome activated by OVA required licensing through TLR2, suggesting that TLR2-NLRP3 axis mediated OVA-allergic airway inflammation.
Figure 3.

OVA-induced activation of NLRP3 inflammasome and decrease of melatonin biosynthesis are TLR2 dependent. (A) NLRP3, mature IL-1β, pro-IL-1β, and caspase1(p20) western blot analysis. (B–D) Relative ratio of NLRP3 to GAPDH, IL-1β to pro-IL-1β, and caspase1(p20) to GAPDH. (E,F) ELISA analysis of IL-1β and IL-18 in BALF. (G) Western blot analysis of AANAT and ASMT in lung tissues. (H,I) Relative ratio of AANAT or ASMT to GAPDH. (J,K) The levels of 5-HT in BALF and melatonin in the lung homogenate were analyzed by ELISA. *p < 0.05, **p < 0.01, ***p < 0.001.
Consequently, we sought to investigate the regulatory networks how TLR2-NLRP3 axis mediated allergic airway diseases. Previous study has shown that TLR9 negatively regulates melatonin production in response to OVA challenge, and this endogenous synthesized melatonin may regulate airway inflammation (22). Here, our present study showed that OVA notably suppressed the protein expression of ASMT but not AANAT in lung tissues (Figures 3G–I), and lowered the level of 5-HT in BALF and melatonin in lung homogenate in WT mice (Figures 3J,K), while these reductions were significantly restored by TLR2 deficiency (Figures 3H–K). These data confirmed that besides TLR9, TLR2, another member of TLRs family suppressed endogenous melatonin biosynthesis in OVA-induced allergic airway inflammation, therefore suggesting that TLR2-NLRP3 -mediated allergic airway inflammation was associated with decreased endogenous melatonin biosynthesis.
The Effect of Melatonin or Luzindole on OVA-Induced Airway Inflammation
Another important question arises whether endogenous melatonin increased by TLR2 deficiency directly suppresses NLRP3 inflammasome activity or feedback controls TLR2-NLRP3 signal. To address this question, melatonin or its receptor antagonist luzindole was applied as illustrated in Figure 4A. First, we found that administration of melatonin significantly attenuated the protein level of OVA-induced TLR2 in WT mice (Figures 4B,C). Meanwhile, melatonin-treated mice showed reductions in leukocyte recruitment and mucus productions compared with vehicle-treated WT mice post OVA challenge (Figures 4D–G). However, administration of luzindole only significantly promoted mucus productions in comparison with vehicle-treated TLR2−/− mice post OVA challenge (Figures 4D–G).
Figure 4.

The effect of melatonin or luzindole on OVA-induced airway inflammation. (A) Protocol of administration of melatonin or luzindole during establishing allergic airway diseases model. (B,C) Protein expression of TLR2 in lung tissues of OVA-challenged WT mice treated with melatonin or not. (D) Lung histopathology based on H&E staining to evaluate inflammatory cell infiltration, arrows indicates infiltrated leukocytes. (E) Mucus secretion was analyzed based on PAS staining, arrow heads indicates goblet cells. (F) Histological scoring of lung inflammation. (G) Quantification of mucus secretion. *p < 0.05, **p < 0.01, ***p < 0.001.
The Effect of Exogenous Melatonin or Luzindole on OVA-Induced Inflammatory Cells Infiltration, IgE, and Th2 Cytokines Production
Moreover, upon OVA challenge, melatonin treatment dramatically reduced the total number or composition of the BALF cellularity except for monocytes (Figures 5A–E), lowered the concentration of IgE in serum (Figure 5F), and decreased the productions of IL-4 and IL-13 in BALF (Figures 5G,H) in comparison with those in vehicle-treated WT mice. Interestingly, no significant difference was found between luzindole-treated and vehicle-treated TLR2−/− mice in BALF cellularity or production of IgE and Th2-associated cytokines following OVA challenge (Figures 5A–C,E,F). We only found significant increase in monocytes infiltration in luzindole-treated TLR2−/− mice (Figure 5D). These results demonstrated that melatonin remarkably promoted resolution of allergic airway inflammation by down-regulating TLR2 signaling. However, blocking the effect of melatonin in TLR2−/− mice by luzindole only aggravated allergen-induced mucus hyper-secretion, but had null effect on leukocyte infiltration and Th2 cytokines production.
Figure 5.

The effect of exogenous melatonin or luzindole on OVA-induced inflammatory cells infiltration, IgE and Th2 cytokines production. (A–E) Analysis of total and differential cells found in BALF of OVA-challenged WT or TLR2−/− mice after melatonin or luzindole treatment, respectively. (F) The level of OVA-specific IgE in serum. (G,H) The levels of IL-4 and IL-13 in BALF. **p < 0.01, ***p < 0.001.
The Effect of Exogenous Melatonin or Luzindole on NLRP3 Inflammasome Activation and Endogenous Melatonin Synthesis
Subsequently, we showed that administration of melatonin dramatically decreased the protein expressions of NLRP3, mature IL-1β and caspase 1(p20), lowered the productions of IL-1β and IL-18 in comparison with those in vehicle-treated WT mice following OVA challenge (Figures 6A–F). Importantly, NLRP3 inflammasome activity was not significantly different between vehicle-treated and luzindole-treated TLR2−/− mice post OVA challenge (Figures 6A–F). These data suggested that melatonin elicited its effect on NLRP3 inflammasome activity via TLR2 signaling, when TLR2 was deficient, blocking the effect of melatonin had null effect on NLRP3 inflammasome activity.
Figure 6.

The effect of exogenous melatonin or luzindole on NLRP3 inflammasome activation and endogenous melatonin synthesis. (A) Representative blots showing the protein expression of NLRP3, mature IL-1β, pro-IL-1β, and caspase1(p20) in lung homogenate. (B–D) Relative band densities of NLRP3 and caspase1(p20) were analyzed by normalizing to GAPDH, relative band densities of IL-1β was analyzed by normalizing to pro-IL-1β. (E,F) The levels of IL-1β, IL-18 in BALF were detected by ELISA. (G) Representative blots showing the levels of AANAT and ASMT in lung homogenate. (H,I) Relative band densities of AANAT and ASMT were analyzed by normalizing to GAPDH. (J,K) The level of 5-HT in BALF and melatonin in the lung homogenate were detected by ELISA. *p < 0.05, **p < 0.01, ***p < 0.001.
Finally, we found that treatment of melatonin resulted in dramatic increase in the protein expression of ASMT, marked elevation in the levels of 5-HT and melatonin, while did not affect the AANAT expression in comparing with vehicle-treated WT mice (Figures 6G–K). The protein expression of ASMT in lung tissues, the levels of 5-HT in BALF and melatonin in lung homogenate in melatonin-treated WT mice were comparable to that in vehicle-treated TLR2−/− mice post OVA challenge. However, TLR2−/− mice treated with luzindole showed notably reduced expressions of both AANAT and ASMT (Figures 6G–I), and lowered the level of melatonin in lung homogenate but not 5-HT in BALF in comparison with that of vehicle-treated TLR2−/− mice following OVA challenge (Figures 6J,K). Therefore, our study suggested that administration of melatonin further promoted endogenous melatonin biosynthesis, while treatment of luzindole significant inhibited endogenous melatonin synthesis in TLR2−/− mice.
Discussion
In the present study, we demonstrated that OVA-induced allergic airway inflammation occurred in parallel with increased-expression of TLR2, activation of NLRP3 inflammasome and decreased biosynthesis of melatonin. Deletion of TLR2 effectively alleviated allergic airway inflammation and concomitantly inhibited the activation of NLRP3 inflammasome as well as restored the level of melatonin. Furthermore, exogenous addition of melatonin to OVA-challenged WT mice pronouncedly ameliorated airway inflammation, decreased OVA-induced TLR2 expression and NLRP3 activity, and increased melatonin biosynthesis to similar level as that in OVA-challenged TLR2−/− mice. However, although luzindole significantly reduced the expression of AANAT and ASMT and subsequent level of melatonin in OVA-challenged TLR2−/− mice, it exhibited null effect on OVA-induced airway inflammation and activation of NLRP3, it only aggravated allergen-induced mucus hyper-secretion. These results are the first to show the existence of TLR2-melatonin feedback loop in allergic airway diseases, which regulates NLRP3 inflammasome activity and may represent a mechanism underlying the initiation and persistence of airway inflammation.
An allergic airway disease is usually dominated by a strong Th2 response which can be redirected to Th1 response by regulation of TLRs signaling (4, 31, 32), making TLRs attractive therapeutic targets. During the last decade, accumulating evidence has been focused on the role of TLRs in the pathogenesis of airway inflammatory diseases such as asthma and the possibility of using TLRs-based therapies for asthma (33, 34). To date, TLR2 has been considered as the most relevant to the onset of asthma. Asthmatic patients who ultimately die have increased expression of TLR2 (2), activation of TLR2 promotes Th2-biased immune responses, which may be correlated with the imbalance of Th1/Th2 in asthma (3, 4). In line with these data, our present study demonstrated a significant increase of TLR2 expression in WT mice post OVA challenge, and this increase was accounted from its induced-expression on various cell types. Previous studies also indicate its wide spectral expression (35–37), and reports from our group and others have indicated that TLR2 on macrophages (38), group 2 innate lymphoid cells (39) and epithelial cells (40) may contribute to allergic airway inflammation. Furthermore, the increased expression of TLR2 was accompanied with lung inflammation exacerbation in WT mice post OVA challenge in this study. However, such OVA-induced airway inflammation, including leukocyte recruitment to bronchial, mucus metaplasia in the airways, total number or composition of the BALF cellularity, the level of OVA-specific IgE, as well as IL-4 and IL-13, was notably alleviated in OVA-challenged TLR2−/− mice. Based on these data, TLR2 was considered to mediate allergic airway inflammation, and targeting TLR2 may have therapeutic benefit in allergic airway diseases.
However, the mechanism underlying TLR2 mediation of allergic airway inflammation is still unknown. NLRP3 inflammasome is one of the most extensively characterized NLRs due to its relevance in human inflammatory disorders such as asthma. Invading pathogens including viral or bacterial which commonly associated with asthma exacerbation, have been shown to trigger NLRP3 activation (5, 41). IL-1β is increased in the serum and BALF of human asthmatics, and administration of IL-1β induces airway hyper-reactivity (12, 13). Increased expressions of NLRP3, caspase-1 and IL-1β are found in macrophages as well as neutrophils in sputum of neutrophilic asthma (14). Consistent with these studies, our present study showed that in WT mice, OVA challenge substantially increased protein expression of NLRP3, mature IL-1β and caspase 1(p20) in lung tissues, markedly elevated the levels of NLRP3-associated IL-1β and IL-18 in BALF. However, this effect was only seen in WT mice, but not in TLR2−/− mice. The interaction between TLR2 and NLRP3 inflammasome has been also reported in various cell types. In monocytes (42), macrophages (43, 44), bone marrow-derived DCs (45) and several different cell lines (46, 47), lacking TLR2 failed to upregulate NLRP3 inflammasome as well as its substrate IL-1β. Therefore, our results have suggested that activation of NLRP3 inflammasome induced by OVA requires TLR2 signaling, thus TLR2 may mediate allergic airway inflammation through regulating NLRP3 inflammasome activity.
A question arises now is, by which TLR2 crosstalks with NLRP3 inflammasome and consequently mediates allergic airway inflammation. Melatonin has been reported to exert important immune-modulating effects in allergic airway diseases (19, 20, 48, 49), and the level of melatonin in saliva or serum of asthma patients were significantly lower than those in healthy controls (21, 50, 51). Particularly, it has been shown that TLR9 signaling regulates endogenous melatonin synthesis in allergic airway inflammation (22). Considering TLR2 and TLR9 are structurally similar, we asked whether TLR2 regulates endogenous melatonin synthesis and thereby modulated NLRP3 inflammasome activity. Indeed, the current study demonstrated that TLR2 protein expression was remarkably increased accompanying with decreased expression of melatonin synthetase ASMT and lower level of melatonin in lung homogenate in WT mice post OVA challenge, while deletion of TLR2 significantly rescued melatonin biosynthesis. Since it have been reported that C57BL/6 mice were pineal melatonin-deficient mice (52, 53) or produce much lower melatonin in their pineal gland (17, 54), suggested an extra-pineal melatonin synthesis might compensate for the pineal deficiency (17). Actually, there are results reporting that several extra-pineal tissues, such as thymus, spleen and skin, in C57BL/6 mice can synthesize melatonin (17, 55), our current study further found that another extra-pineal tissue, i.e., lung synthesized melatonin, and this process maybe modulated by TLR2 signal.
On the other hand, melatonin has been evidenced to significantly inhibit airway inflammation (49, 56), and suppress TLR3/4-mediated inflammation in liver injury (23). Most notably, NLRP3 is a novel molecular target for melatonin in murine model of septic response, liver injury and acute lung injury (24–26). Consistent with these findings in animals, melatonin also has been shown to exert inhibitory effect on TLRs including TLR3/4/9 signaling in macrophage (57–59) and NLRP3 inflammasome in epithelial cells (60). Therefore, we asked whether endogenous melatonin, which was regulated by TLR2 signal, alleviated allergic airway inflammation through directly suppressing NLRP3 inflammasome activity or feedback controlling TLR2-NLRP3 signal. Our current study showed that treatment of melatonin notably alleviated OVA-induced airway inflammation in WT mice, which was consistent with previous findings (19, 56). However, we extended previous observations by showing that melatonin treatment strongly inhibited OVA-induced protein expressions of TLR2, NLRP3, mature IL-1β and caspase1(p20), as well as lowered the levels of NLRP3-associated IL-1β and IL-18 in BALF in WT mice, suggesting that melatonin mitigates allergic airway inflammation by inhibiting TLR2 and NLRP3 inflammasome activity. Considering the data shown above that TLR2 signaling regulated melatonin synthesis, we speculated that a TLR2-melatonin feedback loop may exist in allergic airway disease, and melatonin elicits its effect on activation of NLRP3 inflammasome through TLR2 signal. Additionally, we found that exogenous addition of melatonin further increased protein expression of ASMT, as well as elevated the level of 5-HT in BALF and melatonin in lung homogenate in OVA-challenged WT mice. These interesting data suggested that the proven effect of exogenous melatonin in the resolution of inflammation was paralleled by the effect of endogenous synthesized melatonin.
Among the three melatonin receptors, the anti-inflammatory effect of melatonin is reported to be mainly mediated by MT2 melatonin receptor (61). Luzindole is considered as a non-selective MT1/MT2 receptor antagonist, showing 15–26 times higher affinity for MT2 receptor, which pharmacologically blocks the effect of melatonin (62). Therefore, luzindole was used as indicated (28) to further confirm that melatonin elicited its effect on NLRP3 inflammasome activity via TLR2 signal. Our results showed that although administration of luzindole to OVA-challenged TLR2−/− mice significantly reduced the expression of melatonin synthetase AANAT and ASMT, and subsequent level of melatonin, it exhibited null effects on the level of 5-HT in BALF, OVA-induced leukocyte infiltration, the level of IgE, Th2 cytokines production, and NLRP3 inflammasome activity, it only aggravated allergen-induced mucus hyper-secretion. To our knowledge, this is the first study showing that luzindole inhibited lung melatonin synthesis, which may be due to the blockage of 5-HT conversion to melatonin. These combined data suggested that the effect of melatonin on airway inflammation and NLRP3 inflammasome activity requires TLR2 signaling, and melatonin which is modulated by TLR2 signal feedback regulates TLR2 signaling, and subsequently regulates NLRP3 inflammasome activity and airway inflammation. However, the inhibitory effect of melatonin on airway remodeling characterized by mucus hyper-secretion may not require TLR2 signal, because luzindole still deteriorates mucus production even in the absence of TLR2 signal.
In conclusion, the present study demonstrates that a TLR2-melatonin feedback loop regulates the activation of NLRP3 inflammasome in allergic airway inflammation, and suppression of melatonin synthesis by TLR2 activation in turn results in the loss of its inhibitory effect on the TLR2 signaling (Figure 7). This important endogenous regulatory feedback loop may drive the onset of allergic airway inflammation, and melatonin may be a promising therapeutic medicine for airway inflammatory disease such as asthma. The present study used OVA model, which has been the most widely used pre-clinical allergic asthma model and recapitulates many of the hallmarks of allergic asthma in humans, however, bio-models offer other allergen-induced allergic model, such as house dust mite (HDM), Alternaria, papain and IL-33, with each mimicking the major features of human asthma. When OVA, HDM, or Alternaria was given via inhalation or intranasally, each of them can be recognized and sensed by TLRs reportedly, especially TLR2 (40). Similarly, intranasally given IL-33 is reported can induce direct stimulation of TLR2 on ILC2s (39). However, papain, which is homologous of HDM-derived Der p1, activates innate immune responses in a manner distinct from that of PAPMs and can induce production of an alarmin IL-33 (63). We hypothesize our findings may be also applicable to other allergens–induced allergic airway inflammation models in which TLR2 is activated by allergen. However, the true fact needs future study to discover.
Figure 7.
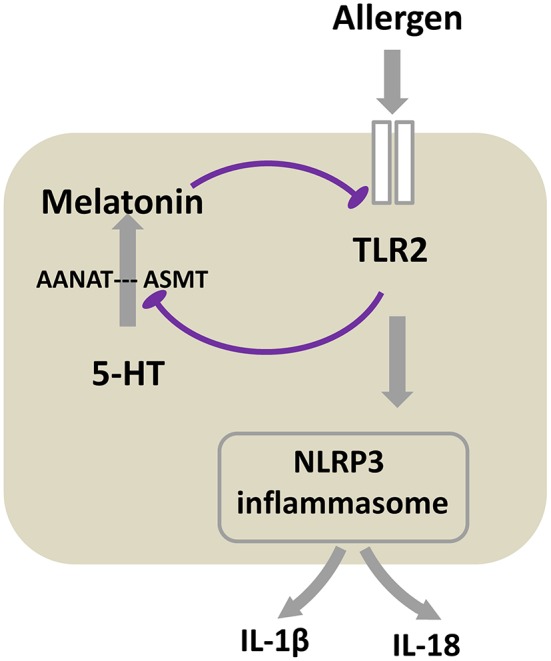
Proposed schematic of the mechanism that TLR2-melatonin feedback loop regulates the activation of NLRP3 inflammasome in allergic airway inflammation. After TLR2 is activated by allergen, it subsequently induces NLRP3 inflammasome activation. Meanwhile, TLR2 inhibits the expression of melatonin synthetase ASMT, accordingly blocks 5-HT converting to melatonin, ultimately leading to the decreased level of melatonin. This inhibition of melatonin synthesis results in the loss of its inhibitory effect on the TLR2 signaling. The result of the cycle is the persistent of allergic airway inflammation.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
This study was carried out in accordance with the recommendations outlined in Animal Care and Use Committee guideline of Anhui Medical University. The protocol was approved by the Animal Care and Use Committee guideline of Anhui Medical University.
Author Contributions
H-MW designed and carried out most of the experiments, analyzed the data, and wrote the manuscript. C-CZ, Q-MX, and JX established the murine model of allergic airway disease, done western blotting, and analyzed the data. G-HF designed the experiment and revised the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by grants from the National Natural Science Foundation of China (Nos. 81770032, 81570034, 81300027), Foundation for Distinguished Young Scholars of the First Affiliated Hospital of Anhui Medical University, Program for the Youth Distinguished Talents of Anhui Medical University, Doctoral research foundation of the First Affiliated Hospital of Anhui Medical University (No.3101005002442), and Anhui Key Lab of Geriatric molecular medicine.
References
- 1.Aryan Z, Rezaei N. Toll-like receptors as targets for allergen immunotherapy. Curr Opin Allergy Clin Immunol. (2015) 15:568–74. 10.1097/ACI.0000000000000212 [DOI] [PubMed] [Google Scholar]
- 2.Ferreira DS, Annoni R, Silva LF, Buttignol M, Santos AB, Medeiros MC, et al. Toll-like receptors 2, 3, and 4 and thymic stromal lymphopoietin expression in fatal asthma. Clin Exp Allergy. (2012) 42:1459–71. 10.1111/j.1365-2222.2012.04047.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Chen Q, Chu C, You H, Jin M, Zhao X, et al. Ovalbumin-induced experimental allergic asthma is Toll-like receptor 2 dependent. Allergy Asthma Proc. (2014) 35:e15–20. 10.2500/aap.2014.35.3735 [DOI] [PubMed] [Google Scholar]
- 4.Redecke V, Hacker H, Datta SK, Fermin A, Pitha PM, Broide DH, et al. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. J Immunol. (2004) 172:2739–43. 10.4049/jimmunol.172.5.2739 [DOI] [PubMed] [Google Scholar]
- 5.Im H, Ammit AJ. The NLRP3 inflammasome: role in airway inflammation. Clin Exp Allergy. (2014) 44:160–72. 10.1111/cea.12206 [DOI] [PubMed] [Google Scholar]
- 6.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. 10.1016/j.cell.2014.04.007 [DOI] [PubMed] [Google Scholar]
- 7.Brusselle GG, Provoost S, Bracke KR, Kuchmiy A, Lamkanfi M. Inflammasomes in respiratory disease: from bench to bedside. Chest. (2014) 145:1121–33. 10.1378/chest.13-1885 [DOI] [PubMed] [Google Scholar]
- 8.Lee S, Suh GY, Ryter SW, Choi AM. Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. Am J Respir Cell Mol Biol. (2016) 54:151–60. 10.1165/rcmb.2015-0231TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- 10.Allen IC, Jania CM, Wilson JE, Tekeppe EM, Hua X, Brickey WJ, et al. Analysis of NLRP3 in the development of allergic airway disease in mice. J Immunol. (2012) 188:2884–93. 10.4049/jimmunol.1102488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Besnard AG, Guillou N, Tschopp J, Erard F, Couillin I, Iwakura Y, et al. NLRP3 inflammasome is required in murine asthma in the absence of aluminum adjuvant. Allergy. (2011) 66:1047–57. 10.1111/j.1398-9995.2011.02586.x [DOI] [PubMed] [Google Scholar]
- 12.Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI. Cytokines in symptomatic asthma airways. J Allergy Clin Immunol. (1992) 89:958–67. 10.1016/0091-6749(92)90218-Q [DOI] [PubMed] [Google Scholar]
- 13.Lee TH, Song HJ, Park CS. Role of inflammasome activation in development and exacerbation of asthma. Asia Pac Allergy. (2014) 4:187–96. 10.5415/apallergy.2014.4.4.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J. (2014) 43:1067–76. 10.1183/09031936.00105013 [DOI] [PubMed] [Google Scholar]
- 15.Koch KN, Hartung ML, Urban S, Kyburz A, Bahlmann AS, Lind J, et al. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J Clin Invest. (2015) 125:3297–302. 10.1172/JCI79337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiao Y, Wang P, Qi J, Zhang L, Gao C. TLR-induced NF-kappaB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. (2012) 586:1022–6. 10.1016/j.febslet.2012.02.045 [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Corvera A, Cerrillo I, Molinero P, Naranjo MC, Lardone PJ, Sanchez-Hidalgo M, et al. Evidence of immune system melatonin production by two pineal melatonin deficient mice, C57BL/6 and Swiss strains. J Pineal Res. (2009) 47:15–22. 10.1111/j.1600-079X.2009.00683.x [DOI] [PubMed] [Google Scholar]
- 18.Sutherland ER, Martin RJ, Ellison MC, Kraft M. Immunomodulatory effects of melatonin in asthma. Am J Respir Crit Care Med. (2002) 166:1055–61. 10.1164/rccm.200204-356OC [DOI] [PubMed] [Google Scholar]
- 19.Shin IS, Park JW, Shin NR, Jeon CM, Kwon OK, Kim JS, et al. Melatonin reduces airway inflammation in ovalbumin-induced asthma. Immunobiology. (2014) 219:901–8. 10.1016/j.imbio.2014.08.004 [DOI] [PubMed] [Google Scholar]
- 20.Shin IS, Shin NR, Park JW, Jeon CM, Hong JM, Kwon OK, et al. Melatonin attenuates neutrophil inflammation and mucus secretion in cigarette smoke-induced chronic obstructive pulmonary diseases via the suppression of Erk-Sp1 signaling. J Pineal Res. (2015) 58:50–60. 10.1111/jpi.12192 [DOI] [PubMed] [Google Scholar]
- 21.Fei GH, Liu RY, Zhang ZH, Zhou JN. Alterations in circadian rhythms of melatonin and cortisol in patients with bronchial asthma. Acta Pharmacol Sin. (2004) 25:651–6. [PubMed] [Google Scholar]
- 22.Wu HM, Shen QY, Fang L, Zhang SH, Shen PT, Liu YJ, et al. JNK-TLR9 signal pathway mediates allergic airway inflammation through suppressing melatonin biosynthesis. J Pineal Res. (2016) 60:415–23. 10.1111/jpi.12323 [DOI] [PubMed] [Google Scholar]
- 23.Kang JW, Koh EJ, Lee SM. Melatonin protects liver against ischemia and reperfusion injury through inhibition of toll-like receptor signaling pathway. J Pineal Res. (2011) 50:403–11. 10.1111/j.1600-079X.2011.00858.x [DOI] [PubMed] [Google Scholar]
- 24.Cao Z, Fang Y, Lu Y, Tan D, Du C, Li Y, et al. Melatonin alleviates cadmium-induced liver injury by inhibiting the TXNIP-NLRP3 inflammasome. J Pineal Res. (2017) 62. 10.1111/jpi.12389 [DOI] [PubMed] [Google Scholar]
- 25.Garcia JA, Volt H, Venegas C, Doerrier C, Escames G, Lopez LC, et al. Disruption of the NF-kappaB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-alpha and blocks the septic response in mice. FASEB J. (2015) 29:3863–75. 10.1096/fj.15-273656 [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Li X, Grailer JJ, Wang N, Wang M, Yao J, et al. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J Pineal Res. (2016) 60:405–14. 10.1111/jpi.12322 [DOI] [PubMed] [Google Scholar]
- 27.Locke NR, Royce SG, Wainewright JS, Samuel CS, Tang ML. Comparison of airway remodeling in acute, subacute, and chronic models of allergic airways disease. Am J Respir Cell Mol Biol. (2007) 36:625–32. 10.1165/rcmb.2006-0083OC [DOI] [PubMed] [Google Scholar]
- 28.Qian ZX, Liu RY, Zhou L. Efects of melatonin and its receptor antagonist luzindole on airway inflammation and remodeling in a mouse asthma model. Chin J Respir Crit Care Med. (2006) 5:117–21. 10.3969/j.issn.1671-6205.2006.02.012 [DOI] [Google Scholar]
- 29.Camateros P, Tamaoka M, Hassan M, Marino R, Moisan J, Marion D, et al. Chronic asthma-induced airway remodeling is prevented by toll-like receptor-7/8 ligand S28463. Am J Respir Crit Care Med. (2007) 175:1241–9. 10.1164/rccm.200701-054OC [DOI] [PubMed] [Google Scholar]
- 30.Koltsida O, Karamnov S, Pyrillou K, Vickery T, Chairakaki AD, Tamvakopoulos C, et al. Toll-like receptor 7 stimulates production of specialized pro-resolving lipid mediators and promotes resolution of airway inflammation. EMBO Mol Med. (2013) 5:762–75. 10.1002/emmm.201201891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hennessy EJ, Parker AE, O'Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. (2010) 9:293–307. 10.1038/nrd3203 [DOI] [PubMed] [Google Scholar]
- 32.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. (2007) 13:552–9. 10.1038/nm1589 [DOI] [PubMed] [Google Scholar]
- 33.Dong Z, Xiong L, Zhang W, Gibson PG, Wang T, Lu Y, et al. Holding the inflammatory system in check: TLRs and their targeted therapy in asthma. Mediators Inflamm. (2016) 2016:2180417. 10.1155/2016/2180417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farrokhi S, Abbasirad N, Movahed A, Khazaei HA, Pishjoo M, Rezaei N. TLR9-based immunotherapy for the treatment of allergic diseases. Immunotherapy. (2017) 9:339–46. 10.2217/imt-2016-0104 [DOI] [PubMed] [Google Scholar]
- 35.GeurtsvanKessel CH, Lambrecht BN. Division of labor between dendritic cell subsets of the lung. Mucosal Immunol. (2008) 1:442–50. 10.1038/mi.2008.39 [DOI] [PubMed] [Google Scholar]
- 36.Hoppstadter J, Diesel B, Zarbock R, Breinig T, Monz D, Koch M, et al. Differential cell reaction upon Toll-like receptor 4 and 9 activation in human alveolar and lung interstitial macrophages. Respirat Res. (2010) 11:124. 10.1186/1465-9921-11-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koller B, Kappler M, Latzin P, Gaggar A, Schreiner M, Takyar S, et al. TLR expression on neutrophils at the pulmonary site of infection: TLR1/TLR2-mediated up-regulation of TLR5 expression in cystic fibrosis lung disease. J Immunol. (2008) 181:2753–63. 10.4049/jimmunol.181.4.2753 [DOI] [PubMed] [Google Scholar]
- 38.Wang M, Wang L, Fang L, Li S, Liu R. NLRC5 negatively regulates LTA-induced inflammation via TLR2/NF-kappaB and participates in TLR2-mediated allergic airway inflammation. J Cell Physiol. (2019) 234:19990–20001. 10.1002/jcp.28596 [DOI] [PubMed] [Google Scholar]
- 39.Ishii T, Muroi M, Horiguchi K, Tanamoto KI, Nagase T, Yamashita N. Activation through toll-like receptor 2 on group 2 innate lymphoid cells can induce asthmatic characteristics. Clin Exp Allergy. (2019) 49:1624–32. 10.1111/cea.13490 [DOI] [PubMed] [Google Scholar]
- 40.Lv J, Yu Q, Lv J, Di C, Lin X, Su W, et al. Airway epithelial TSLP production of TLR2 drives type 2 immunity in allergic airway inflammation. Eur J Immunol. (2018) 48:1838–50. 10.1002/eji.201847663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim RY, Pinkerton JW, Essilfie AT, Robertson AA, Baines KJ, Brown AC, et al. Role for NLRP3 inflammasome-mediated, IL-1beta-dependent responses in severe, steroid-resistant asthma. Am J Respir Crit Care Med. (2017) 196:283–97. 10.1164/rccm.201609-1830OC [DOI] [PubMed] [Google Scholar]
- 42.Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. (2013) 191:4337–47. 10.4049/jimmunol.1300298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. 10.4049/jimmunol.0901363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naganuma Y, Takakubo Y, Hirayama T, Tamaki Y, Pajarinen J, Sasaki K, et al. Lipoteichoic acid modulates inflammatory response in macrophages after phagocytosis of titanium particles through Toll-like receptor 2 cascade and inflammasomes. J Biomed Mater Res. (2016) 104:435–44. 10.1002/jbm.a.35581 [DOI] [PubMed] [Google Scholar]
- 45.Koch KN, Muller A. Helicobacter pylori activates the TLR2/NLRP3/caspase-1/IL-18 axis to induce regulatory T-cells, establish persistent infection and promote tolerance to allergens. Gut Microbes. (2015) 6:382–7. 10.1080/19490976.2015.1105427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kasimsetty SG, DeWolf SE, Shigeoka AA, McKay DB. Regulation of TLR2 and NLRP3 in primary murine renal tubular epithelial cells. Nephron Clin Pract. (2014) 127:119–23. 10.1159/000363208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Zhang M, Jiang N, Zhang A. Sodium phenylbutyrate ameliorates inflammatory response induced by Staphylococcus aureus lipoteichoic acid via suppressing TLR2/NF-kappaB/NLRP3 pathways in MAC-T cells. Molecules. (2018) 23:3056. 10.3390/molecules23123056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calvo JR, Gonzalez-Yanes C, Maldonado MD. The role of melatonin in the cells of the innate immunity: a review. J Pineal Res. (2013) 55:103–20. 10.1111/jpi.12075 [DOI] [PubMed] [Google Scholar]
- 49.Martins E, Jr, Ligeiro de Oliveira AP, Fialho de Araujo AM, Tavares de Lima W, Cipolla-Neto J, Costa Rosa LF. Melatonin modulates allergic lung inflammation. J Pineal Res. (2001) 31:363–9. 10.1034/j.1600-079X.2001.310412.x [DOI] [PubMed] [Google Scholar]
- 50.Fei GH, Liu RY, Zhang ZH, Zhou JN. Relationships between melatonin and cortisol and the status of disease in patients with bronchial asthma. Chin J Tuberc Respir Dis. (2003) 26:679–82. [PubMed] [Google Scholar]
- 51.Gumral N, Caliskan S, Ozguner F, Kaleli S, Akkaya A, Yilmaz H, et al. Melatonin levels and enzymatic antioxidant defense system decrease in blood of patients with bronchial asthma. Toxicol Ind Health. (2009) 25:411–6. 10.1177/0748233709106625 [DOI] [PubMed] [Google Scholar]
- 52.Ebihara S, Marks T, Hudson DJ, Menaker M. Genetic control of melatonin synthesis in the pineal gland of the mouse. Science. (1986) 231:491–3. 10.1126/science.3941912 [DOI] [PubMed] [Google Scholar]
- 53.Roseboom PH, Namboodiri MA, Zimonjic DB, Popescu NC, Rodriguez IR, Gastel JA, et al. Natural melatonin ‘knockdown’ in C57BL/6J mice: rare mechanism truncates serotonin N-acetyltransferase. Brain Res Mol Brain Res. (1998) 63:189–97. 10.1016/S0169-328X(98)00273-3 [DOI] [PubMed] [Google Scholar]
- 54.Maestroni GJ, Conti A, Pierpaoli W. Role of the pineal gland in immunity: II. Melatonin enhances the antibody response via an opiatergic mechanism. Clin Exp Immunol. (1987) 68:384–91. [PMC free article] [PubMed] [Google Scholar]
- 55.Kobayashi H, Kromminga A, Dunlop TW, Tychsen B, Conrad F, Suzuki N, et al. A role of melatonin in neuroectodermal-mesodermal interactions: the hair follicle synthesizes melatonin and expresses functional melatonin receptors. FASEB J. (2005) 19:1710–2. 10.1096/fj.04-2293fje [DOI] [PubMed] [Google Scholar]
- 56.Shin IS, Park JW, Shin NR, Jeon CM, Kwon OK, Lee MY, et al. Melatonin inhibits MUC5AC production via suppression of MAPK signaling in human airway epithelial cells. J Pineal Res. (2014) 56:398–407. 10.1111/jpi.12127 [DOI] [PubMed] [Google Scholar]
- 57.Huang SH, Cao XJ, Wei W. Melatonin decreases TLR3-mediated inflammatory factor expression via inhibition of NF-kappa B activation in respiratory syncytial virus-infected RAW264.7 macrophages. J Pineal Res. (2008) 45:93–100. 10.1111/j.1600-079X.2008.00560.x [DOI] [PubMed] [Google Scholar]
- 58.Xia MZ, Liang YL, Wang H, Chen X, Huang YY, Zhang ZH, et al. Melatonin modulates TLR4-mediated inflammatory genes through MyD88- and TRIF-dependent signaling pathways in lipopolysaccharide-stimulated RAW264.7 cells. J Pineal Res. (2012) 53:325–34. 10.1111/j.1600-079X.2012.01002.x [DOI] [PubMed] [Google Scholar]
- 59.Xu X, Wang G, Ai L, Shi J, Zhang J, Chen YX. Melatonin suppresses TLR9-triggered proinflammatory cytokine production in macrophages by inhibiting ERK1/2 and AKT activation. Sci Rep. (2018) 8:15579. 10.1038/s41598-018-34011-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Liu X, Bai X, Lin Y, Li Z, Fu J, et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J Pineal Res. (2018) 64:e12249. 10.1111/jpi.12449 [DOI] [PubMed] [Google Scholar]
- 61.Dubocovich ML, Markowska M. Functional MT1 and MT2 melatonin receptors in mammals. Endocrine. (2005) 27:101–10. 10.1385/ENDO:27:2:101 [DOI] [PubMed] [Google Scholar]
- 62.Dubocovich ML. Luzindole (N-0774): a novel melatonin receptor antagonist. J Pharmacol Exp Ther. (1988) 246:902–10. [PubMed] [Google Scholar]
- 63.Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An interleukin-33-mast cell-interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. (2015) 43:175–86. 10.1016/j.immuni.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.