

Abstract
Huntington’s disease (HD) is caused by an autosomal dominant polyglutamine expansion mutation of Huntingtin (HTT). HD patients suffer from progressive motor, cognitive, and psychiatric impairments, along with significant degeneration of the striatal projection neurons (SPNs) of the striatum. HD is widely accepted to be caused by a toxic gain-of-function of mutant HTT. However, whether loss of HTT function, because of dominant-negative effects of the mutant protein, plays a role in HD and whether HTT is required for SPN health and function are not known. Here, we delete Htt from specific subpopulations of SPNs using the Cre-Lox system and find that SPNs require HTT for motor regulation, synaptic development, cell health, and survival during aging. Our results suggest that loss of HTT function in SPNs could play a critical role in HD pathogenesis.
Graphical Abstract
In Brief
Burrus et al. show that striatal projection neurons require Huntingtin, the gene mutated in Huntington’s disease, for normal synaptic connectivity, regulated gene expression, and neuronal survival with aging. Loss of Huntingtin from striatal neurons recapitulates several features of Huntington’s disease pathology, an important consideration for therapies non-specifically targeting Huntingtin expression.
INTRODUCTION
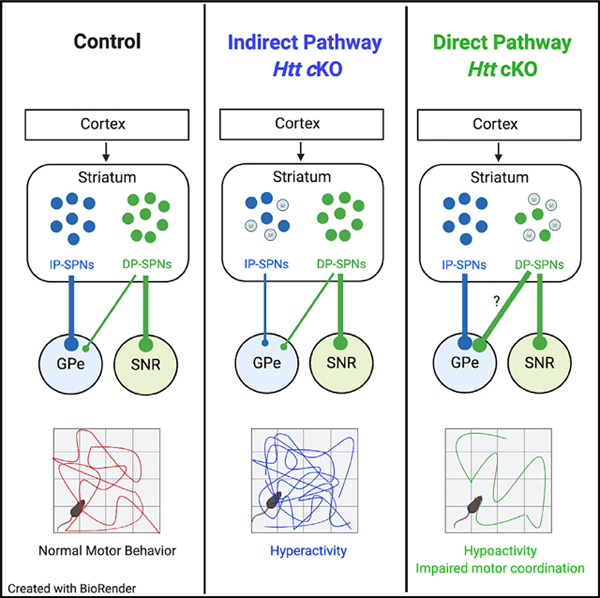
Huntington’s disease (HD) is a fatal, inherited neurodegenerative disorder with motor, psychiatric, and cognitive symptoms that typically emerge in midlife. The hallmark of HD is the progressive death of striatal projection neurons (SPNs) (Vonsattel et al., 1985). SPNs, which comprise >90% of the cells in the striatum, are GABAergic output neurons that are divided into two groups: the direct pathway (DP) and indirect pathway (IP) SPNs (DP-SPNs and IP-SPNs, respectively). Both SPN subtypes receive extensive glutamatergic inputs from the cortex and thalamus, and dopaminergic inputs from the ventral tegmental area and substantia nigra pars compacta. However, they differ with regard to their principal synaptic targets and their dopamine receptor expression. IP-SPNs project exclusively to the globus pallidus (GPe) and express the dopamine receptor D2 (Smith et al., 1998). DP-SPNs express the dopamine receptor D1 and project primarily to the substantia nigra pars reticulata (SNR) and entopeduncular nucleus, but also send collaterals to the GPe to a lesser extent (Cazorla et al., 2014). When stimulated, DP-SPNs promote movement and IP-SPNs inhibit movement (Alexander and Crutcher, 1990; Durieux et al., 2009; Kravitz et al., 2010). The correct balance in the activities of these SPN pathways is essential for coordinated voluntary motor function, and dysfunction of these pathways is linked to many movement disorders, including HD (Albin et al., 1989; Kravitz et al., 2010).
HD is caused by an autosomal dominant CAG repeat expansion in the first exon of the Huntingtin (HTT) gene (ENSG00000197386), which results in enlargement of a polyglutamine stretch near the N terminus of the HTT protein (Macdonald et al., 1993). HTT is proposed to act as a signaling scaffold for cellular processes, including autophagy, vesicle transport, and mitotic spindle orientation (Rui et al., 2015; Caviston et al., 2007; Gauthier et al., 2004; Godin et al., 2010). Presence of more than 39 CAG repeats causes HD, and the number of CAG repeats and age of disease onset are inversely correlated (Duyao et al., 1993). Despite the known genetic cause, the mechanisms of SPN degeneration in HD remain unclear.
Many mouse models expressing mutant HTT (mHTT) display features similar to HD, such as dysregulated motor function, changes in SPN gene expression, and progressive neurodegeneration (McKinstry etal., 2014; Luthi-Carteret al., 2000; Hodgson et al., 1999). Interestingly, patients and mouse models show abnormal cortical and striatal synaptic connectivity preceding motor symptom onset, mHTT aggregation, and SPN loss (Deng et al., 2013; Milnerwood and Raymond, 2007; Unschuld et al., 2012). However, the mechanisms driving synapse and circuit dysfunction in HD are not yet known.
Given the autosomal dominant nature of the HD-causing mutation, it has long been thought that SPN death in HD is mainly caused by toxic “gain-of-function” of mHTT. Although evidence for neuronal toxicity of mHTT is extensive, there are also strong indications for dominant-negative “loss-of-function” contributions of mHTT to HD pathology (Arrasate et al., 2004; Cattaneo et al., 2005). For instance, wild-type HTT is important for cell health and viability, because deleting Htt in the mouse central nervous system leads to aberrant synaptic connectivity, cellular stress, neuroinflammation, and neuronal death (McKinstry et al., 2014; Dragatsis et al., 2000, 2018; Mehler et al., 2019). On the other hand, wild-type HTT is neuroprotective and can shield neurons against mHTT toxicity (Leavitt et al., 2006). However, whether HTT is specifically required for SPN development, connectivity, and survival has not yet been determined. To address this critical knowledge gap, we deleted Htt from murine SPNs and found that HTT loss in SPNs leads to motor dysfunction with concurrent changes in SPN synaptic connectivity and function. Loss of HTT in SPNs also altered gene expression and nuclear morphology, preceding aging-dependent SPN loss and reactive gliosis. Taken together, these results demonstrate that HTT loss in SPNs disrupts the development, connectivity, and survival of these neurons during aging, recapitulating several key features of HD. Therefore, loss-of-function mechanisms may play important roles in the death and dysfunction of SPNs in HD.
RESULTS
Conditional Deletion of Htt in IP-SPNs and DP-SPNs
To investigate the role of HTT in SPN connectivity and health, we used the Cre-Lox system to conditionally delete Htt from IP-SPN and DP-SPN subpopulations in mice (Figures 1A and 1B). IP-SPNs were targeted by using Adora2A-Cre (A2A-Cre) transgenic mice, and DP-SPNs by using D1-Cre transgenic mice (Gerfen et al., 2013). To delete Htt from SPNs, we crossed Cre(Tg/Tg) Htt(+/−) and Htt(f/f) mice (Dragatsis et al., 2000), producing litter-mate offspring that were Cre(Tgm) Htt(f/+) (control) or Cre(Tgm) Htt(f/−) (conditional knockout [cKO]) (Figure 1B). A Cre-reporter Rosa-(STOP)f/+-TdTomato transgene (RTM) was also present in all offspring to visualize Cre-expressing cells. cKO mice had both Htt alleles deleted in SPNs but were heterozygous for Htt in all other cell types. Control mice were heterozygous for Htt in SPNs but had both Htt alleles in all other cells. The Htt(f/−) allele is needed to effectively delete both copies of Htt by Cre recombination and reduce mRNA levels, because Htt expression is upregulated to wild-type levels in Htt(+/−) neurons (McKinstry et al., 2014). Importantly, Htt heterozygosity in mice or humans does not affect neuronal health, survival, or behavior (Ambrose et al., 1994; Duyao et al., 1995).
Figure 1. Conditional Deletion of Htt in Indirect and Direct Pathway SPNs.
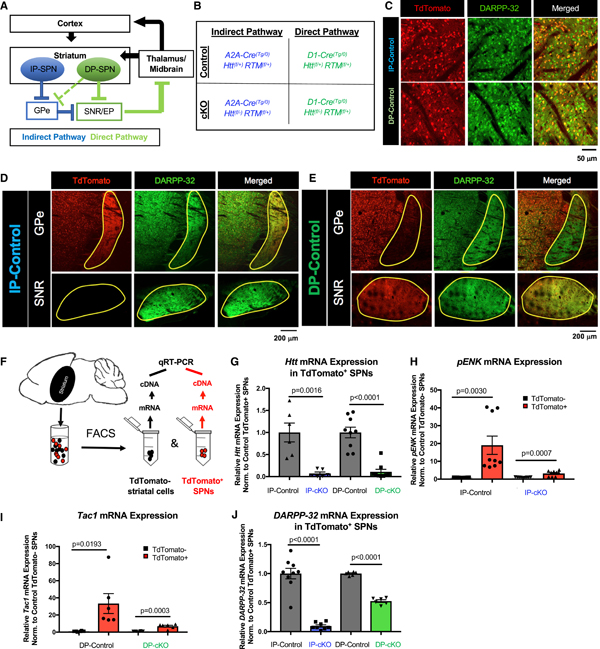
(A) Schematic of the basal ganglia circuit controlling motor function. Arrows indicate excitatory synaptic connections; blunt ends indicate inhibitory synaptic connections. DP-SPN, direct pathway striatal projection neuron; EP, entopeduncular nucleus; GPe, globus pallidus externus; IP-SPN, indirect pathway striatal projection neuron; SNR, substantia nigra pars reticulata.
(B) Breeding scheme for SPN-specific Htt cKO mice. Htt is deleted in IP-SPNs using the A2A-Cre transgene and in DP-SPNs using the D1-Cre transgene in combination with the floxed Htt allele. All mice have a Rosa-(STOP)f/+-TdTomato (RTM) allele, which drives the expression of TdTomato in Cre-expressing cells.
(C) Cre-reporter TdTomato is expressed in SPN cell bodies. A2A-Cre (upper) and D1-Cre (lower) result in TdTomato expression in ~50% of DARPP-32+ SPNs.
(D) A2A-Cre is expressed by SPNs that extend to the GPe, but not to the SNR.
(E) D1-Cre is expressed by SPNs that extend axons to SNR and to a lesser extent to the GPe.
(F) Schematic of fluorescence-activated cell sorting (FACS) approach. The striatum is dissected from 2-month-old mice, and FACS is used to separate TdTomato+ SPNs from TdTomato− cells. qRT-PCR is used to analyze gene expression after mRNA isolation/cDNA synthesis.
(G) Htt mRNA expression, as measured by qPCR, is reduced in IP-cKO SPNs compared with IP-control SPNs (n = 2 experiments, samples run in triplicate, unpaired two-way t test, t = 4.296, degrees of freedom [df] = 10, p = 0.0016). Htt mRNA expression is also reduced in DP-cKO SPNs compared with DP-control SPNs (n = 3 experiments, samples run in triplicate, unpaired two-way t test, t = 6.663, df = 16, p < 0.0001; graph displays mean ± SEM).
(H) Quantification of pENK mRNA expression in 2-month-old IP-cKOs and IP-controls (n = 3 experiments, samples run in triplicate; IP-control TdTomato versus IP-control TdTomato+ unpaired two-way t test, t = 3.497, df = 16, p = 0.0030; IP-cKO TdTomato− versus IP-cKO TdTomato+ unpaired two-way t test, t = 4.202, df = 16, p = 0.0007; graph displays mean ± SEM).
(I) Quantification of Tac1 mRNA expression in 2-month-old DP-cKOs and DP-controls (n = 2–3 experiments, samples run in triplicate; DP-control TdTomato− versus DP-control TdTomato+ unpaired two-way t test, t = 2.785, df = 10, p = 0.0193; DP-cKO TdTomato− versus DP-cKO TdTomato+ unpaired two-way t test, t = 5.433, df = 16, p = 0.0003; graph displays mean ± SEM).
(J) Quantification of DARPP-32 mRNA expression in 2-month-old IP-cKO IP-SPNs versus IP-control, and DP-cKO DP-SPNs versus DP-control (n = 2–3 experiments, samples run in triplicate; IP-control TdTomato+ versus IP-cKO TdTomato+ unpaired two-way t test, t = 9.239, df = 15, p < 0.0001; DP-control TdTomato+ versus DP-cKO TdTomato+ unpaired two-way t test, t = 17.73, df = 10, p < 0.0001; graph displays mean ± SEM).
See Figures S1 and S2.
In line with previous reports on A2A-Cre or D1-Cre mice (Zhang et al., 2006; Bateup et al., 2010; Gerfen et al., 2013), Cre (TdTomato) expression was confined to ~50% of DARPP-32-expressing (DARPP-32+) SPN cell bodies (Figure 1C). A2A-Cre-expressing SPNs exclusively extended their axonal projections to the GPe, and no TdTomato signal was observed in the SNR (Figure 1D), whereas D1-Cre-driven TdTomato expression was primarily in axons extending to the SNR (Figure 1E, bottom). Some axonal TdTomato signal was observed in the GPe of D1-Cre mice, because DP-SPNs send axon collaterals through the GPe (Figure 1E, top; Cazorla et al., 2014). A2A-Cre expression was restricted to IP-SPNs; however, D1-Cre-expressing cells were present in the cortices of D1-Cre mice, because subsets of cortical neurons express the dopamine receptor D1 (Vijayraghavan et al., 2017). We found that ~15% of layer 2/3, ~15% of layer 4, and ~40% of layer 5/6 NeuN+ neurons express D1-Cre in the M1 motor cortex, which harbors pyramidal neurons that project to dorsal striatum that is affected in HD (Figures S1A–S1C). However, there were no differences in the proportion of D1-Cre+ cortical neurons between genotypes (Figures S1B and S1C).
We used western blot to analyze striatal HTT protein expression of 2-month-old cKO mice and their controls to determine whether HTT expression was sufficiently reduced by A2A-Cre and D1-Cre. As expected, there was significantly less HTT protein expressed in cKO striata compared with controls (Figures S1D–S1F). Consistent with the cortical D1-Cre expression, HTT protein expression was also reduced in DP-cKO cortical lysates compared with DP-controls (Figures S1G and S1H).
We next used fluorescence-activated cell sorting (FACS) to separate TdTomato+ SPNs (DP-SPNs in DP-cKO, IP-SPNs in IP-cKO) from the remaining TdTomato− cells from the striata of 2-month-old mice for quantitative PCR (qPCR) analysis (Figure 1F). Htt expression was dramatically reduced in cKO SPNs compared with corresponding controls (Figure 1G). To verify that the cells we sorted were bona fide SPNs, we analyzed the expression of SPN marker genes pENK and Tac1, which are enriched in IP-SPNs or DP-SPNs, respectively (Lobo et al., 2006). As expected, TdTomato+ IP-SPNs were enriched for pENK compared with TdTomato− cells, and TdTomato+ DP-SPNs were enriched for Tac1 in both controls and cKOs (Figures 1H and 1I). However, we observed a substantial reduction in the degree of enrichment of pENK in cKO IP-SPNs and of Tac1 in cKO DP-SPNs, compared with their controls (Figures 1H and 1I). pENK protein expression, measured by fluorescence intensity of immunostaining, was also reduced in IP-SPNs of IP-cKO mice and in the IP-SPNs of DP-cKO mice, suggesting both cell-autonomous and non-cell-autonomous effects of Htt deletion on the gene expression of IP-SPNs (Figures S2A–S2D). Intriguingly, pENK and Tac1 expression are both diminished in HD patients and model mice (Luthi-Carter et al., 2000; Hodges, et al., 2006).
DARPP-32, a key downstream integrator of dopaminergic signaling cascades, is a pan-SPN marker that is downregulated in HD (Bibb et al., 2000). DARPP-32 mRNA expression was strikingly reduced in cKO SPNs of both genotypes compared with control SPNs (Figure 1J). DARPP-32 protein expression, measured as fluorescence intensity per cell, was also diminished (Figures S2E–S2H). IP-cKO caused a reduction in DARPP-32 protein expression both in IP-SPNs lacking HTT and their HTT-expressing DP-SPN neighbors, showing a non-cell-autonomous effect of IP-cKO on DARPP-32 expression (Figures S2E and S2F). However, DARPP-32 protein expression in DP-cKO mice was reduced only in DP-SPNs (Figures S2G and S2H).
CTIP, a nuclear protein involved in cell proliferation and transcriptional regulation, is another pan-SPN marker and is upregulated in HD mice (Langfelder et al., 2016). We found that CTIP protein expression was slightly increased in cKO IP-SPNs compared with controls (Figures S2I and S2J). We also found a trending increase in CTIP expression in neighboring DP-SPNs in IP-cKOs (Figures S2I and S2J). On the other hand, CTIP expression in DP-cKOs was reduced both in DP-SPNs and neighboring IP-SPNs relative to DP-controls (Figures S2K and S2L). Taken together, these results show that Htt deletion disrupts SPN-specific gene expression, reminiscent of gene expression changes seen in HD.
Huntingtin Is Not Required for Initial SPN Survival
Wild-type HTT plays critical roles in cell health and viability. Global knockout of Htt in mice is embryonically lethal, and loss of HTT in progenitor cells leads to impairments in cell proliferation, migration, and survival (Godin et al., 2010; Elias et al., 2015; Tong et al., 2011; Mehler et al., 2019; Nasir et al., 1995). Furthermore, Htt cKO in mouse cortical neurons leads to synaptic dysfunction, cell stress and neuroinflammation, and neuronal death (Dragatsis et al., 2000; McKinstry et al., 2014). However, whether wild-type Htt is required for SPN genesis or survival is unknown.
We counted TdTomato+ SPNs in 2-month-old cKO mice and their respective controls. To do so, we immunostained three coronal brain sections per mouse spanning the anterior-to-posterior striatum (bregma 1.345 to −0.755 mm) for TdTomato and DARPP-32. Our imaging strategy allows us to analyze SPN density along the striatal dorsal-to-ventral axis. Using a confocal microscope with an automated stage, we acquired images from identical regions by maintaining a standardized displacement from the corpus callosum (dorsal-to-ventral axis, y-displacement) and the lateral ventricle (lateral-to-medial axis, x-displacement) (Figure 2A, top). All TdTomato+ (Cre+) SPNs were counted, resulting in >7,500 Cre+ cells counted per genotype (Figure 2A, bottom).
Figure 2. Htt Is Not Required for Initial SPN Survival.
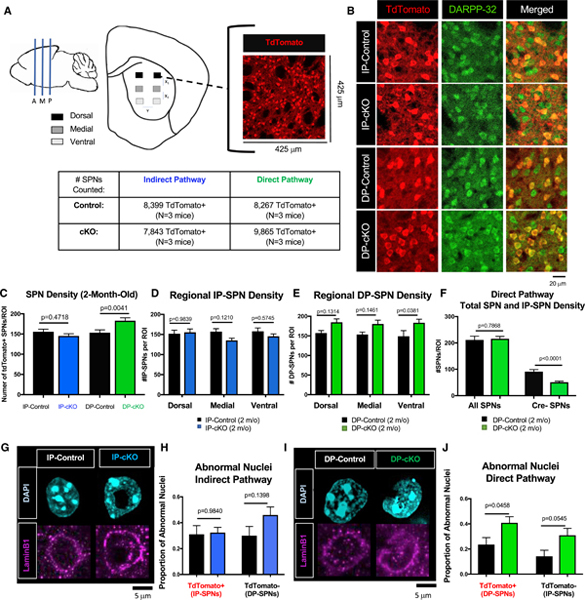
(A) Schematic of unbiased SPN counting strategy. Three coronal slices spanning anterior, medial, and posterior striatum were collected and stained for TdTomato and DARPP-32. Two images were acquired from the dorsal, medial, and ventral striatum, with displacement (x1, x2, y) between images kept constant across slices. Number of cells counted/mice analyzed is displayed in the table.
(B) Representative images of 2-month-old IP-control, IP-cKO, DP-control, and DP-cKO striatum stained for TdTomato (red, TdTomato/Cre+ SPNs) and DARPP-32 (green, all SPNs).
(C) There is no difference in IP-SPN density in 2-month-old IP-cKO mice compared with IP-controls, whereas there are more DP-SPNs in DP-cKOs relative to DP-controls (n = 6 images/mouse, 3 mice/genotype, one-way ANOVA with Sidak’s multiple comparisons test F(3,104) = 6.081, p values displayed on graph; graph displays mean ± SEM).
(D) There is no effect of region on IP-SPN density in 2-month-old IP-cKOs versus IP-controls (n = 6 images/animal, 3 animals/genotype, two-way ANOVA genotype by region interaction F(2,102) = 1.447, p = 0.2401, Sidak’s multiple comparisons test for within-region comparisons, p values displayed above; graph displays mean ± SEM).
(E) There is no effect of region on DP-SPN density in 2-month-old DP-cKOs compared with DP-controls (n = 6 images/animal, 3 animals/genotype, two-way ANOVA genotype by region interaction F(2,102) = 0.09598, p = 0.9086, Sidak’s multiple comparisons test for within-region comparisons, p values displayed above; graph displays mean ± SEM).
(F) There is no difference in the total density of SPNs (DARPP-32+ cells) in DP-cKOs compared with controls (n = 2–3 mice/genotype, 6 images/mouse, unpaired two-way t test, t = 0.2724, df = 37, p = 0.7868), but there is a significant reduction in the number of Cre−/DARPP-32+ SPNs (n = 2–3 mice/genotype, 6 images/mouse, unpaired two-way t test, t = 5.073, df = 37, p < 0.001; graph displays mean ± SEM).
(G) Representative Airyscan images of SPN nuclei from the dorsal striatum of 2-month-old IP-controls and IP-cKOs stained for DAPI (cyan) and Lamin B1 (magenta).
(H) There is no difference in the proportion of abnormal nuclei of TdTomato+ IP-SPNs or TdTomato− DP-SPNs in the dorsal striatum of 2-month-old IP-cKOs compared with IP-controls (n = 3 mice/genotype, 3 images/mouse, one-way ANOVA F(3,44) = 1.572, Sidak’s multiple comparisons test for within-region comparisons, p values displayed above; graph displays mean ± SEM).
(I) Representative Airyscan images of SPN nuclei from the dorsal striatum of 2-month-old DP-controls and DP-cKOs stained for DAPI (cyan) and Lamin B1 (magenta).
(J) There is a greater proportion of abnormal TdTomato+ DP-SPN nuclei and a trending increase in abnormal nuclei of TdTomato− IP-SPN nuclei in the dorsal striatum of 2-month-old DP-cKOs compared with DP-controls (n = 3 mice/genotype, one-way ANOVA F(3,44) = 4.725, Sidak’s multiple comparisons test for within-region comparisons, p values displayed above; graph displays mean ± SEM).
We found no difference in the density of IP-SPNs in 2-month-old IP-cKO mice compared with IP-controls, and there was no effect of region on IP-SPN density (Figures 2B and 2D). However, we observed more DP-SPNs in 2-month-old DP-cKO mice compared with DP-controls (Figures 2C and 2E). To determine whether the density of IP-SPNs in DP-cKO mice was also affected, we counted all DARPP-32+ SPNs of DP-cKO and control mice. We found no difference in the total DARPP-32+ SPN density, indicating that DP-cKOs had significantly fewer IP-SPNs (Cre−/DARPP-32+ cells) relative to DP-controls (Figure 2F). This result suggests that HTT is required for controlling the genesis or differentiation of SPNs.
While counting cells, we observed irregularly shaped nuclei of cKO SPNs that had apparent holes and/or invaginations. Invaginated nuclei are seen in human HD patient and mouse model neurons (Roos and Bots, 1983; Kutscher and Shaham, 2017; Davies et al., 1997; Goldman et al., 2004). Abnormal nuclear structure has also been associated with developmentally programmed cell death (Abraham et al., 2007). Nuclear architecture is intimately related to transcriptional regulation, and abnormal nuclear structure may lead to dysregulated gene expression. In line with our findings of abnormal SPN gene expression, we asked whether Htt deletion in IP-SPNs or DP-SPNs leads to a greater frequency of SPN nuclear crenellation.
To identify abnormal nuclei, we stained control and cKO dorsal striatum for TdTomato to label Cre+ SPNs and DAPI to visualize nuclei (Figures 2G and 2I). Nuclei were hand-scored as either normal (nuclei with round, uniform morphology) or abnormal (nuclei with membrane invaginations, membrane crinkling, or “holes”) by experimenters blind to genotype. Normal and abnormal nuclei were observed in SPNs of all genotypes, including controls. We did not observe a difference in the proportion of abnormal nuclei in 2-month-old IP-cKO SPNs compared with controls (Figures 2G and 2H). However, we did find a robust increase in the proportion of abnormal DP-SPN nuclei in 2-month-old DP-cKO mice and a trending increase in crenellations in their IP-SPN neighbors (Figures 2I and 2J). In sum, loss of HTT does not cause SPN loss in early adulthood, but HTT plays a role in regulating the initial number of DP-SPNs formed. The abnormal nuclear structure we observed in DP-cKO SPNs may contribute to the dysregulated gene expression we also found in these cells (see Figures 1H–1J and S2).
Loss of Huntingtin in SPNs Impairs Motor Function
Because SPNs are an integral part of the basal ganglia circuit controlling motor function, we asked whether loss of HTT in IP-SPNs or DP-SPNs affects mouse motor behavior using the open field test (OFT) (Pogorelov et al., 2005) to examine the gross motor activity of 2-month-old cKO mice and their age-matched controls. Compared with 2-month-old IP-controls, IP-cKO mice were hyperactive in the OFT. IP-cKOs traveled significantly longer distances throughout the 60-min testing window, leading to a greater cumulative distance traveled and more time spent moving (Figures 3A and 3B). The difference in distance traveled was significant only during the first 30 min of testing, but mice spent more time moving during both the first and last 30 min of testing (Figures S3A and S3B). IP-cKOs did not perform more stereotypic episodes, nor did they spend more time in the margins of the OFT, suggesting that their hyperactivity is not due to increased anxiety (Figures S3E and S3F).
Figure 3. Htt Is Required in SPNs for Normal Motor Function.
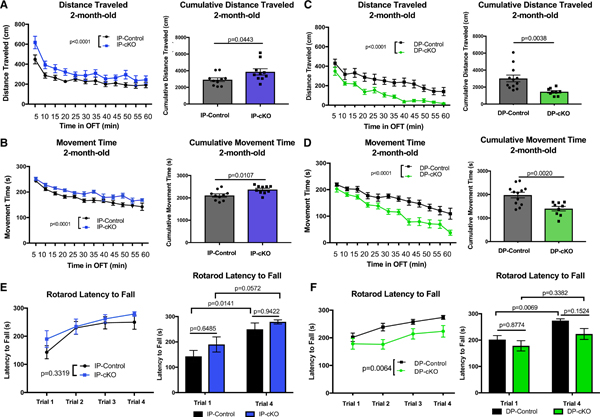
(A) Left: 2-month-old IP-cKOs were hyperactive in the open field test (OFT) compared with IP-controls (n = 10 animals/genotype, analysis of covariance [ANCOVA] with Tukey’s post hoc testing, mean squared error [MSE] = 14,481, df = 237, p < 0.0001). Right: IP-cKOs traveled a greater cumulative distance over the 60-min test window (n = 10 animals/genotype, unpaired two-way t test, t = 2.163, df = 18, p = 0.0443; graph displays mean ± SEM).
(B) Left: 2-month-old IP-cKOs spent more time moving throughout the OFT compared with IP-controls(n = 10 animals/genotype, ANCOVA with Tukey’s post hoc testing, MSE = 721.69, df = 237, p < 0.0001). Right: IP-cKOs spent more time moving cumulatively over the 60-min test window (n = 10 animals/genotype, unpaired two-way t test, t = 2.848, df = 18, p = 0.0107; graph displays mean ± SEM).
(C) Left: 2-month-old DP-cKOs were hypoactive in the OFT compared with DP-controls (n = 9–12 animals/genotype, ANCOVA with Tukey’s post hoc testing, MSE = 13,815, df = 249, p < 0.0001). Right: DP-cKOs traveled a shorter cumulative distance over the 60-min test window (n = 9–12 animals/genotype, unpaired two-way t test, t = 3.299, df = 19, p = 0.0038; graph displays mean ± SEM).
(D) Left: 2-month-old DP-cKOs spent less time moving throughout the OFT compared with DP-controls (n = 9–12 animals/genotype, ANCOVA with Tukey’s post hoc testing, MSE = 1,588.9, df = 249, p < 0.0001). Right: DP-cKOs spent less time moving cumulatively over the 60-min test window(n = 9–12animals/genotype, unpaired two-way t test, t = 3.583, df = 19, p = 0.0020; graph displays mean ± SEM).
(E) Left: 2-month-old IP-cKOs did not significantly differ from IP-controls on the ART latency to fall (n = 10 animals/genotype, two-way repeated-measures ANOVA of genotype F(1,18) = 0.9943, p = 0.3319). Right: IP-cKOs did not differ from controls on trial 1 or trial 4 latency to fall, and both genotypes displayed improvement between trials 1 and 4 (two-way ANOVA of genotype F(1,36) = 2.708, p = 0.1085; Sidak’s multiple comparisons test p values displayed above; graph displays mean ± SEM).
(F) Left: 2-month-old DP-cKOs had a shorter latency to fall on the ART (n = 9–12 animals/genotype, two-way repeated-measures ANOVA of genotype F(1,19) = 9.380, p = 0.0064). Right: DP-cKOs did not significantly improve between trials 1 and 4 (two-way ANOVA of genotype F(1,38) = 5.641, p = 0.0227; Sidak’s multiple comparisons test p values displayed above; graph displays mean ± SEM).
See Figure S3.
Conversely, Htt deletion in DP-SPNs led to hypoactivity in the OFT. Two-month-old DP-cKOs traveled shorter distances and spent less total time moving compared with DP-controls (Figures 3C and 3D). There was also a trending reduction in the number of stereotypic episodes performed by DP-cKOs, mirroring their overall reduced activity level (Figure S3E). The hypoactivity of DP-cKO mice was much more pronounced during the last 30 min of the OFT. These results suggest that DP-cKOs are not as deficient in their ability to initiate adequate motor activity at baseline (Figures S3C and S3D), but instead they lack the capacity and/or motivation to sustain activity throughout the test. DP-cKO mice did spend slightly more time in the margins of the OFT compared with controls, which may be explained by their overall hypoactivity and increased time spent immobile (Figure S3F).
Next, we examined motor coordination in cKO and control mice using the accelerating rotarod task (ART) (Wang et al., 2016b). Htt deletion in IP-SPNs did not cause any detectable deficits in motor coordination or learning on the ART at 2 months old (Figure 3E). However, DP-cKOs performed significantly worse on the ART compared with DP-controls (Figure 3F). Also, DP-cKOs did not display significant improvement in latency to fall between trials 1 and 4, indicating that Htt deletion in DP-SPN may impair motor learning (Figure 3F, right). In sum, both SPN subtypes require HTT for the proper control of movement. Deleting Htt in IP-SPNs results in hyperactive locomotion without any significant motor coordination impairment, whereas Htt loss in DP-SPNs leads to hypoactive locomotion along with impaired coordination. These findings suggest that HTT is required in SPNs for the proper function of the basal ganglia circuit controlling movement.
IP-SPNs Require Huntingtin for Proper Synaptic Connectivity and Function
Given that Htt deletion in both SPN subtypes disrupted the motor behavior of 2-month-old mice, we asked whether loss of Htt in SPNs alters the synaptic connectivity of these neurons. To do so, we analyzed the number and function of inhibitory synapses in the GPe of 2-month-old IP-cKO mice using immunohistochemical and electrophysiological approaches (Figure 4A). To count GABAergic synapses in the GPe, we stained sagittal brain sections containing the GPe (lateral to midline, 1.725 to 2.40 mm) for vesicular GABA transporter (VGAT) and gephyrin, a pair of pre- and post-synaptic markers that are specific to inhibitory synapses (Figure 4B). We quantified the colocalization of VGAT and gephyrin as a measure of the number of GABAergic synapses in the central GPe. VGAT and gephyrin are located in different subcellular compartments (axon versus dendrite) and appear “colocalized” only at synaptic junctions because of their extremely close proximity. Details of this quantification method have been previously described, and techniques such as electron microscopy and electrophysiology have been used to verify this method (Eroglu et al., 2009; Stogsdill et al., 2017; Ippolito and Eroglu, 2010). We found a dramatic (~50%) reduction in GABAergic synapse number in the GPe of 2-month-old IP-cKO mice compared with controls (Figure 4C). We found no difference in the number of inhibitory synapses in the SNR of 2-month-old IP-cKO mice compared with controls, suggesting that deletion of Htt in IP-SPNs affects only GPe inhibitory synapses (Figures S4A and S4B). The appearance of colocalized VGAT/gephyrin puncta in these analyses was not due to chance, because randomization of the puncta by rotating the gephyrin channel 90° eliminated the vast majority of colocalized puncta (Figures S4C–S4F).
Figure 4. IP-SPNs Require Htt for Proper Synaptic Connectivity and Function.
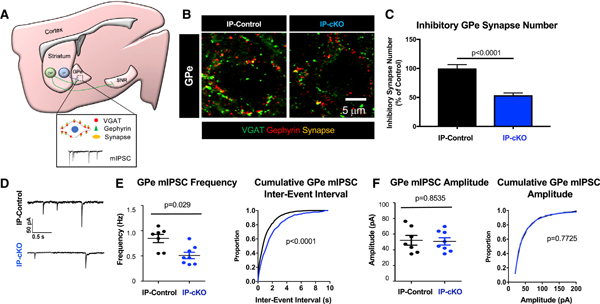
(A) Diagram of GPe synapse analysis approach of IP-controls and IP-cKOs. The GPe is stained for VGAT and gephyrin. “Colocalization” of VGAT and gephyrin marks GABAergic inhibitory synapses. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded from medial GPe neurons.
(B) Representative images of inhibitory synapses in the GPe of 2-month-old IP-controls and IP-cKOs stained for VGAT (green) and gephyrin (red).
(C) Inhibitory synapse numbers are reduced in the GPe of 2-month-old IP-cKO mice compared with controls (n = 3–4 replicates/animal, 3 animals/genotype, 110 total images analyzed, nested ANOVA by genotype, F = 44.26, df = 1, p < 0.0001; graph displays mean ± SEM).
(D) Sample mIPSC traces from 2-month-old IP-control and IP-cKO medial GPe neurons.
(E) Left: the mean frequency of GPe mIPSCs is reduced in 2-month-old IP-cKOs compared with IP-controls (n = 7–8 cells per genotype, unpaired two-way t test, p = 0.0056, t = 3.309, df = 13; graph displays mean ± SEM). Right: the distribution of GPe mIPSC inter-event interval cumulative probability differs between 2-month-old IP-cKOs and IP-controls (Kolmogorov-Smirnov test, p < 0.0001).
(F) Left: there is no difference in the mean amplitude of GPe mIPSCs of 2-month-old IP-cKOs compared with IP-controls (n = 7–8 cells per genotype, unpaired two-way t test, p = 0.8535, t = 0.1883, df= 13; graph displays mean ± SEM). Right: the distribution of GPe mIPSC amplitude cumulative probability does not differ between 2-month-old IP-cKOs and IP-controls (Kolmogorov-Smirnov test, p = 0.7725).
See Figure S4.
Next, we measured miniature inhibitory postsynaptic currents (mIPSCs) of central GPe neuronsfrom 2-month-old IP-cKOs and IP-controls. In line with the decrease in structural synapse number, the frequency of mIPSCs was significantly reduced in 2-month-old IP-cKO mice compared with controls, whereas mIPSC amplitude did not differ (Figures 4D–4F). Taken together, these results show that IP-cKO reduces the number and function of GPe inhibitory synapses, and suggest that reduced GPe inhibition by IP-SPNs drives the hyperactive motor behavior of IP-cKO mice.
Loss of Huntingtin in DP-SPNs Leads to Increased Inhibition in the SNR and Enhanced Collateral Connections to the GPe
Given that DP-cKO mice display hypoactive motor behavior, we asked whether loss of Htt decreases inhibitory synapse numbers and/or activity in the SNR. Quantification of inhibitory synapses (VGAT+/gephyrin+) in the dorsal SNR (lateral to midline, 1.95 to 1.525 mm) of 2-month-old mice revealed no significant difference in inhibitory synapse numbers of DP-cKOs compared with controls (Figures 5A–5C). Furthermore, when we recorded mIPSCs from the dorsal SNRs of 2-month-old mice, mIPSC frequency was increased in DP-cKOs compared with DP-controls (Figures 5D and 5E). There was no difference in the mean amplitude of SNR mIPSCs in DP-cKOs, although the cumulative distributions were significantly different between genotypes (Figure 5F). Multiple factors can drive increased mIPSC frequency, including increased inhibitory synapse numbers or greater size of the readily releasable pool of vesicles. Given that we did not detect a change in the neuroanatomical number of inhibitory synapses in the SNR, the greater mIPSC frequency we observed is likely caused by an increase in release probability at these terminals.
Figure 5. DP-cKO Leads to Increased Inhibition in the SNR and Enhanced GPe Synapses.
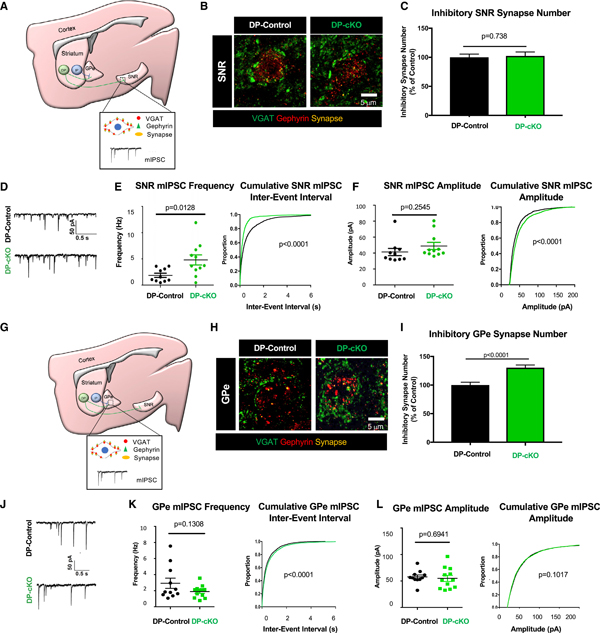
(A) Diagram of SNR synapse analyses of DP mice. GABAergic synapse staining with presynaptic VGAT (green) and postsynaptic gephyrin (red) in dorsal SNR neurons. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded from dorsal SNR neurons.
(B) Representative images of inhibitory synapses in the SNR of 2-month-old DP-control and DP-cKO mice stained for VGAT (green) and gephyrin (red).
(C) Quantification of SNR inhibitory synapses of 2-month-old DP-controls and DP-cKOs (n = 3 replicates/animal, 3 animals/genotype, 90 images analyzed, nested ANOVA by genotype, F = 0.11292, df = 1, p = 0.73769; graph displays mean ± SEM).
(D) Sample traces of mIPSCs from 2-month-old DP-control and DP-cKO dorsal SNR neurons.
(E) Left: quantification of the mean frequency of mIPSCs (n = 10–11 cells per genotype, unpaired two-way t test, p = 0.0128, t = 2.749, df = 19, graph displays frequencies from individual neurons as well as mean ± SEM). Right: cumulative probability plots of inter-event interval (Kolmogorov-Smirnov test, p < 0.0001).
(F) Left: quantification of the mean amplitude of mIPSCs (n = 10–11 cells per genotype, unpaired two-way t test, p = 0.2545, t = 1.175, df = 19, graph displays frequencies from individual neurons as well as mean ± SEM). Right: cumulative probability plots of mIPSC amplitude (Kolmogorov-Smirnov test, p < 0.0001).
(G) Diagram of GPe synapse analysis approach. GABAergic synapse staining with presynaptic VGAT (green) and postsynaptic gephyrin (red) in the GPe. mIPSCs were recorded from neurons in the medial GPe.
(H) Representative images of inhibitory synapses in the GPe of 2-month-old DP-control and DP-cKO mice stained for VGAT (green) and gephyrin (red).
(I) Quantification of inhibitory synapses in the GPe of 2-month-old DP-controls and DP-cKOs (n = 3 replicates/animal, 3 animals/genotype, 90 images analyzed, nested ANOVA by genotype, F = 20.462, df = 1, p < 0.0001; graph displays mean ± SEM).
(J) Sample traces of mIPSCs from 2-month-old DP-control and DP-cKO GPe neurons.
(K) Left: quantification of the mean frequency of GPe mIPSCs (n = 11–12 cells per genotype, unpaired two-way t test, p = 0.1308, t = 1.572, df = 21, graph displays frequencies from individual neurons as well as mean ± SEM). Right: cumulative probability plots of inter-event interval (Kolmogorov-Smirnov test, p < 0.0001).
(L) Left: quantification of the mean amplitude of GPe mIPSCs (n = 11–12 cells per genotype, unpaired two-way t test, p = 0.6941, t = 0.3987, df = 21, graph displays frequencies from individual neurons as well as mean ± SEM). Right: cumulative probability plots of the mIPSC amplitude (Kolmogorov-Smirnov test, p = 0.1017).
See Figure S5.
The alterations in the GABAergic synapse number and function that we observed within the SNR and GPe of DP-cKOs do not fully explain the hypoactive motor behavior of DP-cKO mice. One reason for this could be that the SNR may not receive inhibitory inputs solely from DP-SPNs. Thus, to isolate the specific functional properties of DP-SPN synapses within the SNR, we used optogenetics to specifically activate DP-SPN terminals upon stimulation with blue light. Two-month-old DP-cKO and control mice received a single intrastriatal injection of a Cre-dependent channelrhodopsin virus (AAV-LSL-ChR2) (Figure S5A). Acute slices containing the SNR were prepared 3 weeks later, and evoked inhibitory postsynaptic currents (eIPSCs) of dorsal SNR neurons were recorded while blue light bursts were applied to the SNR (Figure S5B). We found a trending reduction in paired pulse ratio between DP-cKOs and controls (Figure S5C). Additionally, DP-cKO response kinetics differed significantly between genotypes. Specifically, the decay time and width of eIPSCs were reduced in DP-cKOs, with no change in the rise time (Figures S5D–S5F). The reduced decay and width kinetics could be caused by multiple factors, such as decreased presynaptic GABA reuptake or altered postsynaptic GABA receptor subunit expression.
Approximately 60% of DP-SPNs send collateral projections through the GPe (Figure 1E). Interestingly, we found significantly more GPe inhibitory synapses of 2-month-old DP-cKOs compared with age-matched DP-controls (Figures 5G–5I). However, when we recorded mIPSCs from the GPe of DP-cKOs, the mIPSC frequency and amplitudes were similarto those recorded from DP-controls (Figures 5J–5L). Rotation analysis of GPe and SNR synapses substantially reduced the number of colocalized puncta, confirming that VGAT/gephyrin colocalization in these samples was not random (Figures S5G–S5J).
Taken together, these results show that, unlike IP-cKO, loss of Htt in DP-SPNs does not decrease the number of inhibitory synapses formed at their principal target, the SNR. Instead, DP-cKO enhances inhibitory synaptic activity in the SNR and leads to increased inhibitory synapse numbers in the GPe either by increasing DP-SPN bridging collateral synapses or by enhancing IP-SPN connectivity.
Impaired Motor Function Due to SPN Htt Deletion Is Maintained with Aging
We next investigated the function of HTT in the SPNs in middle-aged mice (10 months old), analogous to when many human HD patients begin displaying severe motor symptoms and SPN degeneration (Walker, 2007). Similar to 2-month-old mice, 10-month-old IP-cKO mice were hyperactive in the OFT. IP-cKO mice traveled a greater distance and spent more time moving compared with controls (Figures 6A and 6B). IP-cKO mice traveled a greater distance only during the first 30 min of the OFT, although they spent more time moving during both the first and last 30 min of the test (Figures S6A and S6B).
Figure 6. Behavioral Deficits in Htt cKO Mice Persist with Aging.
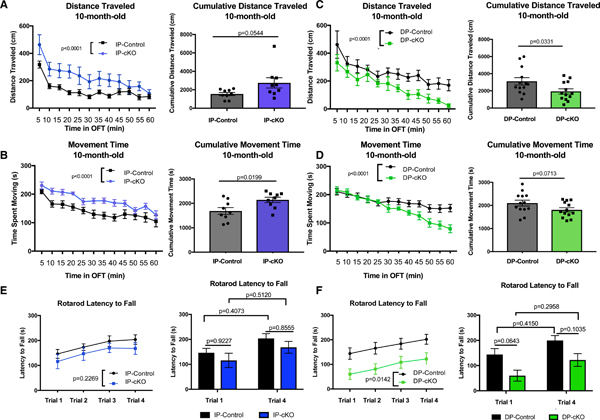
(A) Left: 10-month-old IP-cKOs were hyperactive in the open field test (OFT) compared with IP-controls (n = 9 animals/genotype, analysis of covariance [ANCOVA] with Tukey’s post hoc testing, MSE = 14,106, df =213, p< 0.0001). Right: IP-cKOs displayed a trending increase in cumulative distance traveled over the 60-min test window (n = 9 animals/genotype, unpaired two-way t test, t = 2.075, df = 16, p = 0.0544; graph displays mean ± SEM).
(B) Left: 10-month-old IP-cKOs spent more time moving compared with IP-controls (n = 9 animals/genotype, ANCOVA with Tukey’s post hoc testing, MSE = 1,311.2, df = 213, p < 0.0001). Right: IP-cKOs spent more cumulative time moving compared with controls (n = 9 animals/genotype, unpaired two-way t test, t = 2.587, df = 16, p = 0.0199; graph displays mean ± SEM).
(C) Left: 10-month-old DP-cKOs were hypoactive in the OFT compared with IP-controls (n = 13 animals/genotype, ANCOVA with Tukey’s post hoc testing, MSE = 22207, df = 309, p < 0.0001). Right: DP-cKOs traveled a cumulative shorter distance compared with controls (n = 13 animals/genotype, unpaired two-way t test, t = 2.261, df = 24, p = 0.0331; graph displays mean ± SEM).
(D) Left: 10-month-old DP-cKOs spent less time moving in the OFT compared with IP-controls (n = 13 animals/genotype, ANCOVA with Tukey’s post hoc testing, MSE = 1839.7, df = 309, p < 0.0001). Right: DP-cKOs displayed a trending decrease in cumulative time spent moving compared with controls (n = 13 animals/genotype, unpaired two-way t test, t = 1.887, df = 24, p = 0.0713; graph displays mean ± SEM).
(E) Left: 10-month-old IP-cKOs did not differ from IP-controls on ART latency to fall (n = 9 animals/genotype, two-way repeated-measures ANOVA of genotype F(1,16) = 1.579, p = 0.2269). Right: IP-cKOs did not differ from controls on trial 1 or trial 4 latency to fall, and neither group displayed statistically significant improvement between trials 1 and 4 (two-way ANOVA of genotype F(1,32) = 2.128, p = 0.1544; Sidak’s multiple comparisons test p values displayed above; graph displays mean ± SEM).
(F) Left: 10-month-old DP-cKOs had a significantly shorter latency to fall on the ART (n = 9 animals/genotype, two-way repeated-measures ANOVA of genotype F(1,24) = 6.986, p = 0.0142). Right: DP-cKOs displayed a trending reduction in latency to fall during trials 1 and 4 compared with controls, although neither group displayed significant improvement between trials 1 and 4 (two-way ANOVA of genotype F(1,48) = 12.97, p = 0.0007; Sidak’s multiple comparisons test p values displayed above; graph displays mean ± SEM).
See Figure S6.
Conversely, 10-month-old DP-cKOs were hypoactive in the OFT, traveling a shorter distance and spending less time moving compared with controls (Figures 6C and 6D). DP-cKOs displayed normal levels of locomotor activity during the first part of the OFT, but their activity levels dropped off sharply during the final half of the test (Figures S6C and S6D). Neither 10-month-old IP-cKO nor DP-cKO mice differed from controls in terms of their stereotypic episodes (Figure S6E). However, IP-cKO mice spent less time in the margins of the open field, potentially reflecting their increased activity and reduced time spent still in any one area (Figure S6F).
Consistent with their performance at 2 months old, 10-month-old IP-cKO mice did not display significant impairments on the ART (Figure 6E). However, 10-month-old DP-cKOs were severely impaired in motor coordination on the ART (Figure 6F). In sum, we found that the motor dysfunctions of IP-cKO and DP-cKO mice are maintained and intensified with aging.
Huntingtin Deletion Leads to SPN Loss with Aging
To determine whether loss of Htt in IP-SPNs or DP-SPNs affects SPN survival with aging, we used our cell counting strategy outlined in Figure 2 to count >6,100 TdTomato+ and >10,000 DARPP32+ SPNs per genotype in 10-month-old striata (Figure 7A). We observed a significant reduction in the density of IP-SPNs in 10-month-old IP-cKO mice compared with IP-controls (Figures 7B and S7A). The total density of DARPP-32+ SPNs in IP-cKOs was reduced, an effect driven by the reduction in IP-SPN density (Figure S7B). We also found a dramatic reduction in DP-SPN density in 10-month-old DP-cKO mice compared with DP-controls (Figure 7B). DP-SPN density was reduced across the dorsal, medial, and ventral striatum (Figures S7C and S7D). These findings show that HTT is required in SPNs for neuronal survival during aging. It is possible that the cell death observed in DP-cKOs is stronger than in IP-cKOs because D1-Cre also deletes Htt in a subset of cortical neurons (Figures S1A–S1C), exacerbating the loss-of-function phenotypes in these mice.
Figure 7. Htt Is Required for SPN Survival with Aging.
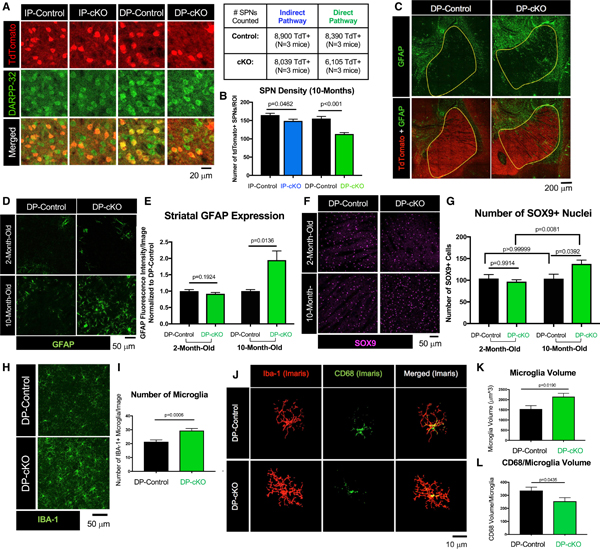
(A) Representative images of striatum from 10-month-old IP-control, IP-cKO, DP-control, and DP-cKO mice stained for TdTomato (red, Cre+ SPNs) and DARPP-32 (green, all SPNs). Number of cells counted per mice analyzed is displayed in the table.
(B) There are fewer IP-SPNs in 10-month-old IP-cKOs compared with IP-controls, and fewer DP-SPNs in 10-month-old DP-cKOs compared with DP-controls (n = 3 mice/genotype, 6 images/mouse, one-way ANOVA with Sidak’s multiple comparisons test, F(3,104) = 21.3, p values above; graph displays mean ± SEM).
(C) Representative tile scans of 10-month-old DP-control and DP-cKO brains stained for GFAP (green) and TdTomato (red). Yellow boundary demarcates the striatum.
(D) Representative close-up images of 2- and 10-month-old DP-control and DP-cKO dorsal striatum stained for GFAP (green).
(E) GFAP fluorescent intensity (integrated density) was increased in the striatum of 10-month-old DP-cKO mice (2 month-old: n = 4 images/mouse, 3 mice/genotype, unpaired two-way t test, t = 1.345, df = 22, p = 0.1924; 10 month-old: n = 2–3 images/mouse, 2–3 mice/genotype, unpaired two-way t test, t = 2.889, df = 12, p = 0.0136; graph displays mean ± SEM).
(F) Representative images of 2- and 10-month-old DP-control and DP-cKO striatum stained for SOX9 (magenta).
(G) There are more SOX9+ astrocytes in 10-month-old DP-cKO striatum compared with DP-controls, with no difference at 2 months old (n = 4 images/animal, 3 animals/genotype, one-way ANOVA, F(3, 43) = 4.671, p = 0.0065, Sidak’s multiple comparisons test for within-region comparisons, p values above; graph displays mean ± SEM).
(H) Representative images of 10-month-old DP-control and DP-cKO striatum stained for Iba-1 (green).
(I) There are more microglia in the striatum of 10-month-old DP-cKO mice relative to DP-controls (n = 3 mice/genotype, 3–5 images/mouse, unpaired two-way t test, t = 3.885, df = 26, p = 0.0006; graph displays mean ± SEM).
(J) Imaris 3D surface reconstructions of 10-month-old DP-control and DP-cKO striatal microglia stained for IBA-1 (red) and CD68 (green).
(K) Microglia volume is increased in 10-month-old DP-cKO mice (n = 2 mice/genotype, 5–10 microglial cells/mouse, unpaired two-way t test, t = 2.501, df = 26, p = 0.0190; graph displays mean ± SEM).
(L) CD68 volume per microglia of 10-month-old DP-cKOs was reduced relative to DP-controls (n = 3 mice/genotype, 8–15 microglia analyzed per mouse, unpaired two-way t test, t = 3.534, df = 18, p = 0.0024; graph displays mean ± SEM).
See Figure S7.
We next sought to determine the mechanism by which DP-SPNs die in DP-cKOs with aging because the cell loss phenotype at 10 months of age was substantially stronger in these mice. First, we stained 2- and 10-month-old DP-control and DP-cKO dorsal striatum for cleaved caspase-3 (CC3), which is highly upregulated by cells undergoing apoptosis (Figure S7E). Although sparse CC3 puncta were detected in the striatum of 10-month-old mice, there was no difference in CC3 expression between genotypes, suggesting that Htt cKO does not cause SPNs to die via apoptosis (Figure S7F). Interestingly, how SPNs die in HD is not yet known, but apoptosis is not thought to be the main cause (Hickey and Chesselet, 2003).
Reactive astrogliosis is classically observed in the striatum of HD patients when SPNs are dying, and the degree of gliosis directly correlates with the severity of HD pathology (Vonsattel et al., 1985). Thus, we stained 2- and 10-month-old DP brains for glial fibrillary acidic protein (GFAP), which is expressed at low levels in striatal astrocytes but is upregulated with reactivity, and for SRY-Box transcription factor 9 (SOX9), a marker for astrocytic nuclei in adult mouse CNS (Figures 7C–7F) (Khakh, 2019; Sun et al., 2017). We found a striking increase in GFAP expression in 10-month-old DP-cKO striatum compared with controls, with no difference between genotypes at 2 months old (Figures 7C–7E). Similarly, there was a substantial increase in the number of SOX9+ astrocytes in the striatum of 10-month-old DP-cKOs compared with controls, with no difference present between genotypes at 2 months old (Figures 7F and 7G).
Reactive astrogliosis and neuronal loss are often accompanied by alterations in microglia number, morphology, and phagocytic activity in HD brains (Sapp et al., 2001). We labeled microglia with microglial marker ionized calcium-binding adapter molecule 1 (IBA-1) in DP-cKO and control brains, and found significantly more microglia in the dorsal striatum of 10-month-old DP-cKOs compared with controls (Figures 7H and 7I). Next, we stained the same tissue for CD68, which marks the phagocytic compartment of microglia, and used Imaris Software to create 3D surface reconstructions of individual microglia and the CD68 within them (Figure 7J). We found that the average microglia cell volume was increased in 10-month-old DP-cKOs, concurrent with a slight reduction in CD68 volume within DP-cKO microglia cells (Figures 7K and 7L). Taken together, these results show that loss of Htt in DP-SPNs leads to non-apoptotic cell death and reactive gliosis reminiscent of HD brains.
DISCUSSION
Although HD is caused by a well-characterized CAG repeat mutation of HTT, the cellular and molecular mechanisms of HD pathogenesis remain elusive. Here, we show that Htt loss in SPNs leads to dysfunctional motor behavior with concurrent abnormal synaptic connectivity, nuclear morphology, and changes in SPN gene expression. Importantly, Htt deletion leads to SPN loss with aging and reactive gliosis, all reminiscent of HD neuropathology. Thus, our findings show that Htt loss in SPNs mimics key features of HD, offering insights into functions of HTT in the striatum and providing strong evidence that loss-of-function mechanisms contribute to SPN pathology in HD.
HTT Is Required in SPNs for the Integrity of the Basal Ganglia Circuit
Synaptic dysfunction is a feature of many neurodegenerative diseases, including HD. Many individuals carrying the HD mutation have abnormal brain circuit function that is evident well before motor symptom onset (Unschuld et al., 2012). Several mouse models of HD display aberrant synaptic connectivity preceding both motor dysfunction and neuronal loss (McKinstry et al., 2014; Milnerwood and Raymond, 2007; Deng et al., 2013). Here, we found that selective loss of Htt in IP-SPNs or DP-SPNs disrupts synaptic connectivity, suggesting that Htt loss-of-function in SPNs may also contribute to the synaptic dysfunction in HD. IP-cKO dramatically reduced the number and function of inhibitory synaptic connections onto their target GPe. Behaviorally, IP-cKO mice were hyperactive, likely due to reduced inhibition of the GPe. This is intriguing because loss of GPe inhibition is predicted to be the cause of several symptoms associated with early HD, including hyperkinesia and chorea (Deng et al., 2004; Reiner et al., 1988). We also found more inhibitory synapses within the GPe of DP-cKOs, potentially arising from an increase in GPe bridging collaterals formed by DP-SPNs. Taken together, our findings indicate that loss of Htt alters GPe connectivity of SPNs, albeit in opposite directions. Further work is needed to determine why the SPN subtypes and their synaptic connections are differentially affected by Htt deletion and whether these differences underlie their differing susceptibilities to Htt loss.
SPNs Require HTT for the Maintenance of Cell Health and Gene Expression
Htt deletion from SPNs adversely affects SPN health and function long before neurodegeneration occurs. For instance, Htt cKO alters the expression of SPN-enriched genes, including pENK and DARPP-32. Changes in SPN gene expression have been observed in HD patients and many HD models (Hodges et al., 2006; Mazarei et al., 2010; Luthi-Carter et al., 2000; Bibb et al., 2000; Ament et al., 2017; Langfelder et al., 2016). DARPP-32, which was strongly downregulated both in cKO SPNs and in HD, is a downstream regulator of dopaminergic signaling in SPNs. Intriguingly, cKO of DARPP-32 in IP-SPNs or DP-SPNs causes motor behavior deficits (hyperactivity versus hypoactivity, respectively), similar to what we observed here (Bateup et al., 2010). Therefore, the motor dysfunction caused by Htt cKO in SPNs may be partly mediated by DARPP-32 reduction in these cells.
How does Htt loss alter SPN gene expression? HTT interacts with several transcriptionally active proteins, such as SP-1 and REST/dynactin p150Glued (Zhai et al., 2005; Shimojo, 2008). HTT may also modulate transcription by binding directly to DNA(Benn et al., 2008). In addition, HTT interacts with Polycomb Repressive Complex 2 (PRC2) subunits, which catalyze histone H3K27 methylation. Embryos lacking HTT have disrupted PRC2-mediated regulation of gene expression (Seong et al., 2010). Similar to our findings in Htt cKOs, PRC2 loss-of-function in SPNs leads to neurodegeneration (Von Schimmelmann et al., 2016). Therefore, Htt loss in SPNs may disrupt PRC2-dependent regulation of SPN-specific genes.
Our findings also suggest that SPNs require HTT for maintaining proper nuclear structure. Nuclear invaginations, impaired nucleocytoplasmic transport, and disrupted nuclear pore complexes are associated with HD (Kutscher and Shaham, 2017; Grimaetal., 2017). Tightly regulated nuclear structure and function is critical for transcriptional integrity. Nuclear crenellation is also a feature of developmentally programmed linker cell death in C. elegans, which is controlled by a polyQ-containing protein (Abraham et al., 2007). The death of the linker cell, which regulates male reproductive system organization, occurs independently of classic apoptotic pathways. Nuclear invagination is also apparent in dying cells in HD brains, suggesting that changes to the nuclear structure may be related to a non-apoptotic degenerative mechanism (Kutscher and Shaham, 2017).
Nuclear invaginations also occur transiently in response to synaptic activity (Wittmann et al., 2009). For instance, activity-dependent nuclear infolding plays a role in enhancing the relay of calcium signals to the nucleus. Future work is needed to discern whether abnormalities in nuclear structure are a direct consequence of Htt loss or are a byproduct of synaptic dysfunction induced by SPN wiring errors, and to determine whether they underlie abnormal gene expression and/or degeneration observed in cKO SPNs.
Loss of Htt in SPNs Partially Recapitulates Several HD-like Phenotypes
The precise mechanisms of SPN death in HD remain unknown. We found that Htt loss in either SPN subtype disrupts motor function and drives aging-related neuronal loss accompanied by reactive gliosis. Therefore, three main aspects of HD pathology, abnormal motor function, reactive gliosis, and SPN death, can be partially produced by Htt loss-of-function in SPNs. Previous studies have shown that loss of Htt in forebrain neurons (Dragatsis et al., 2000) or cortex (Dragatsis et al., 2018) leads to widespread cell death, reduced cortical volume, and abnormal motor behavior. However, the pattern of degeneration does not resemble the striatum-specific SPN loss seen in HD. Deleting Htt from cells of subpallidal lineage using Gsx2-Cre causes hyperlocomotion during early adulthood and aging-dependent neuronal loss (Mehler et al., 2019). However, Gsx2-Cre is not specific for SPNs; all progenitors from the lateral ganglionic eminence are targeted, including oligodendrocytes and interneurons.
Synaptic dysfunction in the cortico-striatal axis is an early event in HD and likely an important trigger for HD pathology (Unschuld et al., 2012). Furthermore, HTT expression is higher in cortical pyramidal neurons rather than the SPNs, where it functions to control synapse development (McKinstry et al., 2014; Fusco et al., 1999). Therefore, it is possible that cortical D1-Cre expression may contribute to the severity of phenotypes of DP-cKO mice. HTT loss in D1-Cre+ cortical neurons could alter their functional connectivity onto SPNs, which may contribute to synaptic circuit and/or behavioral phenotypes of DP mice and exacerbate the neuronal health issues caused by HTT loss in DP-SPNs.
The toxic gain-of-function and dominant-negative loss-of-function mechanisms of mHTT are not mutually exclusive, and both mechanisms likely contribute to SPN degeneration and other aspects of HD pathology. mHTT, especially in the soluble form, is toxic and can accelerate the rate of cell death (Arrasate et al., 2004). However, overexpressing wild-type HTT in cells also expressing mHtt slows cell death, suggesting a neuroprotective role for HTT (Leavitt et al., 2006). Therefore, it is likely that synaptic dysfunction in cortico-striatal circuits and cellular stress from protein aggregation synergize with the loss-of-function-like effects of mHTT in SPNs to produce the complex HD phenotypes.
Implications for HD Therapeutic Strategies
Our findings have significant implications for HD therapeutics, because many strategies seeking to cure HD aim to silence Htt expression. One such therapeutic currently in clinical trials (IONIS-HTTRx, ClinicalTrials.org: NCT03342053) employs an intrathecal antisense oligonucleotide to non-selectively target the human HTT gene and reduce HTT expression in HD mutation carriers (Rodrigues and Wild, 2018). Although several studies in rodent and non-human primate models have shown that Htt knockdown in adult animals is not harmful (Wang et al., 2016a; McBride et al., 2011; Boudreau et al., 2009), one study found that Htt deletion in the adult mouse leads to brain atrophy, extensive reactive gliosis, and progressive motor symptoms (Dietrich et al., 2017). Our findings show that wild-type HTT plays an important role in both IP-SPNs and DP-SPNs, and preserving this function is critical for maintaining striatal health and function. As such, therapeutic strategies that selectively reduce the expression of mHTT (e.g., Li et al., 2019) are needed to provide the greatest benefit to HD patients.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Cagla Eroglu (cagla.eroglu@duke.edu). This study did not generate new unique reagents. All stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In Vivo Experiments
To conditionally inactivate the Huntingtin (Htt) gene, we used previously described alleles of Htt: afloxed allele Htttm2Szi (hereafter will be referred to as Httflox,) and a null allele (Htt−) (Dragatsis et al., 2000). To conditionally silence Htt in IP-SPNs, we utilized the Adenosine A2a receptor (Adora2a) Cre mouse line (hereafter, A2A-Cre(Tg/Tg) mice) developed by Nathaniel Heintz and Charles Gerfen (a kind gift from Dr. Marc Caron of Duke University). We chose this Cre line because it has been shown to successfully induce recombination specifically in IP-SPNs, and it avoids the wider expression profile of Cre lines that are based on the D2 receptor promoter (Gerfen et al., 2013). To identify Cre-expressing cells, we crossed the Httflox mice to the Gt(ROSA)26Sortm2(CAa-TdTomato>Fawa mouse line (a kind gift from Dr. Fan Wang of Duke University) that expresses TdTomato upon Cre recombination (hereafter, TdTomatoflox). Experimental breeding pairs were as follows: Htt(+/−);A2A-cre(rg/Tg x Htt(flox/flox);TdTomato(flox/flox). Control mice were Htt(flox/+);A2A-Cre(Tg/0);TdTomato(flox/0), and IP-SPN Htt conditional deletion mice (IP-cKOs) were Htt(flox/−);A2A-Cre(Tg/Tg);TdTomato(flox/0). To conditionally silence Htt in the DP-SPNs, we utilized the Dopamine Receptor Drd1-Cre mouse line (hereafter, D1-Cre(Tg/Tg) mice). This Cre mouse line has been successfully used to recombine alleles in DP-SPNs (Gerfen et al., 2013). Experimental breeding pairs were as follows: Htt(+/−);D1-cre(rg/0) x Htt(flox/flox);TdTomato(flox/flox). Control mice were Htt(flox/+);D1-Cre(Tg/0);TdTomato(flox/0), and DP-SPN conditional deletion mice (DP-cKOs) were Htt(flox/−;D1-Cre(Tg/0);TdTomato(flox/0). All the mice used (IP-Control, IP-cKO, DP-Control, and DP-cKO) have a mixed C57Bl6/129 background. The age of the mice used for each experiment is reported in the Results section and related figure captions. Mice of both sexes were utilized for all experiments.
All experiments involving mice were conducted in accordance with the guidelines set forth by the National Institute of Health’s Guide for the Care and Use of Laboratory Animals. All protocols were approved in advance of their implementation by Duke University’s Institutional Animal Care and Use Committee (Assurance number A3195–01). Mice were group-housed (N = 2–5 mice per cage) in standard housing conditions with a 12-hour light/dark cycle. All efforts were made to minimize distress and suffering endured by the mice during the procedures performed for this study. For tissue collection, mice were deeply anesthetized with 200 mg/kg Avertin and after brain death was confirmed, were intracardially perfused with Tris-Buffered Saline (TBS, 25 mM Tris-base, 135 mM NaCl, 3 mM KCl, pH 7.6) supplemented with 7.5 μM heparin, followed with 4% paraformaldehyde (PFA) in TBS.
For viral injections, mice were anesthetized with 3L/min Isoflurane and small craniotomies were made over the injection sites. 0.4 μL of virus was delivered unilaterally to dorsolateral striatum via a Nanoject II (Drummond Scientific) at a rate of 0.1 μL/min. The injection pipette was held in place for 10 minutes following injection and then slowly removed. Mice were closely monitored postoperatively and were allowed ~3 weeks recovery after viral injection before additional experiments were performed.
METHOD DETAILS
Flow Cytometry and qRT-PCR
TdTomato+ and TdTomato− cells were flow sorted from 2-m/o IP-Control, IP-cKO, DP-Control, and DP-cKO mice, similar to previously described methods (Stogsdill et al., 2017). Briefly, animals were anesthetized by intraperitoneal injection of 200 mg/kg avertin, decapitated after brain death, and brains resected in DPBS (Thermo-Fisher). Striata were quickly microdissected under a dissection scope and chopped into < 1mm3 chunks. Striata were digested with papain (Worthington; 7.5U/mL) with DNaseI diluted in DPBS for 45min at 33–34°C. Digested tissues were triturated in 5 mL DPBS with a P1000 pipette and centrifuged at 900 g for 5 minutes at room temperature. The remaining pellet was triturated in 8 mL lo-ovomucoid (Worthington) with a P1000 pipette to create a single-cell suspension and then centrifuged at 1100 g for 10 min at room temperature. The pelleted cells were resuspended in 2–4 mL panning buffer (DPBS with BSA and insulin) and passed through a 40 μm filter to strain out large clumps of cells. Cells were kept on ice until sorting. Cell sorting was performed on a BD DiVa sorter (BD Biosciences) to separate TdTomato+ and TdTomato− cells. Cells were sorted directly into RNeasy Mini Kit Lysis Buffer RLT (QIAGEN).
Total RNA was isolated from flow cytometry-sorted TdTomato+ and TdTomato− cells. The RNeasy Mini Kit (QIAGEN) was used to extract RNA, following manufacturer instructions. RNA concentration was estimated using a NanoDrop 2000 (Thermo Scientific). Extracted RNA was reverse transcribed with the High Capacity cDNA Synthesis Kit (Applied Biosciences) to create cDNA per manufacturer’s protocol. qPCR was performed with equal volumes of cDNA from IP-Control, IP-cKO, DP-Control, and DP-cKO samples. cDNA from each sample was mixed with Power SYBR Green PCR Master Mix (Applied Biosystems), and exon-skipping primers (see the complete list of primers below) from IDT-DNA. All samples were run in triplicate on the ABI 7300 cycler (Applied Biosystems) for each primer set. A no-cDNA sample (water only) served as a negative control. To ensure the integrity and specificity of each primer set, an R2 correlation based upon a serial dilation (1:1,1:10 and 1:100) of the pooled cDNA samples was generated for each set. For all primer sets used, the R2 value fell between 0.90 and 0.99. qPCR products were also run on a 2% TBE + ethidium bromide gel and all produced a single band of the predicted sequence size. After running the qPCR reaction, the average transcript Ct value obtained for each sample was normalized to the Ct of its own GAPDH level (ΔCt). For comparison of transcript expression between genotypes, ΔCt values were normalized to the relevant control levels (which was set to 1). Unpaired two-tailed t testing was then used to compare transcript expression between the genotypes.
Sequences of Primers Used for qRT-PCR (5′- > 3′):
HTT Forward: CAGGTCCGGCAGAGGAAC HTT Reverse: CATAGCGATGCCCAAGAGTT pENK Forward: GTTGTCTCCCGTTCCCAGTA pENK Reverse: GACAGCAGCAAACAGGATGA Tac1 Forward: TCGATGCCAACGATGATCTA Tac1 Reverse: AGCCTTTAACAGGGCCACTT DARPP-32 Forward: CCCAAAGTCGAAGAGACCCA DARPP-32 Reverse: CCGAAGCTCCCCTAACTCATC
Western Blot
Brains from 2-m/o IP-Control, IP-cKO, DP-Control, and DP-cKO mice were isolated after mice were anesthetized with 200 mg/kg Avertin and perfused intracardially with Tris-Buffered Saline (TBS, 25 mM Tris-base, 135 mM NaCl, 3 mM KCl, pH 7.6) supplemented with 7.5 μM heparin. The cortex and striata of each brain were dissected and flash-frozen in liquid nitrogen. Frozen tissue was homogenized in RIPA buffer (Thermo-Fisher) with cOmplete protease inhibitor (Roche) + 2mM sodium orthovanadate + 10 mM sodium fluoride. Tissue was dounced 10–15 times on ice and the liquid homogenate was transferred to a chilled Eppendorf tube. The homogenate was rocked at 4°C for 20 minutes, then centrifuged at 15,000 rpm for 10 minutes at 4°C. The resulting supernatant was decanted and protein concentration was measured using the micro BCA protein assay kit according to manufacturer’s protocol (Thermo-Fisher). The samples were then mixed with 2xLaemmli buffer + beta-mercaptoethanol (1:200) and were warmed at 70°C for 10 minutes. For Western Blotting for HTT protein, 50 μg of total protein/well was loaded into 4%–15% stain-free polyacrylamide gels (BioRad) and resolved by SDS-PAGE. A BioRad Imager was then used to activate the gel and total adjusted protein expression per lane was measured using ImageLab Software (BioRad). Protein was then transferred onto an Immobilin-FL PVDF membrane (Millipore) which had been activated by a 5-minute methanol wash. Blots were rinsed in PBS containing 0.01 % Tween-20 and then blocked in 50% Odyssey blocking buffer (Li-Cor) in PBS for one hour at room temperature before incubating with primary antibody (1:1000 mouse anti-HTT, Millipore 2166) in blocking buffer overnight at 4°C. Fluorescence-conjugated secondary antibodies (Li-Cor) were diluted (1:5000) in 50% Odyssey blocking buffer (Li-Cor) in PBS and western blots were incubated with secondary antibodies at room temperature for 2 hours. Detection was performed using the Li-Cor Odyssey System. Tissue from 3–4 mice per genotype was used. The intensities of protein bands were quantified using ImageStudioLite Software. HTT band intensities in each lane were normalized to the total adjusted protein per lane. The relative intensities of cKO lysates were normalized to the relevant control lysates. Statistical differences in protein levels between genotypes were assessed using unpaired two-way t testing.
Immunohistochemistry
For immunohistochemical studies (IHC), mice were perfused intracardially with Tris-Buffered Saline (TBS, 25 mM Tris-base, 135 mM NaCl, 3 mM KCl, pH 7.6) supplemented with 7.5 mM heparin, followed with 4% paraformaldehyde (PFA) in TBS. The brains were dissected and fixed overnight in 4% PFA in TBS at 4°C. The following day, brains were rinsed 3 times with TBS and then cryoprotected with 30% sucrose in TBS at 4°C. After cryoprotection, brains were embedded in a 2:1 mixture of 30% sucrose in TBS:OCT (Tissue-Tek). Brains were cryosectioned using a Leica CM3050S cryostat.
The following procedure was utilized for IHC studies using anti-RFP, anti-CTIP, anti-pENK, anti-DARPP-32, anti-VGAT, anti-Gephryin, anti-CD68, anti-TdTomato, anti-IBA-1, anti-SOX9, and anti-LaminB1. Free-floating sections were washed 3 times for 10 minutes with TBS with 0.2% Triton X-100 (Roche) and blocked in 10% Normal Goat Serum (NGS; Jackson ImmunoResearch) with 0.2% Triton X-100 in TBS for 1 hour at room temperature. Primary antibodies (see table below) were diluted in 5% NGS in TBS with 0.2% Triton X-100. Sections were incubated overnight at 4°C with primary antibodies and washed three times for 10 minutes with TBS the following morning. Secondary Alexa-fluorophore conjugated antibodies (Invitrogen) were diluted (1:300) in 5% NGS in TBS with 0.2% Triton X-100, and sections were incubated with secondary antibodies for 2 hours at room temperature, protected from light. After incubation, sections were washed three times for 10 minutes in TBS and mounted with Vectashield with DAPI (Vector Laboratories). Images were acquired on confocal laser-scanning microscopes (Leica SP8, Zeiss LSM 710, or Olympus Fluoview 3000). The microscope used for each experiment was selected based on experimental need and only one microscope was used for each experiment to ensure consistency.
Primary Antibodies used for Immunohistochemistry
| Antibody Name | Vendor/Catalog Number | Dilution for IHC |
|---|---|---|
| Rabbit anti-RFP | Rockland Immunochemicals 600–401-379 | 1:2000 |
| Rat anti-DARPP-32 | R&D MAB4320 | 1:1000 |
| Guinea pig anti-VGAT | Synaptic Systems 131004 | 1:1000 |
| Rabbit anti-Gephryin | Synaptic Systems 147002 | 1:1000 |
| Rat anti-CD68 | 3ioLegend 137002 | 1:1000 |
| Chicken anti-IBA1 | Synaptic Systems 234006 | 1:1000 |
| Rabbit anti-TdTomato | Kerafast EST203 | 1:1500 |
| Rabbit anti-LaminB1 | ProteinTech 12987–1-AP | 1:1000 |
| DAPI | ThermoFisher D1306 | 1:10,000 |
| Rabbit anti-pENK | Invitrogen 30928 | 1:1000 |
| Rat anti-CTIP2 | Abcam ab18465 | 1:1000 |
Cell Number Quantifications
SPN Number Quantification:
Following 4% PFA perfusion and sucrose cryoprotection as described above, brains were cryosectioned into 20 μm coronal slices beginning at the anterior striatum and proceeding through the posterior striatum (Bregma 1.345mm to −0.755mm). Sections were stained with rabbit anti-RFP and rat-DARPP-32, as described in detail above. For each mouse, three brain sections were chosen for analysis: one containing the anterior striatum (~Bregma 1mm), one containing the medial striatum (~Bregma 0.0mm), and one containing the posterior striatum (~Bregma −0.5mm). For each of the three selected brain sections, six images were acquired at various preselected points along the dorsal-ventral axis of the striatum. The dorsal striatum was identified by its anatomical relationship to the corpus callosum in the coronal plane. Once the dorsal striatum was identified, the coordinates of the Zeiss LSM-710 automated microscope stage were set to x = 0 and y = 0 and an image was acquired. The automated stage was then moved 200 μm in the x-direction with no y-displacement to take a second image in the dorsal region. For images of the medial striatum, the stage was moved ventrally (450 μm in the y-direction) and two images were acquired with an x-displacement of 200 μm. For images of the ventral striatum, a y-displacement of 250 μm from the medial striatum was used and a 150 μm x-displacement was employed between images. Z stack images spanning 8.64 μm were acquired using the 20x dry objective (1.08 μm between optical slices, 8 optical slices total). Maximum intensity Z-projections were created using the Max-Z Projection plugin in ImageJ. DARPP-32 fluorescence was used to identify SPNs. Cre-expressing SPNs were identified by expression of TdTomato. Comprehensive counting of all TdTomato+ and DARPP-32+ SPNs was performed on all acquired images using the Cell Counter plugin in ImageJ. The experimenter was blind to genotype during image acquisition as well as during cell counting. Three animals/genotype/age were analyzed. Data was analyzed with a one-way ANOVA followed by Sidak’s multiple comparisons test.
Cortical Neuron Quantification:
20 μm sagittal slices containing the M1 motor cortex of 2- and 10-m/o DP-Control and DP-cKO were stained for TdTomato (RFP) and DARPP-32, as described in detail above. The 20x dry objective of an Olympus FluoView 3000 confocal microscope was used to acquire and stitch three tile scan images per mouse of the M1 motor cortex spanning from the pia to the dorsal striatum. ImageJ was used to create maximum z-projections of the stitched tile scans. ROIs of Layer 2/3, Layer 4, and Layer 5&6 were created for each image, and the area of each ROI was recorded. The ImageJ Analyze Particles Plugin was used to quantify the number of TdTomato+ and NeuN+ in each image, and the accuracy of this method was verified by hand-counting using the Cell Counter Plugin. The experimenter was blind to genotype during image acquisition as well as during cell counting. One-way ANOVA with Sidak’s multiple comparisons test was used to test for statistical differences between the proportion of TdTomato+/NeuN+ cortical neurons between genotypes.
Quantification of Astrocytes and Microglia Numbers: Sections containing the striatum were stained for either IBA-1 or SOX9 (see Primary Antibodies Table above). Confocal z stacks of the dorsal striatum were acquired using the 20x dry objective on an Olympus FluoView 3000 microscope. A minimum of four images were acquired per mouse, and at least three mice were analyzed per genotype. All cell bodies of microglia and astrocytes were counted in each image using ImageJ’s Cell Counter Plugin. The experimenter was blind to genotype during image acquisition as well as during counting. Unpaired two-way t testing was used to determine statistical difference between genotypes.
Fluorescence Intensity Quantifications
For each experiment involving quantification of immunohistochemical fluorescent intensity, confocal z stacks of the striatum were acquired using either the 20x dry or the 60x oil objective on an Olympus FluoView 3000 microscope holding gain and laser power constant across all images. Max-Z projections were created using ImageJ and ROIs were drawn around cells. The area and integrated density of the fluorescent signal within each ROI was measured and recorded, with experimenter blinded to genotype. Fluorescence intensity was calculated as integrated density/ROI area. Statistical analysis was conducted using one-way ANOVA with Sidak’s multiple comparisons test on the average of all integrated density/ROI area measurements per image.
Nuclear Morphological Analysis
20 μm sagittal sections containing the striatum were stained for RFP, LaminB1, and DAPI (see primary antibody table above). Confocal z stacks of the striatum were acquired using the 60x oil objective on an Olympus FluoView 3000 microscope, with 3 images acquired per mouse brain. An experimenter blind to genotype analyzed the nuclei of all RFP+ SPNs within an image and identified each nucleus as either “normal” or “abnormal.” Nuclei were recorded as “abnormal” if they contained at least one or more of the following features: a single large membrane invagination, a “hole” within the DAPI staining, or significant membrane rippling/jaggedness. The TdTomato-nuclei (IP-SPNs of DP animals, and DP-SPNs of IP animals) were also assessed in the same fashion. The proportion of abnormal nuclei was computed, and genotype differences were determined after unblinding using a one-way ANOVA with Sidak’s test for multiple comparisons. At least 3 2-m/o mice were used per genotype.
Behavior
Open Field Test :
Mice were placed individually into the Open Field arena (AccuScan Instruments, Columbus, OH) and spontaneous motor activity was monitored over 60 minutes as described (Pogorelov et al., 2005). Horizontal activity or locomotion was measured as the distance traveled and time spent moving within the entire open field. Time spent in the open field margins was also monitored. Stereotypical activity was measured as repetitive beam-breaks less than 1 s.
Rotarod Test :
Balance and coordination were evaluated on the accelerating (4–40 rpm) rotarod (Med-Associates, St. Albans, VT) as described (Wang et al., 2016b). Mice were tested over 4 successive 5-minute trials which were separated by 30-minute inter-trial intervals. Trials were terminated at 300 s, or when the mouse fell from the rod. Latency to fall was recorded for each mouse.
GABAergic Synapse Quantification
20 μm sagittal brain sections were stained with pre- (VGAT) and post-synaptic (gephyrin) marker pairs adapted from the protocols described in Ippolito and Eroglu (2010) (3–4 animals/genotype). Sections were selected based on anatomical features to contain the brain region of interest (i.e., GPe or SNR). 5.1 μm-thick confocal images (optical section depth 0.34 μm, 15 sections/scan, imaged area/scan = 13514 μm2) were acquired at 60x magnification on an Olympus FluoView 3000 microscope within the anatomical region of interest. Maximum projections of 3 consecutive optical sections (corresponding to 1.02 μm total depth) were generated using ImageJ. The Puncta Analyzer Plugin (available upon request; c.eroglu@cellbio.duke.edu) for ImageJ was used to count the number of colocalized synaptic puncta. At least 5 maximum projections per brain, from 2–4 brain sections per animal, were analyzed using a Nested ANOVA. Rotation analysis was used for each GABAergic synapse quantification experiment to verify that colocalized puncta (i.e., synapses) were not due to random chance. For these analyses, the gephyrin channel for each image was rotated 90 degrees using ImageJ and then was re-merged with the non-rotated VGAT channel. The Puncta Analyzer Plugin was used to quantify the number of VGAT, gephyrin, and colocalized puncta of each image. Puncta numbers of the rotated images were normalized to puncta numbers of the original, non-rotated images. One-way ANOVA was used to assess differences between the number of puncta counted in the rotated versus the original images.
Electrophysiology
Miniature Inhibitory Postsynaptic Currents:
For whole-cell patch-clamp recordings, brains from mice (N = 3–5 per condition) were removed quickly into ice-cold solution bubbled with 95% O2%–5% CO2 containing the following (in mM): sucrose (194), NaCl (30), KCl (2.5), MgCl2 (1), NaHCO3 (26), NaH2PO4 (1.2), and D-glucose (10). After 3 – 4 minutes the brains were blocked and sagittal slices were taken at 250 μm. During the recovery period (30 minutes) the slices were kept at 35.5°C with oxygenated artificial cerebrospinal fluid (aCSF) solution containing the following (in mM): NaCl (124), KCl (2.5), CaCl2 (2), MgCl2 (1), NaHCO3 (26), NaH2PO4 (1.2), and D-glucose (10). Internal solution for the pipette (3–5 MΩ) contained (in mM): CsCl (130), potassium gluconate (30), CaCl2 (0.1), EGTA (1), HEPES (10), magnesium ATP (2), and sodium GTP (0.2) with pH adjusted to 7.2 with KOH and osmolarity set to 300 mosM. All recordings were performed with the MultiClamp 700B amplifier (Molecular Device). Signals were filtered at 10 kHz and digitized at 20 kHz with the Digidata 1440A digitizer (Molecular Device). During recording, slices were maintained under continuous perfusion of aCSF at 28 – 29°C with 2–3 mL/min flow rate. In the whole-cell configuration (series resistance < 20 MΩ), we recorded miniature IPSCs (mIPSCs) on the cell bodies of GPe neurons with 1 μM TTX, 50 μM APV, and 50 μM DNQX in the bath solution in voltage-clamp mode (cells held at −70 mv). The amplitudes and frequencies of mIPSCs were analyzed using peak detection software in pCLAMP10 (Molecular Devices).
Optogenetics:
Ten 2-m/o mice were used in the optogenetic experiments (DP-Control, n = 5; DP-cKO, n = 5). Adeno-associated viral vectors were used for Cre-dependent expression of the excitatory channelrhodopsin (pAAV-EFIa-DIO-hChR2(H134R)-EYFP, Duke Viral Core, titer >1 × 1012 particles/mL). 0.4 μL of virus was delivered unilaterally via a Nanoject II (Drummond Scientific) at a rate of 0.1 μL/min to the following coordinates relative to Bregma: AP: + 0.4 mm, ML: − 2.0 mm, DV: 2.0 mm. Slices were generated for paired-pulse experiments ~3 weeks after viral injection. Slices were stimulated with 470-nm light generated from an LED assembly (Thor Labs) focused through a × 40 objective (Olympus). During recordings, paired light flashes (1 ms) were delivered at 50, 100, and 150 ms interval with an LED current driver (Thor Labs). Power density was ~5 mW/mm2.
Microglial Analysis using Imaris Software
20 μm sagittal brain sections containing the striatum of 10-m/o DP-Control and DP-cKO mice were stained for IBA-1 and CD68 (see primary antibodies table). Confocal z stacks of the dorsal striatum were acquired using the 60x oil objective on an Olympus FluoView 3000 microscope. Imaris Software 9.0.0 was used to create surface reconstructions of all whole, individual microglia within each image. A surface reconstruction was also generated for CD68 localized specifically within the microglial cell reconstruction. The average microglia volume and average CD68 volume per microglial cell were calculated per image and unpaired two-way t testing was used to query differences between genotypes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses of the quantified data were done using the appropriate statistical test for each kind of analyses. Details for each can be found above within the sections describing each individual method. The names of the tests and the number of mice used per analysis can also be found in the Results section and/or within the figure captions. Statistica (StatSoft, OK) and Prism7 (GraphPad Software) were used for all statistical analyses. Unless otherwise specifically stated, all data are expressed as mean ± SEM and significance is defined as p < 0.05. The exact statistics and p values are given for each analysis within the text.
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-RFP | Rockland Immunochemicals | Cat#600–401-379; RRID: AB_2209751 |
| Rat anti-DARPP-32 | R&D Systems | Cat#MAB4230; RRID: AB_2169021 |
| Guinea pig anti-VGAT | Synaptic Systems | Cat#131004; RRID: AB_887873 |
| Chicken anti-Iba-1 | Synaptic Systems | Cat#234006; RRID: AB_2619949 |
| Rabbit anti-TdTomato | Kerafast | Cat#EST203; RRID: AB_2732803 |
| Rabbit anti-LaminB1 | ProteinTech | Cat#12987–1-AP; RRID: AB_2136290 |
| Rabbit anti-pENK | ThermoFisher Scientific | Cat#30928; RRID: AB_1958811 |
| Rabbit anti-CTIP2 | Abcam | Cat# ab18465; RRID: AB_2064130 |
| DAPI | ThermoFisher Scientific | Cat#D1306; RRID: AB_2629482 |
| Rabbit anti-Gephryin | Synaptic Systems | Cat#147002; RRID: AB_2619838 |
| Rat anti-CD68 | BioLegend | Cat#137002; RRID: AB_2044004 |
| Critical Commercial Assays | ||
| RNeasy Mini RNA Isolation Kit | QIAGEN | 74134 |
| High Capacity cDNA Reverse Transcription Kit | ThermoFisher Scientific | 4368814 |
| Experimental Models: Organisms/Strains | ||
| D1-Cre(Tg/0) Htt(f/+) RTM(f/+) DP-Control Mice | This Paper | N/A |
| D1-Cre(Tg/0) Htt(f/-) RTM(f/+) DP-cKO Mice | This Paper | N/A |
| A2A-Cre(Tg/0) Htt(f/+) RTM(f/+) IP-Control Mice | This Paper | N/A |
| A2A-Cre(Tg/0) Htt(f/-) RTM(f/+) IP-cKO Mice | This Paper | N/A |
| A2A-Cre(Tg/Tg) Mice | Gift from Caron Laboratory, Duke University; originally a gift from Gerfen Laboratory, NIMH | MGI: 5051776 |
| D1-Cre(Tg/Tg) Mice | Jackson Laboratory | MGI: 3836633 |
| RTM(f/f) Mouse: Gt(ROSA)26Sortm2(CAG-TdTomato)Fawa | Gift from Wang Laboratory, Duke University | MGI_ MGI: 5305341 |
| Htt(f/f) Mice: Htttm2Szi | Gift from Zeitlin Laboratory, University of Virginia (Dragatsis et al., 2000). | MGI: 2177755 |
| Oligonucleotides | ||
| Htt Forward Primer: CAGGTCCGGCAGAGGAAC | This Paper | N/A |
| Htt Reverse Primer: CATAGCGATGCCCAAGAGTT | This Paper | N/A |
| pENK Forward Primer: GTTGTCTCCCGTTCCCAGTA | This Paper | N/A |
| pENK Reverse Primer: GACAGCAGCAAACAGGATGA | This Paper | N/A |
| Tac1 Forward Primer: TCGATGCCAACGATGATCTA | This Paper | N/A |
| Tac1 Reverse Primer: AGCCTTTAACAGGGCCACTT | This Paper | N/A |
| DARPP-32 Forward Primer: CCCAAAGTCGAAGAGACCCA | This Paper | N/A |
| DARPP-32 Reverse Primer: CCGAAGCTCCCCTAACTCATC | This Paper | N/A |
| Software and Algorithms | ||
| Statistica | StatSoft | http://www.statsoft.com/Produ cts/STATISTICA-Features |
| Prism7 | GraphPad | https://www.graphpad.com/scientiflc-software/prism/ |
| Imaris 9.0.0 | Oxford Instruments | https://imaris.oxinst.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
Highlights.
Loss of Htt in striatal neurons recapitulates key features of Huntington’s disease
Striatal neurons require Htt for survival during aging
Deletion of Htt from striatal neurons disrupts synaptic connectivity
ACKNOWLEDGMENTS
This work was supported by the NIH (grant RO1 NS096352 to C.E., grant RO1 NS090914 to S.Z., grant RO1 NS094754 to H.H.Y., and grant F31 NRSA 106777-01 to C.J.B.), a Duke Chancellor’s Discovery Award (to C.E. and H.H.Y.), and a Ruth K. Broad Biomedical Research Foundation’s Research Award for Graduate Students (to C.J.B. and S.U.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.069.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abraham MC, Lu Y, and Shaham S (2007). A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev. Cell 72, 73–86. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, and Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 72, 366–375. [DOI] [PubMed] [Google Scholar]
- Alexander GE, and Crutcher MD (1990). Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 73, 266–271. [DOI] [PubMed] [Google Scholar]
- Ambrose CM, Duyao MP, Barnes G, Bates GP, Lin CS, Srinidhi J, Baxendale S, Hummerich H, Lehrach H, Altherr M, et al. (1994). Structure and expression of the Huntington’s disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat. Cell Mol. Genet. 20, 27–38. [DOI] [PubMed] [Google Scholar]
- Ament SA, Pearl JR, Grindeland A, St Claire J, Earls JC, Kovalenko M, Gillis T, Mysore J, Gusella JF, Lee JM, et al. (2017). High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum. Mol. Genet. 26, 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, and Finkbeiner S (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 437, 805–810. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ, Fisone G, Nestler EJ, and Greengard P (2010). Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc. Natl. Acad. Sci. USA 107, 14845–14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, Clark TW, Bouzou B, and Cha JH (2008). Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 28, 10720–10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, and Greengard P (2000). Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. USA 97, 6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, and Davidson BL (2009). Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol.Ther. 17, 1053–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, and Tartari M (2005). Normal huntingtin function: an alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 6, 919–930. [DOI] [PubMed] [Google Scholar]
- Caviston JP, Ross JL, Antony SM, Tokito M, and Holzbaur EL (2007). Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 104, 10045–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, de Carvalho FD, Chohan MO, Shegda M, Chuhma N, Rayport S, Ahmari SE, Moore H, and Kellendonk C (2014). Dopamine D2 receptors regulate the anatomical and functional balance of basal ganglia circuitry. Neuron 81, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, and Bates GP (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. [DOI] [PubMed] [Google Scholar]
- Deng YP, Albin RL, Penney JB, Young AB, Anderson KD, and Reiner A (2004). Differential loss of striatal projection systems in Huntington’s disease: a quantitative immunohistochemical study. J. Chem. Neuroanat. 27, 143–164. [DOI] [PubMed] [Google Scholar]
- Deng YP, Wong T, Bricker-Anthony C, Deng B, and Reiner A (2013). Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol. Dis. 60, 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich P, Johnson IM, Alli S, and Dragatsis I (2017). Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 13, e1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, and Zeitlin S (2000). Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 26, 300–306. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Dietrich P, Ren H, Deng YP, Del Mar N, Wang HB, Johnson IM, Jones KR, and Reiner A (2018). Effect of early embryonic deletion of huntingtin from pyramidal neurons on the development and long-term survival of neurons in cerebral cortex and striatum. Neurobiol. Dis. 111,102–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux PF, Bearzatto B, Guiducci S, Buch T, Waisman A, Zoli M, Schiffmann SN, and de Kerchove d’Exaerde A (2009). D2R striatopallidal neurons inhibit both locomotor and drug reward processes. Nat. Neurosci. 12, 393–395. [DOI] [PubMed] [Google Scholar]
- Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M, et al. (1993). Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 4, 387–392. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, et al. (1995). Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269, 407–410. [DOI] [PubMed] [Google Scholar]
- Elias S, McGuire JR, Yu H, and Humbert S (2015). Huntingtin Is Required for Epithelial Polarity through RAB11A-Mediated Apical Trafficking of PAR3-aPKC. PLoS Biol. 13, e1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, et al. (2009). Gabapentin receptor a25–1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, and Reiner A (1999). Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington’s disease. J. Neurosci. 19, 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, and Saudou F (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, and Heintz N (2013). GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80, 1368–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F, Dragatsis I, et al. (2010). Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 67, 392–406. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, and Collins FS (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 101, 8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer JC, et al. (2017). Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 94, 93–107.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey M, and Chesselet M (2003). Apoptosis in Huntington’s disease. Prog. Neuropsychopharmacol. Biol. Psych. 27, 255–265. [DOI] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. (2006). Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 15, 965–977. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, et al. (1999). A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23, 181–192. [DOI] [PubMed] [Google Scholar]
- Ippolito DM, and Eroglu C (2010). Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J. Vis. Exp 45, 2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS (2019). Astrocyte-Neuron Interactions in the Striatum: Insights on Identity, Form, and Function. Trends Neurosci. 42, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, and Kreitzer AC (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutscher LM, and Shaham S (2017). Non-apoptotic cell death in animal development. Cell Death Differ. 24, 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. (2016). Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 19, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, and Hayden MR (2006). Wild-type huntingtin protects neurons from excitotoxicity. J. Neurochem. 96, 1121–1129. [DOI] [PubMed] [Google Scholar]
- Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T, Ma L, Gao C, Yang Y, Sun Y, et al. (2019). Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 575, 203–209. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, and Yang XW (2006). FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat. Neurosci. 9, 443–452. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, et al. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9, 1259–1271. [DOI] [PubMed] [Google Scholar]
- Macdonald M, Ambrose C, Duyao M, Myers R, Lin C, Srinidhi L, Barnes G, Taylor S, James M, Groot N, et al. ; The Huntington’s Disease Collaborative Research Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Mazarei G, Neal SJ, Becanovic K, Luthi-Carter R, Simpson EM, and Leavitt BR (2010). Expression analysis of novel striatal-enriched genes in Huntington disease. Hum. Mol. Genet. 19, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, and Davidson BL (2011). Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol.Ther. 19, 2152–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinstry SU, Karadeniz YB, Worthington AK, Hayrapetyan VY, Ozlu MI, Serafin-Molina K, Risher WC, Ustunkaya T, Dragatsis I, Zeitlin S, et al. (2014). Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. 34, 9455–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehler MF, Petronglo JR, Arteaga-Bracho EE, Gulinello ME, Winchester ML, Pichamoorthy N, Young SK, DeJesus CD, Ishtiaq H, Gokhan S, and Molero AE (2019). Loss-of-huntingtin in medial and lateral ganglionic lineages differentially disrupts regional interneuron and projection neuron subtypes and promotes Huntington’s disease-associated behavioral, cellular and pathological hallmarks. J. Neurosci. 39, 1892–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, and Raymond LA (2007). Corticostriatal synaptic function in mouse models of Huntington’s disease: early effects of huntingtin repeat length and protein load. J. Physiol. 585, 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, and Hayden MR (1995). Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81, 811–823. [DOI] [PubMed] [Google Scholar]
- Pogorelov VM, Rodriguiz RM, Insco ML, Caron MG, and Wetsel WC(2005). Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene. Neuropsychopharmacology 30, 1818–1831. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, and Young AB (1988). Differential loss ofstriatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. USA 85, 5733–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues FB, and Wild EJ (2018). Huntington’s Disease Clinical Trials Corner: February 2018. J. Huntingtons Dis. 7, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos RA, and Bots GT (1983). Nuclear membrane indentations in Huntington’s chorea. J. Neurol. Sci. 61, 37–47. [DOI] [PubMed] [Google Scholar]
- Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, David G, Sun Y, Stimming EF, Bellen HJ, et al. (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17, 262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, and DiFiglia M (2001). Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 60, 161–172. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong IS, Woda JM, Song JJ, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee JM, Wheeler VC, Walz T, et al. (2010). Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 19, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M (2008). Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150 Glued. J. Biol. Chem. 283, 34880–34886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, and Bolam JP (1998). Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 86, 353–387. [DOI] [PubMed] [Google Scholar]
- Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji RR, and Eroglu C (2017). Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 551, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Cornwell A, Li J, Peng S, Osorio MJ, Aalling N, Wang S, Benraiss A, Lou N, Goldman SA, and Nedergaard M (2017). SOX9 Is an Astrocyte-Specific Nuclear Marker in the Adult Brain Outside the Neurogenic Regions. J. Neurosci. 37, 4493–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Ha TJ, Liu L, Nishimoto A, Reiner A, and Goldowitz D (2011). Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J. Neurosci. 31, 14794–14799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unschuld PG, Joel SE, Liu X, Shanahan M, Margolis RL, Biglan KM, Bassett SS, Schretlen DJ, Redgrave GW, van Zijl PC, et al. (2012). Impaired corti-costriatal functional connectivity in prodromal Huntington’s Disease. Neurosci. Lett. 514, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan S, Major AJ, and Everling S (2017). Neuromodulation of Prefrontal Cortex in Non-Human Primates by Dopaminergic Receptors during Rule-Guided Flexible Behavior and Cognitive Control. Front. Neural Circuits 11,91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schimmelmann M, Feinberg PA, Sullivan JM, Ku SM, Badimon A, Duff MK, Wang Z, Lachmann A, Dewell S, Ma’ayan A, et al. (2016). Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 19, 1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, and Richardson EP Jr. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577. [DOI] [PubMed] [Google Scholar]
- Walker FO (2007). Huntington’s disease. Lancet 369, 218–228. [DOI] [PubMed] [Google Scholar]
- Wang G, Liu X, Gaertig MA, Li S, and Li XJ (2016a). Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc. Natl. Acad. Sci. USA 113, 3359–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, et al. (2016b). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7, 11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann M, Queisser G, Eder A, Wiegert JS, Bengtson CP, Hellwig A, Wittum G, and Bading H (2009). Synaptic activity induces dramatic changes in the geometry of the cell nucleus: interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J. Neurosci. 29, 14687–14700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai W, Jeong H, Cui L, Krainc D, and Tjian R (2005). In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell 123, 1241–1253. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang L, Jiao H, Zhang Q, Zhang D, Lou D, Katz JL, and Xu M (2006). c-Fos facilitates the acquisition and extinction of cocaine-induced persistent changes. J. Neurosci. 26, 13287–13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.