

Abstract
Vaccine development has progressed significantly and has moved from whole microorganisms to subunit vaccines that contain only their antigenic proteins. Subunit vaccines are often less immunogenic than whole pathogens; therefore, adjuvants must amplify the immune response, ideally establishing both innate and adaptive immunity. Incorporation of antigens into biomaterials, such as liposomes and polymers, can achieve a desired vaccine response. The physical properties of these platforms can be easily manipulated, thus allowing for controlled delivery of immunostimulatory factors and presentation of pathogen-associated molecular patterns (PAMPs) that are targeted to specific immune cells. Targeting antigen to immune cells via PAMP-modified biomaterials is a new strategy to control the subsequent development of immunity and, in turn, effective vaccination. Here, we review the recent advances in both immunology and biomaterial engineering that have brought particulate-based vaccines to reality.
Introduction
Vaccination has proven to be one of the greatest medical interventions. Vaccines educate and prime our innate and adaptive defenses, mounting an immune response that can effectively clear many pathological agents, including viruses and bacteria. The immune system works by first capturing these agents, subsequently amplifying a complex network of specialized cells that are adept at clearing pathogens, and ultimately creating an immunological memory of that agent in the event of future exposure. Clearly, such a powerful defense strategy relies on intricate interactions and timely functional responses that involve effective scanning, processing, differentiation and memory to discriminate self from invaders. These are hallmark features of successful immunity and the basis of the success of prophylactics that exploit the natural defenses of the body to eradicate disease. However, with viruses and pathogens that can actively evade immunity, or with conditions, such as cancer, that can subvert normal host defense responses, novel approaches to manipulate immune responses in a predictable and tunable fashion are urgently needed. In addition, the need for higher safety profiles has led research to move away from more traditional formulation strategies, such as live–attenuated and killed–inactivated preparations towards the use of subunit vaccines, which require the addition of substances, such as adjuvants, to enhance their efficacy, thus requiring new formulation strategies (Box 1).
Box 1. Currently licensed vaccine formulations.
Live attenuated
Attenuated vaccines are pathogens that have been repeatedly passaged in cell culture, often in a non-human cell line, until the virus or bacterium is no longer pathogenic. Some examples of attenuated vaccines include include measles, mumps, and rubella (MMR), and Bacille Calmette Guerin. A live attenuated vaccine is not an option against pathogens that have limited growth in culture, such as hepatitis C virus or HPV, which propagate poorly in cell systems [123,124]. Attenuated vaccines can be administered in a single dose by various routes of administration (oral, nasal, or intramuscular), and, because these vaccines closely resemble a natural infection, they are potent activators of antibody (i.e. humoral) and cell-mediated immune responses. Although attenuated vaccines generally succeed at generating a robust immune response, live attenuated vaccines might result in disease if administered to immunocompromised patients [125]. In some rare instances, attenuated vaccines revert to virulence [126].
Killed inactivated
Inactivation of live pathogens by heat or chemical treatment is a safer model for widespread vaccination than attenuated vaccines. Inactivated vaccines are typical for influenza, polio and hepatitis A viruses. Similar to attenuated vaccines, inactivated vaccines present many target antigens in the context of the microbe, providing signals for the immune system that would be present during infection. However, because the pathogen is unable to replicate or invade host cells, multiple doses, or ‘boosters’, must be administered, and the resultant immune response is generally in the form of neutralizing antibodies – not antigen-specific cytotoxic T lymphocytes (CTLs). That is, inactivated vaccines successfully elicit a humoral response yet, for the most part, not a cellular immune response. Additionally, as with attenuated vaccines, inactivated vaccines are limited to pathogens that can be cultured in vitro.
Pathogen fragments (subunit vaccines)
The final class of vaccines includes those for tetanus and diphtheria toxins and HPV. Subunit vaccines are fragments of pathogens, such as toxins, structural units, and other components, that are typically targets of neutralizing antibodies. However, antigens purified from pathogens often lack immunogenicity, experience poor uptake and processing rates by APCs, and generally exhibit short half-lives in vivo [127]. Thus, an adjuvant is included in vaccine formulations to augment the immune response to these antigens and to minimize dose.
The term ‘biomaterial vaccines’ is a general descriptor for artificial or natural substrates, such as synthetic polymers, lipids, peptides or other macromolecules, that can function as vaccine building blocks. These are attractive because they offer control over the physical and chemical properties of the material, thereby allowing the addition or subtraction of antigen, adjuvant, and target molecules for immune recognition (Box 2). The major advantage of such systems is that they can be ‘tuned’ to optimize the magnitude and direction of a vaccine response. Thus, unlike conventional adjuvants, such as alum, which historically has been shown to elicit strong humoral responses (high antibody titers), biomaterial particulate systems offer the potential to produce humoral and cellular immune responses. A strong T-cell-mediated or mucosal immune response is equally important for effective vaccination, and materials that can be tailored to induce cellular and humoral immunity via parenteral and mucosal routes are emerging as important tools for the development of new vaccines.
Box 2. Benefits of vaccine delivery vehicles.
Several groups are developing pathogen-mimicking vaccines using nano- and micro-scale delivery vehicles, such as liposomes and polymeric particles, by which antigens enter and activate APCs. Particulate systems might overcome limitations of clinically approved adjuvants because they:
Promote cell-mediated immunity
Typically, exogenous antigen, once endocytosed, is loaded on major histocompatibility complex (MHC) class II molecules and presented to CD4+ T cells. However, particles can be engineered to escape endosomal and lysosomal compartments after APC internalization [128,129] and subsequently enter the cytosol. Here, encapsulated antigen-peptide epitopes can access MHC class I molecules in a process known as cross-presentation, which promotes CD8+ T cell activation and elicits a robust cell-mediated response.
Are easily modulated
Pathogens constantly emerge and change; therefore, an ideal vaccine vehicle is easily modified to generate immune responses of the appropriate magnitude and direction against evolving pathogens. Synthetic delivery systems can be tailored to control antigen release rate [130,131], as well as the valency [130–133] and amount of antigen. Additionally, the circulation time of the vaccine vehicle can be controlled along with the type and density of the targeting ligand.
Minimize dose and eliminate booster immunizations
Vaccine particulates can display antigen in bulk, mimic physiological antigen presentation, and create much higher local antigen concentrations than soluble antigen. Additionally, encapsulated antigen is protected from degradation, which allows it to remain intact for interaction with APCs. This attribute, in combination with the ability of particulates to deliver antigen to specific cell types, allows for a significant decrease in the amount of antigen necessary per dose. Furthermore, many particulates release their antigen payload over a tunable period of time; this controlled release may be optimized to prime and boost immune cells in a single dose [134,135].
Enable mucosal administration
Particulate systems enable parenteral and mucosal administration because encapsulation and other functionalization strategies protect antigen from harsh conditions in the gastrointestinal tract and allow transport across mucous membranes [136–138]. Oral and intranasal vaccination methods are simple and painless and evoke higher patient compliance. Additionally, mucosal vaccination generates IgA protection at these tissues, which provides a first line of defense against many pathogens.
Well-suited for global distribution
Mucosal vaccination with particles can obviate the need for trained medical personnel, whereas single dose regimens are attractive for developing regions where many vaccine recipients cannot return for boosters. Many of these systems can be freeze dried and stored in a stable solid state, which eliminates the need for refrigeration and reduces storage and transportation costs.
Such biomaterials, typically particulates that have been classically used as drug delivery carriers, can combine the antigen of interest and the ligands that direct those antigens to antigen-presenting cells (APCs), such as B cells, macrophages and dendritic cells (DCs) (Figure 1a). Of the main types of APCs, DCs are unique in their ability to activate naïve T cells (Figure 1b) potently, thereby acting as the primary initiators of antigen-specific immune responses [1]. Therefore, the priming of DCs with an antigen effectively controls whether subsequent immunity will develop, and therefore, whether effective vaccination can be achieved. When possible, vaccine design strategy should take this principle into account.
Figure 1.
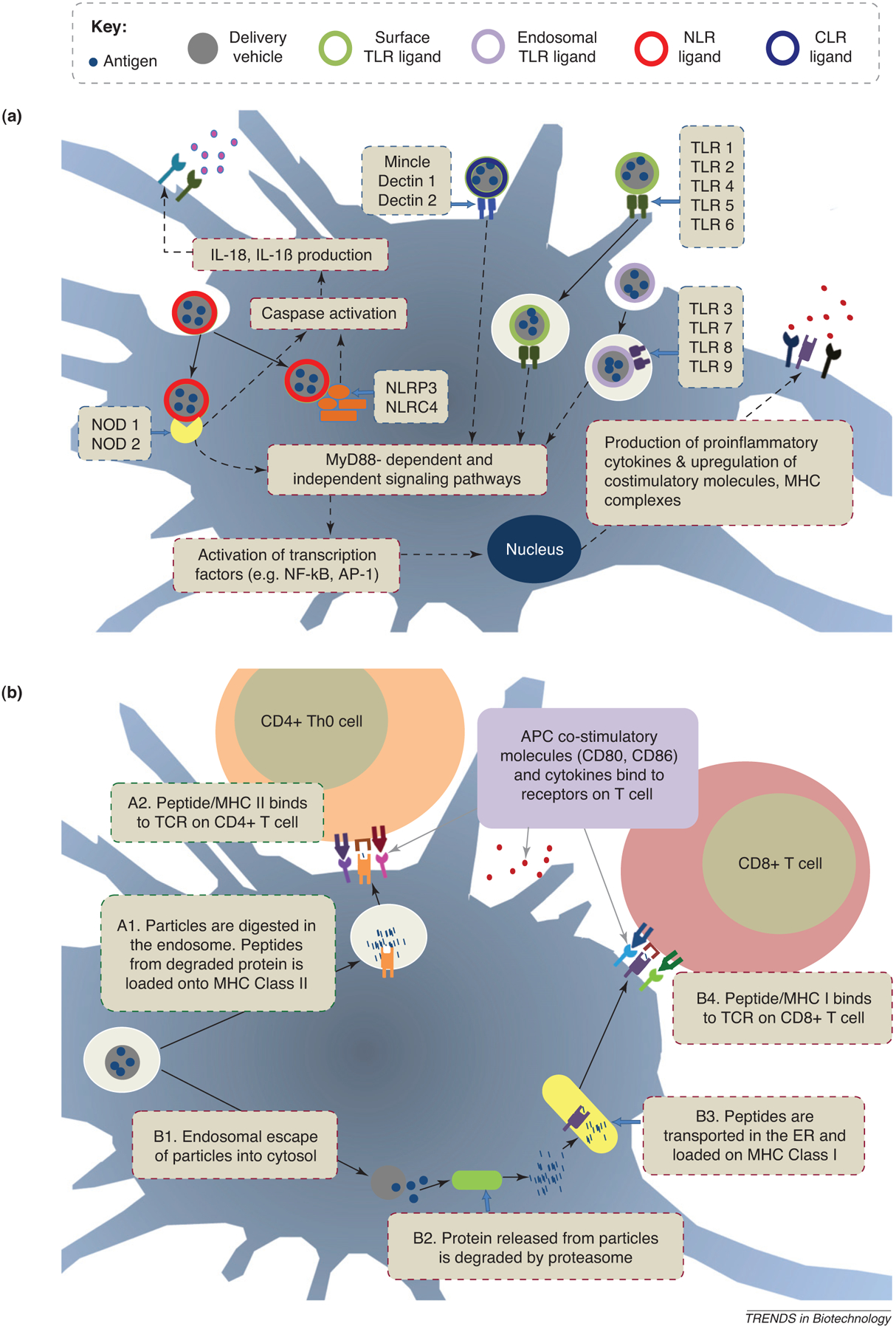
Particles with ligands that target TLRs, NLRs, or CLRs. (a) Signaling. Nano- or microparticles encapsulate antigenic components or incorporate agonists for surface or endosomal TLRs, intracellular NLRs, or membrane-bound CLRs. Particle components promote the production of proinflammatory cytokines and often the expression of co-stimulatory molecules and MHC complexes, thus leading to increased vaccine efficacy. (b) Antigen presentation. Once endocytosed, particles might be degraded in the endosome and access the MHC class II pathway, thereby activating CD4+ T cells (Pathway A). Alternatively, particles might escape the endosome so that encapsulated antigen can be processed in the cytosolic space and be presented via MHC class I to CD8+ T cells (Pathway B).
One relevant biological feature of DCs is their ability to sense conditions under which antigen is encountered, which initiates a process of DC maturation. Using receptors for microbial and inflammatory products, DCs respond to antigen exposure in different ways, depending on the nature of the pathogen encountered (e.g. virus, bacterium, or protozoan). Pattern recognition receptors (PRRs) are membrane-bound or secreted microbial sensory receptors that include Toll-like receptors (TLRs), Nod-like receptors (NLRs), and C-type lectin receptor (CLRs). PRRs on DCs recognize conserved molecular patterns on microbes known as pathogen-associated molecular patterns (PAMPs) (Figure 1a). This information is transmitted to T cells via altered cytokine release, expression of co-stimulatory molecules, such as CD40 and CD80, and the upregulation of integrins for adhesion, such as CD11c, by the DC at the time of antigen presentation in the lymph nodes (Figure 1b). By targeting DCs through their PRRs, novel vaccine biomaterials can quantitatively enhance the delivery of antigen and general immune responses, while qualitatively controlling the nature of the immune response (Table 1).
Table 1.
PRR-targeted particles for vaccination
| PRR | Ligand | Delivery vehicle | Antigen | Refs. |
|---|---|---|---|---|
| TLR 1/2 | Pam3CAG | Liposome | Hemagglutinin peptide | [49] |
| TLR 2/6 | Pam2CAG | [49] | ||
| TLR 3 | Poly (I:C) | PLGA MP | N/A | [53] |
| TLR 3 | Poly (I:C) | Polyketal MP | Ovalbumin | [54] |
| TLR 3 | Poly (I:C) | Liposome | Ovalbumin | [55] |
| TLR 4 | LPS | Liposome | HBsAg | [75] |
| TLR 4 | LPS | Gelatin MP | BSA | [78] |
| TLR 4/NLRP3 | LPS | PLGA NP | Ovalbumin, WNV envelope protein | [79] |
| TLR 4 | Lipid A | Liposome | Malaria antigen | [121] |
| TLR 4 | MPLA | Liposome (Stimuvax) | N/A | Merck |
| TLR 4 | MPLA | Liposome (AS01) | N/A | GSK |
| TLR 4 | MPLA | PLGA NP | MUC1 lipopeptide | [83] |
| TLR 4 | MPLA | PLGA NP | HBcAg | [84] |
| TLR 5 | Flagellin | Polyanhydride NP | Ovalbumin | [89] |
| TLR 5 | Flagellin | Polypropylene sulfide NP | Ovalbumin | [90] |
| TLR 7 | 3M019 | Liposome | Ovalbumin | [61] |
| TLR 9 | Plasmid DNA | Liposome | HBsAg | [65] |
| TLR 9 | CpG ODN | Liposome | Hemagglutinin, Neuraminidase, HBsAg | [68] |
| TLR 9 | CpG ODN | Liposome | Ovalbumin | [69,66] |
| TLR 9 | CpG ODN | Gelatin NP | N/A | [72] |
| TLR 9 | CpG ODN | PLGA MP | p55 gag, gp120 env (HIV-1) | [69] |
| TLR 9 | CpG ODN | Alginate-coated chitosan NP | HBsAg | [70] |
| TLR 9 | CpG ODN | PLGA NP | WNV envelope protein | [73] |
| Mincle | TDM | CAF01 | Mycobacterial antigens | [101] |
| Dectin-1 | β-Glucan | β-Glucan | Ovalbumin | [99] |
| NOD2 | MDP | Liposome | HBsAg | [122] |
| NOD2 | MDP | Liposome | Mycobacterial antigens | [117] |
| NOD2 | MDP | Gelatin MP | BSA | [78] |
Abbreviations: BSA, bovine serum albumin; HBsAg/HBcAg, Hepatitis B surface/core antigen; NP, nanoparticle; MP, microparticle; WNV, West Nile virus.
In this review, we discuss some of the most common biomaterial elements – liposomes, solid biodegradable polymers, and natural polymers – used in vaccine applications, starting with alum. We note that, in most cases, biomaterials used in these applications are particulate in nature. This is particularly interesting because viruses and pathogens that elicit or subvert immune responses are, in essence, small particles endowed with the ability to interact with or avoid immune cells in various ways; for example, vaccinia virus inhibits cytokines at the site of infection, and HIV targets immune cell surface molecules, such as DC-SIGN [2]. Much has been learned about their individual strategies, and this new biological information is currently guiding the design of novel, rational, biomaterial-based vaccine technologies. The types of known PRRs and their PAMP ligands are also reviewed, emphasizing what has been achieved with regard to engineering these interactions in biomaterial vaccine formulations. We conclude with a few perspectives regarding the future of vaccine design and how the emerging understanding of pathogen recognition and danger recognition signals that signal immune activation are influencing a paradigm shift in the design of biomaterials for vaccines.
Biomaterial elements: from the old to the new
Alum
Aluminum salts, which broadly include aluminum hydroxide, aluminum phosphate, and potassium aluminum sulfate (commonly referred to as alum), are the oldest adjuvants and have been used clinically for over 60 years. Antigens are adsorbed to or form a precipitate with aluminum salts, which elicit a long-lasting and robust humoral response when administered parenterally. Several vaccines, including diphtheria, tetanus, and pertussis (DTP), hepatitis A and B, anthrax, and rabies, all contain aluminum salts [3]. With the long history and extensive use of aluminum salts, it is surprising that only recently have some of the mechanisms behind the effects of aluminum adjuvants been elucidated. They are thought to work in part by a depot effect, which can persist at the injection site, and by stimulating a multiprotein complex called the inflammasome [4,5], which is responsible for the activation of inflammatory processes. The inflammasome is an intracellular complex of proteins composed of cytoplasmic PRRs, the apoptosis-associated speck-like (ASC) adaptor protein, and proinflammatory cytokine-releasing caspases. Alum administration has been shown to promote the release of the endogenous danger signal, uric acid, from necrotic cells, and has been implicated in inflammasome activation [6,7]. These studies have provided insight into the mechanisms behind the potency of alum as an adjuvant and further support the concept that combinations of signals (in this case a danger signal with an antigen) might be necessary for effective vaccination.
Liposomal carriers
Liposomes, first proposed as vaccine adjuvants over 35 years ago [8], are spherical vesicles formed by a hydrated phospholipidbilayer that surrounds an aqueous core. Since liposomes possess hydrophobic and hydrophilic compartments, they have been useful for the delivery of a wide array of candidate antigens and immunostimulatory molecules with different physiochemical properties. Liposomes can be prepared as unilamellar or multilamellar vesicles, and their size can be easily adjusted from tens of nanometers to several microns in diameter [9] – an important consideration for the intracellular delivery of an antigen or the creation of a vaccine depot. Although liposomes can vary widely in their preparation, they are mainly composed of amphiphilic lipids, typically neutral lipids like phosphatidylcholine (PC), and cholesterol. It is also important to note that the composition of the liposome itself can affect the immune response; therefore, lipids can function as adjuvants. For example, cationic liposomes have induced elevated CTL levels over anionic and neutral formulations [10]. Liposomes can be surface-modified by covalently coupling the phospholipid head groups to reactive groups on targeting molecules [11]. Liposomes can be prepared with materials that are very similar to the composition of mammalian cell membranes, and are therefore generally considered biocompatible and relatively non-toxic. One attractive feature is the ease of preparation under buffered conditions, which facilitates incorporation of proteins without denaturation; however, classical drawbacks have been poor control over levels of encapsulated protein and sustainable release of associated antigen.
Synthetic, solid, biodegradable particle carriers
Synthetic polymer particles offer distinct advantages over liposomal-based systems. In addition to increased stability, by varying the polymer composition of the particle and morphology, one can effectively tune in a variety of controlled release characteristics, allowing moderate constant doses constant doses over prolonged periods of time. This capability for sustained release of an encapsulated or incorporated, protected antigen can potentially abrogate the booster requirement. For example, one oral dose of hepatitis B antigen entrapped in polyester polymer particles can yield a long-term protection equivalent to three doses of injected antigen [12]. This single booster effect has also been reported for tetanus [13] and diphtheria toxoid [14,15].
The development of biodegradable solid particles began with the realization that a wide variety of compounds – hydrophilic or hydrophobic, small molecule, protein, or gene – could be conjugated to or encapsulated within a matrix of biodegradable polymers and slowly released over time with the degradation of the polymer and/or diffusion out of the matrix. Biodegradable nanoparticles have, for the most part, been engineered from materials that are already known to be biocompatible and often commonly used in medicine for other purposes. These polymers can be designed to degrade through physiological processes (e.g. hydrolysis or enzymatic degradation), and leave behind biocompatible monomers. Therefore, one immediate benefit is that of negligible toxicity.
These delivery vehicles have been fabricated from a variety of materials, for example, polyesters, polyanhydrides and polyacrylates. Various polyanhydrides have been utilized as drug delivery platforms, including vaccine delivery systems [16]. Recently, polyanhydrides have been found to elicit Th1 responses by acting as agonists for TLRs 2, 4 and 5 [17]. Polyacrylate particles, which are highly stable under aqueous conditions and degrade enzymatically, have demonstrated efficacy as mucosal vaccine carriers [18]. Perhaps the most widely used are the aliphatic polyesters, specifically the hydrophobic poly(lactic acid) (PLA), the more-hydrophilic poly(glycolic acid) (PGA), and their copolymer, poly(lactic-co-glycolic acid) (PLGA). The physiological compatibility of PGA, PLA and PLGA has been established for safe use in humans; these materials have a history of >30 years in various human clinical applications [19,20]. The clinical translational potential of polyester particles has been an attractive feature for their use in several targeted vaccine formulations. PLGA has been used in vaccine applications for nearly two decades [21] and has been explored for mucosal immunization [22]. Varying the molecular weight and the ratio of PLA to PGA allows for tunable, sustained release of protected antigen [23]. This control over antigen liberation, ranging from days to months, can abrogate the need for a vaccine boost [24]. Furthermore, PLGA surface properties can be easily modified by the introduction of functional groups. For example, functional fatty acids have been incorporated into the particles during the formation process, thereby facilitating the addition of targeting modules to the particle surface [25]. In this way, the particles not only encapsulate antigens, but also target antigens to APCs in conjunction with other adjuvants that are required for eliciting the desired type of immune response.
Natural polymers
Other materials fabricated from natural polymers have been proposed and tested in vaccine applications, including chitosan, alginate and gelatin. Chitosan is the deacylated form of chitin, a positively charged polysaccharide of glucosamine and N-acetylglucosamine, and is naturally found in the exoskeleton of crustaceans and insects. It is a nontoxic, biodegradable polymer that has traditional applications in pharmacology, dentistry, orthopedics, ophthalmology, and surgery. Chitosan is degraded by lysozyme in vivo, and its products can be incorporated into metabolic pathways or excreted [26]. The free amino groups on this polymer introduce mucoadhesive properties in formulated particulates, which have been applied in oral and nasal vaccine delivery [27].
Other adjuvant polymers include alginate and gelatin. Alginate, a thickening and gelling agent used in the food industry, is a polysaccharide containing mannuronic and guluronic acid [28]. Enzymes that are capable of degrading alginate are not present in the body; therefore, alginate particles can persist for several months and release their contents by diffusion [29]. Gelatin protein, a hydrolyzed form of collagen degraded by proteolytic enzymes, features a high density of amino acids that are useful for antigen conjugation or target coupling (e.g. lysine), making it an attractive substrate for vaccine delivery applications. Gelatin is FDA-approved as a component of plasma expanders; however, concerns regarding allergic responses to gelatin-incorporating vaccines, such as MMR and DTP, have raised concerns regarding the clinical translation of some formulations [30].
Bioconjugation to the carrier
An important feature of antigen-carrying biomaterial substrates for vaccination is the ability to direct the antigenic load to APCs by targeting PRRs. Immobilization of PAMPs, which can be proteins, carbohydrates or nucleic acids, onto these substrates might be achieved by physical adsorption or chemical conjugation to the biomaterial. Physical adsorption is simple, especially in the absence of functional groups for chemical conjugation. However, physical adsorption of PAMPs relies on weak intermolecular interactions, such as electrostatic, hydrophobic or hydrogen bonding. These are transient interactions and have several drawbacks, including instability in the physiological milieu and poor control over the density and orientation of the displayed ligand. Thus, chemical conjugation through covalent bonding between the surface and the ligand of interest has been the primary means for coupling PAMPs to biomaterials. Given the ability to tune the availability of functional groups and the site of conjugation, chemical techniques offer more control over PAMP presentation. A comprehensive review of the various conjugation methods is outside the scope of this review, and the reader is instead referred to recent reviews [31–33]. Here, we discuss the most common chemistries to achieve this purpose (Figure 2).
Figure 2.
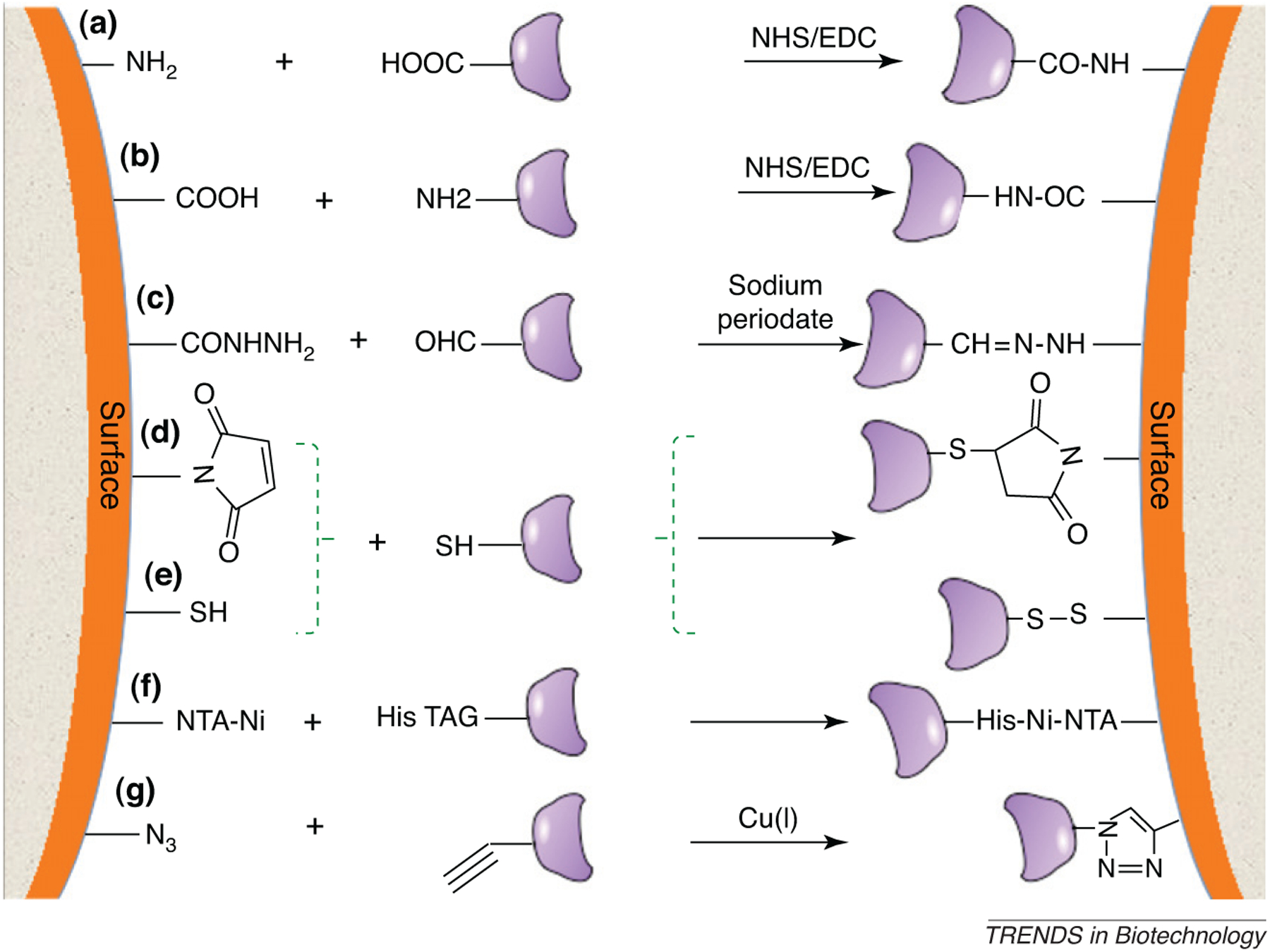
Schematic diagram showing a variety of possible chemistries for conjugation of PAMP moieties to functionalized substrates. (a,b) Covalent coupling of an amine (-NH2)-functionalized surface or PAMP to a carboxylate (-COOH) functional group, as mediated by NHS/carbodiimide reactive intermediate chemistry. (c) Carbohydrate PAMPs with unreactive hydroxylate groups might be oxidized with an agent such as sodium periodate, which yields a reactive aldehyde that can be conjugated to a dihydrazide-modified surface. The resultant hydrazone linkage is very stable. (d,e) Thiol-modified PAMPs might be linked to a maleimide-functionalized surface through selective reaction or a thiol-modified surface. (f) Histidine-tagged molecules can selectively bind to Ni chelated by tetradentate nitrilotriacetic acid: a procedure widely used for affinity separations. (g) An example of click chemistry; an azide (N3)–alkyne cycloaddition catalyzed by a Cu(I) catalyst results in the formation of a stable triazole linkage at room temperature.
The most ubiquitous chemical conjugation method is via amines and carboxylates, which are available on peptides, proteins and many carbohydrates. Amine coupling reactions in common practice introduce N-hydroxysuccinimide (NHS) esters onto the protein or surface by modification with a mixture of NHS and carbodiimides; these esters then react spontaneously with amines and other nucleophilic groups on the ligand to form covalent linkages [33]. Carboxylates can also be activated by similar methods utilizing carbodiimides in the presence of hydrazide to form a reactive intermediate that effectively couples with amines. Finally, some proteins and many peptides have only a single cysteine residue, which enables site-specific labeling with thiol-reactive surfaces. In many cases, sulfhydryl groups might be exploited for efficient coupling to surfaces, either by formation of disulfide linkages or by coupling to thiol-reactive maleimide groups (Figure 2).
More recently, ‘click chemistry’ has gained attention as a versatile means for coupling molecules to surfaces [34]. In the most widely used strategy, the molecule of interest is incorporated with an azide or an alkyne, and the surface is functionalized with a complementary alkyne or azide, respectively. The ensuing azide–alkyne reaction (Figure 2) ‘clicks’ a molecule into place on the surface in the presence of a copper catalyst. The important features of this chemistry that make it attractive are the efficiency, the specificity of the linkage, and the small size of the coupling groups.
Regardless of the methodology, two specific challenges with bioconjugation remain. First, serum proteins might mask the efficacy of an immobilized PAMP, and this non-specific interaction could compromise the ability to target PRRs. Therefore, inertness of the substrate is a key factor that controls unwanted adsorption. Polymer substrates that are intrinsically inert to non-specific protein adsorption or the addition of polymers, such as poly(ethylene glycol) are widely used strategies to mediate this effect. Second, the density of substrate attachment might play a significant role. Although the concept of immobilizing PAMPs is attractive for development of new vaccines, little is known regarding how the density of PAMPs on a substrate might alter an immune response. Clustering of PRRs by multivalent presentation of activating ligands might have unintended consequences. For example, it has been shown that crosslinking of the mannose receptor on the surface of monocyte-derived dendritic cells induces the production of anti-inflammatory signals, such as interleukin (IL)-10, and antagonizes pro-inflammatory responses that are important for vaccine responses [35]. Similarly, we have demonstrated that, depending on the density of antibodies on nanoparticles that target the C-type lectin DEC205, differential immune responses are triggered [36]. Therefore, any strategy for ligand immoblization on the surface of biomaterials must take into account the availability of ligand for targeting and its density.
PRR ligands
Many pathogen surface molecules that are necessary for structure, host cell entry, and mobility are evolutionarily conserved; as a result, these molecules are the ligands for PRRs in our innate immune system and have been exploited for the development of therapeutic and prophylactic vaccines. PRRs can associate agonists of these receptors to biomaterials, thus resulting in effective targeting and activation of cells of the immune system (Figure 1).
PRRs can be expressed intracellularly, presented extracellularly, or even secreted. Generally, they function by recognition of PAMPs, which triggers intracellular signaling cascades and ultimately results in the expression and upregulation of inflammatory effector molecules, co-stimulatory molecules, cytokines and/or reactive oxygen species. Extracellular and intracellular PRRs currently comprise four main families: TLRs, RIG-1-like receptors (RLRs), CLRs and the NLRs [37]. Although the use of TLR agonists represents a majority of these systems, we also discuss the expanding field of NLR and CLR-agonist inclusive vaccines (Table 1).
TLRs
TLRs were first discovered based on their homology with the Drosophila protein Toll, which is required for dorsoventral patterning during embryonic development. Later, it was found that Toll induces the production of the antimicrobial protein drosomycin, which suggests a role in immune responses. Homologs of Toll have since been identified in several species, ranging from plants to mammals, and the first human TLR was reported in 1994 [38]. The physiological relevance of these proteins in mammals was not identified until 1998, when it was shown that mice susceptible to infection with Gram-negative bacteria possess a mutation in their Tlr4 gene. This suggests that the role of TLR4 is to sense the presence of pathogens through lipopolysaccharide (LPS), a TLR4 ligand that is expressed on Gram-negative bacteria [39]. Since this initial discovery, 13 mammalian TLRs have been identified, among which 10 are functional TLRs in humans and 12 in mice; TLR1–9 are conserved in both species [40].
TLRs are type I transmembrane proteins, which means that they possess an extracellular N terminus. However, the defining features of all TLRs is the presence of an intracellular Toll/IL-1 receptor (TIR) domain and an extracellular leucine-rich repeat (LRR) associated with pathogen sensing [41]. The LRR region consists of β-strands and α-helices, which run parallel to the same axis, and produce a non-globular, horseshoe-shaped molecule. More importantly, the LRR is associated with interactions between the receptor and its ligands [42]. The TIR domain is highly conserved and forms complexes with the TIR-containing adaptor molecules myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like protein (Mal/TIRAP), TIR domain-containing adaptor protein inducing interferon β (TRIF), and TRIF-related adaptor molecule (TRAM) [43]. In addition to these stimulatory adaptors, sterile-α and HEAT-Armadillo motifs (SARM) have been identified as the first TIR-containing adaptors to have an inhibitory role [44]. MyD88 is utilized by all TLRs, except for TLR3, which signals through TRIF, and TLR4 has the ability to recruit either MyD88 or TRIF [45].
Multiple pathways can potentially be activated by TLR signaling, including nuclear factor (NF)-κB, mitogen-activated protein kinase (MAPK) and type I interferon regulatory factors. Activation of these pathways ultimately induces the translocation of transcription factors into the nucleus, which in turn regulates the expression of pro-inflammatory genes, including those for cytokines, chemokines, co-stimulatory molecules, and antimicrobial peptides [41]. In general, TLRs can be broadly classified based on their cellular localization; for example, on the cell surface (TLR1,2,4–6) or intracellularly on the endosomal surfaces (TLR3,7–9).
Recognition of surface proteins:
TLR1, TLR2, TLR6 Lipoproteins, which are prevalent in bacteria and have a hydrophobic lipid chain that anchors the protein to the cellular membrane, constitute one example of a TLR ligand. Triacylated lipoproteins are recognized by TLR2 dimerized with TLR1 [46,47], whereas diacylated lipoproteins are generally recognized by TLR2 dimerized with TLR6 [48]. Palmitoyl chain (Pam) diacylated and triacylated lipopeptides (Pam2CAG and Pam3CAG, respectively) have been incorporated into liposomes and shown to be effective at maturing human DCs [49]. Similarly, functionalized Pam3C has been incorporated into a chitosan derivative for vaccine applications [50]. Currently, an ongoing phase I clinical trial is evaluating a lipopeptide-incorporating therapeutic vaccine for HIV-1 ().
Recognition of nucleic acids:
TLR3, TLR7–9 Motifs in pathogen genetic material comprise another class of PRR ligands that have been exploited to construct new vaccines and therapeutics. Viral double-stranded RNA (dsRNA) is thought to be recognized by endosomal TLR3 and the RLRs RIG-1 and melanoma differentiation-associated protein 5 (MDA-5) during viral replication [51,52]. Extracellular dsRNA is inefficient at crossing membrane barriers, providing a clear motivation for coupling with particulates for delivery to APCs. Polyriboinosinic:polyribocytidylic acid, or poly(I:C), is a synthetic analog of dsRNA that is composed of mismatched strands of inosinic acid opposing cytidylic acid. Poly(I:C) has been evaluated as an adjuvant for 40 years and has been integrated into biodegradable microparticles [53,54] and liposomes [55], thereby enabling DC maturation and subsequent CD8+ T cell activation. Small interfering RNAs have also been shown to activate TLR3, regardless of sequence [56].
TLR7 and TLR8 recognize single-stranded RNA (ssRNA). The synthetic imidazoquinoline drug compounds imiquimod and resiquimod target TLR7. TLR7 is expressed on plasmacytoid DCs and B cells, but not macrophages [57]. This differential expression is of interest because ssRNA, unlike other TLR ligands, is present in all human somatic cells, and non-reactive macrophages in the periphery are necessary to clear debris from autologous dead cells. Conversely, TLR8 is mainly expressed on macrophages, monocytes, and myeloid DCs [58,59]. Very few studies have explored the particulate delivery of TLR7 or TLR8 agonists, although imiquimod has been included in liposome-forming lipid implants, liposomes and PLGA microparticles for vaccine applications [60,61]. Aldara, a 5% imiquimod topical cream, has been licensed for human use for the treatment of warts and other viral epithelial conditions [62], thus paving the way for further vaccines incorporating TLR7 and TLR8 ligand adjuvants.
A common element in viral and bacterial, but not human, DNA is the CpG (cytosine–phosphate–guanine) motif. Sequences rich in these nucleosides that are framed by two 5′ purines and two 3′ pyrimidines are recognized by TLR9 [63], which is primarily expressed on B cells and plasmacytoid DCs in humans. These CpG oligodeoxynucleotides (ODNs) were first encapsulated within liposomes in an effort to deliver effectively antisense ODNs to target DNA or RNA for regulation of gene expression [64]. CpG DNA has also been incorporated into liposomes [65–68] and biodegradable particles [69–72] to create delivery vehicles that promote cell-mediated responses. Additionally, we have shown that biodegradable PLGA nanoparticles loaded with the recombinant envelope protein antigen from the West Nile virus and surface-modified with CpG elicit humoral and cellular responses, thus providing protection against the live virus [73]. Animals immunized with this system show robust Th1-biased humoral responses compared with predominantly Th2-biased responses with the adjuvant aluminum hydroxide. Immunization with CpG ODN-modified nanoparticles has resulted in more circulating effector-like T cells and higher activity of antigen-specific lymphocytes, compared with unmodified nanoparticles or aluminum hydroxide alone [73]. Four clinical trials for CpG ODNs as adjuvants in malaria vaccines have been completed to date (, , , ). CpG is also being used in combination adjuvant systems that incorporate several PRR agonists within one delivery system to boost efficacy. For example, AS15 incorporates CpG, monophosphoryl lipid A (MPLA) and QS21 (saponin derivative) within a liposomal delivery system and has been tested for its efficacy in therapeutic cancer vaccines ().
Recognition of surface carbohydrates:
TLR4 Like surface proteins, surface carbohydrates are often the targets of PRRs. One such ubiquitous carbohydrate is the bacterial endotoxin LPS – a major component of the cell wall of Gram-negative bacteria and a ligand for TLR4, which is widely expressed on monocytes, myeloid DCs, granulocytes, and intestinal epithelial cells. LPS is an amphiphilic molecule that comprises a strain-specific side-chain known as the O-antigen, a core polysaccharide, and conserved lipid tails in the lipid A region. LPS has been incorporated into liposomes [74–77] and gelatin particles [78] for vaccine delivery, which results in high titers of antigen-specific antibodies. Our recent work with LPS immobilized on the surface of PLGA particles loaded with antigen has demonstrated that LPS-modified particles are preferentially internalized by DCs compared to uncoated nanoparticles; furthermore, the system, when administered to mice, elicits potent humoral and cellular immunity against a model antigen, ovalbumin, and the antigen derived from the West Nile envelope protein [79]. It has also been shown that wild type macrophages pulsed with LPS-modified nanoparticles produce pro-inflammatory cytokine IL-1β, consistent with inflammasome activation. Furthermore, inflammasome activity from LPS-modified particles requires the rupture of lysosomal compartments and their relocation into the cytoplasm of the cell [79,80]. The generality of this vaccination approach has been demonstrated in the encapsulation of a recombinant West Nile virus envelope protein into LPS-modified particles, which protects against a murine model of West Nile encephalitis [79].
LPS is an endotoxin, and its use as a PAMP in vaccine construction is therefore problematic owing to this toxicity. Thus, MPLA, a derivative of lipid A from Salmonella minnesota, is considered a safer TLR4 agonist, and hence, preferable for vaccine applications. MPLA adjuvants are less toxic than LPS and have different immunological effects [81,82]; however, in some cases, MPLA adjuvants are less effective than LPS [76]. MPLA coupling to liposomes and PLGA nanoparticles has been explored for vaccine applications [76,83,84]. The success of this method has propelled the development of two MPLA–liposomal vaccine formulations: Stimuvax is currently in phase III clinical trials for non-small cell lung carcinoma (, ), and AS01 is in phase I/II/III for malaria (, ). AS04, which is alum and MPLA, has recently been used as an adjuvant in Cervarix® (GlaxoSmithKline), a human papilloma virus (HPV) vaccine.
Recognition of flagellin:
TLR5 The PRR TLR5 is expressed by several cell types, including monocytes, DCs, epithelial cells, granulocytes and B cells [85]. TLR5 recognizes a conserved epitope of flagellin, the major protein of bacterial flagella [86,87]. Cytosolic flagellin in Salmonella-infected macrophages is recognized by another PRR called IPAF (NLRC4) and has been shown to activate the IPAF inflammasome, independent of TLR recognition [88]. Therefore, the incorporation of flagellin with biomaterials as adjuvants provides the potential to activate two PRR systems, TLRs and NLRs, which further enhances immune responses. Flagellin-loaded, bio-adhesive polyanhydride nanoparticles have been shown to yield a potent humoral response to a model antigen [89]. In addition, flagellin has been conjugated to polypropylene sulfide nanoparticles, and this system has been shown to promote Th1 type systemic and mucosal immune responses [90]. The use of flagellin as an adjuvant has also been recently tested in human clinical trials (, , ) and is a prospective candidate for future applications with vaccine delivery vehicles (, , ).
CLRs
PRRs also include the transmembrane lectins, such as the CLRs on the surface of DCs and other APCs. These receptors recognize sugar moieties, including mannose, fucose, and glucan expressed on the surface of mycobacteria, fungi and viruses. CLR engagement leads to internalization, processing and presentation of pathogens. In addition, several CLRs, including Mincle and DC-SIGN have the ability to trigger the NF-κB pathway, which ultimately promotes the secretion of chemokines and cytokines from APCs [91]. The field of antibody-mediated targeting of antigens to DCs has been pioneered through several CLRs, including DEC-205 [92], langerin [93] and DC-SIGN [94]. The prototypical CLR, DEC-205, has been the most widely studied DC target molecule for the induction of immune responses against model and vaccine antigens. Indeed, chimeric antibodies that consist of antigen fused to the C terminus of the heavy chain of anti-DEC-205 monoclonal antibody have been shown to deliver antigen efficiently to late endosomal/lysosomal processing compartments for presentation by class II MHC and CD1 molecules [95]. Targeting of DEC-205 has also been shown to promote cross-presentation of antigen by DCs, which results in CTL responses. As a result of the significant enhancement in CD4+ and CD8+ T cell responses, DEC-205-targeted vaccination is a promising way to increase vaccine efficacy.
Several studies have demonstrated that targeting antigen to DCs via CLRs improves receptor-mediated endocytosis and antigen presentation. For example, mannosylated liposomes that bind the mannose receptor on DCs are more readily endocytosed by human DCs than non-targeted liposomes [96]. Microparticles conjugated to antigen and that present the β-1,3 glucan, a fungal ligand recognized by the CLR Dectin-1 [97], are readily endocytosed by macrophages and elicit high antigen-specific antibody titers in mice after intradermal and oral vaccination [98].
In additionto the use of antibodiesto target CLRs, the use of whole β-glucan particles, derived from Saccharomyces cerevisiae cell walls has also been reported [99]. These particles have an inherent ability to stimulate Dectin-1 owing to the high levels of 1,3-β-glucans, thus allowing them to activate DCs directly. They have been shown to induce primarily Th1 and Th17 responses in vivo [99]. These β-glucan particles have also been reported to alter the response of immune cells, including DCs and human monocytes, to TLR agonists [100]. It highlights the fact that, although simultaneous activation of several PRRs offers an effective method of boosting responses, it might also completely alter the type of response generated; this should be considered when incorporating PRR agonists into biomaterials.
A novel adjuvant system, known as CAF01, has recently been shown to activate the CLR Mincle [101]. CAF01 consists of N,N′-dimethyl-N,N′-dioctadecylammonium (DDA), which is a synthetic amphiphilic lipid that spontaneously forms liposomes when in an aqueous environment, and the synthetic form of the trehalose 6,6′-dimycolate (TDM) glycolipid component of the mycobacterial cell wall known as trehalose 6,6′-dibehenate [102,103]. The ligation of Mincle is central to its ability to drive potent Th1 and Th17 responses [101]. A clinical trial is currently underway to investigate the safety and efficacy of this novel biomaterial in healthy adults ().
NLRs
NLRs are found within the cytosol of cells; once activated via ligand sensing, they undergo conformational changes that allow them to form oligomers, which trigger different downstream events depending on the NLR. For example, nucleotide oligomerization domain (NOD)1 and NOD2 recruit other signaling proteins that ultimately trigger the MAPK and NF-κB pathways resulting in the secretion of proinflammatory cytokines, chemokines and antimicrobial peptides, such as α and β defensins [104]. Other NLRs, such as NLRP3, form multiprotein complexes that promote activation of the proinflammatory caspases, which results in the secretion of IL-1β [105,106]. NLRs were initially discovered using a bioinformatic database when searching for other proteins that contain known LRRs [107]. These proteins comprise three distinct regions: the LRR; the central NOD; and the N-terminal domain, which can consist of a pyrin domain (PYD), a caspase recruitment domain (CARD), or a baculovirus inhibitor of apoptosis repeat (BIR) domain. The LRR is thought to play a similar role in ligand binding in NLRs as it does in TLRs [108].
NLRs can be subdivided into several families based on the class of their N-terminal effector domain: NLRA proteins that contain an acidic N terminus; NLRB proteins that contain a BIR domain; NLRC proteins that contain an N-terminal CARD domain; NLRP proteins that contain a PYD; and NLRX proteins that contain N termini with no known homology to currently identified proteins [108–110]. The most widely studied and most relevant NLRs with respect to biomaterials are the NLRP and NLRC proteins.
The NLRP subfamily is characterized by the presence of an N-terminal PYD and, with 14 members, it is one of the largest subfamilies of NLRs. One of its most infamous members, NLRP3, was initially discovered as cryopyrin, the protein product of a gene that is segregated in family members with Muckle–Wells syndrome and familial cold autoinflammatory syndrome [106,111,112]. NLRP3 forms part of a mulitprotein complex, known as the inflammasome, which activates caspase-1 and results in the secretion of bioactive IL-1β [7]. More recently, NLRP3 had been implicated in the adjuvant activities of several widely used biomaterials, including alum, PLGA microparticles and chitosan [4–6,79,113]. The ability of these biomaterials to activate NLRP3 is usually dependent on the presence of a TLR agonist, which demonstrates the existence of crosstalk and dependency among PRR systems. Injection of biomaterial alone is sufficient to activate the inflammasome and promote IL-1β secretion in vivo in the absence of exogenous TLR stimuli. This suggests that biomaterials also have the ability to synergize with endogenous danger signals that can potentially be released by dying, damaged or stressed cells [5].
In addition to the NLRPs, the NLRC family of proteins, mainly NOD2 and IPAF, have also been targeted using biomaterials. Lipophilic analogs of the peptidoglycan bioactive unit N-acetyl muramyl dipeptide, recognized primarily by NOD2 [114] and NLRP3 [115], have been packaged into liposomal and gelatin particulate formulations [78,116,117], thereby inducing antigen-specific antibody and Th1-biased cellular responses. As mentioned previously, NLRC4 or IPAF can recognize flagellin in a TLR-independent manner, which has led to its incorporation into biomaterials [89]. However, the role of NLRC4 in the adjuvant properties of flagellin remains to be investigated.
Future perspectives
Our current understanding of the biology of immune activation combined with construction of new biomaterials is progressing rapidly. Biomaterial systems are attractive for several reasons; not only are they capable of encapsulating a wide range of antigens, but they can also support the addition of modules to enable selective targeting to and uptake by desired cell types. Biomaterial systems allow the introduction of specific immunomodulators to guide the immune response and other elements that will deliver internalized antigens to appropriate intracellular compartments.
Targeting of PRRs is a central feature of many adjuvant systems. For example, it has recently been shown that the intracellular NLRP3 is activated by several classical adjuvants, including aluminum hydroxide, chitosan and saponins [4,113]. Knowledge of PRRs can be exploited in combination with biomaterial engineering to create new, optimized vaccine systems endowed with signals that trigger immunity in predictable and tunable ways. Although this review focuses on PRRs that recognize microbial pathogens, it is important to note that other components of the immune system can be targeted or encapsulated into biomaterials. The complement system, for example, represents a multi-protein cascade and has the ability to promote phagocytosis and subsequent destruction of extracellular pathogens or to induce cytotoxicity once activated; therefore, incorporating complement-activating ligands into biomaterials is an attractive method of enhancing vaccine efficacy [118].
Cytokines have also been incorporated into biomaterials, which allows these systems to mimic infection and to activate the immune system through the release of endogenous factors. A prime example of this is the incorporation of Granulocyte-macrophage colony-stimulating factor (GM-CSF) into a macroporous PLG matrix, which promotes the migration of DCs towards the scaffold [119]. In addition, the immobilization of melanoma tumor lysates onto these GM-CSF-releasing scaffolds has shown a reduced rate of tumor development in mice [119]. Alternatively, biomaterials can simultaneously recruit DCs and T cells to the site of antigen presentation to promote the interaction of these cell types, which represents a key step in immune activation [120].
Considering the bright future and promise of these biomimetic systems, it is not a surprise that considerable attention is now focused on how different nanomaterials and synthetic biomaterials generally interact with the body. Before advanced clinical trials can proceed, intensive studies are needed into the in vivo trafficking patterns or effects of these systems on the immune system. Questions awaiting rigorous investigation include:
How are antigenic memory responses shaped in the presence of these materials?
What is the long-term effect on the regulatory T cell pool?
What subsets of cells are activated beyond antigen-specific T cells?
Additionally, we must determine the toxicity of biomaterials and if chronic inflammation with these systems is an unforeseen hazard. Overall, key factors necessary to activate effectively the immune system can be engineered into biomaterials, which provides an attractive template for the next generation of vaccines.
References
- 1.Steinman RM (2001) Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. Mt. Sinai J. Med., N.Y 68, 160–166 [PubMed] [Google Scholar]
- 2.Geijtenbeek TB et al. (2000) DC-SIGN, a dendritic cell-specific HIV1-binding protein that enhances trans-infection of T cells. Cell 100, 587–597 [DOI] [PubMed] [Google Scholar]
- 3.Baylor NW et al. (2002) Aluminum salts in vaccines—US perspective. Vaccine 20 (Suppl. 3), S18–23 [DOI] [PubMed] [Google Scholar]
- 4.Eisenbarth SC (2008) Use and limitations of alum-based models of allergy. Clin. Exp. Allergy 38, 1572–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharp FA et al. (2009) Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A 106, 870–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kool M et al. (2008) Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol 181, 3755–3759 [DOI] [PubMed] [Google Scholar]
- 7.Martinon F et al. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 [DOI] [PubMed] [Google Scholar]
- 8.Allison AG and Gregoriadis G (1974) Liposomes as immunological adjuvants. Nature 252, 252. [DOI] [PubMed] [Google Scholar]
- 9.Copland MJ et al. (2005) Lipid based particulate formulations for the delivery of antigen. Immunol. Cell Biol 83, 97–105 [DOI] [PubMed] [Google Scholar]
- 10.Nakanishi T et al. (1999) Positively charged liposome functions as an efficient immunoadjuvant in inducing cell-mediated immune response to soluble proteins. J. Control Release 61, 233–240 [DOI] [PubMed] [Google Scholar]
- 11.Hansen CB et al. (1995) Attachment of antibodies to sterically stabilized liposomes: evaluation, comparison and optimization of coupling procedures. Biochim. Biophys. Acta 1239, 133–144 [DOI] [PubMed] [Google Scholar]
- 12.Nellore RV et al. (1992) Evaluation of biodegradable microspheres as vaccine adjuvant for hepatitis B surface antigen. J. Parenter. Sci. Technol 46, 176–180 [PubMed] [Google Scholar]
- 13.Singh M et al. (1997) Immunogenicity and protection in small-animal models with controlled-release tetanus toxoid microparticles as a single-dose vaccine. Infect. Immun 65, 1716–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh M et al. (1992) Immunogenicity studies on diphtheria toxoid loaded biodegradable microspheres. Int. J. Pharm 85, R5–R8 [Google Scholar]
- 15.Singh M et al. (1998) Controlled release microparticles as a single dose diphtheria toxoid vaccine: immunogenicity in small animal models. Vaccine 16, 346–352 [DOI] [PubMed] [Google Scholar]
- 16.Kipper MJ et al. (2006) Single dose vaccine based on biodegradable polyanhydride microspheres can modulate immune response mechanism. J. Biomed. Mater. Res. A 76, 798–810 [DOI] [PubMed] [Google Scholar]
- 17.Tamayo I et al. (2010) Poly(anhydride) nanoparticles act as active Th1 adjuvants through Toll-like receptor exploitation. Clin. Vaccine Immunol 17, 1356–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaman M et al. (2010) Synthetic polyacrylate polymers as particulate intranasal vaccine delivery systems for the induction of mucosal immune response. Curr. Drug Deliv 7, 118–124 [DOI] [PubMed] [Google Scholar]
- 19.Jiang W et al. (2005) Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv. Drug Deliv. Rev 57, 391–410 [DOI] [PubMed] [Google Scholar]
- 20.Bramwell VW et al. (2005) Particulate delivery systems for biodefense subunit vaccines. Adv. Drug Deliv. Rev 57, 1247–1265 [DOI] [PubMed] [Google Scholar]
- 21.O’Hagan DT et al. (1991) Controlled release microparticles for vaccine development. Vaccine 9, 768–771 [DOI] [PubMed] [Google Scholar]
- 22.Gupta PN et al. (2007) M-cell targeted biodegradable PLGA nanoparticles for oral immunization against hepatitis B. J. Drug Target 15, 701–713 [DOI] [PubMed] [Google Scholar]
- 23.Sesardic D and Dobbelaer R (2004) European union regulatory developments for new vaccine adjuvants and delivery systems. Vaccine 22, 2452–2456 [DOI] [PubMed] [Google Scholar]
- 24.Gupta RK et al. (1998) Poly(lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Adv. Drug Deliv. Rev 32, 225–246 [PubMed] [Google Scholar]
- 25.Fahmy TM et al. (2005) Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials 26, 5727–5736 [DOI] [PubMed] [Google Scholar]
- 26.Senel S and McClure SJ (2004) Potential applications of chitosan in veterinary medicine. Adv. Drug Deliv. Rev 56, 1467–1480 [DOI] [PubMed] [Google Scholar]
- 27.van der Lubben IM et al. (2001) Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur. J. Pharm. Sci 14, 201–207 [DOI] [PubMed] [Google Scholar]
- 28.Wee S and Gombotz WR (1998) Protein release from alginate matrices. Adv. Drug Deliv. Rev 31, 267–285 [DOI] [PubMed] [Google Scholar]
- 29.Gao C et al. (2009) Preparation and controlled degradation of oxidized sodium alginate hydrogel. Polym. Degrad. Stab 94, 1405–1410 [Google Scholar]
- 30.Kelso JM et al. (1993) Anaphylaxis to measles, mumps, and rubella vaccine mediated by IgE to gelatin. J. Allergy Clin. Immunol 91, 867–872 [DOI] [PubMed] [Google Scholar]
- 31.Di Marco M et al. (2010) Overview of the main methods used to combine proteins with nanosystems: absorption, bioconjugation, and encapsulation. Int. J. Nanomed 5, 37–49 [PMC free article] [PubMed] [Google Scholar]
- 32.Veronese FM and Morpurgo M (1999) Bioconjugation in pharmaceutical chemistry. Farmaco 54, 497–516 [DOI] [PubMed] [Google Scholar]
- 33.Hermanson G (2008) Bioconjugate Techniques, Elsevier [Google Scholar]
- 34.Nwe K and Brechbiel MW (2009) Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother. Radiopharm 24, 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chieppa M et al. (2003) Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J. Immunol 171, 4552–4560 [DOI] [PubMed] [Google Scholar]
- 36.Bandyopadhyay A et al. (2011) The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials 11, 3094–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferwerda G et al. (2010) The role of Toll-like receptors and C-type lectins for vaccination against Candida albicans. Vaccine 28, 614–622 [DOI] [PubMed] [Google Scholar]
- 38.Nomura N et al. (1994) Prediction of the coding sequences of unidentified human genes. I. The coding sequences of 40 new genes (KIAA0001-KIAA0040) deduced by analysis of randomly sampled cDNA clones from human immature myeloid cell line KG-1. DNA Res. 1, 27–35 [DOI] [PubMed] [Google Scholar]
- 39.Poltorak A et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 40.Kawai T and Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 41.Kawai T and Akira S (2009) The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol 21, 317–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobe B and Kajava AV (2001) The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol 11, 725–732 [DOI] [PubMed] [Google Scholar]
- 43.Bortoluci KR and Medzhitov R (2010) Control of infection by pyroptosis and autophagy: role of TLR and NLR. Cell. Mol. Life Sci.: CMLS 67, 1643–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carty M et al. (2006) The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol 7, 1074–1081 [DOI] [PubMed] [Google Scholar]
- 45.O’Neill LA and Bowie AG (2010) Sensing and signaling in antiviral innate immunity. Curr. Biol 20, R328–333 [DOI] [PubMed] [Google Scholar]
- 46.Shimizu T et al. (2007) Triacylated lipoproteins derived from Mycoplasma pneumoniae activate nuclear factor-kappaB through toll-like receptors 1 and 2. Immunology 121, 473–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeuchi O et al. (2002) Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol 169, 10–14 [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi O et al. (2001) Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol 13, 933–940 [DOI] [PubMed] [Google Scholar]
- 49.Espuelas S et al. (2005) Effect of synthetic lipopeptides formulated in liposomes on the maturation of human dendritic cells. Mol. Immunol 42, 721–729 [DOI] [PubMed] [Google Scholar]
- 50.Heuking S et al. (2009) Toll-like receptor-2 agonist functionalized biopolymer for mucosal vaccination. Int. J. Pharm 381, 97–105 [DOI] [PubMed] [Google Scholar]
- 51.Alexopoulou L et al. (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738 [DOI] [PubMed] [Google Scholar]
- 52.Loo YM et al. (2008) Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol 82, 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wischke C et al. (2009) Poly(I:C) coated PLGA microparticles induce dendritic cell maturation. Int. J. Pharm 365, 61–68 [DOI] [PubMed] [Google Scholar]
- 54.Heffernan MJ et al. (2009) The stimulation of CD8+ T cells by dendritic cells pulsed with polyketal microparticles containing ion-paired protein antigen and poly(inosinic acid)-poly(cytidylic acid). Biomaterials 30, 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaks K et al. (2006) Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J. Immunol 176, 7335–7345 [DOI] [PubMed] [Google Scholar]
- 56.Kariko K et al. (2004) Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J. Immunol 172, 6545–6549 [DOI] [PubMed] [Google Scholar]
- 57.Krieg AM (2007) The toll of too much TLR7. Immunity 27, 695–697 [DOI] [PubMed] [Google Scholar]
- 58.Bekeredjian-Ding I et al. (2006) T cell-independent, TLR-induced IL12p70 production in primary human monocytes. J. Immunol 176, 7438–7446 [DOI] [PubMed] [Google Scholar]
- 59.Hornung V et al. (2002) Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol 168, 4531–4537 [DOI] [PubMed] [Google Scholar]
- 60.Myschik J et al. (2008) Immunogenicity of lipid sustained release implants containing imiquimod, alpha-galactosylceramide, or Quil-A. Pharmazie 63, 686–692 [PubMed] [Google Scholar]
- 61.Johnston D et al. (2007) TLR7 imidazoquinoline ligand 3M-019 is a potent adjuvant for pure protein prototype vaccines. Cancer Immunol. Immunother 56, 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slade HB et al. (1998) Imiquimod 5% cream (Aldara). Expert Opin. Investig. Drugs 7, 437–449 [DOI] [PubMed] [Google Scholar]
- 63.Hemmi H et al. (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 [DOI] [PubMed] [Google Scholar]
- 64.Akhtar S et al. (1991) Interactions of antisense DNA oligonucleotide analogs with phospholipid membranes (liposomes). Nucleic Acids Res. 19, 5551–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gursel M et al. (1999) Immunoadjuvant action of plasmid DNA in liposomes. Vaccine 17, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 66.Gursel I et al. (2001) Sterically stabilized cationic liposomes improve the uptake and immunostimulatory activity of CpG oligonucleotides. J. Immunol 167, 3324–3328 [DOI] [PubMed] [Google Scholar]
- 67.Li WM et al. (2001) Enhanced immune response to T-independent antigen by using CpG oligodeoxynucleotides encapsulated in liposomes. Vaccine 20, 148–157 [DOI] [PubMed] [Google Scholar]
- 68.Joseph A et al. (2002) Liposomal immunostimulatory DNA sequence (ISS-ODN): an efficient parenteral and mucosal adjuvant for influenza and hepatitis B vaccines. Vaccine 20, 3342–3354 [DOI] [PubMed] [Google Scholar]
- 69.Singh M et al. (2001) Cationic microparticles are an effective delivery system for immune stimulatory cpG DNA. Pharm. Res 18, 1476–1479 [DOI] [PubMed] [Google Scholar]
- 70.Borges O et al. (2008) Immune response by nasal delivery of hepatitis B surface antigen and codelivery of a CpG ODN in alginate coated chitosan nanoparticles. Eur. J. Pharm. Biopharm 69, 405–416 [DOI] [PubMed] [Google Scholar]
- 71.Fischer S et al. (2009) Concomitant delivery of a CTL-restricted peptide antigen and CpG ODN by PLGA microparticles induces cellular immune response. J. Drug Target 17, 652–661 [DOI] [PubMed] [Google Scholar]
- 72.Zwiorek K et al. (2008) Delivery by cationic gelatin nanoparticles strongly increases the immunostimulatory effects of CpG oligonucleotides. Pharm. Res 25, 551–562 [DOI] [PubMed] [Google Scholar]
- 73.Demento SL et al. (2010) TLR9-targeted biodegradable nanoparticles as immunization vectors protect against West Nile encephalitis. J. Immunol 185, 2989–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petrov AB et al. (1994) Non-specific modulation of the immune response with liposomal meningococcal lipopolysaccharide: role of different cells and cytokines. Vaccine 12, 1064–1070 [DOI] [PubMed] [Google Scholar]
- 75.Jain V et al. (2008) Enhancement of T-helper type I immune responses against hepatitis B surface antigen by LPS derivatives adjuvanted liposomes delivery system. J. Drug Target 16, 706–715 [DOI] [PubMed] [Google Scholar]
- 76.Arigita C et al. (2005) Well-defined and potent liposomal meningococcal B vaccines adjuvated with LPS derivatives. Vaccine 23, 5091–5098 [DOI] [PubMed] [Google Scholar]
- 77.Petrov AB et al. (1992) Toxicity and immunogenicity of Neisseria meningitidis lipopolysaccharide incorporated into liposomes. Infect. Immun 60, 3897–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Franz J et al. (1998) Adjuvant efficacy of gelatin particles and microparticles. Int. J. Pharm 168, 153–161 [Google Scholar]
- 79.Demento SL et al. (2009) Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine 27, 3013–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hornung V et al. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salkowski CA et al. (1997) Lipopolysaccharide and monophosphoryl lipid A differentially regulate interleukin-12, gamma interferon, and interleukin-10 mRNA production in murine macrophages. Infect. Immun 65, 3239–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Henricson BE et al. (1993) Dissociation of lipopolysaccharide (LPS)inducible gene expression in murine macrophages pretreated with smooth LPS versus monophosphoryl lipid A. Infect. Immun 61, 2325–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elamanchili P et al. (2007) Pathogen-mimicking” nanoparticles for vaccine delivery to dendritic cells. J. Immunother 30, 378–395 [DOI] [PubMed] [Google Scholar]
- 84.Chong CS et al. (2005) Enhancement of T helper type 1 immune responses against hepatitis B virus core antigen by PLGA nanoparticle vaccine delivery. J. Control Release 102, 85–99 [DOI] [PubMed] [Google Scholar]
- 85.Honko AN and Mizel SB (2005) Effects of flagellin on innate and adaptive immunity. Immunol. Res 33, 83–101 [DOI] [PubMed] [Google Scholar]
- 86.Hayashi F et al. (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103 [DOI] [PubMed] [Google Scholar]
- 87.Smith KD et al. (2003) Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat. Immunol 4, 1247–1253 [DOI] [PubMed] [Google Scholar]
- 88.Franchi L et al. (2006) Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol 7, 576–582 [DOI] [PubMed] [Google Scholar]
- 89.Salman HH et al. (2009) Immunoadjuvant capacity of flagellin and mannosamine-coated poly(anhydride) nanoparticles in oral vaccination. Vaccine 27, 4784–4790 [DOI] [PubMed] [Google Scholar]
- 90.Stano A et al. (2010) PPS nanoparticles as versatile delivery system to induce systemic and broad mucosal immunity after intranasal administration. Vaccine 29, 804–812 [DOI] [PubMed] [Google Scholar]
- 91.Geijtenbeek TB and Gringhuis SI (2009) Signalling through C-type lectin receptors: shaping immune responses. Nat. Rev. Immunol 9, 465–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bozzacco L et al. (2007) DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc. Natl. Acad. Sci. U.S.A 104, 1289–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheong C et al. (2007) Production of monoclonal antibodies that recognize the extracellular domain of mouse langerin/CD207. J. Immunol. Methods 324, 48–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singh SK et al. (2009) Targeting glycan modified OVA to murine DC-SIGN transgenic dendritic cells enhances MHC class I and II presentation. Mol. Immunol 47, 164–174 [DOI] [PubMed] [Google Scholar]
- 95.Bonifaz LC et al. (2004) In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med 199, 815–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Irache JM et al. (2008) Mannose-targeted systems for the delivery of therapeutics. Expert Opin. Drug Deliv 5, 703–724 [DOI] [PubMed] [Google Scholar]
- 97.Goodridge HS et al. (2009) Beta-glucan recognition by the innate immune system. Immunol. Rev 230, 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berner VK et al. (2008) Conjugation of protein antigen to microparticulate beta-glucan from Saccharomyces cerevisiae: a new adjuvant for intradermal and oral immunizations. Appl. Microbiol. Biotechnol 80, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 99.Huang H et al. (2010) Robust stimulation of humoral and cellular immune responses following vaccination with antigen-loaded beta-glucan particles. MBio 1, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang H et al. (2009) Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and toll-like receptor agonists. Infect. Immun 77, 1774–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schoenen H et al. (2010) Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol 184, 2756–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davidsen J et al. (2005) Characterization of cationic liposomes based on dimethyldioctadecylammonium and synthetic cord factor from M. tuberculosis (trehalose 6,6(-dibehenate)-a novel adjuvant inducing both strong CMI and antibody responses. Biochim. Biophys. Acta 1718, 22–31 [DOI] [PubMed] [Google Scholar]
- 103.Korsholm KS et al. (2007) The adjuvant mechanism of cationic dimethyldioctadecylammonium liposomes. Immunology 121, 216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Franchi L et al. (2009) Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev 227, 106–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agostini L et al. (2004) NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity 20, 319–325 [DOI] [PubMed] [Google Scholar]
- 106.Manji GA et al. (2002) PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NFkappa B. J. Biol. Chem 277, 11570–11575 [DOI] [PubMed] [Google Scholar]
- 107.Ting JP et al. (2010) How the noninflammasome NLRs function in the innate immune system. Science 327, 286–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ting JP and Davis BK (2005) CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu. Rev. Immunol 23, 387–414 [DOI] [PubMed] [Google Scholar]
- 109.Ting JP et al. (2008) The NLR gene family: a standard nomenclature. Immunity 28, 285–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tattoli I et al. (2008) NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 9, 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aganna E et al. (2002) Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 46, 2445–2452 [DOI] [PubMed] [Google Scholar]
- 112.Hoffman HM et al. (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat. Genet 29, 301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li H et al. (2008) Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J.Immunol 181, 17–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tanabe T et al. (2004) Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 23, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Martinon F et al. (2004) Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr. Biol 14, 1929–1934 [DOI] [PubMed] [Google Scholar]
- 116.Jain V et al. (2009) Well-defined and potent liposomal hepatitis B vaccines adjuvanted with lipophilic MDP derivatives. Nanomed.: Nanotechnol. Biol. Med 5, 334–344 [DOI] [PubMed] [Google Scholar]
- 117.Sridevi K et al. (2003) Reversal of T cell anergy in leprosy patients: in vitro presentation with Mycobacterium leprae antigens using murabutide and Trat peptide in liposomal delivery. Int. Immunopharmacol 3, 1589–1600 [DOI] [PubMed] [Google Scholar]
- 118.Hubbell JA et al. (2009) Materials engineering for immunomodulation. Nature 462, 449–460 [DOI] [PubMed] [Google Scholar]
- 119.Ali OA et al. (2009) Infection-mimicking materials to program dendritic cells in situ. Nat. Mater 8, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stern E et al. (2009) Spatiotemporal control over molecular delivery and cellular encapsulation from electropolymerized micro- and nanopatterned surfaces. Adv. Funct. Mater 19, 2888–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alving CR et al. (1992) Liposomes containing lipid A as a potent nontoxic adjuvant. Res. Immunol 143, 197–198 [DOI] [PubMed] [Google Scholar]
- 122.Jain V et al. (2009) Well-defined and potent liposomal hepatitis B vaccines adjuvanted with lipophilic MDP derivatives. Nanomedicine 5, 334–344 [DOI] [PubMed] [Google Scholar]
- 123.Bartenschlager R (2006) Hepatitis C virus molecular clones: from cDNA to infectious virus particles in cell culture. Curr. Opin. Microbiol 9, 416–422 [DOI] [PubMed] [Google Scholar]
- 124.Aaltonen LM et al. (1998) A novel method to culture laryngeal human papillomavirus-positive epithelial cells produces papillomatype cytology on collagen rafts. Eur. J. Cancer 34, 1111–1116 [DOI] [PubMed] [Google Scholar]
- 125.Kotton CN (2008) Vaccination and immunization against travelrelated diseases in immunocompromised hosts. Expert Rev. Vaccines 7, 663–672 [DOI] [PubMed] [Google Scholar]
- 126.Naylor CJ and Jones RC (1994) Demonstration of a virulent subpopulation in a prototype live attenuated turkey rhinotracheitis vaccine. Vaccine 12, 1225–1230 [DOI] [PubMed] [Google Scholar]
- 127.Xiang SD et al. (2006) Pathogen recognition and development of particulate vaccines: does size matter? Methods 40, 1–9 [DOI] [PubMed] [Google Scholar]
- 128.Beaudette TT et al. (2009) In vivo studies on the effect of coencapsulation of CpG DNA and antigen in acid-degradable microparticle vaccines. Mol. Pharm 6, 1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shen H et al. (2006) Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 117, 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kanchan V et al. (2009) Memory antibody response from antigen loaded polymer particles and the effect of antigen release kinetics. Biomaterials 30, 4763–4776 [DOI] [PubMed] [Google Scholar]
- 131.Howland SW and Wittrup KD (2008) Antigen release kinetics in the phagosome are critical to cross-presentation efficiency. J. Immunol 180, 1576–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aguado MT and Lambert PH (1992) Controlled-release vaccines—biodegradable polylactide/polyglycolide (PL/PG) microspheres as antigen vehicles. Immunobiology 184, 113–125 [DOI] [PubMed] [Google Scholar]
- 133.Espuelas S et al. (2008) Influence of ligand valency on the targeting of immature human dendritic cells by mannosylated liposomes. Bioconjug. Chem 19, 2385–2393 [DOI] [PubMed] [Google Scholar]
- 134.Coombes AG et al. (1999) Biodegradable lamellar particles of poly(lactide) induce sustained immune responses to a single dose of adsorbed protein. Vaccine 17, 2410–2422 [DOI] [PubMed] [Google Scholar]
- 135.Katare YK et al. (2003) Potentiation of immune response from polymer-entrapped antigen: toward development of single dose tetanus toxoid vaccine. Drug Deliv. 10, 231–238 [DOI] [PubMed] [Google Scholar]
- 136.Yuki Y and Kiyono H (2009) Mucosal vaccines: novel advances in technology and delivery. Expert Rev. Vaccines 8, 1083–1097 [DOI] [PubMed] [Google Scholar]
- 137.Neutra MR and Kozlowski PA (2006) Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol 6, 148–158 [DOI] [PubMed] [Google Scholar]
- 138.Vajdy M et al. (2004) Mucosal adjuvants and delivery systems for protein-, DNA-and RNA-based vaccines. Immunol.CellBiol 82, 617–627 [DOI] [PubMed] [Google Scholar]