


Abstract
We here describe a technique termed STRIDE (SensiTive Recognition of Individual DNA Ends), which enables highly sensitive, specific, direct in situ detection of single- or double-strand DNA breaks (sSTRIDE or dSTRIDE), in nuclei of single cells, using fluorescence microscopy. The sensitivity of STRIDE was tested using a specially developed CRISPR/Cas9 DNA damage induction system, capable of inducing small clusters or individual single- or double-strand breaks. STRIDE exhibits significantly higher sensitivity and specificity of detection of DNA breaks than the commonly used terminal deoxynucleotidyl transferase dUTP nick-end labeling assay or methods based on monitoring of recruitment of repair proteins or histone modifications at the damage site (e.g. γH2AX). Even individual genome site-specific DNA double-strand cuts induced by CRISPR/Cas9, as well as individual single-strand DNA scissions induced by the nickase version of Cas9, can be detected by STRIDE and precisely localized within the cell nucleus. We further show that STRIDE can detect low-level spontaneous DNA damage, including age-related DNA lesions, DNA breaks induced by several agents (bleomycin, doxorubicin, topotecan, hydrogen peroxide, UV, photosensitized reactions) and fragmentation of DNA in human spermatozoa. The STRIDE methods are potentially useful in studies of mechanisms of DNA damage induction and repair in cell lines and primary cultures, including cells with impaired repair mechanisms.
INTRODUCTION
Decades of studies on mechanisms of DNA damage and repair have led to the development of a number of techniques for the detection of various types of DNA lesions. The most sensitive, but indirect and not fully specific (1,2) techniques of microscopy-based in situ detection of double- or single-strand breaks (DSBs or SSBs) are immunofluorescent staining for phosphorylated histone H2AX (γH2AX) (3) or recruited repair factors like 53BP1 (4), RAD51 (5) or XRCC1 (6,7). These methods, although relatively sensitive, involve two assumptions: (i) that the repair machinery has been deployed at the site of damage and (ii) that the DNA lesion is located exactly at the center of the microscopically detectable focus consisting of the recruited repair factors. However, accumulation of repair factors at non-break sites can also occur; thus, false positive results are possible (8). Also, the center of the repair focus may be positioned at a distance from the lesion (9,10). Direct in situ detection of the presence and determining the spatial position of DNA breaks (i.e. by a chemical reaction at exposed DNA ends) are therefore essential. The two existing techniques that can be used for direct microscopy detection of DNA breaks in situ, namely terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL, for DSBs) or the nick translation (NT, for SSBs) assay (11,12), usually rely on labeling of accessible DNA ends by procedures that include immunolabeling. Since immunofluorescent detection is limited by typical problems, including low signal-to-noise ratio and various levels of nonspecific and uneven staining (13,14), sensitivity of these methods does not permit unambiguous detection of the presence and precise location of individual DNA breaks by fluorescence microscopy.
Several new sophisticated, sensitive genome-wide techniques such as BLESS (15), BLISS (16), i-BLESS (17), GUIDE-seq (18) and DSBCapture (19) that can map DSBs to specific genomic loci throughout a cell population have become available in recent years. However useful these ensemble genomic DNA methods are proving to be, the microscopy toolbox remains very limited (7,11) and does not offer the levels of specificity and sensitivity that approach those of the aforementioned methods. Attempts to develop experimental systems to visualize in situ single broken DNA ends have been made (20). These methods, however, enable detection of DSBs only at predetermined sites in the genome.
Here, we present a method abbreviated STRIDE (SensiTive Recognition of Individual DNA Ends), with its two independent variants, which offers unprecedented sensitivity, specificity and ability to reveal precisely the spatial location of single- and double-strand DNA breaks in the nuclei of fixed cells by fluorescence microscopy. This robust tool can detect a DNA break in any nuclear location.
In the course of this study, and to assess the sensitivity of STRIDE, we developed a unique strategy based on CRISPR/Cas9, which enables simultaneous labeling of a specific genomic locus and induction of one or several closely spaced double-strand cleavages or single-strand nicks at this site in the genome.
MATERIALS AND METHODS
Cell culture and cell treatment: sperm cells
HeLa, human U2OS cells and skin fibroblasts were used, and cultured under standard conditions. Human sperm cells (obtained from FertiMedica Clinic, Warsaw) were attached to poly-l-lysine-coated coverslips. Technical details of cell culture and other methods are available in Supplementary Data at NAR Online.
dSTRIDE (detection of DSBs)
After cell fixation, BrdU was incorporated into DNA ends using terminal deoxynucleotidyl transferase (TdT) (Phoenix Flow Systems, AU: 1001) and detection and fluorescence enhancement was achieved by applying the procedure described in detail in Figure 1 and Supplementary Materials and Methods (Supplementary Figure S2).
Figure 1.

Detecting double-strand DNA breaks by dSTRIDE. Schematic representation of subsequent major steps leading to fluorescent labeling of free DNA ends at the site of a DSB, in fixed cells, by the dSTRIDE technique: (1) enzymatic conjugation of nucleotide analogues to DNA ends; (2) attaching primary antibodies of two types (from different hosts), both directed against the incorporated nucleotide analogues, at the concentrations ensuring proximity between the attached antibodies of different types; (3) attaching secondary antibodies with conjugated oligonucleotides to the primary antibodies; (4) hybridizing connector oligonucleotides to two closely located antibody-bound oligonucleotides and ligating them (not shown) to form circular DNA template; (5) RCA reaction—oligonucleotides of one of the antibodies act as a primer for DNA polymerase; (6) synthesizing concatemeric sequences attached to the oligonucleotides on the other antibody by DNA polymerase; and (7) hybridizing short fluorescently labeled oligonucleotides to the amplicon (see ‘Materials and Methods’ section, and Supplementary Figures S1 and S2).
sSTRIDE (detection of SSBs)
All four biotinylated and unmodified deoxynucleoside triphosphates were mixed at a ratio of 3:1 and allowed to be incorporated at sites of single-strand DNA breaks by Escherichia coli DNA polymerase I (Pol I). Detection of the incorporated nucleotide analogues and amplification of the fluorescence signals were achieved as described in Supplementary Materials and Methods (Supplementary Figures S1 and S2).
Induction of DNA breaks with a CRISPR/Cas9 system
In the experimental system we used specific combination of the guide RNAs and the co-expressed fluorescently tagged SpCas9 allowed fluorescent labeling of the targeted repetitive sequence on the long arm of chromosome 3, and simultaneous cutting or nicking DNA in this genomic region. Clusters of DNA cuts or nicks were induced within the repetitive sequence, while individual cuts or nicks were induced at a specific locus immediately adjacent to this repetitive sequence (within distance that is not resolved by standard optical microscopy).
Four types of DNA damage were induced by CRISPR/Cas9: several closely located nuclease-induced double-strand cuts (which we call ‘clusters of cuts’ for brevity), individual cuts, clusters of nickase-induced nicks and individual nicks.
In order to induce clusters of double-strand cuts, CRISPR/Cas9 nuclease with appropriate guide RNAs (which decreased the nuclease activity of Cas9), targeted to a specific repetitive sequence in the long arm of chromosome 3, was used. A cluster of DSBs was thus induced. At the same time, DNA-bound SpCas9–3XGFP served as a fluorescent label for this genomic locus.
Individual cut on chromosome 3 was induced by targeting enzymatically active SpCas9–gRNA complex to a selected locus near the repetitive sequence on chromosome 3, as described in detail in Supplementary Materials and Methods. GFP–Cas9 bound to a single-copy genomic locus would be very difficult to detect by fluorescence confocal microscopy. Thus, we labeled fluorescently the immediately adjacent genomic repetitive sequence by targeting it with numerous 3XGFP-tagged Cas9 molecules. Targeting was achieved by using truncated gRNA, which promoted binding of Cas9–3XGFP to this repetitive sequence but yielded the DNA-bound Cas9–gRNA complex inactive, that is the nuclease activity was abrogated, and no DNA cuts were induced in the repetitive sequence. The physical distance between the labeled repetitive sequence and the site of an individual DNA lesion was small (on the order of a few nanometers); thus, their positions were not resolved by microscopy and the fluorescence signature of Cas9–3XGFP bound to the repetitive sequence marked also the position of an individual cut in the image of the whole nucleus.
In order to induce clusters of nicks (single-strand DNA breaks), CRISPR/Cas9n (a mutated Cas9 protein, exhibiting nickase activity) with appropriate guide RNA, targeted to the above-mentioned specific repetitive sequence in the long arm of chromosome 3, was used. A number of SSBs were induced in this chromosome region. As in the case of clusters of cuts, also in the case of nicks DNA-bound SpCas9n–3XGFP served as a fluorescent label for this genomic locus.
Individual nick was induced by targeting enzymatically active Cas9n–gRNA complex to a selected genomic locus near the repetitive sequence on chromosome 3, as described in detail in Supplementary Materials and Methods. As in the case of Cas9-induced cut, also SpCas9n–3XGPP inducing individual nick would be very difficult to detect; thus, we used the same strategy—we targeted multiple 3XGFP-tagged Cas9 molecules to the adjacent repetitive sequence. Targeting was achieved by using short gRNA, which promoted binding of Cas9n–3XGFP to this repetitive sequence but deprived the DNA-bound Cas9n–gRNA of nickase activity.
In control experiments designed to verify the ability of Cas9 to label the targeted genomic sequences, Cas9–3XGFP or Cas9n–3XGFP was targeted to a selected genomic locus by short (truncated) guide RNAs. In this case, Cas9 or Cas9n was able to bind to DNA but unable to induce nicks or cuts. In these experiments, DNA-bound Cas9–3XGFP or Cas9n–3XGFP was readily detected as fluorescent foci.
Plasmid construction (Supplementary Table S1), SpCas9 or SpCas9n variants and guide RNA labels and sequences (Supplementary Table S2), transfection procedures, detailed description of this experimental system and other relevant information on methods are available in Supplementary Data at NAR Online.
RESULTS
STRIDE principle
The detection of endogenous (spontaneous) or induced single- or double-strand DNA breaks by STRIDE consists of three major steps: (i) conjugation of deoxynucleotide analogues to exposed free DNA ends (3′-OH) by TdT or Pol I (step 1, Figure 1, and Supplementary Figure S1); (ii) specific recognition of the incorporated nucleotides at DNA ends by a mixture of two (or more if appropriate) different antibodies (at carefully optimized concentrations resulting in antibodies of both types bound side by side at the target) against this target molecule (steps 2 and 3, Figure 1, and Supplementary Figure S1); and finally (iii) fluorescence signal enhancement based on rolling circle amplification (RCA) reaction (as optimized for typical proximity ligation assay) and detection of the amplified DNA by hybridization with fluorescently labeled oligonucleotides (steps 4–7, Figure 1, and Supplementary Figure S1). The combination of direct reaction with DNA ends, binding of antibodies to the conjugated nucleotide analogues and RCA reaction (occurring only with two adjacent antibodies of different types located close to each other) followed by hybridization of numerous fluorescent probes results in strong signal amplification and near-zero signal background in microscopy images where even individual DNA breaks are represented by bright, readily detectable fluorescent signals (Supplementary Figure S2). The critical point of the method is the use of two antibodies raised in different hosts, directed against a chain of identical components, yet avoiding the use of fluorescently labeled secondary antibodies. This strategy makes it possible to take advantage of the high signal amplification offered by the proximity ligation assay, and avoiding typical background signal always associated with the detectable nonspecific binding of the antibodies in immunofluorescence assays. Very low or undetectable background and a lack of nonspecific signals are preconditions for high-sensitivity detection of individual molecular events like DNA breaks, and these criteria are met by STRIDE (for further discussion of the pertinent technical issues and variants of the technique, see ‘Materials and Methods’ section, and Supplementary Figures S1 and S2).
Sensitivity and break-type recognition of STRIDE: detection of Cas9-induced DNA cuts and nicks
To test sensitivity of a new method, it is first necessary to induce a known and controllable low number of DNA breaks in the nucleus to be detected. DNA lesions occurring all throughout the nucleus can be induced by a number of methods, including exposure of the whole cell to UV, ionizing radiation or certain DNA-damaging drugs. In these cases, spatial positions of the induced DNA lesions cannot be predicted, nor can such a damage be distinguished from endogenous DNA breaks. DNA damage can be induced in a defined nuclear region by exposing a selected site within the nucleus to a focused beam of visible light, in the presence or absence of DNA-bound photosensitizers (microirradiation (6,21,22)). In all these approaches, the type of DNA damage (oxidative, DNA breaks of various types), and the number and the exact spatial or genomic positions of the induced lesions are not under full experimental control. Thus, we developed CRISPR/Cas9-based experimental system capable of inducing well-defined damage in the form of SSBs or DSBs, without base modifications or oxidative damage, at a targeted locus in the genome. Using specially adapted CRISPR/Cas9, we in situ induced small clusters or individual, double-strand or single-strand DNA breaks resulting from the cleavage activity of nuclease SpCas9, or the nicking activity of the non-target strand-cleaving nickase version of SpCas9 with HNH domain deactivated by introduction of a point mutation H840A (23,24) We used engineered guide RNAs with nuclease-active SpCas9 tagged with 3XGFP or 3XmCherry, or nickase-active SpCas9n tagged with 3XGFP (25). They were employed to induce DNA lesions and simultaneously visually detect the sites of their induction inside cell nuclei. We targeted a subtelomeric region of the long arm of chromosome 3 that contains a repetitive sequence that is unique to this site and this chromosome (26) (note that U2OS cells used here are most likely karyotypically heterogeneous and were shown to possess four copies of chromosome 3 (26)). Subsequently, we detected the Cas9-induced DNA damage by sSTRIDE or dSTRIDE.
Clusters of DSBs induced by CRISPR/Cas9 were readily detected by dSTRIDE (Figure 2A–C). The foci representing the target-bound Cas9 itself (Figure 2A) and the dSTRIDE signal (Figure 2B) colocalize (Figure 2C, seen also in magnified views and profiles of fluorescence signals in panels c-1 and c-2) confirming that Cas9-induced DSBs were detected by dSTRIDE. The expected accumulation of the repair factor 53BP1, and phosphorylation of histone H2AX, were also observed at the sites of accumulation of Cas9 (data not shown). In parallel, we tested the capacity of TUNEL, the only available method of direct in situ detection of DSBs, to detect these Cas9-induced DSBs. Figure 2, panels A–C versus D–F, shows a comparison between fluorescence signals from these clusters of DSBs as detected by dSTRIDE and TUNEL in nuclei of cells in which the subtelomeric repeats were cleaved by Cas9. Distinct dSTRIDE foci were readily observed (Figure 2B and C), whereas no signal was detected with TUNEL (Figure 2E and F), despite the fact the Cas9 was clearly present at the targeted loci in both cases (Figure 2A and D, presented also in magnified views and profiles of fluorescence signals in panels f-1 and f-2). We also compared the sensitivity of dSTRIDE and TUNEL to detect DNA ends generated by DNase I (Figure 3) again revealing that dSTRIDE displayed the greater sensitivity (Figure 3D and H). We conclude that TUNEL did not have the sensitivity required to detect either clusters of closely localized DNA breaks induced by Cas9 or the low level damage induced by DNase I.
Figure 2.
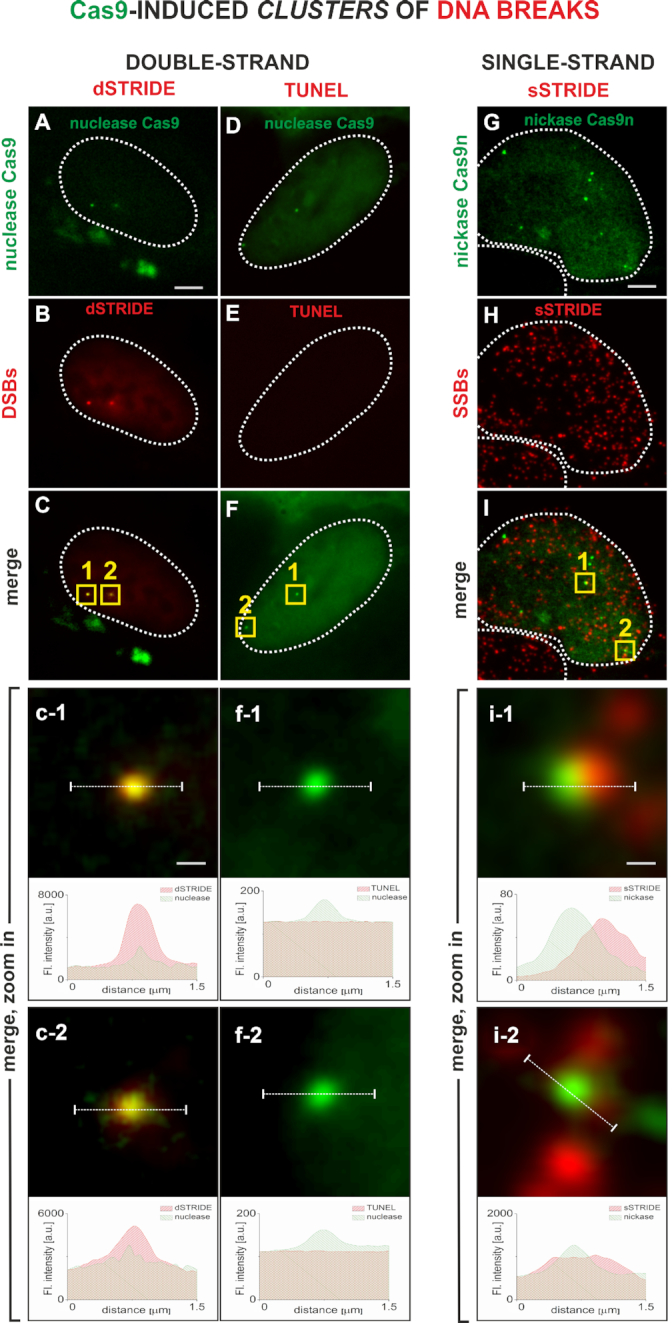
Imaging clusters of double- or single-strand DNA breaks by STRIDE. DNA lesions were induced by Cas9 in a unique region of chromosome 3 that contains chromosome-specific repetitive sequence (Supplementary Figure S5). The genomic loci targeted by Cas9 are detected as green fluorescent foci due to the presence of many chromatin-bound 3XGFP-labeled Cas9 molecules (25). Distinct fluorescent dSTRIDE or sSTRIDE foci colocalize with the sites of Cas9 or Cas9n accumulation, confirming the ability of STRIDE to detect clusters of DNA lesions localized on the long arm of chromosome 3. TUNEL assay does not have sensitivity to detect Cas9-induced DNA lesions. Scale bars: 5 and 0.3 μm (zoom in). Contours of nuclei are marked with white lines. Detection of DNA-bound Cas9 and clusters of Cas9 nuclease-induced DSBs: (A) green foci representing Cas9–3XGFP nuclease accumulated in genomic loci on chromosome 3; (B) red foci of dSTRIDE signal, associated with clusters of double-strand DNA breaks; (C) merging of Cas9 nuclease and dSTRIDE signals demonstrating the ability to detect the presence and spatial positions of DNA breaks in the region of accumulation of Cas9 on chromosome 3; and (c-1, c-2) magnified views of the overlapping foci of Cas9 and dSTRIDE (yellow squares in panel C) at the two intranuclear locations, and the corresponding fluorescence profiles showing relative positions of these foci. Clusters of double-strand DNA breaks induced in the two CRISPR-targeted loci by Cas9–3XGFP (green) are not detected by TUNEL assay (red): (D) green foci representing Cas9–3XGFP accumulated in genomic loci on chromosome 3; (E) weak red background TUNEL signal present throughout the nucleus, but no red foci or areas of higher fluorescence intensity can be detected at the sites of Cas9 accumulation; (F) merging of Cas9–3XGFP and TUNEL signals; and (f-1, f-2) magnified views of the two Cas9 nuclease accumulation foci (panel F, yellow squares), showing no TUNEL signal at the sites of DNA lesions induced by Cas9, and the corresponding fluorescence profiles showing the positions of Cas9 foci and the absence of TUNEL signal at these sites. Detection of DNA-bound Cas9n and clusters of Cas9n nickase-induced SSBs: (G) green foci representing Cas9n–3XGFP nickase accumulated at genomic loci on chromosome 3; (H) numerous red foci of sSTRIDE signal, associated with numerous endogenous, and Cas9n-induced, single-strand DNA breaks; (I) merging of Cas9n (in the region of accumulation of Cas9n on chromosome 3) and sSTRIDE signals (representing DNA ends), demonstrating the ability to identify Cas9n-induced single-strand DNA breaks among many endogenous SSBs; and (i-1, i-2) magnified views of the overlapping foci of Cas9n and sSTRIDE (yellow squares in panel I) at the intranuclear locations, where SSBs were induced by Cas9n. Corresponding fluorescence profiles show relative positions of these foci.
Figure 3.

dSTRIDE is more sensitive than TUNEL. To validate the dSTRIDE method and compare the sensitivity of dSTRIDE (A–D) with TUNEL (E–H), free DNA ends were induced in fixed HeLa cells by exposure to DNase at low (0.2 U) or high (4 U) concentration, and detected using either method. For both techniques, treatment of cells with DNase I, leading to induction of numerous DNA ends, resulted in significant fluorescence signals in the treated nuclei when compared to untreated cells (C, G—where only DNA breaks resulting from low-level endogenous damage are to be expected). (A-C, E-G) Representative fluorescence confocal images (and magnified views) of untreated and DNase I-treated HeLa cells in which DNA ends were detected by dSTRIDE (A–C) or TUNEL (E–G) are shown. For both assays, treatment of cells with a high concentration of DNase I (4 U) (A, E) resulted in a higher signal in the nuclei than in untreated cells (0 U) (C, G). However, only for dSTRIDE a difference between cells treated with a low concentration of DNase I (0.2 U) (B) and untreated cells (0 U; only endogenous damage) (C) was detectable—the number of detected DSBs was significantly higher in DNase-treated cells. For TUNEL assay, no significant fluorescent signal was observed in samples treated with a low concentration of DNase I (0.2 U; low number of induced DNA ends) (F) when compared to untreated cells (0 U, i.e. exclusively endogenous damage) (G). Scale bars: 5 μm. (D, H) Box plots representing the results of analysis of microscopic images of DNA breaks. For TUNEL (H) assay, the signal, due to its characteristic blurriness, was the mean gray value of pixels (fluorescence intensity) within the nucleus (note that individual lesions or groups of lesions were not detectable). For dSTRIDE (D), the number of foci (representing individual lesions or their clusters) in each nucleus was determined. The bottom of each box is the 25th percentile, and the top is the 75th percentile. The solid horizontal line represents the mean value, and the whiskers represent standard error of the mean (SEM). An independent two-sample t-test has shown that the difference in mean values between samples treated with a low concentration of DNase I (0.2 U; low number of induced DNA ends) and untreated samples (0 U; only endogenous DNA damage) was statistically significant only in dSTRIDE (D) (P-value = 2.8 × 10−7).
We then investigated whether the clustered DNA nicks produced by nickase Cas9n can be detected by sSTRIDE (Figure 2G–I). In this case, sSTRIDE was also shown to be very sensitive. As anticipated, colocalization of sSTRIDE signals with Cas9n was also observed, demonstrating that the clusters of SSBs induced by Cas9n were readily detected by sSTRIDE (Figure 2G–I). Numerous endogenous SSBs are known to be present in cultured cells and such lesions were also readily detected by sSTRIDE (Figure 2H) (XRCC1, a repair factor involved in SSB repair, was also recruited to Cas9n-induced and endogenous damage, as expected; data not shown). The fact that sSTRIDE can detect spontaneous SSBs adds to its potential applications (see ‘Discussion’ section).
We further found that STRIDE is capable of detecting not only clusters of breaks, but even individual double- or single-strand DNA breaks induced at the sites of CRISPR/Cas9 accumulation (Figure 4A–F). As expected, in a control experiment, when the actions of Cas9 or Cas9n were silenced by the use of truncated guide RNAs (see ‘Materials and Methods’ section for strategy) resulting in no cleavages, there was no colocalization between dSTRIDE and Cas9, or sSTRIDE and Cas9n foci (in this case, STRIDE signals represented only endogenous DNA breaks; see also Supplementary Figure S3). When active Cas9/gRNA or Cas9n/gRNA complexes were deployed, an individual DNA cut or nick was induced, and detected by STRIDE. Figure 4 shows CRISPR/Cas9-induced individual DSBs or SSBs detected by dSTRIDE (Figure 4A–C; see also quantitative analysis of the number of foci in Supplementary Figure S3A) and sSTRIDE (Figure 4D–F, Supplementary Figure S3B), respectively. To the best of our knowledge, the images in Figure 4 are the first examples of direct fluorescence-based microscopy in situ detection of individual DNA ends induced by CRISPR/Cas9.
Figure 4.
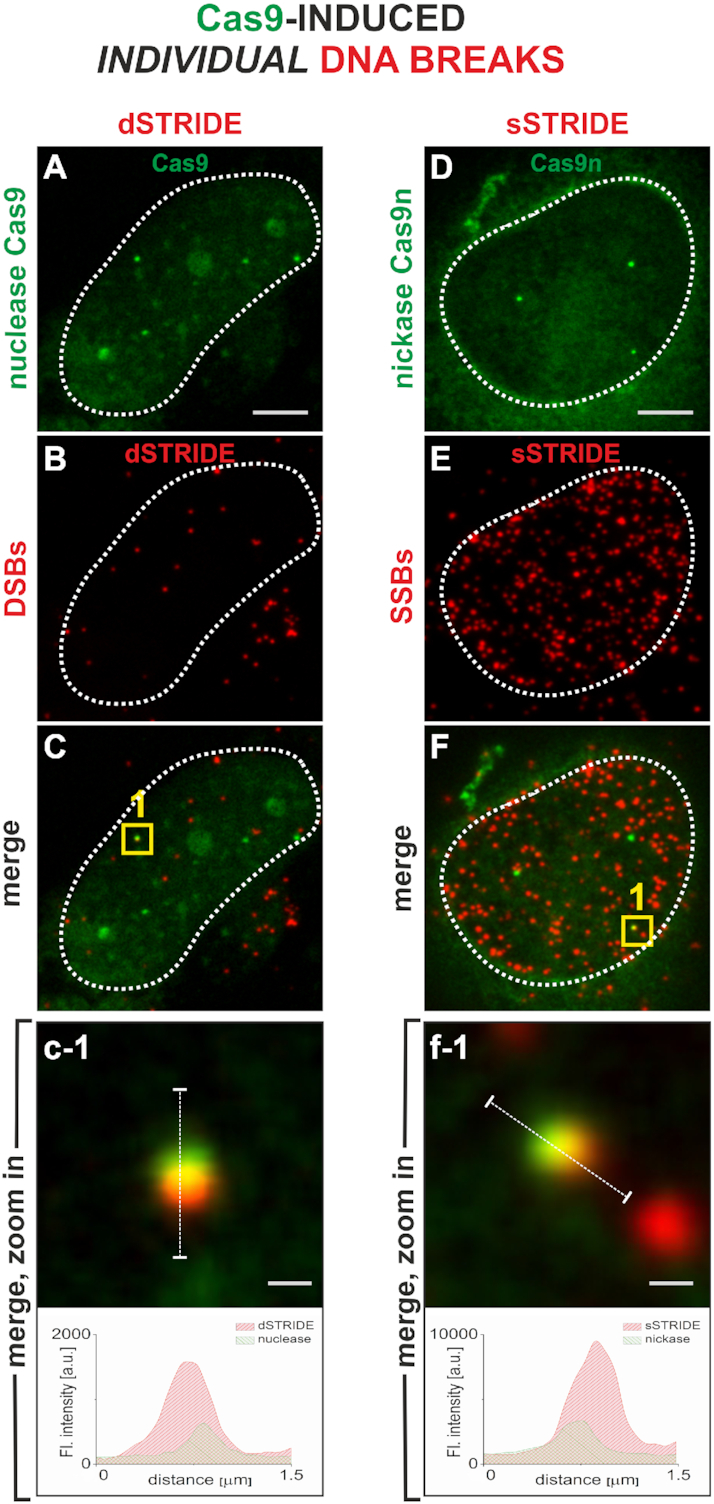
High sensitivity of STRIDE demonstrated by imaging individual single- or double-strand DNA breaks. The individual DSBs and SSBs were induced by targeting a fluorescently labeled nuclease (Cas9, green) (A) or nickase (Cas9n, green) (D) to a unique sequence on chromosome 3 (Supplementary Figure S5; details in ‘Materials and Methods’ section). Scale bars: 5 and 0.3 μm (zoom in). Contours of nuclei are marked with white lines. sSTRIDE and dSTRIDE are both capable of detecting individual CRISPR/Cas9- and CRISPR/Cas9n-induced double- or single-strand DNA breaks, as shown by colocalization of the green (Cas9) and red (STRIDE) foci (C, F), yielding yellow areas. (A–C) Individual DSB detected by dSTRIDE (A: Cas9, green; B: dSTRIDE, red; C: merging of both signals; c-1: a magnified view of the damage site, yellow square in panel C). (D–F) Individual SSB detected by sSTRIDE (D: Cas9n, green; E: sSTRIDE, red; F: merging of both signals; f-1: a magnified view of the damage site, yellow square in panel F).
Detecting endogenous and induced DNA breaks
The mechanisms of damage induction and the type of resulting damage vary substantially among different damaging stimuli. Thus, we tested the capacity of STRIDE to detect DNA breaks arising throughout the nucleus from the action of various agents, in cells of various types and chromatin of different levels of compaction.
As shown in Figure 5, STRIDE was found to be capable of detecting low levels of endogenous double-strand DNA breaks in fibroblasts obtained from 3- and 60-year-old donors and maintained in primary cell cultures (Figure 5A and B), with a higher number in the 60-year-old donor (Figure 5B and C), as would be expected based on the existing indirect evidence (27). STRIDE was also capable of measuring differences between the numbers of doxorubicin-induced double-strand DNA breaks in cells from 3- versus 60-year-old donor, with a significantly higher number in the latter (Figure 5C). It is to be noted that the cell-to-cell variation in the numbers of DNA breaks induced with doxorubicin was greater in the cells from the older donor.
Figure 5.
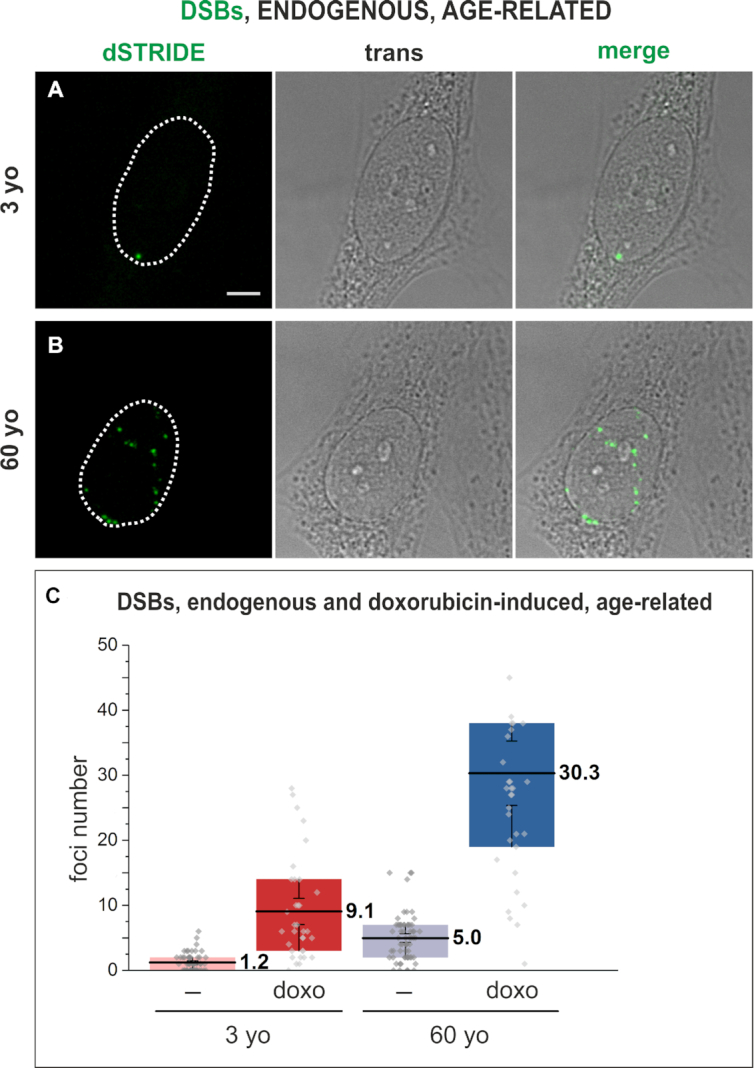
Imaging DNA breaks, endogenous or experimentally induced by doxorubicin, in human fibroblasts from a 3- or 60-year-old donor in primary cultures. Fluorescence signals marking DSBs detected by dSTRIDE (green) in fibroblasts (transmitted light images) from a 3- (A) or 60-year-old (B) donor. Scale bar: 5 μm. (C) The numbers of endogenous and doxorubicin-induced double-strand DNA breaks detected in fibroblasts from 3- and 60-year-old donors, demonstrating a higher number and a larger range of numbers of endogenous DNA lesions in cells from an older donor, and showing a higher number of lesions induced by the drug in cells of an elderly individual. Box plots represent the results of analysis of microscopic images of DNA breaks in which the number of foci (representing individual lesions or their clusters) in each nucleus was determined. The bottom of each box is the 25th percentile, and the top is the 75th percentile. The solid horizontal line represents the mean value, and the whiskers represent SEM.
We further observed that dSTRIDE detected DNA breaks induced by various conditions and damaging treatments that are known to result in various levels and types of DNA damage. DNA breaks in cells undergoing spontaneous apoptosis showed very high levels of dSTRIDE signal, as expected (Supplementary Figure S4). DNA breaks induced in cells of an established cell line by the radiomimetic antitumor antibiotic bleomycin (Figure 6), chemical factors including hydrogen peroxide (Figure 7A), topoisomerase inhibitor topotecan (Figure 7B) and by exposure to UV (Figure 7C) were also readily detected by dSTRIDE, regardless of the nature of the factor inducing damage.
Figure 6.
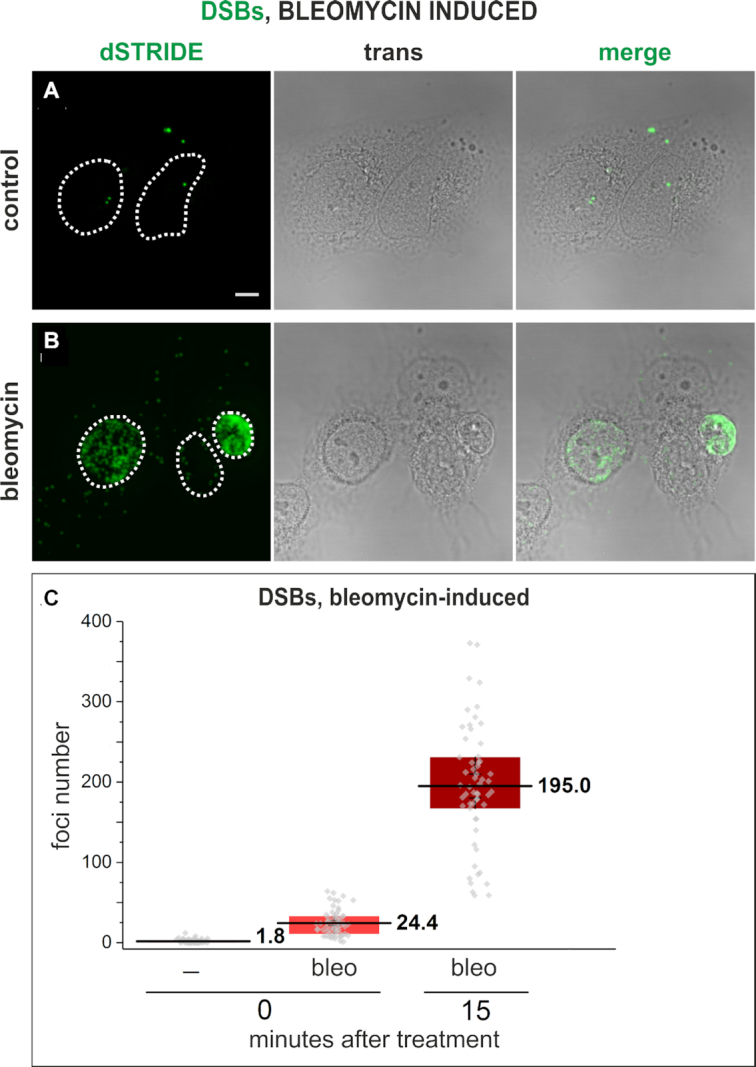
Imaging DNA breaks induced in U2OS cells in an in vitro culture, following exposure to a radiomimetic antitumor antibiotic bleomycin. Control cells, showing only low-level endogenous DNA damage (A) and cells treated with bleomycin (B) that show signs of heavy damage or apoptosis (the cell nucleus presented on the right-hand side of the image) (dSTRIDE fluorescence signals, transmitted light image showing cell morphology and an overlay of both images). Scale bar: 5 μm. (C) Quantitative analysis of the numbers of DSBs in control cells, and in cells exposed to bleomycin for 30 min and fixed and stained for STRIDE immediately (labeled with 0) or 15 min (labeled with 15) after exposure to bleomycin, showing an increase of the number of DNA lesions during 15 min after exposure to the drug. Box plots represent the results of analysis of microscopic images of DNA breaks in which the number of foci (representing individual lesions or their clusters) in each nucleus was determined. The bottom of each box is the 25th percentile, and the top is the 75th percentile. The solid horizontal line represents the mean value, and the whiskers represent SEM.
Figure 7.

Imaging DNA breaks induced in HeLa cells in an in vitro culture, by various damaging agents. Double-strand DNA breaks induced by exposure to hydrogen peroxide (A), topoisomerase inhibitor topotecan (B) and ultraviolet light (C), and detected by dSTRIDE are shown. STRIDE signals (green), DAPI-stained nuclei, transmitted light images and merged images (with DAPI in blue) are shown. Scale bar: 5 μm.
dSTRIDE also detected DNA breaks induced by laser microirradiation in a selected region of the nucleus. This approach is often used in basic research on DNA repair in order to image the recruitment of repair factors to the site of damage. A standard method is to apply a focused light beam to a small region of the cell nucleus, in the presence of a DNA-bound photosensitizer (21), inducing oxidative damage and DNA breaks (21). Using this method, DSBs in the illuminated area in the cell nucleus were induced, and detected by dSTRIDE (Figure 8A and B). In contrast, TUNEL was not capable of detecting these lesions (Figure 8C and D).
Figure 8.
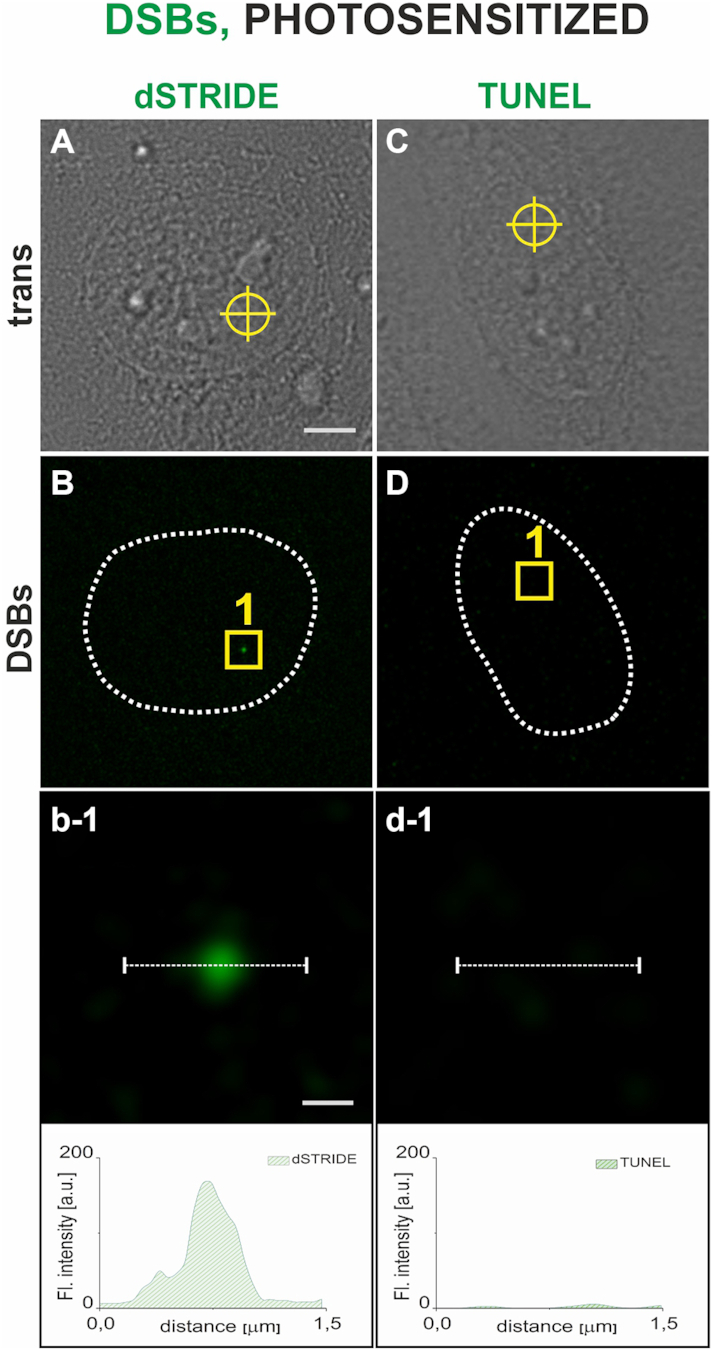
Detection, by dSTRIDE and TUNEL assay, of DNA breaks inflicted in a small selected region of the cell nucleus by photosensitized reactions. DNA-bound ethidium anion was used as a photosensitizer (500 nM ethidium bromide in culture medium) (21); ethidium was excited by local illumination with 488 nm focused laser beam (300 nm diameter). Scale bars: 5 and 0.3 μm (zoom in). Transmitted light image of a cell, with a site at which the laser beam was focused marked by a circle (A), an image showing a fluorescence dSTRIDE signal at the site of damage (B), and a magnified view of the region with the damage site as well as the corresponding fluorescence profile (b-1) demonstrating that dSTRIDE detected the local photosensitized damage. Transmitted light image of a cell showing the site of induction of photosensitized damage (C), fluorescence image (D) and a magnified view as well as the corresponding fluorescence profile (d-1) demonstrating a lack of TUNEL signal at the site of photosensitized damage.
Finally, in order to test versatility of STRIDE in wider domains of biological science we use this method to detect DNA damage in spermatozoa and other materials. Spontaneous DNA fragmentation in sperm cells is an important parameter in human in vitro fertilization and veterinary reproductive science. As shown in Figure 9A and B, both DSBs and SSBs were detected in human sperm cells by STRIDE. In additional assays, spontaneous DNA breaks were detected in lymphocytes as well as formalin-fixed, paraffin-embedded and frozen mouse tissue (data not shown).
Figure 9.
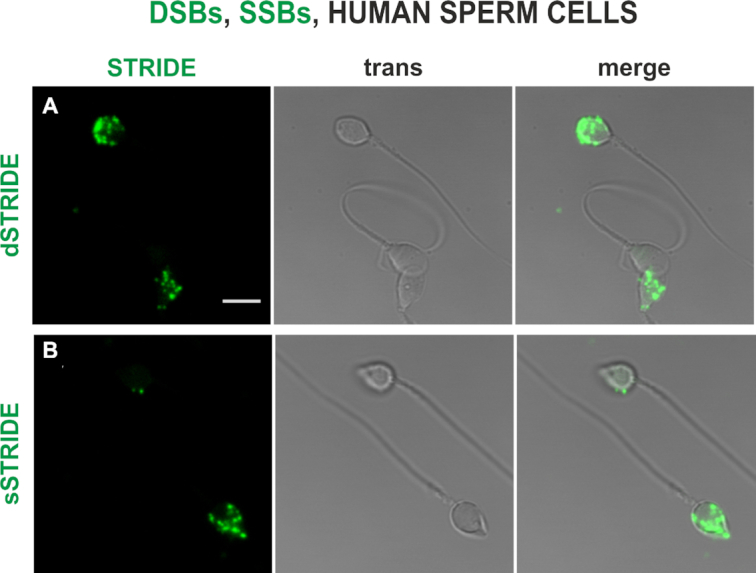
DNA fragmentation in spermatozoa—detecting endogenous levels of DNA damage (SSBs and DSBs) in human sperm cells by dSTRIDE and sSTRIDE. Examples of low and high levels of DNA double-strand (A) and single-strand (B) breaks in sperm cells of a fertility clinic patient are shown (dSTRIDE fluorescence signals, transmitted light and merged images). Scale bar: 5 μm.
DISCUSSION
The data presented here demonstrate that dSTRIDE and sSTRIDE are capable of direct in situ detection of small clusters and even individual single- and double-strand DNA breaks, whether spontaneously occurring (endogenous) or induced experimentally in cellular chromatin by factors and treatments of various types. The STRIDE methods rely on a labeling procedure that maintains the nonspecific background signal at an almost undetectable level relative to the strongly amplified damage-specific fluorescence signal. The STRIDE methods are versatile in that DNA breaks resulting from treatments with various DNA damaging agents and exposures to various damage conditions in cultured cell lines, cells from patients maintained in primary cultures, human spermatozoa, lymphocytes and tissue sections can be readily detected. It thus seems that the potential applications in both basic and clinical research are numerous, as well as in veterinary reproductive science.
In addition, due to their high sensitivity both dSTRIDE and sSTRIDE have considerable potential to become powerful tools in detecting and quantitating individual DNA breaks in the CRISPR-based genome-editing field, inter alia. Not only do these methods allow monitoring the immediate effects of the cleaving and nicking activity of the Cas9 and Cas9n proteins applied in these systems, but they could also be used to assess the level of their specificity and STRIDE can become a supplement to existing methods for detection of off-target events. Combining single-cell fluorescence microscopy with high-throughput platforms might prove to be a very helpful tool in speeding up the process of optimization of genome-editing systems and in their comparative analysis.
Also on both the basic and clinical research fronts, STRIDE may prove to be particularly useful for detecting DNA damage where the mechanisms of DNA repair are impaired by mutations in genes coding key repair factors or by drugs like DDR inhibitors (such as PARP, PARG, ATM and DNA-PK inhibitors). In such cells, no recruitment of some typical repair factors occurs, and H2AX phosphorylation or PARylation may be weak or absent (28). Under such conditions, direct detection of DNA breaks by STRIDE could provide an unambiguous measure of DNA damage. Importantly, STRIDE also bears potential to become useful in the clinic for DNA damage level assessment in patient-derived samples in the form of liquid biopsies or tissue sections. It is also worth noting that phosphorylation of H2AX may occur without the presence of DNA breaks (29). In this case, direct measurements of DNA damage by STRIDE may assist in avoiding overestimates of the level of damage.
As STRIDE makes it possible to detect, precisely localize inside the cell nucleus and quantify individual DNA breaks with sensitivity that was not achievable earlier, we expect it to be useful in various fields of research and diagnostics, such as screening of genotoxic, anticancer drugs and testing their functionality and potency, basic research into mechanisms of DNA editing, DNA damage and repair and aging, in various types of medical diagnostics including infertility and monitoring of environmental genotoxic agents. It is possible to envisage STRIDE being used in high-throughput DNA damage assays for testing of new drugs.
Supplementary Material
Notes
Present address: Hanhui Ma, School of Life Sciences and Technology, ShanghaiTech University, Shanghai 201210, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Polish National Science Center (in part by 2015/16/T/NZ3/00157 to M.K and 2017/27/B/NZ3/01065 to J.D) and the U.S. National Institutes of Health grant (U01DA-040588, part of the N.I.H. 4D Nucleome Initiative) to TP. Funding for open access charge: Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University.
Conflict of interest statement. M.M.K., M.Z., K.S. and J.D. are the listed inventors on a patent application covering the STRIDE methods, and co-founders of a company intoDNA.
REFERENCES
- 1. Cleaver J.E., Feeney L., Revet I.. Phosphorylated H2Ax is not an unambiguous marker for DNA double-strand breaks. Cell Cycle. 2011; 10:3223–3224. [DOI] [PubMed] [Google Scholar]
- 2. Rybak P., Hoang A., Bujnowicz L., Bernas T., Zarębski M., Darzynkiewicz Z., Dobrucki J.. Low level phosphorylation of histone H2AX on serine 139 (γH2AX) is not associated with DNA double-strand breaks. Oncotarget. 2016; 139:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M.. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998; 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 4. Guo X., Bai Y., Zhao M., Zhou M., Shen Q., Yun C.-H., Zhang H., Zhu W.-G., Wang J.. Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Res. 2018; 46:689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tarsounas M., Davies D., West S.C.. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene. 2003; 22:1115–1123. [DOI] [PubMed] [Google Scholar]
- 6. Solarczyk K.J., Kordon M., Berniak K., Dobrucki J.W.. Two stages of XRCC1 recruitment and two classes of XRCC1 foci formed in response to low level DNA damage induced by visible light, or stress triggered by heat shock. DNA Repair (Amst.). 2016; 37:12–21. [DOI] [PubMed] [Google Scholar]
- 7. Galbiati A., Beauséjour C., d’Adda di Fagagna F.. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell. 2017; 16:422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ziani S., Nagy Z., Alekseev S., Soutoglou E., Egly J.M., Coin F.. Sequential and ordered assembly of a large DNA repair complex on undamaged chromatin. J. Cell Biol. 2014; 206:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bekker-Jensen S., Lukas C., Melander F., Bartek J., Lukas J.. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J. Cell Biol. 2005; 170:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kordon M.M., Szczurek A., Berniak K., Szelest O., Solarczyk K., Tworzydło M., Wachsmann-Hogiu S., Vaahtokari A., Cremer C., Pederson T. et al.. PML-like subnuclear bodies, containing XRCC1, juxtaposed to DNA replication-based single-strand breaks. FASEB J. 2019; 33:2301–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gorczyca W., Bruno S., Darzynkiewicz R., Gong J., Darzynkiewicz Z.. DNA strand breaks occurring during apoptosis—their early in situ detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int. J. Oncol. 1992; 1:639–648. [DOI] [PubMed] [Google Scholar]
- 12. Gorczyca W., Gong J., Darzynkiewicz Z.. Detection of DNA strand breaks in individual apoptic cells by the in situ terminal deoxynucleotidyl transferase and nick translation assays. Cancer Res. 1993; 53:1945–1951. [PubMed] [Google Scholar]
- 13. Schnell U., Dijk F., Sjollema K.A., Giepmans B.N.G.. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods. 2012; 9:152–158. [DOI] [PubMed] [Google Scholar]
- 14. Manning C.F., Bundros A.M., Trimmer J.S.. Benefits and pitfalls of secondary antibodies: why choosing the right secondary is of primary importance. PLoS One. 2012; 7:e38313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crosetto N., Mitra A., Silva M.J., Bienko M., Dojer N., Wang Q., Karaca E., Chiarle R., Skrzypczak M., Ginalski K. et al.. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods. 2013; 10:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan W.X., Mirzazadeh R., Garnerone S., Scott D., Schneider M.W., Kallas T., Custodio J., Wernersson E., Li Y., Gao L. et al.. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017; 8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Biernacka A., Zhu Y., Skrzypczak M., Forey R., Pardo B., Grzelak M., Nde J., Mitra A., Kudlicki A., Crosetto N. et al.. i-BLESS is an ultra-sensitive method for detection of DNA double-strand breaks. Commun. Biol. 2018; 1:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V, Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. et al.. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR–Cas nucleases. Nat. Biotechnol. 2015; 33:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lensing S.V., Marsico G., Hänsel-Hertsch R., Lam E.Y., Tannahill D., Balasubramanian S.. DSBCapture: in situ capture and sequencing of DNA breaks. Nat. Methods. 2016; 13:855–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soutoglou E., Dorn J.F., Sengupta K., Jasin M., Nussenzweig A., Ried T., Danuser G., Misteli T.. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007; 9:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zarebski M., Wiernasz E., Dobrucki J.. Recruitment of heterochromatin protein 1 to DNA repair sites. Cytometry A. 2009; 75:619–625. [DOI] [PubMed] [Google Scholar]
- 22. Solarczyk K.J., Zarębski M., Dobrucki J.W.. Inducing local DNA damage by visible light to study chromatin repair. DNA Repair (Amst.). 2012; 11:996–1002. [DOI] [PubMed] [Google Scholar]
- 23. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ran F.A., Hsu P.D., Lin C.-Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y. et al.. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 154:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma H., Naseri A., Reyes-Gutierrez P., Wolfe S.A., Zhang S., Pederson T.. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3002–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma H., Tu L.C., Naseri A., Huisman M., Zhang S., Grunwald D., Pederson T.. CRISPR–Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016; 214:529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anglada T., Repulles J., Espinal A., LaBarge M.A., Stampfer M.R., Genesca A., Martin M.. Delayed γH2AX foci disappearance in mammary epithelial cells from aged women reveals an age-associated DNA repair defect. Aging (Albany, NY). 2019; 11:1510–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schuhwerk H., Bruhn C., Siniuk K., Min W., Erener S., Grigaravicius P., Kruger A., Ferrari E., Zubel T., Lazaro D. et al.. Kinetics of poly(ADP-ribosyl)ation, but not PARP1 itself, determines the cell fate in response to DNA damage in vitro and in vivo. Nucleic Acids Res. 2017; 45:11174–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tu W.-Z., Li B., Huang B., Wang Y., Liu X.-D., Guan H., Zhang S.-M., Tang Y., Rang W.-Q., Zhou P.-K.. γH2AX foci formation in the absence of DNA damage: mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013; 587:3437–3443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.