

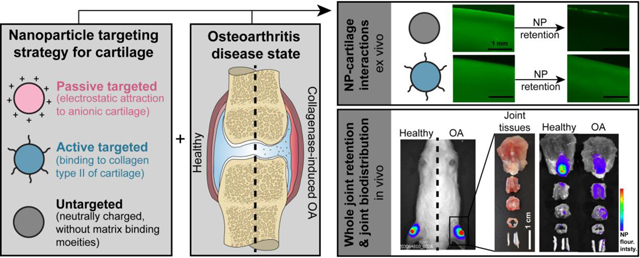
Abstract
Understanding intra-articular biodistribution is imperative as candidate osteoarthritis (OA) drugs become increasingly site-specific. Cartilage has been identified as opportunistic for therapeutic intervention, but poses numerous barriers to drug delivery. To facilitate drug delivery to cartilage, nanoscale vehicles have been designed with different features that target the tissue’s matrix. However, it is unclear if these targeting strategies are influenced by OA and the associated structural changes that occur in cartilage. The goal of this work was to study the effectiveness of different cartilage-targeting nanomaterials with respect to cartilage localization and retention, and to determine how these outcomes change in OA. To address these questions, a nanoparticle (NP) system was developed, and the formulation was tuned to possess three distinct cartilage-targeting strategies: (1) passive targeting cationic NPs for electrostatic attraction to cartilage, (2) active targeting NPs with binding peptides for collagen type II, and (3) untargeted neutrally-charged NPs. Ex vivo analyses with bovine cartilage explants demonstrated that targeting strategies significantly improved NP associations with both healthy and OA-like cartilage. In vivo studies with collagenase-induced OA in rats revealed that disease state influenced joint biodistribution for all three NP formulations. Importantly, the extent of cartilage accumulation for each NP system was affected by disease differently; with active NPs, but not passive NPs, cartilage accumulation was increased in OA relative to healthy knees. Together, this work suggests that NPs can be strategically designed for site-specific OA drug delivery, but the biodistribution of the NPs are influenced by the disease conditions into which they are delivered.
Keywords: drug delivery, targeting, cartilage, osteoarthritis, intra-articular
Graphic abstract:
1. Introduction
Osteoarthritis (OA) is a degenerative joint disease that leads to low quality of life and debilitation. OA is estimated to impact over 60–70% of the population over the age of 65 [1], and projections indicate that this already widespread disease will increase in prevalence with time [2]. Progression of OA involves a complex orchestration of multiple joint tissues including the cartilage, a connective tissue that lines diarthrodal joints. Upon insult, cartilage contributes to the inflammatory environment in the joint and undergoes degenerative changes that progressively worsen due to its limited capacity to repair [1,3]. As such, cartilage is an attractive target tissue for disease intervention. Existing OA treatments only work to manage the symptoms of OA, but numerous drugs are under investigation as potential disease modifying OA drugs (DMOADs) to slow or reverse OA damage. Many DMOADs focus on preventing cartilage matrix degradation, protecting chondrocytes, and improving cartilage matrix synthesis [4,5]. To be therapeutically effective, drugs must accumulate within their target at efficacious concentrations, in this case the cartilage. For drugs designed to modulate specific biological actions within cartilage, it also may be advantageous to reduce off-target accumulation in other joint tissues, which further emphasizes the need for site-specific drug delivery.
Although cartilage is an important target for DMOADs, the tissue presents numerous barriers to drug transport [6,7]. Chondrocytes are sparsely distributed throughout the extracellular matrix (ECM), making up only 2–5% of the cartilage by weight [1], so cell-specific therapies must be able to penetrate into the depth of cartilage to reach the chondrocytes. Adversely, penetration into cartilage is restricted as this matrix is avascular and densely packed with charged molecules. To help drugs overcome these challenges to transport and to facilitate localization within cartilage, nanoscale drug delivery vehicles have been explored. Nanomaterials, including nanoparticles (NPs), offer design features that promote associations with specific cells and tissues and offer opportunities for sustained release of drugs. An important aspect of nanomaterial design is size. Vehicle diameter in part dictates the ability of materials to penetrate into the dense cartilage ECM [6,8,9], a process for which nanomaterials are particularly apt. Additionally, due to their size, nano-scale vehicles can be internalized by cells, which may be ideal for the activity of certain drugs.
In addition to having sizes suitable for cartilage drug delivery, nanomaterials can be functionalized to target components and/or cells of the cartilage. Nanomaterial targeting is established via physicochemical properties such as size, charge, surface chemistry (passive targeting) or decoration with moieties that interact with unique biomolecules (active targeting). Cartilage is comprised of an organized network of collagen type II, a protein that is both unique to and abundant within cartilage [10,11]. The collagen framework is primarily filled with sulfated glycosaminoglycan-containing proteoglycans, which give the cartilage an anionic charge [12,13]. Targeting strategies for cartilage have leveraged these unique ECM characteristics, including passive targeting of nanomaterials to cartilage via electrostatic attraction to the anionic cartilage matrix. Examples of nanomaterials that passively target the cartilage include cationic globular proteins [14] and dendrimers [15]. Cartilage has also been actively targeted by modifying nanomaterials with binding agents specific to collagen type II. Examples of such active targeting strategies include the addition of collagen type II antibodies on liposomes [16,17] and binding peptides on a variety of nanomaterial vehicles [18,19].
Despite the recent progress in passive and active targeting nanomaterials for cartilage drug delivery, there are still gaps in our understanding of these cartilage targeting strategies. First, the number of studies that evaluate the biodistribution of materials throughout joint tissues is limited - to our knowledge, only one other publication that has quantified nanomaterial biodistribution in joint tissues after intra-articular injection [14]. Additionally, the effect of disease on these targeted nanomaterials has not been studied despite the widely accepted principle that the structure and composition of cartilage changes in OA. OA pathogenesis includes fragmentation and loss of proteoglycans and disruption of the collagen network, which are the primary constituents leveraged for targeted drug delivery. To date, neither the whole joint retention nor biodistribution of cartilage-targeting nanomaterials have been evaluated in diseased joints. Whole joint retention of nanomaterials that do not specifically target the cartilage, on the other hand, has been compared across healthy and surgically-induced OA mice [20] and rats [21–23]. However, biodistribution throughout joint tissues was not within the scope of these studies, so it remains unknown how these materials localized to tissues in healthy or diseased joints. Last, while multiple studies compare an untargeted NP to a targeted NP [16,18,19,24–26], the different types of targeting strategies have yet to be compared within a single study. It remains unclear if or how these strategies perform differently, particularly as cartilage and the joint undergo degenerative changes. Advancing knowledge of nanomaterial biodistribution is needed to progress the field of targeted intra-articular drug delivery for OA.
The objectives of this work were to evaluate the role of disease condition on the ability of targeted nanomaterials to accumulate in cartilage, and to identify the extent to which the different targeting strategies affect biodistribution in the joint. To study these questions, we developed a tunable NP platform that could be functionalized with different targeting moieties while maintaining similar nanomaterial composition and size. Poly(lactic-co-glycolic acid) (PLGA) was used as the base material for this system because of its history of biocompatibility and safety, capability of carrying a wide range of drugs, and ability to functionalize with other polymers, imaging agents [27–30], and targeting agents [31]. Simple modifications of the NP formulation with poly(allylamine) hydrochloride (PAA), a primary amine-containing polymer, provided controllable NP properties and successful incorporation of targeting moieties. Together, this NP system yielded (1) cationic, passive targeting NPs (“passive NPs”) for electrostatic attraction to the anionic matrix, (2) active targeting NPs (“active NPs”) with a collagen type II binding peptide, and (3) neutral, unfunctionalized NPs as an untargeted control (“untargeted NPs”). We hypothesized that passive targeting would be more effective in healthy tissue, when proteoglycan content and therefore electrostatic potential are at their greatest, while active NPs would localize to a greater extent in OA tissue, when the degradation of the ECM more readily facilitates penetration and access to the collagen type II network.
2. Methods
2.1. Nanoparticle Formulation and Functionalization
The tunable NP system was made by single emulsion and solvent evaporation as in previous studies [32], but with different aqueous phase compositions. The organic phase was prepared by dissolving 30 mg/mL of PLGA (50:50, 0.55–0.75 dL/g inherent viscosity, Lactel Absorbable Polymers, Birmingham, AL) in chloroform. The aqueous phase was prepared with 1% polyvinyl alcohol (PVA, 30–70 kDa) and varying percentages of PAA (17.5 kDa) in distilled water. The organic and aqueous phases were combined, chilled on ice, vortexed for 15 seconds, probe sonicated for 2 minutes on ice (QSonica Q500, 38% amplitude, 500 W), and the emulsion was stirred overnight. The NPs were then recovered by 3 rounds of ultracentrifugation (154,980 rcf), between which the pellets were suspended by gentle agitation into 4 mL distilled water and probe sonicated on ice for 45 seconds. After the final wash, the NP suspension was centrifuged (436 rcf) to pellet and separate out potential aggregates. Final NP suspensions were aliquoted with 3% sucrose in distilled water as a cryopreservative, lyophilized, and stored at −80 °C. For all experiments, NPs were constituted, vortexed, and bath sonicated on ice for 15 minutes to fully disperse the suspension prior to use.
Fluorophores were conjugated to the surfaces of prepared NPs through carbodiimide chemistry using manufacturer recommendations for AlexaFluor succinimidyl ester dyes. First, NPs were suspended in phosphate buffered saline (PBS). While stirring at 500 rpm, activated AlexaFluor dyes were slowly added to the NP suspension at a ratio of 10 μg dye:10 mg NP, a ratio identified as generating fluorescently bright NPs and still allowing for subsequent peptide conjugation, and then reacted for 2 hours at room temperature, protected from light. The different AlexaFluor molecules that were used are summarized in Table S1.
For peptide-functionalized NPs, collagen type II binding peptides (WYRGRLK, Biomatik, Inc, Wilmington, DE) were conjugated to the NP surface by carbodiimide chemistry between the peptide’s carboxylic acid terminus and the primary amines from the PAA on the particle surface. The peptide had been acetylated on the N-terminus to prevent cross-reactivity with the free acid terminus. Acetylation and conjugation to the free acid terminus did not impact the binding functionality of the peptide in other studies [18,19]. For conjugation of peptide to NPs, 300 μg of peptide was first prepared in 1 mL BupH MES buffer (0.1 M 2-(N-morpholino) ethanesulfonic acid, 0.9% sodium chloride, pH 4.7). The peptide was activated with 2mM EDC (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide) and 5mM sulfo-NHS (N-hydroxysulfosuccinimide) for 15 minutes while stirring at 500 rpm. The activated peptide was added to the fluorescently tagged NPs and reacted for 2 hours, stirring at 500 rpm at room temperature, protected from light. Conjugated NPs (with AlexaFluor and/or collagen type II binding peptide) were washed by centrifuging twice (154,980 rcf).
2.2. Nanoparticle Characterization
Characterization of NP size and zeta potential were conducted by dynamic light scattering (DLS, NICOMP Z3000, Particle Sizing Systems, Port Richey, FL) for which NPs were constituted to 0.2 mg/mL in distilled water at room temperature. Images of NPs were collected by transmission electron microscopy (TEM, TecnaiTM FEI Spirit, 120kV, ThermoFisher Scientific, Waltham, MA) with NPs suspended in distilled water and coated for 30 seconds with 0.2 μm-filtered uranyl acetate. Particle size by TEM was determined by manually measuring particles in ImageJ (version 1.52a) [33]. Both inner and outer diameters (Figure S2) were selected along the longest axis, every fully visible particle in the image was included in the measurement, and two separate images per particle type were assessed.
The presence of the peptide was detected through the microBCA Protein Assay (ThermoFisher Scientific, Waltham, MA). Briefly, NPs that had been formulated with and without peptide conjugation were plated in a 96 well plate at 0.2 mg NP in 0.15 mL of distilled water plus 150 μL of the assay working reagent (prepared per the manufacturer protocol). The plate was sealed to prevent evaporation and incubated at 37 °C, shaking at 60 rpm, for 2 hours. The plate was returned to room temperature, and absorbance was measured at 562 nm by plate reader.
The presence of primary amines was detected through the ninhydrin assay. Ninhydrin reagent was prepared fresh by dissolving 30 mg of hydrindantin and 200 mg of ninhydrin in 7.5 mL of dimethyl sulfoxide, and diluting this solution in 2.5 mL of 4 M sodium acetate buffer (sodium acetate trihydrate in distilled water, pH balanced to 5.2 with acetic acid). 100 μL suspensions of NPs in distilled water were plated in a 96 well plate, and 75 μL of ninhydrin reagent was added to each well. The plate was covered and incubated for 30 minutes at 80 °C. The reaction was then stopped by cooling to room temperature and adding 100 μL of 50% ethanol in water. The absorbance of each well was measured by plate reader at 596 nm.
2.3. Colloidal Stability
Colloidal stability was conducted by constituting NPs to 0.1 mg/mL in PBS in glass thimbles, and storing at 37 °Con an orbital shaker rotating at 60 rpm, protected from light. At each time point for up to 15 weeks, samples were brought to room temperature, measured by DLS for size, and returned to incubation conditions.
Stability of the colloids was also assessed in synovial fluid, as synovial fluid is capable of destabilizing nanomaterials [32,34,35] and would be present in vivo. AlexaFluor 750-tagged NPs were suspended to 10 mg/mL in PBS and added to bovine synovial fluid at ratios of 4:1, 2:1, 1:1, 1:2, and 1:4 (μL NPs:μL synovial fluid). Different ratios were tested to account for the potentially varying volumes of synovial fluid in the rat joint. These suspensions were mixed by vortexting and checked for gross aggregation, which can be visible to the naked eye. Samples incubated at 37 °C and 60 rpm were visually checked after 24 hours for signs of aggregation.
2.4. Poly(allylamine) and AlexaFluor Stability
Compositional stability of the NPs was studied in sink conditions via dialysis. As worst-case scenario, 5% PAA NPs were used as they have the most amines and conjugated fluorophores, which would likely make them more susceptible to dye loss. AlexaFluor 594-tagged 5% PAA NPs were constituted to 0.5 mg/mL in PBS or synovial fluid and placed in a dialysis device (100 kDa MWCO, Spectrum Labs/Repligen, Waltham, MA). Dialysis was conducted at 37 °C on an orbital shaker at 60 rpm, protected from light. The full dialysate was exchanged at 2, 24, and 72 hours. To determine the percent fluorescence loss, dialysate fluorescence was compared to that of a fresh suspension of NPs, free dye, or dye-PAA that had not undergone dialysis. Control groups included 0.002 mg/mL free AlexaFluor 594 in PBS and 1 mg/mL AlexaFluor 594-PAA conjugate in PBS. The dye-PAA conjugate had been prepared by reacting 0.02 mg AlexaFluor 594 with a 10 mg/mL suspension of PAA in 5 mL PBS for 2 hours at room temperature, then purified by dialysis (2 kDa MWCO, Spectrum Labs/Repligen, Waltham, MA) in water for 1.5 days. The free dye control was not assessed in synovial fluid, since protein-dye interactions could confound results. The PAA-dye control was not quantified in synovial fluid because it visibly aggregated in synovial fluid (this was not observed in NP samples).
2.5. Cyotocompatibility
Cytocompatibility of the NPs was conducted with primary chondrocytes, viable cartilage explants, and primary synoviocytes using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega, Madison, WI). Synoviocytes were studied to assess potential cytotoxic effects of the NPs in the synovium, an off-target tissue where injected substances are generally thought to accumulate. Throughout the studies, media type was matched for the cell type. Synoviocyte media comprised 4.5 g/L glucose Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Chondrocyte media, used for chondrocytes and cartilage, comprised 4.5 g/L glucose DMEM with 10 mM hydroxyethyl piperazineethanesulfonic acid (HEPES), 0.1 mM nonessential amino acids, 0.4 mM proline, 50 mg/L ascorbic acid, 10% FBS, and 1% penicillin-streptomycin. For cytocompatibility of cells in monolayer, chondrocytes and synoviocytes were isolated from bovine juveniles as done previously [32]. P0 chondrocytes and P2 synoviocytes were seeded at 3,000 cells per well in tissue culture treated 96 well plates and allowed to attach for 24 hours at 37 °C and 5% CO2. Suspensions of NPs were prepared by constituting lyophilized 1% PVA + 5% PAA NPs in sterile PBS to 10 mg/mL and diluting NPs to various concentrations in cell media. Cells were incubated with NPs for 24 hours, incubated in fresh media for 48 hours, and finally treated with MTS per manufacture’s protocols to quantify viability.
Cartilage cytocompatibility studies were conducted similarly to those with primary cells. First, cartilage was isolated from bovine juveniles under aseptic conditions and cut to 4 mm diameter × 1–3 mm height biopsy samples with the articular surface intact. These biopsies were incubated with chondrocyte media for 1 day at 37 °C and 5% CO2. Suspensions of NPs with and without fluorescent tags were made by constituting NPs in chondrocyte media. Biopsy samples were incubated with NPs for 24 hours, incubated in fresh media for 2.5 days, and finally treated with MTS reagent for 3 hours.
Detailed methods for cytocompatibility with monolayer cells and cartilage biopsies are included in the supplement. For both studies, wells with neither cells nor NPs were used to determine the background signal.
2.6. Ex Vivo NP Uptake into and Retention in Cartilage
Explants of biopsies were prepared for NP interaction studies as described previously [32]. Briefly, cartilage was isolated from bovine stifles and either maintained in PBS (“healthy”) or, as a structural ex vivo model of OA, enzymatically digested with 0.2% w/v collagenase (type 2, Worthington Biochemical, Lakewood, NJ) for 30 minutes (“OA”). Detailed methods regarding tissue preparation are included in the supplement. Biopsy samples were made from the prepared cartilage sections, whereby 6 mm biopsies were taken from the cartilage and trimmed to a height of 2–3 mm, leaving the articular surface intact.
To determine NP uptake into cartilage, biopsy samples were first placed into a grid that sealed the explant bottom and sides such that NPs would only be exposed to the articular surface. A suspension of fluorescently labeled NPs was prepared to 2.5 mg/mL in PBS and 40 μL was added to the articular surface of each biopsy sample, covering the entire exposed surface. The NPs were incubated on the explants protected from light for 1 hour, after which the NPs were removed and the explants were washed thrice with PBS. The cartilage was then diced with a scalpel into pieces (<1 cm3) and homogenized in 0.5 mL of water. Homogenate was plated in a white 96 well plate and fluorescence was measured by plate reader. Standard curves were generated to account for the differences in fluorescence across NP formulations. For standard curves, cartilage explants were taken from the same knee as that used for experimental samples, diced, homogenized in distilled water with known amounts of NP, and plated and measured as done with the samples.
The release of NPs was then determined using explants that had been loaded at the same time and under the same conditions as those prepared for NP uptake measurements. After NP loading and washing, explants were placed into non-treated 24 well plates with 0.75 mL PBS per well to fully cover each explant. Plates were incubated at 37 °C for 24 hours, protected from light, and sealed to prevent evaporation. After incubation, PBS was collected from each well and plated into a white 96 well plate. Fluorescence at 494 nm excitation/517 nm emission was measured by plate reader. Similarly to that done for NP uptake samples, standard curves in PBS were generated to account for the differences in fluorescence across NP formulations. Here, known masses of NP were added to PBS and plated as done for releasate samples. The mass of NPs retained in each explant (μg) was calculated as (average NP mass in explant after loading for each group) – (mass of released NP in PBS from each explant). Groups included each combination of NP formulation and tissue condition.
For both uptake and release samples, fluorescence imaging was performed on tissue cross sections. Explant cross sections of 1 mm or less were cut with a scalpel and imaged on glass dishes by fluorescence microscopy (AMG EVOS digital inverted microscope). It was observed that NPs consistently appeared dimmer in OA tissues compared to healthy, even when quantitation showed comparable levels of NPs. Therefore, when quantitation indicated similar levels of loading, post-processing image brightness was adjusted for “loaded” images of OA tissue to approximately match that of “loaded” images of healthy tissue. The same adjustment was then applied to the corresponding OA “retained” image.
2.7. OA induction in Rodents
OA was induced in the right knees of 24 skeletally mature Lewis rats (340.9 g ± 30.0 g, 14.4 ± 1.0 weeks old) via collagenase injection as a minimally-invasive rodent model of OA. Rats were obtained from Charles River Laboratories (Wilmington, MA). To induce OA, 500 U of sterile-filtered collagenase (type 2, Worthington Biochemical, Lakewood, NJ) was injected into the right knee at day 0 and 3. At the same times, the contralateral control limb received injections of sterile PBS. The dose amount and schedule were selected based on findings of significant OA-like damage in rats [36]. Detailed methods regarding OA induction are included in the supplement. Animals were monitored for 6 weeks during OA development, at which point they were either injected with NPs for biodistribution (n = 18 total, n = 6 per NP type) or euthanized for histological assessment of joint condition (n = 6 per time point). All methods were conducted with the approval of the University of Florida Institutional Animal Use and Care Committee and in conformance with Association for Assessment and Accreditation of Laboratory Animal Care recommendations on animal research.
2.8. In vivo Joint Biodistribution
Six weeks after OA induction by collagenase, NPs were administered to both healthy (left) and OA (right) knees, fluorescently tracked for retention and biodistribution. Briefly, lyophilized AlexaFluor 750-tagged NPs were prepared to 10 mg/mL in sterile PBS. The NPs were handled in a biosafety cabinet and transferred to an autoclaved septum-sealed glass vessel to preserve sterility, and were constituted on the morning of the injections. Rats were anesthetized and their knees were shaved and cleaned. To determine the baseline fluorescence signal in the knee, rats were imaged prior to injection of NPs by an In Vivo Imaging System (IVIS, Perkin Elmer, Waltham, MA). A 20 μL suspension of NPs was then injected into the intra-articular space, and each leg was flexed-extended 10 times to distribute the injected suspension throughout the joint space. Immediately after injection, each rat was imaged by IVIS. Settings for the IVIS were kept uniform throughout the duration of the study (excitation filter 745 nm, emission filter 800 nm, f/stop 2, photograph height 2.5 cm, medium binning, lamp level high, delay 0). Rats were imaged at regular intervals for 24 hours, then euthanized 48 hours post NP injection by cardiac puncture.
Knees were dissected to isolate major joint tissues and regions: the extensor mechanism (containing the distal quadriceps, quadriceps tendon, patella, patellar ligament, infra-patellar fat pad, and synovium), meniscus, distal femoral head, and proximal tibial plateau. The tibial plateau included the insertion points for the cruciate ligaments and, possibly, the insertion point of the patellar ligament. Additionally, extra-articular tissues were collected, including the medial collateral ligament, lateral collateral ligament, and its neighboring tendon of the lateral long digital extensor muscle origin. For imaging, the bony heads were oriented with the cartilagenous surfaces face up and the extensor mechanism was oriented with the intra-articular surfaces face up. As a control for potential tissue autofluorescence, three naïve knees were dissected and imaged in the same manner as those for NP biodistribution. These animals had not received any NP injections, were healthy, and were within a similar age and size range as the study animals (Lewis males, 16 weeks old, 355.2 g ± 16.6 g).
Semi-quantitative analysis of IVIS data was conducted by drawing regions of interest (ROIs) over the rat knees or knee tissues. The average radiance efficiency (RE, [p/s/cm2/sr] / [μW/cm2]) within each ROI used to normalize fluorescence for ROI surface area, which varied between tissue types. For biodistribution data, average RE for each tissue was converted to a percentage to normalize for differences in fluorescent capabilities across NP formulations. Here, % fluorescence signal = (signal from a single tissue from one knee)/sum(signals from all tissues of that knee), where the signal is an average RE. Exclusion of the ligaments, which exist outside of the joint, from this calculation did not change statistical trends (not shown).
To further study biodistribution within the extensor mechanism, the fat pad was removed from the patellar ligament after the initial IVIS imaging. The fat pad was isolated with surgical scissors, dissecting down to the patellar ligament. The separated tissues were then imaged side-by-side via the IVIS under the same settings used for biodistribution analysis.
2.9. Histology
Histology was performed on cartilage biopsies for the ex vivo assay and on whole rat knees for in vivo disease characterization. Both types of histological samples were not exposed to NPs. Cartilage biopsy samples were fixed in 10% formalin for 24 hours, embedded in paraffin, and sectioned. Sections were stained with Safranin-O/fast green or Masson’s Trichrome. Rat knees were isolated after euthanasia, fixed in 10% formalin for 48 hours, decalcified, embedded in paraffin, and sectioned in the coronal plane to the joint center in 5 μm sections. Sections were stained with Toluidine blue or Safranin-O/fast green. Detailed methods are available in the supplement. Disease was evaluated by grading Toluidine Blue stained slides using the OARSI scoring system [37] by a blinded grader in the GEKO software developed at the University of Florida (version 7) [38].
2.10. Reagent Sources and Equipment
The following reagents were obtained from ThermoFisher Scientific (Waltham, MA): ninhydrin, hydrindantin, dimethyl sulfoxide, AlexaFluor dyes (Life Technologies), Halt Protease Inhibitor cocktail (100X), PBS, EDC (Pierce), sulfo-NHS (Pierce), BupH MES buffer, DMEM (Gibco), nonessential amino acids, FBS, HEPES, penicillin-streptomycin, formalin, formic acid, Cal-Ex decalcifier, fast green, toluidine blue, sodium tetraborate, ethanol, hematoxylin, Iron(III) chloride hexahydrate, hydrochloric acid, Safeclear II, and Permount. The following were obtained from MilliporeSigma (Burlington, MA): PVA, PAA, sodium acetate trihydrate, acetic acid, sucrose, chloroform, Safranin-O, ascorbic acid, and proline. Distilled water was prepared in a Synergy Water Purification System (MilliporeSigma, Burlington, MA). Bovine synovial fluid was obtained from Animal Technologies, Inc. (Tyler, TX). All plate reader measurements were conducted on a SpectraMax M5e (Molecular Devices, San Jose, CA).
2.11. Statistics
All statistical analyses were conducted in GraphPad Prism version 8.0.0. In all figures, * p < 0.05, ** p < 0.01, *** p < 0.001 and NS = not significant. For all analyses, error bars represent standard deviations. Individual analyses are described in each figure caption.
3. Results
3.1. Nanoparticle Formulation and Functionalization
Stable NPs successfully formed with tunable surface properties. In all cases, the amount of PVA, which stabilizes the emulsion, was held constant; in formulations without PVA, stable colloids did not form (not shown). The hydrodynamic diameter of the particles was approximately 180 nm by DLS and was not significantly affected by PAA concentration in the aqueous phase (Figure 1 A). The zeta potential, however, increased with increasing concentrations of PAA, and plateaued at 0.4% PAA (Figure 1 B). To ensure sufficient amines for conjugation of dyes and targeting peptides while maintaining a positive zeta potential, an aqueous phase in excess of 0.4% PAA was used for subsequent studies. Accordingly, the cationic 5% PAA NPs were selected as “passive targeting” NPs in further studies. The 5% PAA NPs were further modified with the collagen type II binding peptide to generate “active targeting” NPs. The particles of neutral zeta potential (0.05% PAA) were selected as “untargeted” NP controls. The characteristics of these three selected particle formulations are summarized in Table 1. By TEM, these NP systems were round with a shell-core appearance characteristic of PLGA NPs coated with polymers and biologically-derived materials (Figure 1 C) [39–41]. The average difference between inner (core) and outer diameters was approximately 12 nm (Table 1, Figure S2).
Figure 1.

NP formulation and characterization. (A) Hydrodynamic diameter and (B) zeta potential of various formulations by DLS. (C) Contrast-enhanced TEM micrographs of the “untargeted” 0.05% PAA NPs (left), passive targeted” 5% PAA NPs (center), and “active targeted” 5% PAA + collagen type II peptide (right) NPs that were selected for further study. TEM scale bars = 100 nm. Statistics: n = 3 and compared via a one-way ANOVA.
Table 1.
Summary of NP formulations. Properties were measured on bare NPs, with no fluorescent tags. For all, n = 3. DLS size is Gaussian-distributed and number-weighted.
| Targeting strategy | Formulation (makeup of aqueous phase ) | Size (DLS) | Size (TEM outer diameter) | Size (TEM core diameter) | PDI (DLS) | Zeta potential |
|---|---|---|---|---|---|---|
| Untargeted (control) | 1% PVA + 0.05% PAA | 195.3 ± 14.8 nm | 79.2 ± 19.9 nm | 70.7 ± 14.3 nm | 0.038 ± 0.020 | −1.2 ± 1.4 mV |
| Passive | 1% PVA + 5% PAA | 172.4 ± 20.5 nm | 106.8 ± 32.9 nm | 91.1 ± 30.2 nm | 0.040 ± 0.007 | 22.7 ± 1.4 mV |
| Active | (1% PVA + 5% PAA NPs) + collagen type II binding peptide | 180.2 ± 8.0 nm | 97.3 ± 43.5 nm | 85.0 ± 36.5 nm | 0.043 ± 0.026 | 20.1 ± 9.1 mV |
As is common with polymeric NP systems [42], some degree of dispersity in NP size was observed, but the mean DLS polydispersity index (PDI) was 0.043 or less for all three formulations. The PDIs of the different NP systems were statistically the same (p > 0.954 via Tukey’s multiple comparisons tests). As is typically encountered with TEM analyses [43], the particle size by TEM was smaller than the average hydrodynamic diameter reported by DLS (Figure 1 C, Table 1). Size distribution analysis by DLS (NICOMP) showed a bimodal distribution of NPs for all three formulations (Figure S3) that corroborated the range of sizes visible by TEM.
After formulation, conjugation of fluorescent AlexaFluor tags to the particle surfaces did not significantly change NP size, likely because of the small size of the AlexaFluor molecule (<1k Da) relative to the NP (Figure 2). With fluorescent tagging, the zeta potential was reduced for passive and active NPs, although this change was only significant for active NPs. This reduction in zeta potential was likely a result of the reduction in amine density upon conjugation with both fluorophore and the peptide. Fluorescent tagging caused all three NP systems to change color to the naked eye and fluoresce as detected by plate reader, including the untargeted NPs, which contain a relatively low density of amine groups for surface conjugation (not shown).
Figure 2.

The effect of fluorescent tagging of PLGA NPs on (A) size and (B) zeta potential. Statistics: n = 3, compared with 2-way ANOVAs with Bonferroni-corrected multiple comparisons tests for plain vs. fluorescently tagged NPs.
The primary amine and peptide incorporation onto the NPs were further confirmed through biochemical assays. Between the three formulations selected for subsequent studies, the ninhydrin assay showed significant increases in the presence of amines from untargeted (0.05% PAA) to targeted NPs (5% PAA ± collagen type II binding peptide) (Figure 3 A). This increased incorporation of PAA was grossly observed by the coloration of the NP pellet (Figure 3 A, inset), as PAA is light brown to the naked eye. Active NPs had a reduction of amines on the particle surface compared to passive NPs, likely as a result of the additional conjugation between the peptide and PAA.
Figure 3.

Confirmation of amines and peptide incorporation into the NP system. (A) Primary amines were detected through the ninhydrin assay. (A, inset) The gross presence of PAA, which is light brown in solution, could be visualized as a change in color in the NP pellets after washing. (B) The presence of peptides in the active NPs (“+col2 peptide”) was confirmed through the microBCA assay after conjugation to passive NPs (“-col2 peptide”). Statistics: n = 3 for all groups and compared by a (A) one-way ANOVA with a Tukey’s multiple comparisons test and (B) student’s paired t-test. col2 = collagen type II binding.
The conjugation of the collagen type II binding peptide to 5% PAA NPs was confirmed by the microBCA assay (Figure 3 B). NPs with peptides had significantly more signal than unfunctionalized 5% PAA NPs, indicating the presence of the peptide. As an additional confirmation of peptide conjugation to the NPs, a FITC-tagged version of the peptide was used and these NPs also changed color and fluoresced, demonstrating successful conjugation (Figure S4).
3.2. Colloidal and Compositional Stability
Across multiple formulations, NPs were colloidally stable in PBS at 37 °C for over two months (Figure 4 A). NPs with PAA, AlexaFluor dye, and the collagen type II binding peptide showed signs of degradation at approximately 10 weeks, as determined by a significant reduction in NP size. NPs without dye maintained their size for 13–15 weeks. The NPs maintained gross colloidal stability in synovial fluid immediately after mixing, without any signs of gelation or sedimentation (Figure 4 B), which have been observed in other cationic polymeric systems [32,34,35]. This stability was observed across a variety of volumetric ratios of NPs to synovial fluid with a majority of NPs visibly still in suspension after 24 hours (Figure S5).
Figure 4.

Particle colloidal and chemical stability. (A) Colloidal stability of the NPs in PBS at 37 °C and 60 rpm, determined by longitudinal DLS measurements. (B) NP suspensions mixed with bovine synovial fluid at 1:1 (volumetric, NP:synovial fluid) as a visualization of gross NP stability. (C) Net change in fluorescence signal for AlexaFluor 594-tagged 5% PAA NPs (“NPs-dye”) over 72 hours of dialysis. SF = synovial fluid. Scale bar = 0.5 cm. Statistics: n = 3 for all assessments and (A) compared by a 2-way ANOVA with a Dunnett’s comparison to the time 0 value for each NP type, (C) 1-way ANOVA with a Tukey’s multiple comparison test.
To ensure that the conjugated fluorophore stayed associated with the NP in PBS and synovial fluid suspensions, dye release during dialysis was assessed using the 5% PAA NPs. After dialyzing in PBS or synovial fluid for 72 hours, about a 20% reduction in NP fluorescence was observed (Figure 4 C), with no statistical difference between PBS and synovial fluid suspensions. In contrast, both free dye and dye-PAA conjugates were able to completely pass through the dialysis membrane during this timeframe. Therefore, the reduction in NP fluorescence may be attributed to some degree of disassociation of the dye or dye-PAA from the NP surface. Overall, NPs demonstrated colloidal stability in PBS and synovial fluid, and a majority (>80%) of the AlexaFluor dyes remained associated with the NPs in physiologically relevant fluids.
3.3. Cytocompatibility
Cationic NPs can be cytotoxic due to their ability to disrupt the cell plasma and vesicular membranes [44]. To ensure that NPs did not have deleterious effects on the joint, viability was assessed using the NP system with the highest zeta potential, the passive NPs. Active NPs, which are functionalized from passive NPs, were not also assessed because the collagen type II binding peptide has demonstrated cytocompatibility in vivo across multiple NP systems [18,19].
Viability assays were conducted on multiple cell types of the joint. For both synoviocytes and chondrocytes, viability was maintained with at most a 24% reduction in synoviocyte viability at 1 mg/mL of 5% PAA NPs (Figure 5 A). Cartilage explants showed robust cytocompatibility with the NPs, with no reduction in viability up to 5 mg/mL NPs (Figure 5 B). AlexaFluor molecules are nontoxic and therefore were not expected to contribute cytotoxic effects, but as an additional precaution, tagged NPs were also tested with cartilage. The inclusion of a fluorescent tag on NPs did not impact cytocompatibility with cartilage (Figure 5 B).
Figure 5.

Cytocompatibility of cationic NPs after 24 hours of incubation with (A) chondrocytes and synoviocytes in monolayer and (B) whole cartilage explants. For cartilage explants, viability was tested with 5% PAA NPs with and without AlexaFluor 750 to verify that tagging did not change cytocompatibility. Statistics: n = 6 for all groups except cartilage control group (n = 12), and differences calculated through Dunnett’s comparisons to the ‘No NP control’.
3.4. Interactions with Cartilage
The effectiveness of NP targeting strategies was evaluated ex vivo by measuring uptake and retention in healthy and disease-mimicking cartilage (Figure 6). Safranin-O staining (red) confirmed the loss of proteoglycans on the articular surface of the OA-mimicked tissue after collagenase treatment (Figure 6 A). These proteoglycans continued to release during the subsequent 24 hour incubation (Figure 6 B). The collagen network, while enzymatically disrupted, was not removed through this treatment, as indicated by Masson’s Trichrome staining (blue). In fact, regions of lower proteoglycan density were correlated with deeper blue staining, potentially related to increased access to the collagen fibers by the Masson’s Trichrome reagents.
Figure 6.

NP interactions with healthy and OA-mimicked cartilage. (A) Histological appearance of tissue at the time of NP loading, and (B) after the 24 hour incubation in PBS. (C) NP loading into cartilage explants after 1 hour of surface-restricted incubation with NPs. (D) Retention of NPs in explants after 24 hours in PBS. Histological samples were not exposed to NPs. Scale bar = 1 mm. Statistics: n = 4–8 explants per group, compared by 2-way ANOVAs with Tukey’s multiple comparisons tests.
In assessing the amount of NPs that entered into the cartilage (referred to as “loading”), differences between NP types and tissue conditions were observed (Figure 6 C). In healthy cartilage, active NPs demonstrated 43% greater uptake compared to untargeted NPs, and comparable levels to passive NPs. Interestingly, in OA tissue, there was a 42% increase in loading of the untargeted NPs to levels comparable to both passive and active NPs, possibly due to increased permeability of the tissue. The loading of passive and active NPs did not significantly change between healthy and OA tissue.
NP targeting strategies strongly influenced the degree to which NPs were retained in the loaded explants after 24 hours. Untargeted NPs were essentially all released from healthy cartilage into the surrounding media after 24 hours, leaving only 2.4% of loaded NPs in the cartilage (Figure 6 D). Both passive and active NPs showed significantly improved NP retention relative to untargeted NPs in healthy cartilage, with active NPs retained at 1.9-fold higher levels than passive NPs. In OA cartilage, the retention of untargeted NPs was increased compared to healthy cartilage and was comparable to passive NPs. The active NPs continued to demonstrate improved retention, which was 3.1-fold higher than the passive NPs. Overall, in both healthy and OA conditions, active NPs had significantly more NP retention compared to the other NP systems, including passive NPs. Visualization of NPs in the cartilage matrix showed nearly full-thickness penetration into the tissue, with accumulation and retention most strongly occurring near the superficial zone (Figure 7).
Figure 7.

Visualization of the particles in explant cross sections via fluorescence microscopy, immediately after NP loading (“Loaded”) and after the 24 hour incubation in PBS (“Retained”). Tissue that had not been exposed to NPs serves as a control for potential cartilage autofluorescence. Note that comparisons should be drawn between “loaded” and “retained” images for given group due to differences in the fluorescent properties of the NPs. Fluorescence microscope objective 4X. Dotted line = cartilage articular surface; arrow = direction from articular surface to deeper zones within the cartilage; green = AlexaFluor 488-tagged NPs. Scale bar = 1 mm.
3.5. OA Animal Model Characterization
For in vivo testing, OA was established in rats via the collagenase-induced model, which was first characterized to identify a time frame for NP administration based on disease progression. Intra-articular injection of collagenase induced structural changes in the joint and proteoglycan loss as early as 1 week post injection (Figure S6). By 6 weeks, the joint exhibited features of early- to mid-stage OA: proteoglycan loss, cartilage fibrillation, and synovial thickening (Figure 8 A, Figure S6 A). The degree of proteoglycan loss as a percentage of total cartilage height was similar between the ex vivo and in vivo OA models, with degeneration occurring in the top 25–50% of the cartilage. Blinded grading of the histological sections indicated that, by 6 weeks, collagenase-treated knees had significantly more total cartilage degeneration and synovial thickness than contralateral control knees (Figure S6 B).
Figure 8.

Short-term joint retention of NPs in healthy (PBS, anatomical left) and OA (collagenase, anatomical right) knees. (A) Histological condition of the medial compartment of knees 6 weeks post OA induction. Tissue sections are animal-matched and stained with Safranin-O/fast green. Images were taken at 4X and (inset) 20X. (B) Representative appearance of IVIS images pre- (“baseline”) and post-injection with AlexaFluor 750-tagged passive NPs. Regions of interest (ROIs) were drawn over each knee as shown for semi-quantitative assessment of the fluorescence, as shown in the baseline image. (C) Fluorescent NP tracking via IVIS. S = synovium, M = meniscus, F = femur, T = tibia, red = proteoglycans (cartilage), green = nonspecific tissue. Black scale bar = 500 μm. White scale bar = 50 μm. Statistics: n = 6 and all six curves analyzed by area under the curve and a 2-way ANOVA with Bonferroni-corrected multiple comparisons tests for each NP type (healthy vs. OA).
To more fully understand the disease induced by this model, additional characterization was performed, including measurements of inflammation and bone structure. After the first injection, animals experienced acute swelling in the collagenase-treated limb (110% of the original knee width), which returned to approximately that of the control diameter within five days (Figure S7). Gross comparisons of bone structure by nanoCT showed mineralization on the collagenase-treated anterior meniscus and femur, particularly on the medial and lateral faces of the trochlea (Figure S7 C), but no obvious osteophytes were observed. Additionally, the diseased tibia was more anteriorly translated than the healthy tibia, an anatomical orientation associated with weakened joint ligaments (Figure S7 D) [45,46], which supported the idea that the treated knees were destabilized by ligament disruption. By CT, collagenase did not significantly change bone microstructure in the tibia (Figure S7 E–F), which agrees with findings of this model in mice [47]. Together, these data confirm that the collagenase injections were successful in initiating OA in rats likely from a combination of inflammation and joint instability, and that a 6 week time point was suitable for studying NP biodistribution in an early-to mid-stage OA model.
3.6. In Vivo Joint Biodistribution
Tracking of NPs in vivo showed localization of NPs to the knees with little to no autofluorescence in animals prior to injection (Figure 8 B). In the 24 hours after injection, the mean NP fluorescent signals were consistently lower in OA knees compared to healthy knees, but this trend was only significant for active NPs (Figure 8 C). Within healthy or OA knees, there were no differences in whole joint retention between the three NP types (p ≥ 0.126 via a Tukey’s multiple comparisons test of area under the curve).
Knees were dissected to determine the biodistribution of the NPs throughout the major joint tissues 48 hours following intra-articular administration (Figure 9). Overall, a majority of NPs were found in the extensor mechanism (Figure 9 B and C). Extensor mechanism accumulation was particularly prevalent in healthy knees, which had 1.7 to 2.0 times more NPs relative to diseased knees for both active and passive NPs. As anticipated, a low proportion of signal (< 10%) was associated with the collateral ligaments, which exist outside of the joint capsule, and no autofluorescence was observed in untreated tissues (Figure 9 D). The combination of cartilaginous tissues of the femur and tibia accounted for 26%, 32%, and 35% of signal for active, passive, and untargeted NPs in healthy joints, respectively. In OA, NPs were generally associated with cartilage to a greater extent, increasing to 46%, 42%, and 36% of signal. However, while both femur and tibia contain cartilage with similar proportions of signal, the spatial distribution of signal within these tissues suggested that there may be value in considering the femur and tibia separately. The signal in the femur was generally evenly distributed along the cartilage-containing trochlea and condyles (Figure 9 B). However, the signals in the tibial plateau (and menisci) were intermittently concentrated at anatomical sites of ligament and soft tissue insertion. Consequently, we used the signal from the femur as the representative metric for cartilage localization.
Figure 9.

Biodistribution of NPs in OA and healthy knees 48 hours post injection. (A) Five groups of tissues were isolated from each knee for biodistribution analysis: EM = extensor mechanism, F = distal femoral head, T = proximal tibial head, M = meniscus, and L = medial and lateral collateral ligaments and tendon of the lateral long digital extensor muscle. (B) Epi-fluorescent IVIS images of each of these tissues, normalized to the same colorimetric scale for each NP system. (C) Semi-quantitative analysis of IVIS images, reported as a percent of signal for each tissue type. Colorimetric scales of IVIS images were normalized across healthy and OA knees for each NP formulation. Scales were set to adjust for baseline differences in fluorescent capacities of the different NP systems (Figure S8). (D) Healthy animals that had not been exposed to NPs were included as a control for potential autofluorescence. Tissue scale bar = 0.5 cm. Statistics: n = 6 for all except no NP controls (n = 3), compared by 2-way ANOVAs with Bonferroni-corrected multiple comparisons tests.
Evaluation of NP biodistribution demonstrated important differences in cartilage localization between healthy and OA knees, particularly in the femoral cartilage (Figure 9 B and C). In healthy knees, a relatively small proportion of NPs localized within the cartilaginous tissues of the femur (12% for active, 14% for passive, and 14% for untargeted NPs). In agreement with our hypothesis, active NPs exhibited a significant increase in NP association with femoral cartilage from 12% in healthy knees to 21% of signal in OA knees. Interestingly, femoral cartilage localization did not change for the passive NPs and significantly increased for untargeted NPs in OA, which were both trends similarly observed in ex vivo studies.
Because a majority of the NP signal was associated with the extensor mechanism, which includes multiple tissues, the distribution of NPs in the extensor mechanism was more closely evaluated by isolating the fat pad from the patellar ligament and imaging again by IVIS (Figure 10). In healthy extensor mechanisms, a majority of NPs resided in the fat pad (88% for active, 74% for passive, and 79% for untargeted NPs). However, NPs in diseased joints were more evenly distributed between fat pad and other extensor mechanism tissues (“surrounding tissues”). For example, active NPs were distributed with 56% in the fat pad and 44% in the surrounding tissues, particularly in the synovial tissue on the periphery of the patella. Overall, biodistribution of all NPs within joint tissues was affected by disease state. Particularly for active and untargeted NPs, OA joints had reduced NP accumulation in the fat pad and increased accumulation in femoral cartilage.
Figure 10.

Biodistribution of NPs in the extensor mechanism. (A) After imaging the extensor mechanism for whole joint biodistribution, the fat pad was isolated from the patellar ligament by dissection, as shown. (B) IVIS fluorescence of separated extensor mechanisms and fat pads. (C) Semi-quantitative analysis of IVIS images for each NP system. Tissue scale bar = 1 cm. Statistics: n = 6, compared by 2-way ANOVAs with Bonferroni-corrected multiple comparisons tests.
4. Discussion
Recent advances in DMOADs have provided promising opportunities for slowing or reversing OA. A majority of DMOADs are designed to act at specific tissues and cells, driving the need to localize these drugs to distinct sites within the joint. As a response to this need, numerous nanoscale delivery systems have been developed with a variety of targeting strategies for cartilage [9]. However, there is a limited understanding of how these targeted systems localize to tissues within the joint, even though site-specific localization may be necessary for some drugs to function therapeutically. Moreover, it is not clear if or to what extent OA progression plays a role in the joint retention and biodistribution of targeted nanomaterials. By gaining a deeper understanding of where nanomaterials go within the joint and how this is influenced by the disease state, opportunities to enhance therapeutic outcomes of emerging drugs via strategic nanomaterial design may be illuminated. Here, a NP system was developed to compare two different targeting strategies: passive targeting through electrostatic attraction and active targeting through binding to type II collagen. These strategies were compared using the same base NP platform, and the effectiveness of cartilage localization was compared between healthy and diseased tissues.
The NP platform used in this study was strategically selected for its versatility and biocompatibility. PLGA-based NPs have a longstanding history as successful drug delivery vehicles for a broad spectrum of drug types, including both hydrophilic and hydrophobic molecules, peptides, and genetic material [48]. PLGA is also renowned for its biocompatibility, controllable biodegradation profiles, and ability to be modified for various biomedical applications [31]. PAA (sometimes referred to as PAH), the source of primary amines for particles in this study, has been utilized for particle formulation in various biomaterial applications. For example, PAA has recently been used as a coating on gold NPs for conjugation of a targeting antibody [49], as a polymer-drug conjugate for pH-sensitive drug delivery to cancer cells [50], and as a polycation for layer-by-layer nano-scale assembly [51,52]. In this study, we demonstrated that PLGA NP properties could be tuned by controlling the concentration of PAA in the aqueous phase during emulsification. Importantly, the amines from the PAA allowed for electrostatic-based cartilage targeting and, even at a neutral zeta potential, surface conjugation with functional molecules. Overall, this NP platform has utility as a tool for studying structure-function relationships, as done here, and as a strategic vehicle for future drug delivery applications in this and other biomedical fields.
For NPs to be effective drug delivery systems, in addition to being colloidally stable and non-toxic, they must have a size that enables them to cross the biological barriers relevant to their intended application. Collagen molecules, the principal structural component of cartilage, in part create the barrier to transport into cartilage and exist with a network mesh size of 50 – 200 nm [6,20,53,54]. The ability to move through this matrix is important for the delivery of most cartilage-specific drugs [7]. NPs in this study met this criterion; NPs were less than 200 nm by DLS, had a core size of 70–90 nm by TEM, and demonstrated penetration into cartilage after 1 hour of surface-restricted incubation on explants. However, it is important to recognize that while the polydispersity indices of these NPs are considered to be narrow by international standards for Dynamic Light Scattering [55,56], NPs were bimodally distributed. Differently-sized NP populations may exhibit differences in interactions with joint tissues and clearance kinetics. In this study, we cannot delineate between the different NP populations, so it remains unknown whether or not the observed tissue interactions are driven by a particular NP population. The goal of this study was to maintain size characteristics while manipulating surface properties; to continue advancing this emerging field of study, cartilage-targeting NPs may be further optimized by probing the effects of NP size on joint biodistribution.
The degree to which NPs enter the cartilage matrix ex vivo was influenced by targeting strategy and disease state. In healthy cartilage, there are rich networks of collagen type II and proteoglycans, which can serve both as a barrier to and target for NPs. The proteoglycan loss in the digested tissue increases ECM porosity, which could facilitate material penetration into the tissue [6]. However, this loss of charged molecules also weakens the electrostatic potential with cationic carriers, decreasing potential interactions with passive NPs. This interplay between ease of penetration and loss of electrostatic potential in OA cartilage was observed in the ex vivo uptake data; untargeted (not electrostatically attracted) NPs had increased uptake into OA tissue, but passive NPs did not. Surprisingly, passive NPs interacted similarly with healthy and OA cartilage despite the pronounced proteoglycan loss associated with the OA tissue. In a previous study of surfactant-containing NPs, cationic PLGA NPs experienced a significant reduction in cartilage interactions with OA cartilage relative to healthy cartilage, which was hypothesized to be a result of a reduction in electrostatic potential [32]. Although we did not observe a similar reduction with the cationic NPs in this study, both studies showed that there were no differences between cationic and non-cationic NP interactions with OA cartilage.
Ex vivo, the role of NP targeting strategies on cartilage interactions was most clearly illuminated by NP retention within the cartilage after loading. There were clear advantages to immobilizing NPs in the cartilage matrix via targeting, particularly in healthy tissues and for active NPs. This finding agrees with literature, where NPs functionalized with the collagen type II binding peptide had significantly improved cartilage extracellular accumulation compared to untargeted controls in healthy tissue [18]. Also in concordance with previous studies using cationic carriers [15,25], a passive targeting approach improved NP associations in healthy cartilage ex vivo. However, in OA tissue, passive NP retention was similar to untargeted NPs, highlighting the impact of proteoglycan loss on electrostatic targeting. Overall, targeting strategy had a strong influence over the NP retention in cartilage ex vivo, and the disease state of cartilage influenced NP-cartilage interactions differently for the three NP systems studied.
While the strength of the ex vivo model is its ability to isolate interactions between NPs and cartilage, an in vivo OA model was used to incorporate the complexity of the whole joint, which contains other tissues with the capacity to compete for NPs. For such studies, the collagenase model, a model that enzymatically destabilizes the joint was used. This model was selected for its longstanding history of use in mice [57–59], its simplicity and cost effectiveness, and its relevance to the ex vivo model, which used the same catabolic enzyme and established a comparable degree of proteoglycan loss. Additionally, this injection-based model is minimally invasive, thereby minimizing the direct disruption of joint tissues. Like all models, however, the collagenase model has limitations. This model exposes the knee to super-physiological levels of collagenase and is an indirect method of inducing joint instability. Clinically, joint instability often results from a traumatic injury [60,61], which is more commonly modeled in rodents by surgical, rather than enzymatic, ligament disruption [62]. However, surgical models also physically disrupt the synovial capsule in order to access the joint space, which could influence particle trafficking. In fact, a recent study demonstrated that a 500 kDa dextran cleared more slowly from surgically-destabilized joints compared to healthy joints [21]. This trend differs from what was observed in this study of collagenase-induced OA, although it is not clear if this disagreement is due to differences in animal model or the properties of injected material. In general, there is little knowledge of how different animal models affect joint clearance and biodistribution of NPs, particularly as NP targeting to cartilage is still in its infancy.
A principal goal of this study was to determine if and how the effectiveness of different NP targeting strategies for cartilage are influenced by disease state, which was studied through whole joint retention and, importantly, biodistribution. Multiple groups have used tissue-specific targeting as a strategy for prolonging whole joint retention [15,19,34,35]. In fact, the same collagen type II binding peptide used in our study has been shown to prolong whole joint retention of a small molecule drug carrier in healthy mice [19]. However, to our knowledge, the comparison of whole joint retention across healthy and diseased joints has only been published twice, using dextran molecules and single-walled carbon nanotubes [20,21]. These studies showed conflicting outcomes whereby some nanomaterials were affected by disease state [21] and some were not [20,21], illustrating the need for clarification and more studies in this field. In our study, no differences in whole joint retention were found between NP types in the first 24 hours post injection, although it is important to note that each NP system has different baseline fluorescent capabilities that may confound this comparison. However, when assessing within each NP type across healthy and OA knees, the extent to which they were retained in the joint did vary. The passive and untargeted NPs were not affected by disease condition, but active NPs had significantly more signal in healthy knees compared to OA knees, suggesting that both NP design and disease state influence the overall retention of NPs after intra-articular injection.
Interestingly, examination of NP biodistribution within the joint revealed differences in tissue localization that were not captured or explained by the whole joint retention data alone. Overall, NPs were more evenly distributed throughout the joint tissues in OA knees, whereas in healthy knees, a predominant proportion of the signal resided in the extensor mechanism. Of particular importance was the significantly increased proportion of active NPs in OA femoral cartilage relative to that in healthy knees, supporting the hypothesis that active NPs would more effectively localize with OA cartilage. This finding suggests that the active NPs likely benefit from improved access to the collagen type II network of diseased cartilage in vivo. This improvement in active NP localization to OA cartilage in vivo was not observed in the ex vivo model, which could be related to the differences in experimental and physiological conditions, such as competition from other joint tissues, increased heterogeneity of cartilage degeneration in vivo, and presence of synovial fluid. Nevertheless, similar patterns with passive NPs were observed ex vivo and in vivo, whereby no improvement to cartilage accumulation was observed in OA tissue relative to healthy tissue. This trend disproved part of our hypothesis that passive NPs would localize to a greater extent in healthy cartilage, and again highlights the trade-off between increased tissue porosity and decreased electrostatic interaction experienced by cationic NPs as cartilage undergoes OA pathogenesis.
This study underscores the value of considering joint biodistribution in addition to whole joint retention when evaluating nanomaterials for targeted intra-articular delivery. In other studies that evaluated whole joint retention of targeted NPs, it has been thought that improvements to retention likely resulted from associations between the NP and the target tissue [14,15,19]. In our study however, active NPs had significantly improved whole joint retention in healthy knees relative to OA knees, yet in healthy knees were primarily associated with off-targets (extensor mechanism), rather than the target (cartilage). Moreover, in OA joints with less whole joint retention, active NPs had improved associations with the targeted cartilage. Even for NPs with the same whole joint retention profiles in healthy in OA knees, the biodistribution within those knees varied. This discordance between retention and biodistribution emphasizes that while whole joint retention is a critical consideration for advancing OA drug delivery, biodistribution needs to also be assessed to determine the degree to which site-specific delivery has been achieved. These considerations would be particularly important for the delivery of biomolecules or drugs designed to act with a specific tissue or cell within the joint.
Biodistribution also showed interesting trends in fat pad localization. This study showed that in healthy tissues, a majority of the NPs were localized within the fat pad, but in OA, the NPs were more evenly distributed between the fat and the synovial tissue. The mechanism driving these fat and synovial interactions is unknown at this time. These changes may be related to the total reduction of NPs in the entire extensor mechanism and the pathological changes that occur in these tissues with OA, such as an increase in synovial vascularization, which may facilitate NP entry into synovial tissues [63]. While the intention of this work was to evaluate the fate of nanomaterials as a potential platform for cartilage-based DMOAD delivery, accumulation in the extensor mechanism is not necessarily undesirable. These findings could be applied to a nanomaterial system intended to deliver drugs targeting OA inflammatory or metabolic mechanisms in the fat pad and synovium, to ultimately address multiple, synergistic disease mechanisms in the different tissues within the joint. Additionally, these findings reinforce the utility of tunable nanoscale systems; NP systems such as the one studied here could serve as a useful platform for probing targeting strategies for other tissue types. For example, the synovium is another tissue for which numerous targeting moieties been developed across a wide range of nanomaterials [9], and there may be value in comparing these strategies across a single base nanomaterial.
This study represents an important first step to understanding the role of disease state and targeting strategies on NP fate in the joint. However, some limitations should be noted regarding this NP system and experimental conditions. For example, NP characterization was primarily conducted in water, even though the measured properties and biological fate of NPs can be influenced by biological media, including synovial fluid [32,42]. Still, the fundamental NP properties were likely playing a key role in biological fate, as the inclusion of amines and/or a binding peptide on the NP surface ultimately elicited changes biodistribution, even across the various ex vivo and in vivo media. Additionally, while it is understood that NPs enter and exit cartilage matrix in a concentration-dependent manner [7], the time scale at which this happens is unknown. Accordingly, the pharmacokinetics of the NPs with respect to cartilage entry or release cannot be determined by the biodistribution data at this single time point. Future work could improve these limitations and expand on these findings by exploring biodistribution at multiple and longer time points, and at more advanced stages of disease. Regardless, this is the first study to spatially show intra-articular tissue localization of NPs in healthy and OA knees, and highlights that joint biodistribution of nanomaterials is influenced by both the presence of disease and nanomaterial targeting strategy.
Last, it is important to consider the translation of nanomaterials into clinical settings. While incorporating targeting moieties into nanomaterials can improve drug delivery outcomes such as retention and biodistribution to target sites, particle functionalization can coincide with greater barriers to translation. Chemical modifications such as peptide conjugation to NP surfaces leads to formidable increases to cost and complexity when considering scale-up for clinical translation [64]. Therefore, moving forward, the additional benefits conferred by a targeting approach need to be scientifically justified in terms of drug efficacy.
In conclusion, this study evaluated NP fate across different cartilage-targeting strategies and disease conditions. NPs were designed to be cationic, a common passive targeting strategy for the anionic cartilage, or to include a binding peptide for collagen type II, an active targeting strategy for the cartilage ECM. These targeted NPs were also compared to neutral NPs of the same size and similar composition, which were considered to represent an untargeted nanomaterial. Ex vivo analyses indicated that targeting strategy strongly influenced the retention of the NPs within the matrix after loading. Here, the inclusion of a targeting strategy improved NP retention in both healthy and OA-mimicked cartilage matrix, with the greatest improvement observed in active NPs. After intra-articular injection in healthy and collagenase-induced OA rat knees, NPs primarily associated with the extensor mechanism, even for cartilage-targeting NPs. However, active NPs showed proportionately more association with femoral cartilage in OA knees compared to healthy knees, indicating that active targeting strategies may be advantageous for drug delivery to diseased cartilage. Due to the influence of both NP design and disease state on NP localization, this study highlights the value of evaluating biodistribution in addition to whole joint retention, and doing so in the disease state in which the nanomaterials are intended to be delivered. These considerations would be particularly important for the delivery of drugs designed to act at unique sites within the joint.
Supplementary Material
Statement of Significance.
As emerging drugs for osteoarthritis are becoming increasingly site-specific, the need for targeted intra-articular drug delivery has evolved. To improve drug delivery to cartilage, targeting strategies for nanomaterials have been developed, but the manner in which these targeted systems accumulate at different sites within the joint remains poorly understood. Moreover, it is unclear how nanomaterial-tissue interactions change in osteoarthritic conditions, as tissue structure and composition change after disease onset. By understanding how nanomaterials distribute within healthy and disease joints, we can advance targeted drug delivery strategies and improve therapeutic outcomes for emerging drugs.
Acknowledgements
This work was supported by the National Institutes of Health [grant number R01AR071335], the University of Florida Clinical and Translational Science Institute, which is supported in part by the NIH National Center for Advancing Translational Sciences [award number UL1 TR001427], and the Longenbaugh Foundation [grant number F023099].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Goldring MB, Update on the biology of the chondrocyte and new approaches to treating cartilage diseases, Best Pract. Res. Clin. Rheumatol 20 (2006) 1003–1025. doi: 10.1016/j.berh.2006.06.003. [DOI] [PubMed] [Google Scholar]
- [2].Hootman JM, Helmick CG, Projections of US prevalence of arthritis and associated activity limitations, Arthritis Rheum. 54 (2006) 226–229. doi: 10.1002/art.21562. [DOI] [PubMed] [Google Scholar]
- [3].Bay-Jensen A-C, Hoegh-Madsen S, Dam E, Henriksen K, Sondergaard BC, Pastoureau P, Which elements are involved in reversible and irreversible cartilage degradation in osteoarthritis?, Rheumatol. Int 30 (2010) 435–442. doi: 10.1007/s00296-009-1183-1. [DOI] [PubMed] [Google Scholar]
- [4].Loeser RF, Goldring SR, Scanzello CR, Goldring MB, Osteoarthritis: A disease of the joint as an organ, Arthritis Rheum. 64 (2012) 1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Janssen M, Mihov G, Welting T, Thies J, Emans P, Drugs and Polymers for Delivery Systems in OA Joints: Clinical Needs and Opportunities, (2014) 799–819. doi: 10.3390/polym6030799. [DOI] [Google Scholar]
- [6].Didomenico CD, Lintz M, Bonassar LJ, Molecular transport in articular cartilage - What have we learned from the past 50 years?, Nat. Rev. Rheumatol 14 (2018) 393–403. doi: 10.1038/s41584-018-0033-5. [DOI] [PubMed] [Google Scholar]
- [7].Bajpayee AG, Grodzinsky AJ, Cartilage-targeting drug delivery: can electrostatic interactions help?, Nat. Rev. Rheumatol 13 (2017) 183–193. doi: 10.1038/nrrheum.2016.210. [DOI] [PubMed] [Google Scholar]
- [8].Ng L, Grodzinsky AJ, Patwari P, Sandy J, Plaas A, Ortiz C, Individual cartilage aggrecan macromolecules and their constituent glycosaminoglycans visualized via atomic force microscopy, J. Struct. Biol 143 (2003) 242–257. doi: 10.1016/j.jsb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- [9].Brown S, Kumar S, Sharma B, Intra-articular targeting of nanomaterials for the treatment of osteoarthritis, Acta Biomater. 93 (2019) 239–257. doi: 10.1016/J.ACTBIO.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Buckwalter JA, Mankin HJ, Grodzinsky AJ, Articular cartilage and osteoarthritis, AAOS Instr. Course Lect 54 (2005) 465–480. doi: 10.1136/ard.51.9.1028-a. [DOI] [PubMed] [Google Scholar]
- [11].Luo Y, Sinkeviciute D, He Y, Karsdal M, Henrotin Y, Mobasheri A, Önnerfjord P, Bay-Jensen A, The minor collagens in articular cartilage, Protein Cell. 8 (2017) 560–572. doi: 10.1007/s13238-017-0377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Palmer AW, Guldberg RE, Levenston ME, Analysis of cartilage matrix fixed charge density and three-dimensional morphology via contrast-enhanced microcomputed tomography, Proc. Natl. Acad. Sci 103 (2006) 19255–19260. doi: 10.1073/pnas.0606406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stanescu R, Leibovich SJ, The negative charge of articular cartilage surfaces. An electron microscopic study using cationized ferritin., J. Bone Jt. Surg 64 (1982) 388–398. doi: 10.2106/00004623-198264030-00009. [DOI] [PubMed] [Google Scholar]
- [14].Bajpayee AG, Scheu M, Grodzinsky AJ, Porter RM, Electrostatic interactions enable rapid penetration, enhanced uptake and retention of intra-articular injected avidin in rat knee joints, J. Orthop. Res 32 (2014) 1044–1051. doi: 10.1002/jor.22630. [DOI] [PubMed] [Google Scholar]
- [15].Geiger BC, Wang S, Padera RF, Grodzinsky AJ, Hammond PT, Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis, Sci. Transl. Med 10 (2018) eaat8800. doi: 10.1126/scitranslmed.aat8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cho H, Stuart JM, Magid R, Danila DC, Hunsaker T, Pinkhassik E, Hasty KA, Theranostic immunoliposomes for osteoarthritis, Nanomedicine Nanotechnology, Biol. Med 10 (2014) 619–627. doi: 10.1016/j.nano.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cho H, Kim BJ, Park S-H, Hasty KA, Min B-H, Noninvasive visualization of early osteoarthritic cartilage using targeted nanosomes in a destabilization of the medial meniscus mouse model, Int. J. Nanomedicine 13 (2018) 1215–1224. doi: 10.2147/IJN.S149375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rothenfluh D, Bermudez H, O’Neil CP, Hubbell J, Biofunctional polymer nanoparticles for intra-articular targeting and retention in cartilage, Nat. Mater 7 (2008) 248–254. doi: 10.1038/nmat2116. [DOI] [PubMed] [Google Scholar]
- [19].Hu HY, Lim NH, Ding-Pfennigdorff D, Saas J, Wendt KU, Ritzeler O, Nagase H, Plettenburg O, Schultz C, Nazare M, DOTAM Derivatives as Active Cartilage-Targeting Drug Carriers for the Treatment of Osteoarthritis, Bioconjug. Chem 26 (2015) 383–388. doi: 10.1021/bc500557s. [DOI] [PubMed] [Google Scholar]
- [20].Sacchetti C, Liu-Bryan R, Magrini A, Rosato N, Bottini N, Bottini M, Polyethylene-glycol-modified single-walled carbon nanotubes for intra-Articular delivery to chondrocytes, ACS Nano. 8 (2014) 12280–12291. doi: 10.1021/nn504537b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mwangi TK, Berke IM, Nieves EH, Bell RD, Adams SB, Setton LA, Intra-articular clearance of labeled dextrans from naive and arthritic rat knee joints, J. Control. Release 283 (2018) 76–83. doi: 10.1016/j.jconrel.2018.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kang ML, Ko JY, Kim JE, Il Im G, Intra-articular delivery of kartogenin-conjugated chitosan nano/microparticles for cartilage regeneration, Biomaterials. 35 (2014) 9984–9994. doi: 10.1016/j.biomaterials.2014.08.042. [DOI] [PubMed] [Google Scholar]
- [23].Kang ML, Kim JE, Il Im G, Thermoresponsive nanospheres with independent dual drug release profiles for the treatment of osteoarthritis, Acta Biomater. 39 (2016) 65–78. doi: 10.1016/j.actbio.2016.05.005. [DOI] [PubMed] [Google Scholar]
- [24].Elsaid KA, Ferreira L, Truong T, Liang A, Machan J, D’Souza GG, Pharmaceutical nanocarrier association with chondrocytes and cartilage explants: Influence of surface modification and extracellular matrix depletion, Osteoarthr. Cartil 21 (2013) 377–384. 10.1016/j.joca.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bajpayee AG, Wong CR, Bawendi MG, Frank EH, Grodzinsky AJ, Avidin as a model for charge driven transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis, Biomaterials. 35 (2014) 538–549. doi: 10.1016/j.biomaterials.2013.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Laroui H, Grossin L, Léonard M, Stoltz JF, Gillet P, Netter P, Dellacherie E, Hyaluronate-covered nanoparticles for the therapeutic targeting of cartilage, Biomacromolecules. 8 (2007) 3879–3885. doi: 10.1021/bm700836y. [DOI] [PubMed] [Google Scholar]
- [27].Wang Y, Ng YW, Chen Y, Shuter B, Yi J, Ding J, Wang S. -c., Feng SS, Formulation of Superparamagnetic Iron Oxides by Nanoparticles of Biodegradable Polymers for Magnetic Resonance Imaging, Adv. Funct. Mater 18 (2008) 308–318. doi: 10.1002/adfm.200700456. [DOI] [Google Scholar]
- [28].Acharya S, Sahoo SK, PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect, Adv. Drug Deliv. Rev 63 (2011) 170–183. doi: 10.1016/J.ADDR.2010.10.008. [DOI] [PubMed] [Google Scholar]
- [29].Sahoo SK, Labhasetwar V, Nanotech approaches to drug delivery and imaging, Drug Discov. Today 8 (2003) 1112–1120. doi: 10.1016/S1359-6446(03)02903-9. [DOI] [PubMed] [Google Scholar]
- [30].Adjei IM, Yang H, Plumton G, Maldonado-Camargo L, Dobson J, Rinaldi C, Jiang H, Sharma B, Multifunctional nanoparticles for intracellular drug delivery and photoacoustic imaging of mesenchymal stem cells, Drug Deliv. Transl. Res 9 (2019) 652–666. doi: 10.1007/s13346-019-00621-6. [DOI] [PubMed] [Google Scholar]
- [31].Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V, PLGA-based nanoparticles: An overview of biomedical applications, J. Control. Release 161 (2012) 505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- [32].Brown S, Pistiner J, Adjei I, Sharma B, Nanoparticle properties for delivery to cartilage: the implications of disease state, synovial fluid, and off-target uptake, Mol. Pharm (2017). doi: 10.1021/acs.molpharmaceut.7b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods 9 (2012) 671–5. http://www.ncbi.nlm.nih.gov/pubmed/22930834 (accessed June 9, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim SR, Ho MJ, Lee E, Lee JW, Choi YW, Kang MJ, Cationic PLGA/eudragit RL nanoparticles for increasing retention time in synovial cavity after intra-articular injection in knee joint, Int. J. Nanomedicine 10 (2015) 5263–5271. doi: 10.2147/IJN.S88363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morgen M, Tung D, Boras B, Miller W, Malfait AM, Tortorella M, Nanoparticles for improved local retention after intra-articular injection into the knee joint, Pharm. Res 30 (2013) 257–268. doi: 10.1007/s11095-012-0870-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Adães S, Mendonça M, Santos TN, Castro-Lopes JM, Ferreira-Gomes J, Neto FL, Intra-articular injection of collagenase in the knee of rats as an alternative model to study nociception associated with osteoarthritis, Arthritis Res. Ther 16 (2014) R10. doi: 10.1186/ar4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gerwin N, Bendele AM, Glasson S, Carlson CS, The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rat, Osteoarthr. Cartil 18 (2010) S24–S34. doi: 10.1016/j.joca.2010.05.030. [DOI] [PubMed] [Google Scholar]
- [38].Kloefkorn HE, Jacobs BY, Xie DF, Allen KD, A graphic user interface for the evaluation of knee osteoarthritis (GEKO): an open-source tool for histological grading, Osteoarthr. Cartil 27 (2019) 114–117. doi: 10.1016/j.joca.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wei X, Ying M, Dehaini D, Su Y, V Kroll A, Zhou J, Gao W, Fang RH, Chien S, Zhang L, Nanoparticle Functionalization with Platelet Membrane Enables Multifactored Biological Targeting and Detection of Atherosclerosis, ACS Nano. 12 (2018) 109–116. doi: 10.1021/acsnano.7b07720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fonte P, Soares S, Costa A, Andrade JC, Seabra V, Reis S, Sarmento B, Effect of cryoprotectants on the porosity and stability of insulin-loaded PLGA nanoparticles after freeze-drying, Biomatter. 2 (2012) 329–39. doi: 10.4161/biom.23246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 10980–5. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gaumet M, Vargas A, Gurny R, Delie F, Nanoparticles for drug delivery: The need for precision in reporting particle size parameters, Eur. J. Pharm. Biopharm 69 (2008) 1–9. doi: 10.1016/j.ejpb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- [43].Diegoli S, Manciulea AL, Begum S, Jones IP, Lead JR, Preece JA, Interaction between manufactured gold nanoparticles and naturally occurring organic macromolecules, Sci. Total Environ 402 (2008) 51–61. doi: 10.1016/J.SCITOTENV.2008.04.023. [DOI] [PubMed] [Google Scholar]
- [44].McConnell KI, Shamsudeen S, Meraz IM, Mahadevan TS, Ziemys A, Rees P, Summers HD, Serda RE, Reduced Cationic Nanoparticle Cytotoxicity Based on Serum Masking of Surface Potential, J. Biomed. Nanotechnol 12 (2016) 154–64. http://www.ncbi.nlm.nih.gov/pubmed/27301181 (accessed May 8, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Patel RM, Brophy RH, Anterolateral Ligament of the Knee: Anatomy, Function, Imaging, and Treatment, Am. J. Sports Med 46 (2018) 217–223. doi: 10.1177/0363546517695802. [DOI] [PubMed] [Google Scholar]
- [46].Domnick C, Raschke MJ, Herbort M, Biomechanics of the anterior cruciate ligament: Physiology, rupture and reconstruction techniques., World J. Orthop 7 (2016) 82–93. doi: 10.5312/wjo.v7.i2.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang H, Lin C, Zeng C, Wang Z, Wang H, Lu J, Liu X, Shao Y, Zhao C, Pan J, Xu S, Zhang Y, Xie D, Cai D, Bai X, Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2, Ann. Rheum. Dis (2018) 1524–1534. doi: 10.1136/annrheumdis-2018-213450. [DOI] [PubMed] [Google Scholar]
- [48].Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM, Nano/micro technologies for delivering macromolecular therapeutics using poly(d,l-lactide-co-glycolide) and its derivatives, J. Control. Release 125 (2008) 193–209. doi: 10.1016/J.JCONREL.2007.09.013. [DOI] [PubMed] [Google Scholar]
- [49].Daems N, Penninckx S, Nelissen I, Van Hoecke K, Cardinaels T, Baatout S, Michiels C, Lucas S, Aerts A, Gold nanoparticles affect the antioxidant status in selected normal human cells, Int. J. Nanomedicine 14 (2019) 4991–5015. doi: 10.2147/IJN.S203546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhou T, Luo T, Song J, Qu J, Phasor–Fluorescence Lifetime Imaging Microscopy Analysis to Monitor Intercellular Drug Release from a pH-Sensitive Polymeric Nanocarrier, Anal. Chem 90 (2018) 2170–2177. doi: 10.1021/acs.analchem.7b04511. [DOI] [PubMed] [Google Scholar]
- [51].Santos AC, Veiga FJ, Sequeira JAD, Fortuna A, Falcão A, Pereira I, Pattekari P, Fontes-Ribeiro C, Ribeiro AJ, First-time oral administration of resveratrol-loaded layer-by-layer nanoparticles to rats – a pharmacokinetics study, Analyst. 144 (2019) 2062–2079. doi: 10.1039/C8AN01998C. [DOI] [PubMed] [Google Scholar]
- [52].Carvalho JA, da Silva Abreu A, Tedesco AC, Junior MB, Simioni AR, Functionalized photosensitive gelatin nanoparticles for drug delivery application, J. Biomater. Sci. Polym. Ed 30 (2019) 508–525. doi: 10.1080/09205063.2019.1580664. [DOI] [PubMed] [Google Scholar]
- [53].Martel-Pelletier J, Boileau C, Pelletier JP, Roughley PJ, Cartilage in normal and osteoarthritis conditions, Best Pract. Res. Clin. Rheumatol 22 (2008) 351–384. doi: 10.1016/j.berh.2008.02.001. [DOI] [PubMed] [Google Scholar]
- [54].Hall BK, Newman S, Cartilage: molecular aspects, CRC Press, Boston, 1991. https://www.crcpress.com/Cartilage-Molecular-Aspects/Hall-Newman/p/book/9780849388170 (accessed November 27, 2018). [Google Scholar]
- [55].Danaei M, Dehghankhold M, Ataei S, Hasanzadeh Davarani F, Javanmard R, Dokhani A, Khorasani S, Mozafari MR, Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems, Pharmaceutics. 10 (2018). doi: 10.3390/pharmaceutics10020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].ISO 22412:2017(en) - Particle size analysis - Dynamic light scattering (DLS), (2017). https://www.iso.org/obp/ui/#iso:std:iso:22412:ed-2:v1:en.
- [57].van der Kraan PM, Vitters EL, van Beuningen HM, van de Putte LB, van den Berg WB, Degenerative knee joint lesions in mice after a single intra-articular collagenase injection. A new model of osteoarthritis., J. Exp. Pathol. (Oxford). 71 (1990) 19 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1998679/ (accessed April 14, 2019). [PMC free article] [PubMed] [Google Scholar]
- [58].Gregory MH, Capito N, Kuroki K, Stoker AM, Cook JL, Sherman SL, A Review of Translational Animal Models for Knee Osteoarthritis, Arthritis. 2012 (2012) 1–14. doi: 10.1155/2012/764621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Little CB, Zaki S, What constitutes an “animal model of osteoarthritis” - the need for consensus?, Osteoarthr. Cartil 20 (2012) 261–267. doi: 10.1016/j.joca.2012.01.017. [DOI] [PubMed] [Google Scholar]
- [60].Carbone A, Rodeo S, Review of current understanding of post-traumatic osteoarthritis resulting from sports injuries, J. Orthop. Res 35 (2017) 397–405. doi: 10.1002/jor.23341. [DOI] [PubMed] [Google Scholar]
- [61].Lieberthal J, Sambamurthy N, Scanzello CR, Inflammation in joint injury and post-traumatic osteoarthritis, Osteoarthr. Cartil 23 (2015) 1825–1834. doi: 10.1016/j.joca.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Poulet B, Models to define the stages of articular cartilage degradation in osteoarthritis development, Int. J. Exp. Pathol 98 (2017) 120–126. doi: 10.1111/iep.12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Evans C, Kraus V, Setton L, Progress in intra-articular therapy, Rheumatol, Nat Rev 10 (2015) 11–22. doi: 10.1038/nrrheum.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A, Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities, Science. 338 (2012) 903–10. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.