


Abstract
G-quadruplex (G4) DNA structures are linked to key biological processes and human diseases. Small molecules that target specific G4 DNA structures and signal their presence would therefore be of great value as chemical research tools with potential to further advance towards diagnostic and therapeutic developments. However, the development of these types of specific compounds remain as a great challenge. In here, we have developed a compound with ability to specifically signal a certain c-MYC G4 DNA structure through a fluorescence light-up mechanism. Despite the compound's two binding sites on the G4 DNA structure, only one of them result in the fluorescence light-up effect. This G-tetrad selectivity proved to originate from a difference in flexibility that affected the binding affinity and tilt the compound out of the planar conformation required for the fluorescence light-up mechanism. The intertwined relation between the presented factors is likely the reason for the lack of examples using rational design to develop compounds with turn-on emission that specifically target certain G4 DNA structures. However, this study shows that it is indeed possible to develop such compounds and present insights into the molecular details of specific G4 DNA recognition and signaling to advance future studies of G4 biology.
INTRODUCTION
G4 DNA structures are noncanonical four stranded secondary DNA structures that can form in certain guanine rich DNA sequences. The central structural unit of G4 DNA structures are four guanines that bind to each other by Hoogsteen hydrogen bonds, forming a G-quartet (1). The G-quartets stack on top of each other to form a G4 DNA structure (2). The guanine rich DNA sequences that form the G4 DNA structure can have different orientations and the loops of random nucleotides that connects these guanine rich sequences can vary in their length, composition, and position thus giving rise to large structural variations among G4 DNA structures (3,4). There are ∼700 000 G4 DNA motifs in the human genome and many of these are evolutionary conserved (5), suggesting that they have important functions. G4 DNA structures has for long been associated to the telomeres at the chromosomal ends and their presence at these locations have been shown in vivo, e.g. using G4 DNA specific antibodies (6–8). In addition to their prevalence in telomeric regions, G4 DNA structures are also suggested to be involved in gene expression, (9,10) such as the regulation of the c-MYC gene (11). MYC proteins are transcription factors that governs the expression of many genes in the human genome, some of which are involved in e.g. apoptosis, oncogenesis, and proliferation (12–14). The expression of the c-MYC gene is estimated to be elevated or deregulated in up to 70% of human cancers (13). MYC is thus a very attractive drug target but it has proven difficult to develop compounds targeting this protein and it has therefore been considered undruggable (15). However, the c-MYC gene contain a G4 DNA motif that form a G4 DNA structure (11), and selective stabilization of this structure is thus an alternative strategy to suppress MYC transcription that has great potential (16,17). In fact, the vast majority of the c-MYC transcriptional activity is controlled by the nuclease hypersensitive element III (NHE III) (18), which is a 27 nucleotide G4-forming sequence of the c-MYC gene also known as Pu27 (19). This sequence can potentially form different G4 DNA structures but the predominant G4 DNA motif involved in c-MYC transcriptional silencing consist of four consecutive guanine sequences within Pu27 (20,21). This 22 nucleotide sequence has been modified with two mutations (G14T/G23T) to adopt the single predominant c-MYC promoter G4 DNA structure, known as Pu22 (22).
The prevalence and biological relevance of G4 DNA structures as well as their links to human diseases has spurred the development of compounds that stabilize G4 DNA structures. This has resulted in many different compound classes with strong binding and stabilization properties that also have good selectivity for G4 DNA structures over other DNA structures (e.g. bisquinolinium compounds, di-indoles, porphyrins, ethidium derivatives, and naphthalene diimide derivatives) (23–29). In general, these compounds have a flat and aromatic backbone that can form strong stacking interactions with the exposed G-tetrad/s on the top and/or bottom of the G4 DNA structure (5′ and/or 3′, respectively) (24,25,30). In addition, ligand induced conformational changes at the 3′ and/or 5′ DNA sequences flanking the G4 DNA structure to sandwich the compound have also proved to be highly important for an efficient binding (27,31–33).
Compounds that bind G4 DNA structures are of interest for potential therapeutic applications but also for their use as chemical research tools to study G4 biology. In particular, there is a strong and ever-growing interest in the development and use of research tools with turn-on emission upon binding to G4 DNA structures (34). In comparison to a potential drug candidate that targets G4 DNA structures, an optimal research tool should signal the presence of a G4 DNA structure by binding to it without affecting its structure and without stabilizing the structure. The probe should thus signal the presence of the specific G4 DNA structure but not affect its existence or appearance to allow native studies of G4 biology. Significant efforts have thus been devoted to the design and synthesis of such derivatives over the last decade which has resulted in some promising examples (34–38). However, considering the vast amount of potential G4 DNA structures in the human genome, it is clear that the specificity between different G4 DNA structures are of utmost importance for the development of valuable research tool compounds that can probe the presence of certain G4 DNA structures in cells under different conditions. This specificity has proven to be very complicated to achieve and predict, likely because all G4 DNA structures share the same core structure and the loops and flanking sequences that differ between the structures are normally hard to target with specific interactions. The successful design of these types of selective fluorescence light-up compounds thus require a deeper understanding of the factors that governs G4 DNA specificity and fluorescence light-up properties.
In this study, we synthesized a set of coumarin-benzothiazoles/benzoimidazoles and investigated both their ability to bind G4 DNA structures and their fluorescence light-up properties. One of the compounds displayed a remarkable selectivity for a certain c-MYC G4 DNA structure. To understand the molecular details of this selectivity, we used computational techniques such as molecular dynamic simulations and confirmed the results with biophysical techniques, i.e. fluorescence/absorbance spectroscopy, circular dichroism, and NMR. Insights gained from these studies and the corresponding chemical derivatives provide both a deeper understanding of how small molecules bind to G4 DNA structures and also show how very subtle G4 DNA structure alterations strongly can affect fluorescence light-up properties. This has implications for the future design and development of compounds targeting G4 DNA structures both as chemical research tools to explore G4 biology and for their further development towards diagnostics and therapeutics.
MATERIALS AND METHODS
Sample preparations for spectroscopy
Oligonucleotide sequences (Supplementary Table S1) were procured from Eurofins Genomics and were used as obtained. The solvents used were of spectroscopic grade. Stock solutions of 4b, 4l and 6b were prepared in DMSO, and the final experimental concentrations of all the compounds were kept in the order of 10−6 M, with <1% DMSO (v/v). The desalted oligonucleotides were dissolved in double distilled water and stored at 4°C. G4 DNA structure was prepared by taking a requisite amount of DNA from the main stock to the buffer solution, and the resulting solutions were folded by heating at 95°C for 5 min. The solutions were then slowly cooled to room temperature and equilibrated overnight at 4°C. All the experiments were carried out using 50 mM KCl, 10 mM KH2PO4 and 1 mM K2EDTA (pH 7.4) at room temperature (25°C).
Screening of compounds by fluorescence spectroscopy
All the compounds have been screened by looking at the fluorescence fold increase or decrease upon binding with the Pu22 c-MYC G4 DNA structure. The fluorescence of 5 μM compound was analyzed in the absence and presence of 10 μM Pu22 G4 DNA in a Jasco FP-6500 spectrofluorometer with a quartz cuvette of 1 cm path length.
Fluorescence titration
Fluorescence spectra were collected using Jasco FP-6500 spectrofluorometer, with a quartz cuvette of 1 cm path length. The slit width was set to 5 nm for excitation and 5 nm for emission beams. Fluorescence titrations were carried out by adding increasing amount of DNA to the cuvette containing fixed concentration of compound (5 μM). Emission spectra were recorded in the range of 500−650 nm for 4l and 6b, and 400−600 nm for 4b, at an excitation wavelength of 480 nm, and 385 nm respectively with successive addition of G4 DNA (0−10 μM). The obtained spectra were further normalized for each compound and DNA to reflect the fold change in intensity as function of DNA concentration. First, the wavelength with maximum intensity at the highest DNA concentration is identified and the spectrum without DNA (0 μM) is scaled to one at this wavelength (normalization wavelength: 480 nm for 4b with Pu22, 486 nm for 4b with Pu24T, 480 nm for 4b with htelo, 480 nm for 4b with duplex, 524 nm for 4l with Pu22, 528 nm for 4l with 5′ΔTGA-Pu22, 524 nm for 4l with 3′ΔTAA-Pu22, 525 nm for 4l with Pu24T, 527 nm for 4l with 5′ΔTGA-Pu24T, 527 nm for 4l with Pu27, 524 nm for 4l with htelo, 529 nm for 4l with duplex, 571 nm for 6b with Pu22, 574 nm for 6b with Pu24T, 571 nm for 6b with htelo and 567 nm for 6b with duplex DNA, Supplementary Figure S6). The obtained scaling factor was next applied on all other spectra (with same compound and varying DNA concentrations) for normalization. The intensity from the normalized spectra at the normalization wavelength were extracted and plotted as a function of DNA concentration. The fold change corresponds to the difference between the intensity of the compounds with DNA compared to that without DNA at the normalization wavelength.
UV−vis titration
The absorption spectra were recorded on a Varian Cary 5000 spectrophotometer using a quartz cuvette of 1 cm path length. During the absorption spectral measurement, the concentration of the compound was kept fixed (15 μM) while varying the concentration of added DNA from 0 to 30 μM. The spectra were normalized in the same way as described for the fluorescence titration above. The maximum intensity at the highest DNA concentration used in the normalization was 406 nm for 4b with Pu22, 430 nm for 4b with Pu24T, 427 nm for 4b with htelo, 429 nm for 4b with duplex, 500 nm for 4l with Pu22, 495 nm for 4l with Pu24T, 471 nm for 4l with htelo, 469 nm for 4l with duplex, 550 nm for 6b with Pu22, 551 nm for 6b with Pu24T, 554 nm for 6b with htelo and 554 nm for 6b with duplex DNA (Supplementary Figure S8).
Circular dichroism spectra
Circular dichroism (CD) spectra of the prefolded G4 DNAs were recorded at room temperature on a JASCO-720 spectropolarimeter (Jasco International Co. Ltd) using a cuvette of 1 mm path length. Each spectrum is the average of three scans over the wavelength range 230–330 nm with an interval of 1 nm at a scan rate of 100 nm/min.
Nuclear magnetic resonance
The G4 DNA stock solutions was prepared by folding 100 μM c-MYC Pu24T in 10 mM potassium phosphate buffer (pH 7.4) and 35 mM KCl by heating to 95°C and slowly cooling to room temperature overnight. 10% D2O and 10% DMSO-d6 was added to the DNA stock solutions, yielding a final DNA concentration of 82 μM. NMR samples were prepared by adding 0.9 μl of 20 mM DMSO stock solutions of 4b, 4l or 6b (1 eq.) to 220 μl of the DNA solution which was then transferred to 3 mm NMR tubes. Samples for the titration series of 4l was prepared in a similar way but with sequential addition of 4l to either Pu22 or Pu24T c-MYC G4 DNA. Control samples with Pu24T c-MYC G4 DNA with and without 10% DMSO was also performed to verify that DMSO did not have a significant effect on the DNA structure. All spectra were recorded at 298 K on a Bruker 850 MHz Avance III HD spectrometer equipped with a 5 mm TCI cryoprobe. Excitation sculpting was used in the 1D 1H experiments, and 256 scans were recorded. Processing was performed in Topspin 3.5 (Bruker Biospin, Germany).
Competition fluorescence displacement experiments
The experiments were performed on a Jasco FP-6500 spectrofluorometer equipped with a temperature controller at 25°C. For this assay, we have chosen Phen-DC3, which is one of the best G-quadruplex stabilizing ligands with end stacking binding mode reported as a competitor to displace 4l. Initially, apparent binding affinity of Phen-DC3 for Pu22 G4-DNA was determined by performing the well-known fluorescence intercalator displacement (FID) assay. 0.25 μM of pre-folded G4-DNA was mixed with 0.50 μM Thiazole orange (TO) and incubated for 2 min and fluorescence spectrum was recorded (λex = 501 nm; λem = 510 − 650 nm). Then Phen-DC3 was added to the mixture stepwise with a 2 min equilibration period, and the fluorescence spectra were recorded. The percentage of TO displacement was calculated from the fluorescence intensity (F) at the emission maxima, using the following equation:
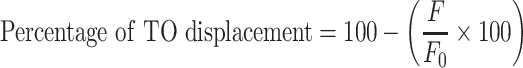 |
where F0 is the initial fluorescence intensity of TO bound to G4-DNA
The percentage of TO displacement was plotted as a function of the concentration of added Phen-DC3 and DC50 is determined. The association binding constant (Ka) of Phen-DC3 was calculated from the following equation using 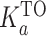 as 5.01 × 106 M−1 (39).
as 5.01 × 106 M−1 (39).
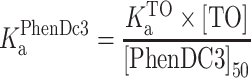 |
Next, similar experiment was performed to displace ligand 4l with Phen-DC3. Ligand 4I (2.5 μM) was added to the pre-folded Pu22 G4 DNA (1.25 μM) and the fluorescence spectra were recorded (λex = 490 nm; λem = 500–650 nm) with stepwise addition of Phen-DC3 which resulted the subsequent quenching. The background subtracted spectra were plotted and DC50 of Phen-DC3 for compound 4l was determined. By following the previous equations, the apparent dissociation binding constant was calculated.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed using a MicroCal Auto-iTC200 instrument (GE Healthcare) at 25°C. All samples were thoroughly degassed while being stirred prior to use. Buffers containing 10 mM potassium phosphate, 100 mM KCl and 1% DMSO (vol/vol) of pH 7.4 was used for the experiments. The sample cell was filled with 10 μM of the ligand which was titrated with 150 μM of pre-folded G4 DNA. The instrumental settings were: reference power 10 μcal·s−1, initial delay 60 s, stirring speed 1000 rpm, spacing 150 s, filter 5 s, injection 0.4 μl (first injection, duration 0.8 s) and 2 μl (subsequent injections, duration 4 s), and high-feedback mode. The experiment was repeated three times with the same results and no heat change was observed during the blank titration (buffer addition to the ligand solution). The obtained binding isotherms were analyzed with MICROCAL PEAQ-ITC analysis software. Because of the complexity of the data, the average of several fittings using a two binding site fitting model was employed to interpret the data and the corresponding apparent dissociation constants were calculated as the average (Supplementary Table S7).
G-Quadruplex complex modeling
For Pu24T and Pu22 c-MYC G4 DNA, structure coordinates were downloaded from PDB with PDB-ID as 2MGN (40) and 1XAV (22), respectively. From the Pu24T structure, the bound ligand was removed. In the next step, three-dimensional structural coordinates of 4b, 4l and 6b were generated using Avogadro package (41). To model the Pu24T c-MYC G4 DNA complexes, docking was performed at the 5′-terminal G-tetrad using Autodock Vina (42) and the two opposite conformations were selected for subsequent molecular dynamics (MD) simulations (Supplementary Figure S1). In case of Pu22, 4l was manually stacked with the 3′-terminal G-tetrad in two conformations using Chimera package (43) such that 4l formed hydrogen bonds with T20 and G22 (Supplementary Figure S12A). Two potassium ions were preserved inside the G-quadruplex channel to maintain its stability during the simulations. To model 4b bound with 5′ΔTGA-Pu22 c-MYC G4 DNA structure, the same binding mode was selected as obtained for the central structure of cluster-2 from the Pu24T-4b complex after MD trajectory clustering. To model 4b bound with 3′ΔTAA-Pu22, the same binding mode was selected as obtained for the central structure of cluster-1 from the Pu22-4b complex after MD trajectory clustering.
Molecular dynamics simulations
All G-quadruplex-compound complexes were prepared for molecular dynamics simulations using GROMACS (44) by placing these at the center of a periodic dodecahedron box and solvating with water molecules. Subsequently, the system was neutralized by adding an excess of 0.100 M KCl using the GROMACS tools. For the DNA, Amber99SB (45) with PARMBSC1 (46) improvements were used as force-field parameters. For water, the TIP3P model (47) was considered while ion parameters were taken from the following reference (48). For 4b, 4l and 6b, at first, their geometry was optimized and ESP was calculated with HF/6-31G* basis set using Gaussian-16 (49) and subsequently the partial atomic charges were calculated with the RESP method using the AmberTools package (50). All four compounds’ force-field parameters were generated from GAFF using the AmberTools package and converted to GROMACS format using acpype script (51). Subsequently, MD simulations were performed using GROMACS package (44) in several stages as presented in the flow-chart (Supplementary Figure S2). Parameter settings for all these stages were previously described in the following reference (27). Length of the MD simulations are summarized in Supplementary Table S2. Subsequently, all trajectories were merged for respective complexes, processed, and further used for the analysis.
Conformational clustering for each compound bound to their corresponding G-Quadruplex DNA structure was performed using our developed gmx_clusterByFeatures tool using PCA based conformational clustering (https://github.com/rjdkmr/gmx_clusterByFeatures). Subsequently, the first 50 frames of each cluster were considered for binding energy calculation using the g_mmpbsa tool (52,53). The exposed fraction of the coumarin rings’ surface area were calculated by dividing the coumarin ring surface area in bound-compound to compound surface area in unbound-compound and these areas were computed using the GROMACS tool sasa. The obtained MD trajectories were visualized and images were rendered using VMD (54).
Computation of absorbance spectra
At first, two planar and perpendicular conformations of 4b were generated and energy minimized using Avogadro. Subsequently, the first six excited states were calculated in implicit solvent water model by semi-empirical DFTB3 and TDDFT methods using the GAMESS package (55). For the DFTB3 method (56), 3OB-3-1 parameters (57,58) were considered while the self-charge-consistent (SCC) approach was employed with third order correction using Hubbard derivatives (56). For SCC convergence, second order SCF (SOSCF) orbital optimization was used. To model the solvent, conductor-like polarizable continuum model (C-PCM) with an iterative solver and solvation model density (SMD) (59) was considered.
To compute absorbance spectra of 4b bound to Pu22 and Pu24T c-MYC G4 DNA, the five conformations nearest to the central structures were extracted from each MD cluster and further energy minimized by the L-BFGS method with a force tolerance of 0.1 kJ mol−1 nm−2 using double precision GROMACS. When the excited states were calculated with similar parameter settings as described above for unbound 4b, >30 excited states were necessary to cover the full spectrum. However, memory consumption increases with an increase in excited states. Therefore, to reduce the memory consumption and the computation time and also to avoid convergence issues, each structure was truncated manually using Pymol (Schrodinger, LLC, The PyMOL Molecular Graphics System, Version 1.8, 2015) as shown in Supplementary Figure S14 and hydrogen atoms were modeled at the truncated position to fulfill the valency. Finally, 50 excited states were calculated with similar parameter setting as described above and the absorbance spectrum was extracted for each truncated structure.
RESULTS AND DISCUSSION
To achieve the desired light-up probes, we designed and synthesized coumarin-benzothaizoles and coumarin-benzoimidazoles with varying electron donating substituents on the coumarin and with or without a permanent charge on the benzothiazole/benzoimidazole to vary the electronic properties of the compounds. The synthesis of these derivatives was accomplished in two-three steps as shown in Figure 1A. First, commercially available substituted ortho-hydroxybenzaldehydes (1a–m) were subjected to Knoevenagel condensation with 2-benzothaizole acetonitrile or 2-benzimidazole acetonitrile in presence of catalytic amounts of piperidine to form iminocoumarin intermediates (2a–m and 3a–b). Subsequent hydrolysis with 2M hydrochloric acid gave the desired coumarin-benzothaizole (4a–n) and coumarin-benzoimidazole (5a–b) in quantitative yields. To investigate the effect of a permanent charge on these derivatives, 4a, l and 5a–b were methylated with methyl iodide under microwave conditions to give the desired quaternised derivatives (6a–d) in quantitative yields.
Figure 1.

(A) Synthesis scheme of the compounds. Reagents and conditions: (i) Piperidine, 2-benzothaizoleacetonitrile/2-benzimidazoleacetonitrile, absolute ethanol, RT, 12 h; (ii) 2 M HCl, ethanol, reflux, 12 h (61–76%); (iii) Pd/C, Hydrogen, MeOH, RT, 12 h (87%); (iv) DCE, Methyl Iodide, 125°C, Microwave, 1 h (87-89%). (B) Fluorescence fold increase of the different compounds (5 μM) in the presence of Pu22 c-MYC G4 DNA structure (10 μM). (C) NMR spectra of Pu24T c-MYC G4 DNA structure alone or after addition of the 3 compounds chosen for further studies; 6b, 4b and 4l. The imino protons that were the most affected by compound binding are marked with asterisks.
All compounds were screened for their fluorescence light-up properties upon interaction with the Pu22 c-MYC G4 DNA structure (Figure 1B) (for DNA sequence, see Supplementary Table S1). Several compounds showed interesting fluorescence properties, 4l and 6b showed a 12-fold fluorescence increase upon binding with Pu22 c-MYC G4 DNA structure, whereas 6a and 5b showed an about 10-fold and 6-fold fluorescence increase, respectively. On the contrary, 4b showed a 4-fold fluorescence decrease upon addition of the Pu22 c-MYC G4 DNA structure. Based on these observed fluorescence light-up/decrease properties, we continued our studies with 4l and 6b, which displayed the strongest fluorescence light-up properties, and 4b that resulted in a fluorescence decrease as a reference compound.
To further study the compounds interactions with G4 DNA we evaluated their ability to bind the Pu24T c-MYC G4 DNA structure using NMR (Figure 1C) (for DNA sequence, see Supplementary Table S1). When no compound is added to the Pu24T G4 DNA structure, the imino protons of the guanines in the G4 structure are clearly visible. However, upon addition of compounds, a clear line broadening or even disappearance of the guanines in the top (5′) G-tetrad is observed, thus suggesting that the compounds bind the 5′-terminal of the Pu24T G4 DNA structure. Compounds 4l and 6b seem to be the strongest binders whereas 4b did not seem to affect the NMR signals from this G4 DNA structure.
According to the NMR observations, the compounds bind to the G-tetrad at the 5′-terminal of Pu24T. However, how these compounds interact with the G4 structure at an atomic scale is not yet clear. To determine the binding conformation and to understand the difference in binding affinity between the compounds, we modeled the three compounds 4b, 4l and 6b at the 5′ face of the Pu24T c-MYC G4 DNA structure (PDB ID: 2MGN) in two different starting conformations (Supplementary Figure S1) and performed MD simulations (Supplementary Table S2 and Figure S2). In all these simulations, the compounds remained at the top of the G-tetrad throughout the simulations. After clustering of the compounds’ conformations from the MD trajectories, we obtained several clusters and considered the three largest, covering 50–70% of the simulation time, for further analysis (Supplementary Table S3). Interestingly, all three compounds preferably bound diagonally on the top G-tetrad in the largest cluster (Figure 2B–D). Additionally, 4l, and 6b proved to bind in different orientations along the G-tetrad's diagonal in the other clusters (Supplementary Figure S3). This was not observed for 4b and in the second and third largest clusters of this compound, it binds along the side of the G-tetrad (Supplementary Figure S3). Furthermore, all compounds formed stacking interaction with the nucleotides flanking the 5′-terminal of the G4 DNA structure (G-2 and A-3) in the major clusters and was thus sandwiched between these flanking nucleotides and the top G-tetrad (Figure 2C and D).
Figure 2.
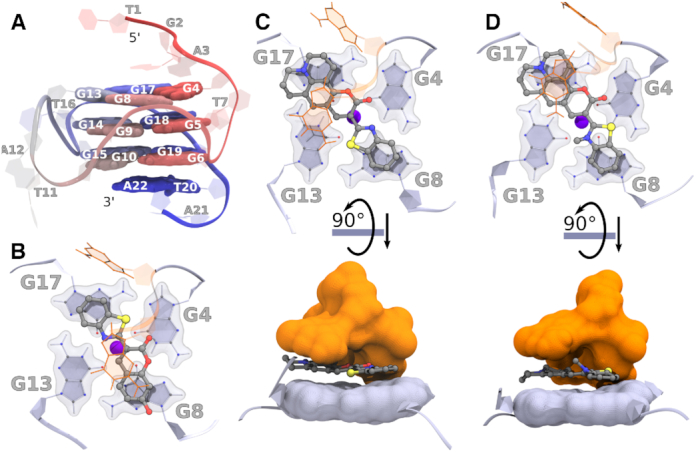
(A) Labeled overview of Pu22 c-MYC G4 DNA showing the structure with residue numbers only for representation purpose. (B–D) Representative binding poses of compounds during MD simulations with the Pu24T c-MYC G4 DNA. The top G4 tetrad (light blue), nucleotides flanking the G4 DNA structure (orange), and compounds (ball-stick model) from the largest cluster's central structure of (B) 4b, (C) 4l, and (D) 6b. The binding energies are tabulated in Supplementary Table S4. The potassium ion (central blue sphere) is also shown. The rotated view of 4l and 6b shows the compounds sandwiched between the top G-tetrad and the flanking nucleotides.
Subsequently, we calculated the binding energy for the three compounds to the Pu24T C-Myc G4 DNA structure using the MM-PBSA method (Supplementary Table S4). The obtained binding energies (order: 4b < 4l < 6b) are in well agreement with the result obtained from the NMR experiments (Figure 1C). Interestingly, the diagonal binding mode is most favourable for all the compounds. 4b is by far the weakest binding ligand and in two of the major clusters it is not interacting with at least one of the four guanine imino-protons in the top G-tetrad (distance over 5 Å) (Supplementary Figure S4), which together likely explains why no NMR shift or broadening was observed for 4b.
When the binding energy was decomposed atom-wise for 4b, 4l and 6b, the binding contribution conversely follow the partial atomic charges (Supplementary Figure S5) suggesting that the compounds’ charge distribution influence their binding. The charge variance for 4b is larger than for 4l and 6b on both the coumarin and the benzothiazole groups (Supplementary Table S5), which reduce the stacking interaction for 4b (Supplementary Table S6). The julolidine group in 4l and 6b result in strong stacking interactions in contrast to the corresponding hydroxyl substituents in 4b (Supplementary Table S6). When the benzothiazole is quaternized, as in 6b, the charge variance is further reduced (Supplementary Figure S5 and Table S5), which increases the stacking interaction of this ligand (Supplementary Table S6). Overall, these results suggest that by reducing the charge variance, the binding affinity increases through increased strength of the stacking interaction with the G-tetrad from below and the flanking sequence from above.
To investigate if the compounds' fluorescence light-up properties are selective to G4 DNA compared to double stranded DNA, or even between G4 DNA structures, we next performed fluorometric titration experiments with different G4 DNA structures and double stranded DNA as control (Figure 3 and Supplementary Figure S6).
Figure 3.
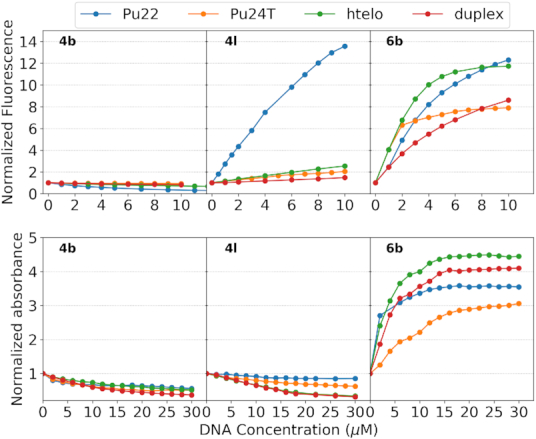
Top panel: Fluorometric titrations of 5 μM 4b, 4l and 6b with increasing concentrations (0-10 μM) of Pu22 c-MYC G4 DNA, Pu24T c-MYC G4 DNA, human telomeric G4 DNA (H-Telo), and duplex DNA, respectively. Detailed spectra for the fluorometric titrations are shown in Supplementary Figure S6. Bottom panel: Spectrophotometric titrations of 15 μM 4b, 4l and 6b with increasing concentrations (0-30 μM) of Pu22 c-MYC G4 DNA, Pu24T c-MYC G4 DNA, human telomeric G4 DNA (H-Telo), and duplex DNA, respectively. Detailed spectra for spectrophotometric titrations are shown in Supplementary Figure S8. The normalization procedure is outlined in the materials and methods section.
In aqueous buffer solution, 4l exhibits emission maxima around 530 nm with low emission intensity. Upon addition of Pu22 c-MYC G4 DNA, we found a significant enhancement (about 12-fold) in the emission intensity of 4l. Interestingly, there was no significant enhancement in the emission intensity of 4l in the presence of Pu24T c-MYC G4 DNA, human telomeric G4 DNA, or duplex DNA. 4l is thus not only selective for G4 DNA over duplex DNA but can also discriminate between very similar G4 DNA structures. Coumarin benzothiazole 6b only differ from 4l in one additional methyl substituent that quaternize the benzothiazole nitrogen, and 6b also exhibits emission maxima at around 530 nm. However, despite the structural similarities, we did not find the same G4 DNA specificity for this compound. Instead, the emission intensity of 6b was both enhanced and red shifted (emission maxima around 580 nm) upon interaction with all G4 DNA structures tested as well as with double stranded DNA (Figure 3). 4b shows emission maxima around 480 nm and, in contrast to the other compounds (4l and 6b), we observed a decrement in emission intensity of 4b without any significant spectral shift for both G4 DNA and double stranded DNA.
The Pu22 c-MYC G4 DNA sequence has two mutations (G14T/G23T) which makes it form the single predominant c-MYC promoter G4 DNA structure. The naturally occurring Pu27 sequence on the other hand, is known to form many different G4 DNA structures (60). The fluorescence light up effect of 4l in the presence of the folded Pu27 c-MYC G4 DNA sequence was 6.5-fold (Supplementary Figure S7A and B), which is in line with the formation of many different parallel G4 DNA structures. The parallel topology of these G4 DNA structures was confirmed using CD spectroscopy (Supplementary Figure S7C). Furthermore, the formation of several different G4 DNA structures from the folded G4 DNA sequence was confirmed using NMR and addition of 4l showed that it interacts with the formed Pu27 c-MYC G4 DNA structures (Supplementary Figure S7D).
We next monitored the changes in the absorption spectra of the compounds in the presence of Pu22 c-MYC G4 DNA, Pu24T c-MYC G4 DNA, human telomeric G4 DNA, and duplex DNA (Figure 3 and Supplementary Figure S8). The absorption spectra of 4l has two bands at 460 nm and 500 nm. In presence of Pu22 c-MYC G4 DNA, the absorption band of the higher wavelength becomes more prominent while for any of the other G4 DNA structures or double stranded DNA, the entire spectra display hypochromic effects. These data thus corroborate that 4l is interacting with the Pu22 c-MYC G4 DNA structure in a different way compared to the other DNA types in this study. In line with the fluorometric titrations, the absorbance spectra of compound 6b is strongly affected by the addition of all DNA types and display a significant decrement in the absorbance at 510 nm along with the generation of an absorption peak at 550 nm. 4b shows hypsochromic shift of the absorption maxima along with hypochromism in the case of Pu22 c-MYC G4 DNA and only hypochromic effects without any significant spectral shift for the other DNA structures.
From the above spectroscopic experiments, it is clear that the fluorescence light-up effect for 4l is much larger for the Pu22 c-MYC G4 DNA structure as compared to that of the Pu24T c-MYC G4 DNA structure. This observation cannot be explained by a 5′ binding mode because the 5′-G-tetrad and the flanking sequences for these two G4 DNA structures are identical (Supplementary Table S4). Binding at the 5′-terminal G-tetrad is thus expected to be the same between the two G4 DNA structures. In contrast, the 3′-terminal G-tetrad and flanking sequence differs considerably (Supplementary Table S1); Pu24T has a triplex (G20, A22, and G23) at the 3′-terminal that prevents compound binding (Figure 4A) and this triplex does not exist in Pu22, which instead has a flexible duplex (T20 and A22) that likely allow ligand binding (Figure 4B). This is in line with our NMR data, which show that 4l does not bind at the 3′-terminal of the Pu24T c-MYC G4 DNA structure (Figure 1C).
Figure 4.

Structure at 3′-terminal end of (A) Pu24T c-MYC G4 DNA structure (PDB ID: 2MGN) and (B) Pu22 c-MYC G4 DNA structure (PDB ID: 1XAV). Tetrads are shown in light blue while the nucleotides flanking the G4 DNA structure are shown in orange as cartoon representation. (C) Conformations of 4l bound at the 3′-terminal of Pu22 c-MYC G4 DNA obtained in the two largest clusters from the MD trajectories. Hydrogen bonds are also shown as blue dashed line. (D) NMR spectra of Pu22 and Pu24T c-MYC G4 DNAs in presence of different amount of 4l. Marked in yellow are the imino-protons least affected by 4l and b (3′-terminal or bottom), m (middle) and t (5′-terminal or top) refer to the different tetrads. Assignment of peaks are adopted from the literature (40,61).
Isothermal Titration Calorimetry (ITC) was next used to probe the dissociation constant for the interaction between the Pu22 and Pu24T c-MYC G4 DNA structures and 4l (Supplementary Figure S9). The respective DNA structures were titrated to 4l giving an apparent dissociation constant (Kd) of 2.3 μM for the Pu22 c-MYC G4 DNA structure and 54 μM for the Pu24T c-MYC G4 DNA structure (the average of several fittings using a two binding site fitting model was used) (Supplementary Figure S9 and Table S7). A second binding event is also suggested although the affinity is very much weaker (33 μM and 194 μM for the Pu22 and Pu24T c-MYC G4 DNA structures, respectively) and the uncertainty is high. To confirm the dissociation constant with the pu22 c-MYC G4 DNA structure and to probe the binding mode, we used the fluorescent properties of 4l and performed a competitive displacement assay using the well-known G4-binding compound Phen-DC3 (39). This data nicely corroborate the ITC data and showed an apparent Kd of 1.7 μM for the Pu22 c-MYC G4 DNA structure (Supplementary Figures S10–S11 and Table S7). This also further confirms the binding mode of 4l as Phen-DC3 bind G4 DNA structures by end stacking and efficient competition would thus not have been possible if 4l had another binding mode. Furthermore, binding of 4l to the Pu22 c-MYC G4 DNA structure did not induce any changes of the topology of the structure even at 8 equivalents of 4l as determined by CD (Supplementary Figure S12), which also is in line with an end stacking binding mode. These data thus suggest that there is a strong first binding of 4l to the G4 DNA structures however, it is not possible to know if this represent a binding to the 5′- or the 3′-terminal of the Pu22 G4 DNA structure or a combination of the two. Considering that the 5′ binding sites of the Pu22 and the Pu24T c-MYC G4 DNA structures are structurally the same, the increased affinity for the Pu22 G4 DNA structure suggest that the 3′-terminal is involved in 4l binding.
To study the possible binding modes at the 3′-terminal of the Pu22 c-MYC G4 DNA structure, we modeled two poses of 4l and performed MD simulations (Supplementary Figure S13A). This showed that 4l seem to be able to bind the 3′-terminal of the Pu22 c-MYC G4 DNA structure. Clustering of the MD trajectories shows that one starting conformation in the largest cluster remained the same during the simulations (Figure 4C and Supplementary Table S8) while the other starting conformation resulted in two different smaller conformational clusters (Figure 4C, Supplementary Figure S13B and Table S8). Binding energy calculations further show that binding energy of 4l is weaker at the 3′-terminal in the two largest clusters (Supplementary Table S6) as compared to the binding energy at the 5′-terminal (Supplementary Table S2), which can be explained by the lack of sandwiching by flanking sequences at the 3′-terminal. Further analysis also showed that 4l forms hydrogen bonds with the flanking nucleotides in all three clusters (Figure 4C and Supplementary Figure S13B).
To confirm these results, we performed NMR titration studies with both the Pu24T and the Pu22 c-MYC G4 DNA structures using 0, 0.1, 0.5, 1 and 2 eq. of 4l compared to G4 DNA (Figure 4D). The results show that 4l is mainly interacting with the 5′ G-tetrad of Pu24T c-MYC G4 DNA structure and even at higher concentrations of compound there is no significant interactions with the 3′-G-tetrad. On the contrary, when 4l is added to the Pu22 c-MYC G4 DNA structure, we observe binding that affect both the 3′- and 5′-G-tetrads even at a 2:1 relationship between G4 DNA and 4l. Importantly, 4l binding to the Pu22 c-MYC G4 DNA structure does not affect the fold of the G4 structure as determined by CD spectroscopy (Supplementary Figure S12). Detailed analysis of 4l binding to the 3′-G-tetrad of Pu22 c-MYC G4 DNA structure reveal that the G-10 and G-15 residues of the 3′-G-tetrad are strongly affected by compound binding whereas G-6 and G-19 residues of the 3′-G-tetrad are almost unaffected by 4l binding (Figure 4D). These results align well with the MD simulations, which show that 4l binds by stacking with the G-10 and G-15 residues while the G-6 and G-19 residues of the 3′-G-tetrad are less affected by 4l binding as they form stacking interactions with the flanking residues A-22 and T-20 (Figure 4C). Furthermore, the A-22 and T-20 residues interact through hydrogen bonds to each other and A-22 also form a hydrogen bond to 4l. Taken together, the MD simulations and NMR experiments show that 4l binds Pu24T c-MYC G4 DNA at only the 5′-G-tetrad and to both the 5′- and the 3′-G-tetrads of the Pu22 c-MYC G4 DNA. However, it is only the interaction with the 3′-G-tetrad of Pu22 c-MYC G4 DNA that cause a strongly amplified fluorescence response. Considering that the G-tetrads normally are very similar, this implies that the DNA sequences flanking the c-MYC G4 DNA structure are involved in these effects.
To further probe the reason behind the observed effects, we computed absorbance spectra of 4l with and without Pu22 c-MYC G4 DNA structure and the Pu24T c-MYC G4 DNA structure in implicit water with semi-empirical DFTB and TDDFT QM method using the GAMESS package. The computationally calculated absorbance of 4l is in well agreement with the experimentally observed spectra with peaks at 460 nm and 510 nm (Figures 5A and Supplementary Figure S8). The computational results suggest that the two peaks are caused by two different conformations, one with the coumarin and the benzothiazole in the same plane at 510 nm and one where they are perpendicular to each other at 460 nm (Figure 5A). When 4l bind to the Pu22 c-MYC G4 DNA structure, there is a strong tilt in the conformational distribution towards the planar conformation of 4l. This can be observed in the experimentally observed absorbance spectra by a strong relative increase of the 510 nm peak compared to the peak at 460 nm (Supplementary Figure S8). The combined computational and experimental absorbance spectra thus suggest that 4l bind to the Pu22 c-MYC G4 DNA structure in a planar conformation which leads to the increased fluorescence.
Figure 5.
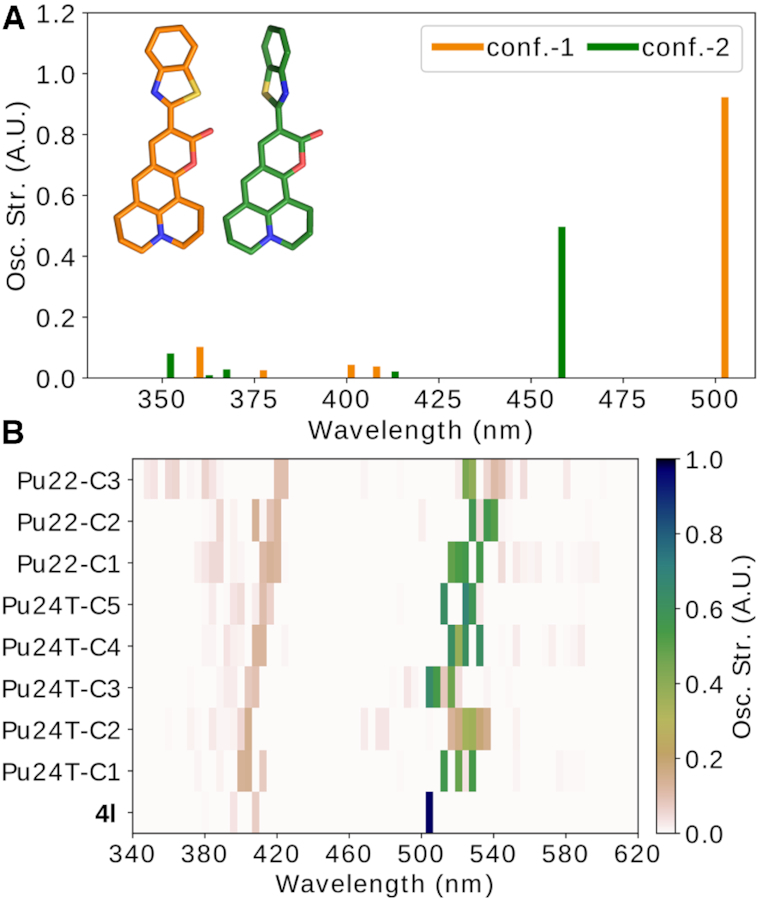
Computed absorbance spectra of 4l with and without Pu24T and Pu22 c-MYC G4 DNA based on the MD simulations. The peak at 460 nm represent 4l in solution and the peak at 510 nm represent 4l bound to DNA, in the MD simulations we only study the bound state. (A) Computed absorbance spectra of unbound 4l in planar (orange) and perpendicular (green) conformations. (B) Computed absorbance spectra of 4l bound to the Pu24T 5′-terminal or the Pu22 3′-terminal structures from the five and three largest clusters obtained from the MD simulations, respectively (see details in the methods section).
To compute the absorbance spectra of 4l with Pu22 or Pu24T c-MYC G4 DNA structure, both G4 DNA structures had to be truncated at the 3′- or 5′-terminal, respectively, to reduce the memory consumption and to avoid convergence issues. Further investigations showed that these truncations did not affect the absorbance peaks (Supplementary Figure S14). The obtained absorbance spectra of 4l bound to the Pu22 or Pu24T c-MYC G4 DNA structure show that the absorbance overall is decreased by 45–50% upon binding (Figure 5B). However, the absorbance decreases by up to 75–80% in the clusters where the coumarin in 4l is less exposed to the water surrounding, i.e. in cluster-2 and cluster-3 of Pu24T and Pu22, respectively (Supplementary Figure S15 and Figure 5B). It is also these clusters that display the strongest binding affinity (Supplementary Table S2, cluster 2, and Supplementary Table S6, cluster 3). These results therefore suggest that the sandwich effect by the flanking sequences increase the binding affinity. However, the sandwich effect also covers most of 4l and reduce its absorbance.
To further investigate the impact of the c-MYC G4 DNA structure's flanking sequences on the fluorescence light-up effect and selectivity of 4l, we deleted the flanking sequences of the confirmed binding areas of 4l (5′-flanking TGA nucleotides of Pu22 (5′ΔTGA-Pu22) and Pu24T (5′ΔTGA-Pu24T) as well as the 3′-flanking TAA nucleotides of Pu22 (3′ΔTAA-Pu22)) and performed fluorometric titrations (Figure 6A and Supplementary Figure S16). Interestingly, this shows that when the 5′-flanking sequence of Pu24T c-MYC G4 DNA structure is deleted, the fluorescence light-up effect is doubled (albeit from a low level, from a two-fold increase to a four-fold increase). This result supports the hypothesis that fluorescence of 4l is partly quenched by the 5′-terminal flanking nucleotides and therefore partly explains the low fluorescence light-up effect observed with Pu24T c-MYC G4 DNA (Figure 3).
Figure 6.
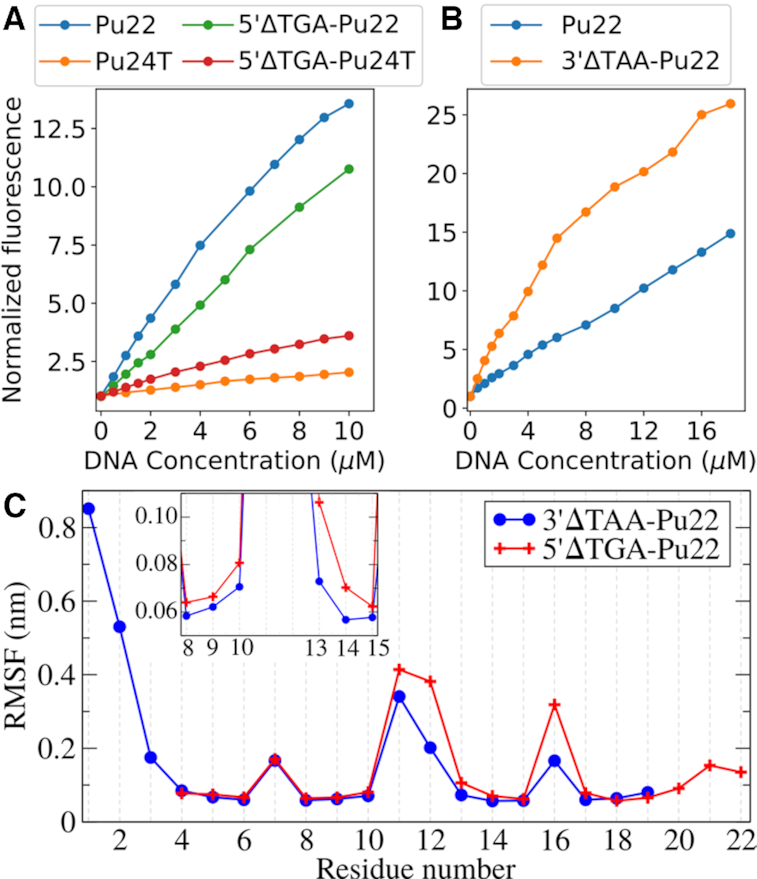
(A) Fluorometric titrations of 5 μM 4l with increasing concentrations of Pu22 c-MYC G4 DNA and Pu24T c-MYC G4 DNA and their truncated 5′ΔTGA structures. (B) Fluorometric titrations of 2.5 μM 4l with increasing concentrations of Pu22 c-MYC G4 DNA and its 3′ΔTAA structure. (C) RMSF as a function of residue number calculated from the largest cluster for Pu22 c-MYC G4 DNA’s truncated structures 5′ΔTGA and 3′ΔTAA bound to 4l. Inset panel shows zoomed view of RMSF for residues between G-8 and G-15.
In contrast to the Pu24T c-MYC G4 DNA structure, the fluorescence intensity of 4l is only slightly affected (from 13 to 11 fold) when the 5′-flanking nucleotides of the Pu22 c-MYC G4 DNA structure is deleted (which is in line with losing the contribution from 4l binding at the 5′-G-tetrad). However, when the 3′-flanking nucleotides of the Pu22 c-MYC G4 DNA structure is deleted, a clear increase in the fluorescence light up effect is observed (Figure 6B and Supplementary Figure S16C). Importantly, the deletions did not affect the oligonucleotides ability to form a G4 DNA structure or induce any major changes in the topology of the structure (Supplementary Figure S17).
When the 5′-terminal flanking nucleotides of the Pu22 c-MYC G4 DNA structure are deleted experimentally, 4l will likely continue to bind to the intact 3′-terminal and this will thus not have a strong impact on the fluorescence light-up effect. However, the strong fluorescence increase of 4l when the 3′-terminal flanking nucleotides are deleted experimentally, suggest that even though the 5′-terminal with intact flanking residues are available for binding, 4l is also binding to the 3′ face of the Pu22 c-MYC G4 DNA structure. Even though this binding interaction likely is significantly weaker because of the absence of interactions with the flanking nucleotides, the overall fluorescence intensity originating from the 3′-bound 4l is still increased thus supporting a fluorescence quenching mechanism related to the nucleotides flanking the G4 DNA structure.
Deletion of the 5′-terminal flanking nucleotides of the Pu24T c-MYC G4 DNA structure results in a fluorescence increase but it is clearly not as powerful as the light up effect of 4l interacting with the 3′ΔTAA-Pu22 c-MYC G4 DNA. This shows that 4l discriminate between the 3′- and the 5′-G-tetrads also with absent flanking nucleotides. To understand the basis for this G-tetrad selectivity, we performed MD simulations of 4l bound to either the 5′ or the 3′-terminal G-tetrad in 5′ΔTGA and 3′ΔTAA variants of Pu22 c-MYC G4 DNA, respectively. These MD simulations complements the experimental data as it only describes 4l when bound to either the 3′- or 5′-terminal of the G4 DNA structure and not the average from the bound/unbound equilibria at both the 3′- and 5′-terminals. Clustering of 4l conformations from the obtained MD trajectories reveal four binding modes along the G-tetrad diagonals (Supplementary Figures S18 and S19). The population of the four binding modes were almost equal at the 5′-terminal (Supplementary Table S9). However, at the 3′-terminal, one binding mode clearly dominated and subsequent binding energy calculations (by MM/PBSA method) show that this binding mode also has the strongest binding affinity of all binding modes at both terminals (Supplementary Table S9). Interestingly, the deletion of the flanking sequences strongly reduced the binding affinity at the 5′-terminal whereas the 3′-terminal was less affected (Supplementary Table S9 versus S4 and S8, respectively), which is in line with the sandwich effect at the 5′-terminal which is not as prevalent at the 3′-terminal. Worth noting is also that the binding affinity of 4l at the 5′-terminal was lowest in the largest conformational cluster from the simulations.
Overall, the experimental data and the simulations suggest that the binding energy/affinity of 4l is less affected at the 3′-terminal compared to the 5′-terminal when the flanking nucleotides are deleted at the respective terminals. Moreover, 4l seems to bind stronger to the 3′-terminal compared to the 5′-terminal when the flanking nucleotides are deleted at the respective terminals. This is surprising considering that G-tetrads normally are very similar in nature and the differences between the 3′- and the 5′-G-tetrads of the c-MYC G4 DNA structure are very small. In fact, when the flanking nucleotides are deleted, the only difference between the G-tetrads is the orientation of the T-11/A-12 loop (Figure 2A). Closer investigations of the simulations show that the differences in binding affinity is correlated with the polar-solvation energy (Supplementary Figure S20A), which in turn is linked to the flexibility of the loops and the guanines in the G4 DNA structure. When the flexibility of the residues was compared (by calculating root mean square fluctuations (RMSF) of the largest cluster), the T-11/A-12 and T-16 loops were more rigid in the 3′ΔTAA-Pu22 c-MYC G4 DNA structure as compared with that of the 5′ΔTGA-Pu22 variant (Figure 6C). Importantly, this observation was confirmed by using a full 1 μs trajectory for RMSF calculation (Supplementary Figure S20B). This larger flexibility of the loops subsequently affects the 5′-terminal G-tetrad flexibility, particularly of G-13 (inset Figure 6C) and G-17. These differences in flexibility is potentially caused by the larger Adenine (A-12) that is connected to G-13 at the 5′-G-tetrad compared to the Thymine (T-11) that is connected to G-10 at the 3′-G-tetrad. This heterogenous flexibility of the loop is thus a plausible explanation for the observed differences in binding and fluorescence light-up of 4l between the 3′- and the 5′-G-tetrads of the c-MYC G4 DNA structure, which also could explain the similar occurrence of four different binding modes at the 5′-G-tetrad compared to the more distinct binding mode at the 3′-G-tetrad. The differences in fluorescence light up between the 3′- and the 5′-G-tetrads can thus be explained in part by a stronger affinity to the 3′-G-tetrad, which is correlated to a difference in flexibility between the two G-tetrads. This flexibility is likely also affecting the fluorescence light-up effect of 4l by influencing its conformation. Indeed, when the angle of 4l was calculated over the simulations it is clear that 4l is present in the planar conformation that is required for the fluorescence to a higher extent at the 3′-G-tetrad compared to the 5′-G-tetrad (Supplementary Figure S20C).
CONCLUSION
G4 DNA structures have over the last years been reported to play important roles in central biological processes that has resulted in a growing interest in G4 DNA structures. With this growing interest comes the need to use small molecules to specifically target G4 DNA structures over other DNA structures in general, and to specifically target certain G4 DNA structures in particular. To further expand the utility of these compounds as research tools, the ability to signal the specific binding of certain G4 DNA structure, e.g. by turn on emission, is highly desired. However, the development of these types of compounds have proven to be really difficult with only very limited examples. Considering the vast amount of G4 DNA structures, it is of utmost importance to understand the molecular details of how small molecules interact with G4 DNA structures, and the factors that govern turn on emission, to improve and facilitate the development of these compounds and thus advance the understanding of G4 biology.
In here, we have designed and synthesized a set of coumarin-benzothiazoles/benzimidazoles and identified one compound, 4l, with ability to bind and specifically fluorescently light-up the pu22 c-MYC G4 DNA structure without perturbing the overall topology of the G4 DNA structure. Very small structural changes in the compounds resulted in large differences in both the compounds' specificity and turn-on emission. To understand the molecular details behind G4 DNA binding, selectivity, and fluorescence light-up effects, we used a combination of computational and spectroscopic techniques. This showed that it is the planar conformation of 4l that is responsible for the fluorescence light-up effect and this is predominating the bound form of the compound. Furthermore, we could show that the compound is able to bind both the 3′- and 5′-terminals of the G4 DNA structure but it is the 3′-terminal binding that results in the strong fluorescence light-up effect. The flanking nucleotides at the 3′- and 5′-terminals of the G4 DNA structure proved to be of key importance for this effect, binding at the 5′-terminal was strongly reduced upon deletion of these flanking sequences whereas the 3′-terminal binding was less affected. Deletion of these flanking nucleotides suggest that the fluorescence and absorbance of 4l in part was quenched by these additional interactions with the flanking nucleotides. However, even with the flanking sequences deleted, we observed a clear difference in fluorescence light-up and binding between the 3′- and the 5′-terminal thus showing that it is possible to discriminate between the structurally similar G-tetrads of G4 DNA structures. This G-tetrad selectivity could be linked to the loops of the G4 DNA structure that resulted in a difference in flexibility between the G-tetrads. This flexibility reduced the binding affinity and reduced the planar conformation of 4l which thus lead to the significant difference in fluorescence light-up effect. Therefore, a rigid and accessible G-tetrad is a prerequisite for the design of this type of fluorescence light-up probes that are based on the enrichment of a planar conformation upon binding the G4 DNA structure. Additional selective interactions such as hydrogen bonds can target the flanking residues although the additional stacking interactions with these flanking residues, which will increase binding affinity (e.g. by a low charge variance), likely will reduce the light-up effect.
Taken together, the insights gained from these studies thus show that the rational design of fluorescence light-up probes with high specificity for a certain G4 DNA structure is challenging as very subtle changes strongly affect the G4 DNA binding and fluorescence light-up abilities. However, we disclose key factors for succeeding with this and show that it is indeed possible to use rational design in the development of these types of probes, which will accelerate their development and thus the possibilities to explore G4 biology.
Supplementary Material
ACKNOWLEDGEMENTS
The MD simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at HPC2N Umeå, Sweden. We also thank the Knut and Alice Wallenberg foundation program NMR for Life (www.nmrforlife.se) for NMR spectroscopy support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swedish Research Council [VR‐NT 2017‐05235]; Wenner-Gren Foundation; Kempe Foundations [SMK-1632]; Åke Wiberg Foundation; S.B. funded by DST, Govt. of India [DST/INSPIRE/04/2014/000217]. Funding for open access charge: Swedish Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gellert M., Lipsett M.N., Davies D.R.. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. U.S.A. 1962; 48:2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sen D., Gilbert W.. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988; 334:364–366. [DOI] [PubMed] [Google Scholar]
- 3. Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S.. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006; 34:5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guedin A., Gros J., Alberti P., Mergny J.-L.. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic. Acids. Res. 2010; 38:7858–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chambers V.S., Marsico G., Boutell J.M., Di Antonio M., Smith G.P., Balasubramanian S.. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. biotechnol. 2015; 33:877–881. [DOI] [PubMed] [Google Scholar]
- 6. Biffi G., Tannahill D., McCafferty J., Balasubramanian S.. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013; 5:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schaffitzel C., Berger I., Postberg J., Hanes J., Lipps H.J., Pluckthun A.. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8572–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu H.Y., Zhao Q., Zhang T.P., Wu Y., Xiong Y.X., Wang S.K., Ge Y.L., He J.H., Lv P., Ou T.M. et al.. Conformation Selective antibody enables genome profiling and leads to discovery of parallel g-quadruplex in human telomeres. Cell Chem. Biol. 2016; 23:1261–1270. [DOI] [PubMed] [Google Scholar]
- 9. Eddy J., Maizels N.. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006; 34:3887–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huppert J.L., Balasubramanian S.. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007; 35:406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simonsson T., Pecinka P., Kubista M.. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998; 26:1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bretones G., Delgado M.D., Leon J.. Myc and cell cycle control. Biochim. Biophys. Acta-Gene Regul. Mech. 2015; 1849:506–516. [DOI] [PubMed] [Google Scholar]
- 13. Dang C.V. MYC on the path to cancer. Cell. 2012; 149:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin C.Y., Loven J., Rahl P.B., Paranal R.M., Burge C.B., Bradner J.E., Lee T.I., Young R.A.. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012; 151:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whitfield J.R., Beaulieu M.E., Soucek L.. Strategies to inhibit myc and their clinical applicability. Front. Cell. Dev. Biol. 2017; 5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brooks T.A., Hurley L.H.. Targeting MYC expression through G-quadruplexes. Genes Cancer. 2010; 1:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Siddiqui-Jain A., Grand C.L., Bearss D.J., Hurley L.H.. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siebenlist U., Hennighausen L., Battey J., Leder P.. Chromatin structure and protein-binding in the putative regulatory region of the c-myc gene in burkitt-lymphoma. Cell. 1984; 37:381–391. [DOI] [PubMed] [Google Scholar]
- 19. Yang D.Z., Hurley L.H.. Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids. 2006; 25:951–968. [DOI] [PubMed] [Google Scholar]
- 20. Seenisamy J., Rezler E.M., Powell T.J., Tye D., Gokhale V., Joshi C.S., Siddiqui-Jain A., Hurley L.H.. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J. Am. Chem. Soc. 2004; 126:8702–8709. [DOI] [PubMed] [Google Scholar]
- 21. Phan A.T., Modi Y.S., Patel D.J.. Propeller-type parallel-stranded g-quadruplexes in the human c-myc promoter. J. Am. Chem. Soc. 2004; 126:8710–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ambrus A., Chen D., Dai J.X., Jones R.A., Yang D.Z.. Solution structure of the biologically relevant g-quadruplex element in the human c-MYC promoter. implications for g-quadruplex stabilization. Biochemistry. 2005; 44:2048–2058. [DOI] [PubMed] [Google Scholar]
- 23. Marchetti C., Zyner K.G., Ohnmacht S.A., Robson M., Haider S.M., Morton J.P., Marsico G., Vo T., Laughlin-Toth S., Ahmed A.A. et al.. Targeting multiple effector pathways in pancreatic ductal adenocarcinoma with a g-quadruplex-binding small molecule. J. Med. Chem. 2018; 61:2500–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duarte A.R., Cadoni E., Ressurreicao A.S., Moreira R., Paulo A.. Design of modular G-quadruplex ligands. ChemMedChem. 2018; 13:869–893. [DOI] [PubMed] [Google Scholar]
- 25. Monchaud D., Teulade-Fichou M.P.. A hitchhiker's guide to G-quadruplex ligands. Org. Biomol. Chem. 2008; 6:627–636. [DOI] [PubMed] [Google Scholar]
- 26. Livendahl M., Jamroskovic J., Ivanova S., Demirel P., Sabouri N., Chorell E.. Design and Synthesis of 2,2′-Diindolylmethanes to Selectively Target Certain G-Quadruplex DNA Structures. Chem. Eur. J. 2016; 22:13004–13009. [DOI] [PubMed] [Google Scholar]
- 27. Prasad B., Jamroskovic J., Bhowmik S., Kumar R., Romell T., Sabouri N., Chorell E.. Flexible versus rigid G-quadruplex DNA ligands – synthesis of two series of bis-indole derivatives and comparison of their interactions with G-quadruplex DNA. Chem. Eur. J. 2018; 24:7926–7938. [DOI] [PubMed] [Google Scholar]
- 28. Dash J., Shirude P.S., Balasubramanian S.. G-quadruplex recognition by bis-indole carboxamides. Chem. Commun. 2008; 3055–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Asamitsu S., Bando T., Sugiyama H.. Ligand design to acquire specificity to intended g-quadruplex structures. Chem. Eur. J. 2019; 25:417–430. [DOI] [PubMed] [Google Scholar]
- 30. Haider S.M., Neidle S., Parkinson G.N.. A structural analysis of G-quadruplex/ligand interactions. Biochimie. 2011; 93:1239–1251. [DOI] [PubMed] [Google Scholar]
- 31. Calabrese D.R., Chen X., Leon E.C., Gaikwad S.M., Phyo Z., Hewitt W.M., Alden S., Hilimire T.A., He F., Michalowski A.M. et al.. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 2018; 9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai J.X., Carver M., Hurley L.H., Yang D.Z.. Solution structure of a 2:1 quindoline-c-myc g-quadruplex: insights into g-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc. 2011; 133:17673–17680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deng N.J., Wickstrom L., Cieplak P., Lin C., Yang D.Z.. Resolving the ligand-binding specificity in c-MYC G-Quadruplex DNA: absolute binding free energy calculations and SPR experiment. J. Phys. Chem. B. 2017; 121:10484–10497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma D.L., Zhang Z.H., Wang M.D., Lu L.H., Zhong H.J., Leung C.H.. Recent developments in G-Quadruplex probes. Chem. Biol. 2015; 22:812–828. [DOI] [PubMed] [Google Scholar]
- 35. Yan J.W., Chen S.B., Liu H.Y., Ye W.J., Ou T.M., Tan J.H., Li D., Gu L.Q., Huang Z.S.. Development of a new colorimetric and red-emitting fluorescent dual probe for G-quadruplex nucleic acids. Chem. Commun. 2014; 50:6927–6930. [DOI] [PubMed] [Google Scholar]
- 36. Doria F., Nadai M., Zuffo M., Perrone R., Freccero M., Richter S.N.. A red-NIR fluorescent dye detecting nuclear DNA G-quadruplexes: in vitro analysis and cell imaging. Chem. Commun. 2017; 53:2268–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yan J.W., Tian Y.G., Tan J.H., Huang Z.S.. Colorimetric and fluorescence detection of G-quadruplex nucleic acids with a coumarin-benzothiazole probe. Analyst. 2015; 140:7146–7149. [DOI] [PubMed] [Google Scholar]
- 38. Zhai Q.Q., Gao C., Ding J.Q., Zhang Y.S., Islam B., Lan W.X., Hou H.T., Deng H., Li J., Hu Z. et al.. Selective recognition of c-MYC Pu22 G-quadruplex by a fluorescent probe. Nucleic Acids Res. 2019; 47:2190–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Largy E., Hamon F., Teulade-Fichou M.P.. Development of a high-throughput G4-FID assay for screening and evaluation of small molecules binding quadruplex nucleic acid structures. Anal. Bioanal. Chem. 2011; 400:3419–3427. [DOI] [PubMed] [Google Scholar]
- 40. Chung W.J., Heddi B., Hamon F., Teulade-Fichou M.-P., Anh Tuan P.. Solution structure of a G-quadruplex bound to the bisquinolinium compound phen-DC3. Angew. Chem. Int. Ed. 2014; 53:999–1002. [DOI] [PubMed] [Google Scholar]
- 41. Hanwell M.D., Curtis D.E., Lonie D.C., Vandermeersch T., Zurek E., Hutchison G.R.. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012; 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trott O., Olson A.J.. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010; 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 44. Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E.. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1–2:19–25. [Google Scholar]
- 45. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C.. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006; 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ivani I., Dans P.D., Noy A., Perez A., Faustino I., Hospital A., Walther J., Andrio P., Goni R., Balaceanu A. et al.. Parmbsc1: a refined force field for DNA simulations. Nat. Methods. 2016; 13:55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 48. Dang L.X. Mechanism and thermodynamics of ion selectivity in aqueous-solutions of 18-crown-6 ether - a molecular-dynamics study. J. Am. Chem. Soc. 1995; 117:6954–6960. [Google Scholar]
- 49. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Petersson G.A., Nakat- suji H. et al.. Gaussian. 2016; 16:Wallingford, CT. [Google Scholar]
- 50. Case D.A., Cerutti D.S., Cheatham T.E.I., Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., Greene D., Homeyer N. et al.. AMBER. 2017; San Francisco: University of California. [Google Scholar]
- 51. Sousa da Silva A.W., Vranken W.F.. ACPYPE - AnteChamber PYthon parser interfacE. BMC Res. Notes. 2012; 5:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A.. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kumari R., Kumar R. Open Source Drug Discovery, C. Open Source Drug Discovery, C. Lynn A.. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014; 54:1951–1962. [DOI] [PubMed] [Google Scholar]
- 54. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 55. Gordon M.S., Schmidt M.W.. Dykstra CE, Frenking G, Kim KS, Scuseria GE. 2005; Amsterdam: Elsevier; 1167–1189.Theory and Applications of Computational Chemistry. [Google Scholar]
- 56. Gaus M., Cui Q., Elstner M.. DFTB3: extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J. Chem. Theory Comput. 2012; 7:931–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gaus M., Goez A., Elstner M.. Parametrization and benchmark of DFTB3 for organic molecules. J. Chem. Theory Comput. 2013; 9:338–354. [DOI] [PubMed] [Google Scholar]
- 58. Gaus M., Lu X., Elstner M., Cui Q.. Parameterization of DFTB3/3OB for sulfur and phosphorus for chemical and biological applications. J. Chem. Theory Comput. 2014; 10:1518–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marenich A.V., Cramer C.J., Truhlar D.G.. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 2009; 113:6378–6396. [DOI] [PubMed] [Google Scholar]
- 60. Phan A.T., Kuryavyi V., Gaw H.Y., Patel D.J.. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 2005; 1:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dutta D., Debnath M., Muller D., Paul R., Das T., Bessi I., Schwalbe H., Dash J.. Cell penetrating thiazole peptides inhibit c-MYC expression via site-specific targeting of c-MYC G-quadruplex. Nucleic Acids Res. 2018; 46:5355–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.