


Abstract
Trypanosoma brucei possesses a highly complex RNA editing system that uses guide RNAs to direct the insertion and deletion of uridines in mitochondrial mRNAs. These changes extensively alter the target mRNAs and can more than double them in length. Recently, analyses showed that several of the edited genes possess the capacity to encode two different protein products. The overlapped reading frames can be accessed through alternative RNA editing that shifts the translated reading frame. In this study, we analyzed the editing patterns of three putative dual-coding genes, ribosomal protein S12 (RPS12), the 5′ editing domain of NADH dehydrogenase subunit 7 (ND7 5′), and C-rich region 3 (CR3). We found evidence that alternatively 5′-edited ND7 5′ and CR3 transcripts are present in the transcriptome, providing evidence for the use of dual ORFs in these transcripts. Moreover, we found that CR3 has a complex set of editing pathways that vary substantially between cell lines. These findings suggest that alternative editing can work to introduce genetic variation in a system that selects against nucleotide mutations.
INTRODUCTION
Trypanosoma brucei is a member of the Kinetoplastea, a group of protozoans characterized by a large network of DNA in their mitochondria known as the kinetoplast (1). The kinetoplast is composed of two types of concatenated circular DNA molecules: maxicircles and minicircles. The maxicircles all encode mitochondrial ribosomal RNAs as well as 18 protein-coding genes, most of which are components of the electron transport chain. The approximately 30–50 identical copies of the maxicircle make up a relatively small proportion of the kinetoplast (2). Most of the DNA network is composed of ∼5000 one kb minicircles, each of which encodes 2–5 small non-coding guide RNAs (gRNAs) (3,4). These gRNAs are used in the process of RNA editing. In T. brucei, RNA editing consists of specific uridine insertion and deletion events that render 12 of the 18 mitochondrially encoded mRNAs translatable (5). The gRNAs act as templates for the large editosome complex which cleaves the mRNA, inserts or deletes the correct number of uridines and then re-ligates the mRNA in an energy intensive process. This is repeated until the mRNA is complementary to the small gRNA. Each gRNA directs edits that generate the anchor region for the next gRNA; thus the RNA editing process is sequentially dependent on correct editing by each gRNA. As editing of some of the extensively edited mRNAs can involve upwards of 40 gRNAs, this renders the process incredibly fragile (6). We hypothesize that such an expensive and fragile process is maintained in response to the unique life cycle of T. brucei.
Trypanosoma brucei is a dixenous parasite, invading the bloodstream of a mammalian host and being transmitted between hosts by bite of a tsetse fly. Once taken up in a blood meal by the tsetse fly, it transitions into the replicating procyclic state in the midgut. The energy T. brucei requires for this replication is gained through metabolism of amino acids (7,8). This is accomplished through use of a portion of the Krebs cycle and the electron transport chain (ETC), thus most of the ATP required is produced by the mitochondria (7–9). This stage of the life cycle is followed by a dramatic bottleneck when the trypanosomes transition from the midgut to the salivary glands of the tsetse fly (10,11). From the salivary glands, trypanosomes are then refluxed into their next mammalian host during a bloodmeal. Once the parasite is deposited into the mammalian bloodstream, it quickly transitions to utilizing glycolysis for its energy generation, removing the requirement for ATP production in the mitochondria (12). While in the mammalian host, T. brucei lives entirely extracellularly. It is frequently subject to attacks by the host's adaptive immune system, and the population evades these attacks through antigenic variation (13). This part of the life cycle can be quite long, with the longest known infection lasting 29 years (14). This life cycle should make T. brucei particularly sensitive to genetic drift, especially for those genes which are not under selection (Krebs cycle and ETC) and should make them extremely vulnerable to Muller's ratchet (the gradual increase of mutational load that eventually leads to extinction) (15–18). One mechanism for protecting small asexual populations is by increasing the severity of the mutations that can occur. If mutations severely impact fitness, mutated individuals are selected out, preventing their fixation (19). Recently, computer modeling studies suggest that small asexual populations can evolve this type of mechanism (termed ‘drift robustness’) in order to maintain fitness (20). The sequential dependence of the RNA editing process implies that the system is inherently fragile to mutations. Even a single point mutation can drastically change the editing pattern and stop the editing process, aborting expression of the protein. Hence, the RNA editing process may operate as a proof-reading system to weed out mutations by making them lethal. This is effective however, only if the mitochondrial genes are under selection. Previously, we showed that many of the mitochondrially pan-edited genes have a distinct mutational bias that is suggestive of dual-coding genes (coding two proteins by overlapping reading frames) (21). The overlapping of ETC genes not under selection in the bloodstream stage with genes that are under selection during this stage of the life cycle, would prevent the accumulation of mutations. As the extensively overlapped genes share most gRNAs, this strategy would ensure that almost all of the genetic material is protected.
Our analyses suggested that of the twelve pan-edited genes in T. brucei, six are potentially dual coding and that the RNA editing system is used to determine which reading frame is accessed. In this study, we deep sequenced the mRNA transcript populations of three putative dual coding genes: ribosomal protein S12 (RPS12), the 5′ editing domain of NADH dehydrogenase subunit 7 (ND7 5′), and C-rich region 3 (CR3), in order to determine if mRNAs with access to multiple open reading frames (ORFs) are found in the mitochondrial transcriptome. Using the previously generated gRNA transcriptomes, we developed a new pipeline which can integrate mRNA sequence data with known gRNA populations. This allowed the construction of detailed editing pathways for each of these genes using two different cell lines, TREU 667 and EATRO 164. In addition, we examined the effect of energy source on the editing process by using two different media, SDM79 and SDM80. In both cell lines, the editing pathway of RPS12 was primarily linear, reflecting the high degree of conservation required for a gene that is essential (22,23). We found no evidence of utilization of the gRNA that provides access to the alternative reading frame (21). In contrast, we did identify transcripts using different reading frames for both CR3 and ND7 5′. This study indicates that RNA editing can be used to access multiple open reading frames using two different methods: in ND7 5′, different gRNAs bring alternate start codons into frame and in CR3, different gRNAs can shift the reading frame of the existing start codon. In addition, while minor cell-line specific editing patterns were observed for RPS12 and ND7, CR3 showed incredible editing diversity, in that the two different cell lines showed very different editing patterns, using different sets of gRNAs to edit the CR3 cryptogene. Furthermore, we found that the energy source available to the parasites did influence overall editing efficiency and the selective use of alternative gRNAs. This suggests that the use of a gRNA-guided editing system can respond to different metabolic conditions and also dramatically increase protein diversity in spite of a rigid and mutationally fragile system.
MATERIALS AND METHODS
T. brucei culture and RNA isolation
Trypanosoma brucei clones from strains EATRO 164 and TREU 667 were used in this study. The TREU 667 cell line was originally isolated from a bovine host in 1966 in Uganda (24), while the EATRO 164 strain was isolated from Alcephalus lictensteini in 1960. The EATRO 164 line was obtained from Dr. K. Vickerman by Dr. Ken Stuart in 1966 (25). Both cell lines were grown in SDM79 and harvested as previously described (26). EATRO 164 cells grown in SDM79 were then gradually transitioned to SDM80 using serial 1:3 dilutions when cells reached a density of at least 5 × 106 cells/ml. SDM80 was prepared as described by Lamour et al. with the exception of using undialyzed FBS, but reducing the amount of FBS added by half (27). This results in the final concentration of glucose being 0.5 mM instead of the 0.15 mM found in the original SDM80 recipe. This concentration is still well below that of SDM79, which has a glucose concentration of 6 mM. Once cells had been acclimated to SDM80, cells were harvested as previously described (26). Mitochondrial vesicles were isolated using differential spins and mitochondrial RNA was then isolated from vesicles as previously described (26).
Preparation, sequencing, and analysis of mRNAs
cDNAs were generated from isolated RNAs using the Applied Biosystems High Capacity cDNA Reverse Transcription Kit. CR3, RPS12 and ND7 5′ editing domain cDNAs were amplified via PCR using the following primers (underlined sequences are gene specific and non-underlined sequences are tag regions used in the deep sequencing reaction:
CR3 5′: ACACTGACGACATGGTTCTACA AGAAATATAAATATGTG
CR3 3′: TACGGTAGCAGAGACTTGGTCT ACAAAAATTATTTGCATACTT
RPS12 5′: ACACTGACGACATGGTTCTACA CTAATACACTTTTG
RPS12 3′: TACGGTAGCAGAGACTTGGTCT AAAAACATATCTTAT
ND7 5′: ACACTGACGACATGGTTCTACA GATACAAAAAAACATGAC
ND7 3′: TACGGTAGCAGAGACTTGGTCT CTTTTATATTCACATAACTTTTCTGTAC
cDNAs were amplified for 30 cycles using a high-fidelity polymerase and gel purified in order to size select against primer dimer products. Amplified cDNAs from EATRO 164 cells grown in SDM79, EATRO 164 cells grown in SDM80, and TREU 667 cells grown in SDM79 were individually barcoded and combined in equal molar amounts. Samples were sequenced in a 2 × 250 bp paired end format (PE250) using an Illumina MiSeq Standard flow cell and 500 cycle reagent cartridge, version 2. This format was chosen in order to ensure complete coverage of sequenced cDNAs, as our largest predicted products would not exceed 500bp. Sequence data was preprocessed as previously described (21). 7.6 million reads were generated and after trimming using FaQCs and trimmomatic, and paired end assembly using PEAR, 86% of reads remained.
Sequence data was then separated by cell line, growth media and gene. The data was then analyzed using a new pipeline and program called SKETCH (Segmentation of Kinetoplast Edited Transcripts to Characterize editing Heterogeneity). This program allowed us to classify mRNAs at the gRNA block editing level and determine which editing patterns were most prominent. For each set of sequences, SKETCH would remove low quality sequences whose sequences containing more than five mismatches to the unedited template, disregarding uridines. In order to classify the editing patterns observed in the mRNA transcripts, SKETCH requires a set of template sequences. Initially, the templates supplied to SKETCH were the conventional fully edited and unedited transcripts. These transcripts were then segmented based on the editing blocks previously defined by the locations of gRNA populations (26). Each transcript was then classified by editing block, with each block being classified as matching unedited, fully edited sequences or being unknown. After the initial characterization of the transcripts, the most abundant unknown sequences for each editing block were then added to the reference pool. Sequences were then reclassified by SKETCH based on the newly added reference sequences. This process was repeated until the most abundant editing patterns were identified. SKETCH code is available on GitHub (https://github.com/laurakirby4/SKETCH). To validate the newly identified editing patterns as true alternatives, the new sequences were screened against the gRNA transcriptome. Identification of gRNAs was accomplished as previously described (26). Briefly, the scoring system awards gRNA points for the longest continuous match to the target and weighs canonical Watson-Crick base pairs more highly than G:U base pairs. gRNAs with scores above a set threshold are then retained. Ideal gRNA matches are defined as gRNAs that are fully complementary to edited sequences and had an anchor binding region consisting of six or more consecutive Watson-Crick base pairs. When ideal gRNA matches were not found, reduced stringency searches were performed (lowered threshold). In some cases, a gRNA with one or two mismatches in the alignment to the edited sequence were the best gRNA matches identified. gRNAs with unpaired nucleotides have been previously identified in the searches of the gRNA transcriptomes for the conventionally edited mRNAs, so these gRNA matches were not excluded (26,28). Identified mRNA sequences with a gRNA match were then considered valid conventional or alternative edits. In the gRNA searches, sequences with identical sequence excluding the poly-U tail addition site and potential 5′ exonuclease activity were combined into single sequence classes as previously described (26). gRNA sequence classes with more than 100 reads were retained, unless gRNAs covering a particular region were relatively rare, in which case, the cut off was lowered.
RESULTS
To confirm that transcripts with access to two reading frames exist in vivo, we analyzed the mRNA transcriptomes for three of the putative dual-coding genes, Ribosomal Protein S12 (RPS12), NADH Dehydrogenase subunit 7 and C-rich Region 3 (CR3). This mRNA deep sequencing data was then used in combination with the sequenced gRNA transcriptomes, to generate precise editing pathway maps. In order to determine how robust the observed editing pathways were, we characterized editing in two different cells lines, TREU 667 and EATRO 164. In addition, to determine how metabolic energy source may influence editing, we analyzed the editing pathways in EATRO 164 cells grown in two different media, SDM79 and SDM80. SDM79 is the standard medium used to grow the procyclic stage parasite. However, it contains 6 mM glucose, and experiments have shown that under these levels of glucose, the procyclic stage can grow in the absence of electron transport chain (ETC) activity (27,29–34). The SDM80 medium was developed to more closely resemble insect gut conditions and has very low glucose concentrations (27). Trypanosome growth in this medium requires ATP production using the ETC (27).
Ribosomal protein subunit 12 (RPS12) is an essential component of the mitochondrial ribosome (22,23,35,36). RPS12 is extensively edited (pan-edited) with 132 Us inserted and 28 Us deleted. Full editing is directed by 12 populations of gRNAs (defined as a group of gRNAs that edit the same region of an mRNA) (26,35). In this analysis, we identify 10 populations, with three of the previously identified populations being combined with other populations that shared a very high amount of overlap. One new population (F) was identified through a search of the gRNA transcriptome under reduced stringency (Figure 1, Supplementary Table S1). Analyses of the canonical editing pattern indicate that there are two long ORFs, and mutational bias analyses indicate that both ORFs may be selected for (21). The longest ORF encodes the RPS12 protein and encompasses a second shorter ORF of unknown function (35). Northern blots revealed that edited RPS12 mRNAs were found in both life cycle stages, however, edited mRNAs were more abundant in bloodstream form than procyclic form trypanosomes (35).
Figure 1.

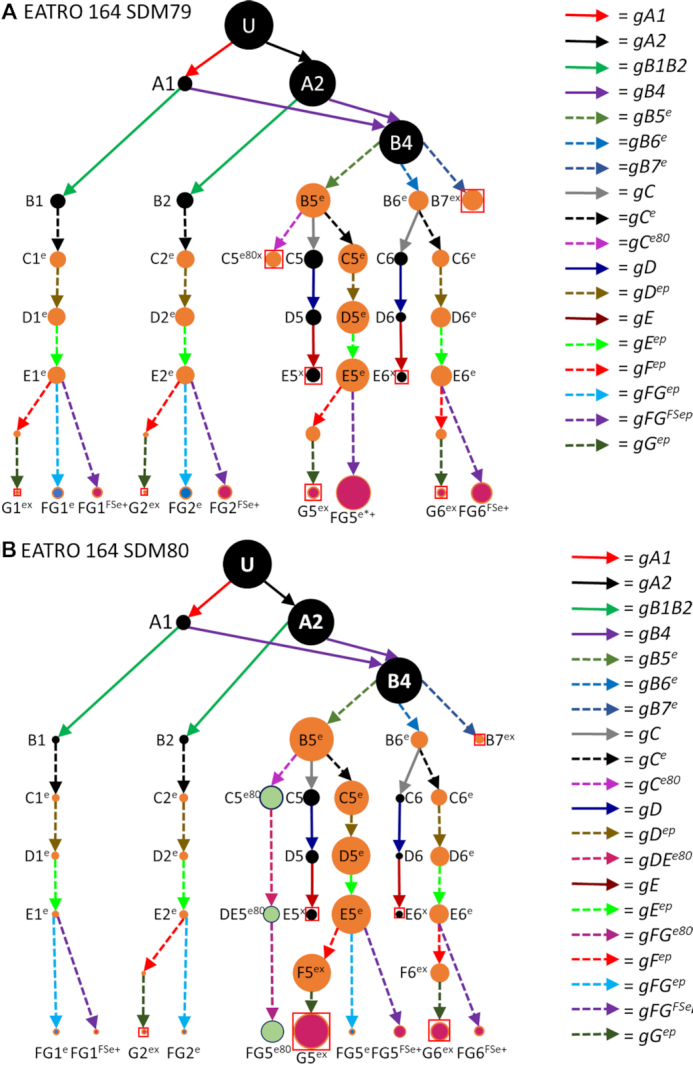
Observed RPS12 editing pathways in the TREU 667 cell line (A) and the EATRO 164 cell line grown in SDM79 (B) and SDM80 (C). U = unedited transcripts. Dot sizes are proportional to the percent of block level edited transcripts using the gRNA indicated. Colored arrows indicate the gRNA population used. Dashed arrows with closed heads represent gRNA populations used in only one cell line (superscript ‘e’ or ‘t’). gRNA names: superscript ‘p’ represent promiscuous gRNAs that can anchor and edit more than one editing block; superscript ‘FS’ indicate gRNAs that introduce a frameshift; * indicates gRNAs that introduce a minor aa change. End point mRNAs with no evidence of further editing are represented by dots enclosed by a red box and the mRNAs are named with a superscript ‘x’. gFp is a promiscuous gRNA that edits both in the D and F editing block of RPS12. Arrows with a boxed ‘m’ represent a gRNA that has mis-anchored.
Because RPS12 is essential, we expected it to have a very robust editing pattern in both cell lines, as well as under both energy conditions. In contrast, neither ND7 or CR3 appear to be essential in the insect stage of the parasite (30). The canonical ND7 has two separate editing domains that are edited independently (37). Interestingly, in EATRO 164 cells, while the 3′ editing domain is fully edited only in the bloodstream life cycle stage, the 5′ editing domain is edited in both life cycle stages (2,37,38). In addition, the mutational bias analyses indicate that only the 5′ editing domain has characteristics indicative of a dual coding gene. The canonically edited CR3 is also a putative Complex I member (ND4L) and is preferentially edited in the BS stage (39,40). Complex I has been shown to be non-essential in both life cycle stages, and other mitochondrially encoded complex I subunits, ND3, ND8 and ND9, have been shown to be preferentially edited in the bloodstream stage (2,30,41–44).
RNA seq data was generated by reverse transcribing mitochondrial RNAs using random primers. For both RPS12 and CR3, transcripts were then selectively amplified using sequence specific primers targeted to the terminal 5′ and 3′ never edited regions as to not bias against any possible editing pattern. For ND7, the 5′ editing domain was selectively amplified using sequence specific primers targeted to the 5′ never edited region and the homology region 3 (HR3) that separates the 5′ and 3′ editing domains (37). The HR3 is a span of 59 nts that is also never edited, hence should not bias the analysis. The targeted transcriptome libraries were generated from TREU 667 cells grown in SDM79 and EATRO 164 cells grown in SDM79 and SDM80. Additionally, for CR3, we generated another library using TREU 667 cell line mRNA by selecting for transcripts of a larger size, instead of taking transcripts of all sizes (SDM79). This allowed us to enrich the library for transcripts that had initiated the editing process. Amplified cDNAs were then gel purified, barcoded and combined in equal molar amounts for sequencings. While the number of total reads obtained did vary by cell line and media used, surprisingly few transcripts were fully edited (canonical AUG + ORF). For both RPS12 and CR3, the majority of reads (>80%) were completely unedited (Table 1). CR3, which has previously been shown to be preferentially edited in the BS stage, had the lowest percentage of fully edited transcripts, with only 0.1–0.2% translatable transcripts detected in both cell lines and under both growth conditions. The high percentage of RPS12 pre-edited (completely unedited) transcripts was surprising. In a previous study of RPS12 using the 29–13 cell line (which is derived from LISTER 427 cells), the authors found only ∼14% pre-edited mRNAs, indicating that the majority of transcripts had initiated the editing process (45,46). While the predominance of pre-edited transcripts may be cell line specific, preferential overamplification of the short unedited transcripts, due to the PCR amplification protocol we used, cannot be ruled out. In this study, the number of transcripts edited through the canonical start codon, was also lower than the 6% found by Simpson et al. (43). While the TREU 667 cells had 2.3% translatable transcripts, the EATRO 164 cells had a surprising low 0.9%. Growth of the EATRO cells in low glucose media (SDM80) did result in a substantial jump in both the number of transcripts that initiated the editing process, and the number of translatable transcripts (∼4-fold increase to 4.2%) (Table 1). This suggests that energy source can substantially influence editing efficiency.
Table 1.
Editing efficiencies of RPS12, ND7 5′ and CR3. TREU 667 enriched refers to the size selection of more fully edited transcripts in the TREU 667 CR3 library
| Transcript | Cell line and media | Total # reads | % unedited | % partial edited | % fully edited |
|---|---|---|---|---|---|
| RPS12 | TREU 667, SDM79 | 787 584 | 89.8% | 7.9% | 2.3% |
| RPS12 | EATRO 164, SDM79 | 846 549 | 92.6% | 6.5% | 0.9% |
| RPS12 | EATRO 164, SDM80 | 1 381 092 | 81.3% | 14.5% | 4.2% |
| ND7 5′ | TREU 667, SDM79 | 1 141 322 | 20.3% | 70.0% | 9.7% |
| ND7 5′ | EATRO 164, SDM79 | 915 610 | 47.0% | 52.8% | 0.2% |
| ND7 5′ | EATRO 164, SDM80 | 313 657 | 27.4% | 72.1% | 0.5% |
| CR3 | TREU 667, SDM79 | 18 832 | 84.9% | 15.0% | 0.1% |
| CR3 | TREU 667 enriched. | 50 589 | 18.1% | 73.1% | 8.8% |
| CR3 | EATRO 164, SDM79 | 348 210 | 93.2% | 6.6% | 0.2% |
| CR3 | EATRO 164, SDM80 | 53 000 | 90.6% | 9.3% | 0.1% |
The ND7 5′ transcriptome analyses differed substantially from both RPS12 and CR3 in that the majority of these transcripts had initiated the editing process (Table 1). The TREU cell line showed the highest editing efficiency with ∼80% of transcripts having initiated editing and 9.7% of the transcripts fully edited and translatable. In contrast, in EATRO cells, only 53% of the transcripts had initiated editing, and a scant 0.2% had completed the editing process. As with RPS12, we did see an increase in editing efficiency when the EATRO parasites were grown in SDM80, with over 70% initiating editing. However, even with the large increase in initiation of the editing process, only a scant 0.5% of transcripts were fully edited (Table 1). The sharp drop in the ability to complete the editing process appears to be due to loss of an optimal gRNA for one region of this transcript (described below). ND7 5′ in T. brucei has also been previously sequenced (again the Lister 427 cell line), with fully edited and pre-edited transcript numbers similar to those found in the TREU cell line (45,47).
Editing cascade and reading frame analyses
In order to determine if the low editing efficiencies were due to any one step in the editing cascades, a full analysis of each editing step was done. For these analyses, we developed a pipeline that used our gRNA database to distinguish true alternative edits from both mis-edited and partially edited transcripts. This pipeline uses two programs, Segmentation of Kinetoplast Edited Transcripts to Characterize Editing Heterogeneity (SKETCH), and the gRNA database search program previously described (26). The SKETCH program analyzes segments of transcripts that are defined by the relative range of coverage of each gRNA population used in conventional editing patterns. Block sequences are compared to both the unedited sequence and the fully edited conventional sequence and then classified into unedited, fully edited and ‘unknown’ blocks. Once the most abundant sequences of all segments are identified, abundant transcripts (>1% of all transcripts) containing ‘unknown’ sequences are used as queries against the gRNA database corresponding to the cell line of the library. If a gRNA is identified that can generate the edit, the sequence is considered a true alternative edit. A valid gRNA match is considered if the gRNA alignment is able to align to the edited sequence, has no gaps or mismatches, and the gRNA has an anchoring region of at least 6 consecutive Watson-Crick base pairs. In some cases, particularly in C-rich regions of the mRNAs, only gRNAs alignments with a small number of mismatches were identified. This observation is consistent with our previous analysis of the gRNA transcriptomes, so in these cases, these gRNAs were also considered plausible (26,28). If no plausible gRNA is identified, the edit is considered a misedit or a junction (which we define as an intermediate in the editing process that does not align to a gRNA), depending on the sequence and the status of other segments on that transcript with this sequence. By examining segments of transcripts independently, we were able to identify both branching and converging editing pathways as well as editing dead-ends (no evidence of any editing beyond that editing block). While other programs capable of analyzing edited kinetoplastid sequences do exist, we opted to create this new software to take advantage of our gRNA transcriptome data (45,47,48).
RPS12 analysis
As expected, the essential RPS12 showed the most robust editing path. In all three analyses, the majority of transcripts used the same series of 10 gRNA populations (Figure 1, circle size is proportional to the % of block edited transcripts using the indicated gRNA). Use of the final gRNA population (gJ) in the cascade lead to only the RPS12 ORF, and we found no evidence of an alternative AUG or frameshift leading to utilization of the second ORF. We do note that there is a downstream start codon, that if translated, would be read in the alternative reading frame (ARF) (For full gRNA sequences and alignments see Supplementary Table S1 and Figure S1). While the editing cascades were relatively straight forward, we did see some minor deviations (Figure 1). Editing of block B could utilize a number of different gRNAs, including several that were used in one cell line only (dashed arrows). gRNAs B1 and B1* are variants of the same gRNA, with gB1* introducing a single amino acid (aa) change (V/Y) (Supplementary Figure S2). In the TREU cell line, a small proportion of transcripts were edited using two gRNAs, gB3t and gB4t, that lead to a distinct editing ‘dead-end’ (dead-end = disruption of the next canonical anchor sequence, and no detection of any further editing). In contrast, editing using gB2FSe (identified in both cell lines, but only observed directing editing in EATRO) did not disrupt the editing cascade. Use of this gRNA variant however, did introduce a frameshift seven amino acids (aa) from the C-terminus (Supplementary Figure S2). Because gB2FSe did not disrupt editing, a significant percentage (5.3% in SDM79 and 7.6% in SDM80) of EATRO translatable RPS12 transcripts did contain the alternative C-terminus (J2 transcripts). This alternative C-terminus was previously reported in the 29-13 strain (45), however, it appears to be absent in the TREU cell line.
A decrease in editing efficiency was seen at the D to E block transition due to the incorrect use of a promiscuous gRNA (gFp guide RNA (Dx)) that disrupted the editing cascade (Table 2). Promiscuous gRNAs (identified with a superscript p) are defined as identified gRNAs that are known to anchor and edit in two different editing blocks. Most of the identified promiscuous gRNAs edit two different mRNA transcripts, and have been previously observed in the alternative editing in MURF2 (49). The gFp gRNA is unusual in that it normally directs editing of the F editing block in RPS12 but can also anchor to and edit the C block. While mis-editing by gFp was limited in the TREU667 cell line (7.1% of D-block edited transcripts), it's use was much more prominent in the EATRO cell line (17.5%), leading to a significant drop in transcripts that could continue past D-block editing. Interestingly, growth in SDM-80 lead to a significant increase in mis-editing by gFp, with over 32.5% of transcripts using gFp incorrectly, resulting in a significant portion of dead-end transcripts. The EATRO cell line had additional minor dead-end pathways at the D to E transition. Misediting by a promiscuous ND7 gRNA (gEep) again disrupted any further editing, and mis-anchoring by the gE guide RNA (marked with box m) also led to the generation of an anchor sequence that could be used by a ND8 gRNA (gFep) disrupting any further editing. Interestingly, the editing efficiency did not drop as transcripts transitioned to the next block of editing (Table 2). In EATRO-SDM80 cells, the editing efficiency at level F is ∼5.6%, and at level G it actually increases to 5.9%. Editing efficiency at the block level is calculated based on the number of transcripts that match any of the fully edited sequences in that block, regardless of the condition of earlier blocks. Analysis of editing intermediates suggests that this increase occurs because transcripts that are edited through block G are capable of being re-edited by alternative gRNAs (gFep). Transcripts being overwritten by an alternative gRNA at the time of the RNA capture, can result in mRNAs that are fully edited at downstream blocks (block G), and only partially edited at the block being overwritten (block F) (Figure 2).
Table 2.
Editing efficiency for each RPS12 gRNA population. Percentages were calculated based on the number of transcripts that had completed each editing level out of the total number of RPS12 transcripts
| Block | Percent complete editing of block | ||
|---|---|---|---|
| TREU 667 (SDM 79) | EATRO 164 (SDM 79) | EATRO 164 (SDM 80) | |
| Initiated editing | 10.2 | 7.4 | 18.7 |
| A | 8.9 | 4.6 | 15.9 |
| B | 6.9 | 4.4 | 14.6 |
| C | 6.0 | 4.2 | 13.5 |
| D | 4.9 | 3.1 | 11.7 |
| E | 4.5 | 2.2 | 6.9 |
| F | 3.8 | 1.4 | 5.6 |
| G | 3.7 | 1.4 | 5.9 |
| H | 3.7 | 1.3 | 5.7 |
| I | 3.4 | 1.2 | 5.4 |
| J | 2.3 | 0.9 | 4.2 |
Figure 2.
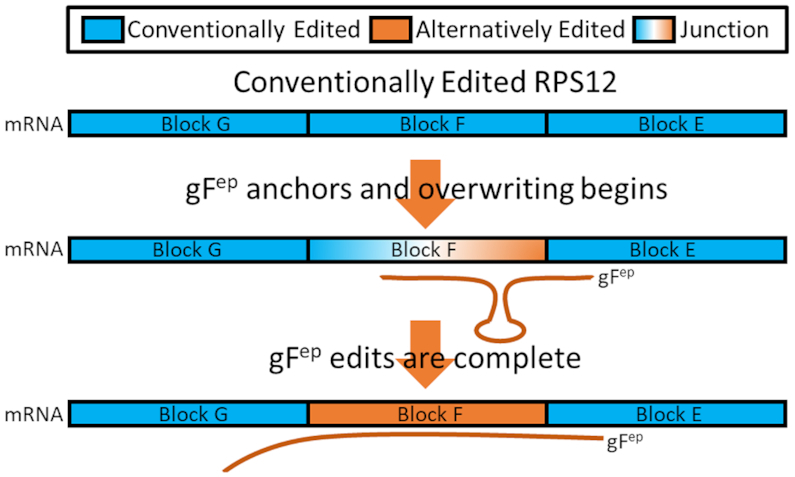
Model of how gFep can overwrite fully edited transcripts to generate observed mRNA transcript sequence patterns. mRNAs are represented by rectangles, with editing blocks labeled. gFep is shown as an orange line. Blue blocks are conventionally edited, orange blocks are alternatively edited, and blocks fading from blue to orange are in an incompletely edited junction state. Overwriting of block F resulting in the decrease in editing efficiency observed at block F as compared to block G (Table 2).
The only other minor variation was the use of the EATRO specific gGe guide that occurs in a highly cytosine-rich region (Figure 3). Previous examinations of the gRNA coverage in this region identified only rare gRNAs with multiple C:A base pairs, alignment mismatches and with gaps between adjacent gRNAs (26,28). While this analysis did extend the identified gRNA population and eliminated the gap region, we did not identify either mRNA sequences or gRNAs that improved the alignment mismatches (Figure 3). The use of alternative base pairs is not unheard of. A study of in vitro deletions found that alternative base pairs such as C:A, C:U, and C:C were tolerated to varying extents (50). Interestingly, this portion of RPS12 encodes the signature sequence, which is nearly universal (75). Use of the gGe variant gRNA results in a single point mutation, substituting a proline in place of a serine within this important sequence.
Figure 3.

Regions with poor gRNA coverage and functionally conserved residues in RPS12. Functionally important aa residues are underlined (72). Pipes (|) indicate Watson/Crick base pairs and colons (:) indicate G/U base pairs. Red highlighted hashtags (#) indicated gaps or mismatches, green highlighted hashtags indicate C:A base pairs. The introduced substitution mutation introduced by use of the gGe gRNA is highlighted in yellow (S>P).
ND7 5′ analysis
Analyses of the ND7 5′ targeted transcriptomes, indicate that full editing of the 5′ domain requires five gRNA populations for both cell lines (Figure 4). Two variants of the terminal population (gE1 and gE2) were identified that resulted in different 5′ terminal editing patterns. Translation of these editing patterns yields two different protein products in two different reading frames (RF). Reading frame one encodes the canonical ND7 protein (E1) and the other (RF3) encodes a putative metabolite transporter (E2) (21). While transcripts for both open reading frames were found in both cell lines, there were notable differences in the populations. The TREU 667 cell line had the highest editing efficiency with over 80% of the transcripts initiating the RNA editing process and ∼9.7% of the transcripts fully edited through Block E (Table 3). Use of the gE1 or gE2 gRNAs appeared to be equally efficient, resulting in nearly equal amounts of RF1 and RF3 fully edited transcripts. A small percentage of transcripts (4.9% of transcripts that completed block E editing) were observed that appeared to be mis-edited by a TREU specific gRNA (gE4t), leading to a dead-end product (no ORF). In addition, gE4t also appeared to be able to overwrite editing directed by gE2, to generate a small number of transcripts that could be translated in RF2 (pink E3t). While the number of ‘dead-end’ pathways were very limited in the TREU cell line, use of the gC guide RNA population appeared to be very inefficient, resulting in a large drop in the percent of Block C-edited transcripts (25.8% drop, Table 3). A mutant gC gRNA (gCFSt), did result in a small percentage of transcripts with a frameshift C-terminus. Interestingly, while 9.1% of C block transcripts used the gCFSt gRNA, only 2.4% of the transcripts that have completed D-block editing come from this minor branch. This suggests that this alternative edit decreases the efficiency of use of the subsequent gRNAs.
Figure 4.

Observed ND7 5′ editing pathways in the TREU 667 cell line (A) and the EATRO 164 cell line grown in SDM79 (B) and SDM80 (C). U = unedited transcripts. For arrow, gRNA and mRNA naming descriptors see Figure 1. + indicates that more than one mRNA form was condensed into this circle to simplify the figure (See Supplementary Figure S4). Condensed forms encode largely the same amino acid sequence with only small variants. Terminal dots are colored blue for reading frame 1, magenta for reading frame 2, or green for reading frame 3. Boxed green dots have no in frame AUG start codon but are translatable into reading frame 3 with the use of an alternative start codon (UUG).
Table 3.
Editing efficiency for each ND7 5′domain gRNA population. Percentages were calculated based on the number of transcripts that had completed each editing level out of the total number of ND7 transcripts
| Block | Percent complete editing of block | ||
|---|---|---|---|
| TREU 667 (SDM 79) | EATRO 164 (SDM 79) | EATRO 164 (SDM 80) | |
| Initiated Editing | 79.7 | 52.4 | 72.6 |
| A | 45.8 | 47.0 | 68.5 |
| B | 44.7 | 45.8 | 66.7 |
| C | 18.9 | 13.4 | 16.9 |
| D | 11.7 | 0.4 | 1.1 |
| E | 9.7 | 0.2 | 0.5 |
In contrast, full editing of the ND7 5′ domain in EATRO 164 was very inefficient. While transcripts were able to initiate the editing process relatively efficiently (∼50 – 70%, dependent on growth medium used), less than 1% of ND7 transcripts were fully edited at level E (Table 3). This appears to be due to the use of several gRNAs used only in EATRO that disrupt further editing. Again, the largest drop in editing efficiency occurred at the B to C-block transition. In addition, the EATRO specific use of gBe, gC1e and gC2e all disrupted the editing cascade (Figure 4). This compounded the editing efficiency problem, with a majority of C-block edited transcripts (47% in SDM79 and 73.4% for SDM80), no longer editing competent. The 5′ end of ND7 has multiple AUG sequences not created by the editing process. Translation predictions of these dead-end transcripts (Bex, C1ex, C2ex) indicate that they do have ORFs that extend through the HR3 region. The Bex transcripts would be translated in the ARF (RF3) but is ten amino acids shorter. Both C1ex and C2ex, translate in the canonical ND7 reading frame, with predicted proteins that are only three amino acids shorter (Supplementary Figure S3). Further drops in efficiency occurred due to an anchor mis-match (A:A) found in the gD guide RNA population (Figure 5). While the gD mutation is also observed in TREU, this cell line contains a sizable population of non-mutated gD guide RNAs (Supplementary Table S2). Editing by the gE4e guide, results in a transcript with no in-frame AUG. However, translation of this transcript (E4ex) in RF3 has no stop codons and we cannot rule out the possibility of a non-canonical START codon.
Figure 5.

Regions with poor gRNA coverage and functionally conserved residues in ND7′5′. Functionally important histidine residues are underlined (51). Pipes indicate Watson/Crick base pairs and colons indicate G/U base pairs. Red highlighted hashtags (#) indicate gaps or mismatches, green highlighted hashtags indicate C/A base pairs.
Similar to RPS12, ND7 5′ has a cytosine-rich region with poor gRNA coverage (Figure 5). This cytosine-rich region contains two conserved residues involved in ND7 function and coincides with the C level of editing in the editing pathways where the largest drop in editing efficiency is observed (Table 3) (51). The gC gRNA population is relatively rare (only 114 reads found in the TREU gRNA transcriptome, and 6185 reads in EATRO) and has 5 nt mismatches with the conventional ND7 sequence (including C:A base pairs; Figure 5).
CR3 analysis
Previous work indicated that C-Rich region 3 is a putative Complex I member, and that in EATRO 164 cells, it is preferentially edited in the Bloodstream stage (39,40). However, CR3 gRNAs are present in both life cycle gRNA transcriptomes, and fully edited transcripts were successfully amplified and sequenced in the TREU 667 procyclic cell line (21,26,28). These studies indicated that multiple forms of the mRNA did exist that used different reading frames suggesting that CR3 is dual-coding and that it is selection of the terminal gRNA that determines which reading frame will be used (21). In this study, we used primers flanking the editing domain in order to analyze the entire CR3 sequence. Interestingly, while 15% of the TREU CR3 transcripts had initiated the editing process, only 2.2% had completed editing by the initiating gA guide RNAs (Table 4). This suggests that the large drop in editing efficiency occurs due to incomplete editing by the block A guides. These gRNAs are fairly abundant, and we see no alignment issues, so it is unclear why editing of Block A is so inefficient (Supplementary Table S3).
Table 4.
Editing efficiencies by block level of CR3. Transcripts whose gRNAs covered two blocks (DEs and FGs) were included in both blocks for this calculation
| Level | Percentage Complete | |||
|---|---|---|---|---|
| TREU 667 | TREU 667 (enriched) | EATRO 164 (SDM 79) | EATRO 164 (SDM 80) | |
| Initiated editing | 15.1 | 81.9 | 6.8 | 9.4 |
| A | 2.2 | 68.3 | 1.9 | 2.3 |
| B | 1.8 | 56.6 | 1.2 | 1.8 |
| C | 1.7 | 54.6 | 0.9 | 1.5 |
| D | 1.6 | 52.0 | 0.6 | 1.1 |
| E | 1.2 | 39.1 | 0.6 | 1.0 |
| F | 0.8 | 22.3 | 0.2 | 0.4 |
| G/FG | 0.4 | 8.8 | 0.2 | 0.3 |
While the percentage of fully edited transcripts was very low (0.2–0.4%, Table 4), we were able to again identify the major 5′ alternative editing patterns that direct translation to either the ORF or to the +1 ARF (RF2). To increase the robustness of the analyses, we also generated a biased CR3 transcriptome, by size selecting for longer transcripts during the amplification process. Analyses of the TREU transcriptome indicates that the full CR3 editing pathway has multiple branches, resulting in a total of 12 major forms of fully edited CR3 (Figure 6). These 12 forms are comprised of three major 5′ editing patterns, paired with any of four different 3′ editing patterns. The two initiating gRNAs identified (gA1 and gA2), direct identical editing patterns except gA2 inserts an additional three U-residues at 1 site (insertion of 1 phenylalanine). The gB guide RNAs all anchor in different areas (Figure 7A, Supplementary Figure S5) and do introduce substantial AA changes near the 3′ end (Figure 7B). However, all gB guide RNAs generate the anchor binding site (ABS) that is recognized by gC, hence all 4 nodes merge to a common sequence guided by gC and gD.
Figure 6.

Observed CR3 editing pathways in the TREU 667 cell line. U = unedited transcripts. For arrow and gRNA naming descriptors see Figure 1. gRNAs with prime designations indicate extensively overlapped gRNAs both needed to cover a single editing block. Terminal dots are colored blue for reading frame 1 or magenta for reading frame 2. Boxed magenta dots have no functional start codon but are translatable into reading frame 2 with the use of an alternative start codon (UUG).
Figure 7.

(A) Four different 3′ end sequences found in the TREU 667 transcriptome for the CR3 transcript. U-residues inserted by editing are indicated by lowercase; different sequences created by the different gRNAs are highlighted in RED. The additional 3 U-residues inserted by gA2 are indicated by dotted underline. Thick underline sequence indicates the anchor binding site (ABS) for the initiating gRNAs (gA1 and gA2). Green = ABS for gB1B2; Blue = ABS for gB3t; Purple = ABS for gB4. (B) CR3 protein sequences for both reading frame 1 (ORF) and reading frame 2 (ARF). Bolded amino acids show sequence variants and shaded sequence shows position of predicted transmembrane domains (73). mRNAs with no functional AUG are named with a superscript ‘x’.
The 5′ end editing patterns begin to diverge after Block D editing. FGtx transcripts (boxed magenta, Figure 6) are generated by the use of two sequential gRNA populations, gEt and gFGtp1. gFGtp1 is a promiscuous gRNA (previously identified as a ND7 gRNA) that spans both the F and G editing blocks. These transcripts were more abundant than both Gt (RF1, blue) and FGt (RF2, magenta), however final editing using this gRNA does not generate an AUG start codon. It has been proposed that trypanosomes can use UUG as an alternative start codon, thus we cannot rule out the possibility that FGtx transcripts can be translated (Figure 7B) (52). Analyses of intermediates suggest that the gE guide (red arrow) can in fact ‘overwrite’ gEt, indicating that a proportion of these may still be re-edited into other forms. Editing via the gE population required an additional gRNA (gEt’) to generate the anchor for either gFt or gFGtp2. Generation of Gt transcripts (canonical CR3) requires 2 additional gRNAs, while FGt (+1 ORF) transcripts are generated by a single gRNA population (gFGtp2), another promiscuous gRNA (CR4).
Surprisingly, when we examined the editing pathways of CR3 in the EATRO 164 libraries, we discovered that while three of the four initial 3′ editing patterns were found in this library, editing beyond those patterns was completely divergent. Use of an alternative gC gRNA (gCe) initiated an editing pathway that used a unique set of gRNAs (Figure 8, Supplementary Figure S6). The divergent pathway did show some superficial similarities to the editing patterns observed in TREU 667 cells. While both the B1 and B2 transcripts could be directly edited by gCe, the B4 transcripts required an additional gRNA to generate the ABS recognized by gCe. In EATRO cells, B4 transcripts could be edited by 3 different gRNAs (gB5e, gB6e and gB7e). While gB7e disrupted editing, both gB5e and gB6e generated the ABS that could be used by either gC or gCe. While the conventional CR3 gC guide RNA (gray arrow) was clearly used by B4 transcripts, we saw no evidence of its use in the B1/B2 pathways, probably due to the low number of transcripts using the gB1/gB2 path. Surprisingly, while transcripts using gC were extended by both gD and gE guide RNAs, no evidence of editing beyond the gE guides was observed, despite the presence of CR3 conventional gRNAs in the EATRO gRNA transcriptome. In contrast, use of the alternative gCe guide RNA population, could be extended by a series of additional guide RNAs, generating transcripts with functional AUG start codons. However, many of the gRNAs used were promiscuous, in that they had been previously identified as gRNAs of other transcripts. As with the TREU editing pathway, we observe transcripts capable of being translated in two reading frames with the FGe mRNAs translating in RF1, and the FGFSe mRNAs translating in RF2 (Figure 8A, Supplementary Figure S7). In addition, the Gex mRNAs, while not having a functional ‘AUG’ do translate into RF2 if the first ‘UUG’ is used. As with ND7, we observed a shift in editing pattern preference when the EATRO 164 cells were changed from SDM79 medium to SDM80. Interestingly, a new fully edited form of CR3 appeared in the EATRO164 SDM80 library only. The gRNA gCe80 is used in the EATRO SDM79 pathway, it dead-ends with this gRNA. Cells grown in SDM80 however, continue this editing pathway with two additional gRNAs, gDEe80 and gFGe80 (Figure 8B, green dots). This mRNA is translatable but produces a distinctly different and shorter protein product (Figure 8A). The protein products of the two different cell lines are highly dissimilar. Using bioinformatics tools to predict the secondary structure of these proteins, we find that the difference is most noticeable in the RF1s of the two cell lines (Figure 9). Interestingly, the RF2s have a very similar predicted secondary structure. This evidence suggests that the two different cell lines are able to use the CR3 transcript with different sets of gRNAs to create distinctly different protein products.
Figure 8.
Observed CR3 editing pathways in the EATRO 164 cell line grown in SDM79 (A) and SDM80 (B). U = unedited transcripts. For arrow and gRNA naming descriptors see Figure 1. Orange dots indicate edited sequence unique to the EATRO 164 cell line. Green dots indicate edited sequence unique to EATRO cells grown in SDM80 medium. Terminal dots are colored blue for reading frame 1 or magenta for reading frame 2. Boxed magenta dots have no functional start codon but are translatable into reading frame 2 with the use of an alternative start codon (UUG). + indicates that more than one mRNA form was condensed into this circle to simplify the figure (see Supplementary Figure S6). Condensed forms encode largely the same amino acid sequence with only small variants.
Figure 9.

Predicted secondary structures of most abundant CR3 predicted proteins. Secondary structure predictions were generated by RaptorX (74–76). Shaded regions indicate predicted transmembrane alpha helices predicted by Phobius (73,77).
DISCUSSION
In this work, we developed a new transcriptome analysis pipeline to fully characterize the editing pathways for three putative dual-coding genes, RPS12, ND7 5′ and CR3. The pipeline uses a new program, SKETCH, in combination with our gRNA database search program (26). Combining these two programs allowed us to separate alternative edits directed by different gRNAs from partially edited or mis-edited transcripts and allowed the precise mapping of the full progression of the editing process. This characterization was done in two different cell lines (TREU 667 and EATRO 164) and under different energy conditions in order to determine the robustness of the editing process. Surprisingly, distinct differences in both editing progression as well as editing efficiency were observed in the two different cell lines. In addition, growth of parasites under different energy conditions also appeared to be able to influence the editing process. A comparison of the two cell lines grown in SDM79 did suggest that overall, the TREU 667 cells were more efficient in editing these three pan-edited transcripts. However, when the EATRO 164 cells were transferred from a high glucose medium (SDM79) to a glucose-restricted medium (SDM80), the number of transcripts that initiated the editing process significantly increased. For RPS12, the increase in editing initiation resulted in a 4-fold increase in the number of fully edited and translatable mRNAs.
Of the transcripts characterized, the essential RPS12 showed the most robust editing progression. Editing of RPS12 is relatively linear, with only a few minor branching alternatives. For this mRNA, the first start codon found on the fully edited transcripts consistently translated into the canonical RPS12 open reading frame and we found no evidence of transcripts that access the alternative reading frame. Previously, we had identified multiple different gRNAs capable of shifting the reading frame of RPS12 (21). The frame-shift gRNA identified in the TREU 667 transcriptome was only moderately less abundant than the gJ population with more than 2600 reads, however we found no evidence of its use. A frame shifting RPS12 gRNA has also been identified as encoded in the Leishmania tarentolae minicircle population, but it is not known if this gRNA is expressed or utilized (53).
In contrast to RPS12, we found distinct evidence that ND7 5′ is dual-coding. In both cell lines, alternative editing by different terminal gRNA variants resulted in transcripts with either RF1 (the canonical ND7) or RF3 (a putative metabolite transporter) linked to the first AUG (21). Interestingly, ND7 5′ has also been sequenced in 29–13 (LISTER 427) cells (45). While that study did not directly state evidence of dual-coding, they did indicate that a large proportion of the fully edited ND7 5′ transcripts had a single nucleotide difference in the 5′ UTR. This difference could very well be the same difference we observe in E2 transcripts that links an upstream AUG to the ARF. When a gene is dual coding, the flexibility of the amino acid composition of both proteins is constrained. This suggests that long-term maintenance of dual coding genes only occurs if the overlap is advantageous to the organism (54). The finding that the ability to access two different reading frames is maintained across at least three different T. brucei cell lines suggests that dual-coding genes must provide an important evolutionary advantage.
While fully edited ND7 5′ transcripts were found in both cell lines, a major difference was observed in the efficiency of the editing process. In TREU 667 cells, over 79% of ND7 5′ transcripts had initiated the editing process and a full 9.7% are fully edited. In contrast, EATRO 164 cells grown under the same conditions (SDM79) had only 52.4% transcripts that initiated editing and a scant 0.2% fully edited. Growth of EATRO 164 cells in SDM80 did substantially increase the number of transcripts that had initiated RNA editing (72.6%), however, no corresponding increase in fully edited transcripts was observed. The major differences in editing efficiency appear to be due to both the use of alternative gRNAs that could disrupt the editing cascade and well as a gRNA mutation that affected the ability of the guide RNA to efficiently anchor. Surprisingly, the gRNAs that disrupt editing in the EATRO cell line are also present in the TREU gRNA transcriptome. It is unclear why we see evidence of their use in only the EATRO cells. It may be that in the TREU cells, these gRNAs are more efficiently used in a different alternatively edited pathway that has yet to be discovered. Characterization of the gRNA transcriptomes identified millions of reads and over ∼64 000 unique gRNA sequences capable of generating conventional editing patterns (26,28). These libraries however, do contain millions of gRNA-like transcripts (correct size, correct transcription start sites and a poly-U tail) that do not match any of the previously published mitochondrial mRNA sequences. This suggests that alternative editing and the coding capacity of the mitochondrial genome may be much greater than originally thought. A full understanding of gRNA selection and use will require the characterization of the entire edited transcriptome. In addition to the large decrease in the efficiency of ND7 5′ editing observed in the EATRO cells, we also saw a distinct shift in the number of fully edited transcripts that translate in RF3, the alternative open reading frame. This alternative protein has been previously hypothesized to be a metabolite transporter as it shares distant homology with a bacterial sugar transporter, SemiSWEET (21).
The most pronounced differences between the two cell lines was observed for the CR3 transcript. In both cell lines, CR3 utilizes a much more complicated editing pathway than either ND7 5′ or RPS12 and the overall efficiency of the editing process is very low. Surprisingly, the number of CR3 transcripts that initiate RNA editing is comparable to the percentage observed for RPS12. However, editing by the initiating gRNA appears to be very inefficient. In TREU cells, while 15.1% of the transcripts initiate editing, only 2.2% are fully edited through the first editing block. A similar drop is also observed in the EATRO cells. The identified gRNA population that initiates editing does not contain any mismatched base pairs and it is unclear why full editing by this gRNA is so inefficient. The canonical CR3 is a putative NADH Dehydrogenase complex I member (ND4L) (39). Editing of Complex 1 members does appear to be developmentally regulated, with early studies performed in EATRO 164 showing edited members being preferentially edited in the bloodstream stage (2,35,37,42,43). However, recent data suggests that complex 1 editing preference may be cell line specific (38). It may be that for most of these transcripts, editing is stalled right after initiation by a transcript specific mechanism. Many proteins involved in RNA editing have been identified to have transcript specific effects through knockdown assays (47,55–67).
Surprisingly, transcripts edited to the canonical CR3 sequence were only observed in the TREU cell line. In this cell line, four different 3′ editing pathways converge to an internal consensus sequence which then diverges again near the 5′ end, generating a possible 12 different ORFs in two different reading frames. In the EATRO cell line, editing initiates with most of the same 3′ gRNAs, but diverges at the internal consensus sequence when they employ a completely different set of gRNAs for full editing. The different editing patterns found in the EATRO cell lines generate a possible 10 different ORFs again in two different reading frames. Because of the utilization of a different set of gRNAs, the variable TREU and EATRO CR3 transcripts are predicted to produce very different protein products with very different structures. Searches were run on various databases in order to determine the putative functions of the many CR3 proteins. Unfortunately, these searches yielded no significant results. The very small percentage of CR3 transcripts that undergo full editing suggests that the protein products may not be made or utilized in this stage of the parasite life cycle. However, we hypothesize that the ability to alternatively edit transcripts may be an important evolutionary mechanism to maintain genetic plasticity. The dual host life cycle of T. brucei leaves it vulnerable to genetic drift especially for the mitochondrial ETC genes which are not under selection during the Bloodstream stage. Previously, we proposed a mechanism that would contribute to the drift robustness of these mitochondrial genes. By overlapping ETC genes not under selection in the bloodstream stage with genes that are under selection during this stage of the life cycle, the accumulation of mutations can be prevented (21). These overlapped genes share most gRNAs, and this strategy ensures that almost all of the genetic material is protected. We also hypothesize that the sequential nature of gRNA use and the sensitivity of the RNA editing process to both mRNA and gRNA mutations can also protect against genetic drift by increasing the deleterious effects of the mutations (20). Increasing the lethality of mutations would insure that deleterious mutations are purged from the population during long periods of growth in the mammalian host (68–70). While the process of RNA editing may weed out mutations by making them lethal, it would also prevent the population from generating beneficial mutations as well. In addition, this strategy would impede the organism's ability to evolve and adapt to new environments. We suggest that alternative edits, such as those seen in the CR3 and others previously observed, generate protein diversity without compromising the genetic information found within the genome (71). The ability of many of the promiscuous gRNAs to generate translatable transcripts suggests a surprisingly robust ability of the RNA editing system to generate protein variation. This use of alternative editing to generate protein variability suggests that RNA editing is a powerful way to balance the protection of genetic information (mutational protection of the mRNA genes) with the need to allow protein diversity and functional adaptation to changing environments.
DATA AVAILABILITY
SKETCH is available for download on GitHub at https://github.com/laurakirby4/SKETCH. The gRNA search program (26) and gRNA transcriptome dataset is available on GitHub at https://github.com/laurakirby4/gRNASearchProgramAndFiles.
Transcriptome data is available through the NCBI’s Sequence Read Archive under sample numbers: SAMN11233338, SAMN11233339, and SAMN11233340.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Ken Stuart Lab for trypanosome cell lines, Hanyou Pan for his work in the collection of the ND7 5′ data, and Chris Adami for thoughtful discussions. We would also like to acknowledge the Elenor L Gilmore Endowed Excellence Award and the Frank Peabody Microbiology Student Research Award for their recognition of LEK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Notes
Present address: Laura E. Kirby, Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R03AI0902 to D.K.]; LEK was supported in part by the Russell B. DuVall Award. Funding for open access charge: Microbiology and Molecular Genetics Department, Michigan State University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Shapiro T.A., Englund P.T.. The structure and replication of kinetoplast DNA. Annu. Rev. Microbiol. 1995; 49:117–143. [DOI] [PubMed] [Google Scholar]
- 2. Priest J.W., Hajduk S.L.. Developmental regulation of mitochondrial biogenesis in Trypanosoma brucei. J. Bioenerg. Biomembr. 1994; 26:179–191. [DOI] [PubMed] [Google Scholar]
- 3. Blum B., Bakalara N., Simpson L.. A model for RNA editing in kinetoplastid mitochondria: ‘Guide’ RNA molecules transcribed from maxicircle DNA provide the edited information. Cell. 1990; 60:189–198. [DOI] [PubMed] [Google Scholar]
- 4. Lukeš J., Wheeler R., Jirsová D., David V., Archibald J.M.. Massive mitochondrial DNA content in diplonemid and kinetoplastid protists. IUBMB Life. 2018; 70:1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Read L.K., Lukeš J., Hashimi H.. Trypanosome RNA editing: the complexity of getting U in and taking U out. Wiley Interdiscip. Rev. RNA. 2016; 7:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aphasizheva I., Aphasizhev R.. U-insertion/deletion mRNA-editing holoenzyme: definition in sight. Trends Parasitol. 2016; 32:144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Weelden S.W.H., Fast B., Vogt A., van der Meer P., Saas J., van Hellemond J.J., Tielens A.G.M., Boshart M.. Procyclic Trypanosoma brucei do not use krebs cycle activity for energy generation. J. Biol. Chem. 2003; 278:12854–12863. [DOI] [PubMed] [Google Scholar]
- 8. van Weelden S.W.H., van Hellemond J.J., Opperdoes F.R., Tielens A.G.M.. New functions for parts of the krebs cycle in procyclic Trypanosoma brucei, a cycle not operating as a cycle. J. Biol. Chem. 2005; 280:12451–12460. [DOI] [PubMed] [Google Scholar]
- 9. Bringaud F., Rivière L., Coustou V.. Energy metabolism of trypanosomatids: Adaptation to available carbon sources. Mol. Biochem. Parasitol. 2006; 149:1–9. [DOI] [PubMed] [Google Scholar]
- 10. Oberle M., Balmer O., Brun R., Roditi I.. Bottlenecks and the maintenance of minor genotypes during the life cycle of Trypanosoma brucei. PLoS Pathog. 2010; 6:e1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abbeele J.V.D., Claes Y., Bockstaele D.V., Ray D.L., Coosemans M.. Trypanosoma brucei spp. development in the tsetse fly: characterization of the post-mesocyclic stages in the foregut and proboscis. Parasitology. 1999; 118:469–478. [DOI] [PubMed] [Google Scholar]
- 12. van Hellemond J.J., Bakker B.M., Tielens A.G.M.. Poole RK. Energy metabolism and its compartmentation in Trypanosoma brucei. Advances in Microbial Physiology. 2005; 50:Academic Press; 199–226. [DOI] [PubMed] [Google Scholar]
- 13. Horn D. Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 2014; 195:123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sudarshi D., Lawrence S., Pickrell W.O., Eligar V., Walters R., Quaderi S., Walker A., Capewell P., Clucas C., Vincent A. et al.. Human african trypanosomiasis presenting at least 29 years after infection—what can this teach us about the pathogenesis and control of this neglected tropical disease?. PLoS Negl Trop Dis. 2014; 8:e3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muller H.J. The relation of recombination to mutational advance. Mutat. Res. Mol. Mech. Mutagen. 1964; 1:2–9. [DOI] [PubMed] [Google Scholar]
- 16. Haigh J. The accumulation of deleterious genes in a population—Muller's Ratchet. Theor. Popul. Biol. 1978; 14:251–267. [DOI] [PubMed] [Google Scholar]
- 17. Poon A., Otto S.P.. Compensating for our load of mutations: freezing the meltdown of small populations. Evolution. 2000; 54:1467–1479. [DOI] [PubMed] [Google Scholar]
- 18. Whitlock M.C. Fixation of new alleles and the extinction of small populations: drift load, beneficial alleles, and sexual selection. Evolution. 2000; 54:1855–1861. [DOI] [PubMed] [Google Scholar]
- 19. Lynch M., Bürger R., Butcher D., Gabriel W.. The mutational meltdown in asexual populations. J. Hered. 1993; 84:339–344. [DOI] [PubMed] [Google Scholar]
- 20. LaBar T., Adami C.. Evolution of drift robustness in small populations. Nat. Commun. 2017; 8:1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirby L.E., Koslowsky D.. Mitochondrial dual-coding genes in Trypanosoma brucei. PLoS Negl. Trop. Dis. 2017; 11:e0005989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cristodero M., Seebeck T., Schneider A.. Mitochondrial translation is essential in bloodstream forms of Trypanosoma brucei. Mol. Microbiol. 2010; 78:757–769. [DOI] [PubMed] [Google Scholar]
- 23. Aphasizheva I., Maslov D.A., Aphasizhev R.. Kinetoplast DNA-encoded ribosomal protein S12. RNA Biol. 2013; 10:1679–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hudson K.M., Taylor A.E.R., Elce B.J.. Antigenic changes in Trypanosoma brucei on transmission by tsetse fly. Parasite Immunol. 1980; 2:57–69. [Google Scholar]
- 25. Agabian N., Thomashow L., Milhausen M., Stuart K.. Structural analysis of variant and invariant genes in Trypanosomes. Am. J. Trop. Med. Hyg. 1980; 29:1043–1049. [DOI] [PubMed] [Google Scholar]
- 26. Koslowsky D., Sun Y., Hindenach J., Theisen T., Lucas J.. The insect-phase gRNA transcriptome in Trypanosoma brucei. Nucleic Acids Res. 2014; 42:1873–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamour N., Rivière L., Coustou V., Coombs G.H., Barrett M.P., Bringaud F.. Proline metabolism in procyclic Trypanosoma brucei is down-regulated in the presence of glucose. J. Biol. Chem. 2005; 280:11902–11910. [DOI] [PubMed] [Google Scholar]
- 28. Kirby L.E., Sun Y., Judah D., Nowak S., Koslowsky D.. Analysis of the Trypanosoma brucei EATRO 164 bloodstream guide RNA transcriptome. PLOS Negl. Trop. Dis. 2016; 10:e0004793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coustou V., Biran M., Breton M., Guegan F., Rivière L., Plazolles N., Nolan D., Barrett M.P., Franconi J.-M., Bringaud F.. Glucose-induced remodeling of intermediary and energy metabolism in Procyclic Trypanosoma brucei. J. Biol. Chem. 2008; 283:16342–16354. [DOI] [PubMed] [Google Scholar]
- 30. Verner Z., Čermáková P., Škodová I., Kriegová E., Horváth A., Lukeš J.. Complex I (NADH:ubiquinone oxidoreductase) is active in but non-essential for procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 2011; 175:196–200. [DOI] [PubMed] [Google Scholar]
- 31. Bochud-Allemann N., Schneider A.. Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. J. Biol. Chem. 2002; 277:32849–32854. [DOI] [PubMed] [Google Scholar]
- 32. Horváth A., Horáková E., Dunajčíková P., Verner Z., Pravdová E., Šlapetová I., Cuninková L., Lukeš J.. Downregulation of the nuclear-encoded subunits of the complexes III and IV disrupts their respective complexes but not complex I in procyclic Trypanosoma brucei. Mol. Microbiol. 2005; 58:116–130. [DOI] [PubMed] [Google Scholar]
- 33. Gnipová A., Panicucci B., Paris Z., Verner Z., Horváth A., Lukeš J., Zíková A.. Disparate phenotypic effects from the knockdown of various Trypanosoma brucei cytochrome c oxidase subunits. Mol. Biochem. Parasitol. 2012; 184:90–98. [DOI] [PubMed] [Google Scholar]
- 34. Kuile B.H. ter. Adaptation of metabolic enzyme activities of Trypanosoma brucei promastigotes to growth rate and carbon regimen. J. Bacteriol. 1997; 179:4699–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Read L.K., Myler P.J., Stuart K.. Extensive editing of both processed and preprocessed maxicircle CR6 transcripts in Trypanosoma brucei. J. Biol. Chem. 1992; 267:1123–1128. [PubMed] [Google Scholar]
- 36. Ramrath D.J.F., Niemann M., Leibundgut M., Bieri P., Prange C., Horn E.K., Leitner A., Boehringer D., Schneider A., Ban N.. Evolutionary shift toward protein-based architecture in trypanosomal mitochondrial ribosomes. Science. 2018; 362:doi:10.1126/science.aau7735. [DOI] [PubMed] [Google Scholar]
- 37. Koslowsky D.J., Bhat G.J., Perrollaz A.L., Feagin J.E., Stuart K.. The MURF3 gene of T. brucei contains multiple domains of extensive editing and is homologous to a subunit of NADH dehydrogenase. Cell. 1990; 62:901–911. [DOI] [PubMed] [Google Scholar]
- 38. Gazestani V.H., Hampton M., Shaw A.K., Salavati R., Zimmer S.L.. Tail characteristics of Trypanosoma brucei mitochondrial transcripts are developmentally altered in a transcript-specific manner. Int. J. Parasitol. 2018; 48:179–189. [DOI] [PubMed] [Google Scholar]
- 39. Duarte M., Tomás A.M.. The mitochondrial complex I of trypanosomatids - an overview of current knowledge. J. Bioenerg. Biomembr. 2014; 46:299–311. [DOI] [PubMed] [Google Scholar]
- 40. Stuart K. The RNA editing process in Trypanosoma brucei. Semin. Cell Biol. 1993; 4:251–260. [DOI] [PubMed] [Google Scholar]
- 41. Surve S., Heestand M., Panicucci B., Schnaufer A., Parsons M.. Enigmatic presence of mitochondrial complex I in Trypanosoma brucei bloodstream forms. Eukaryot. Cell. 2012; 11:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Souza A.E., Myler P.J., Stuart K.. Maxicircle CR1 transcripts of Trypanosoma brucei are edited and developmentally regulated and encode a putative iron-sulfur protein homologous to an NADH dehydrogenase subunit. Mol. Cell Biol. 1992; 12:2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Souza A.E., Shu H.H., Read L.K., Myler P.J., Stuart K.D.. Extensive editing of CR2 maxicircle transcripts of Trypanosoma brucei predicts a protein with homology to a subunit of NADH dehydrogenase. Mol. Cell Biol. 1993; 13:6832–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Read L.K., Wilson K.D., Myler P.J., Stuart K.. Editing of Trypanosoma brucei maxicircle CR5 mRNA generates variable carboxy terminal predicted protein sequences. Nucleic Acids Res. 1994; 22:1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simpson R.M., Bruno A.E., Bard J.E., Buck M.J., Read L.K.. High-throughput sequencing of partially edited trypanosome mRNAs reveals barriers to editing progression and evidence for alternative editing. RNA. 2016; 22:677–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wirtz E., Leal S., Ochatt C., Cross G.M.. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999; 99:89–101. [DOI] [PubMed] [Google Scholar]
- 47. Carnes J., McDermott S., Anupama A., Oliver B.G., Sather D.N., Stuart K.. In vivo cleavage specificity of Trypanosoma brucei editosome endonucleases. Nucleic Acids Res. 2017; 45:4667–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gerasimov E.S., Gasparyan A.A., Kaurov I., Tichý B., Logacheva M.D., Kolesnikov A.A., Lukeš J., Yurchenko V., Zimmer S.L., Flegontov P.. Trypanosomatid mitochondrial RNA editing: dramatically complex transcript repertoires revealed with a dedicated mapping tool. Nucleic Acids Res. 2018; 46:765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tylec B.L., Simpson R.M., Kirby L.E., Chen R., Sun Y., Koslowsky D.J., Read L.K.. Intrinsic and regulated properties of minimally edited trypanosome mRNAs. Nucleic Acids Res. 2019; 47:3640–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lawson S.D., Igo R.P., Salavati R., Stuart K.D.. The specificity of nucleotide removal during RNA editing in Trypanosoma brucei. RNA. 2001; 7:1793–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baradaran R., Berrisford J.M., Minhas G.S., Sazanov L.A.. Crystal structure of the entire respiratory complex I. Nature. 2013; 494:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hensgens L.A., Brakenhoff J., De Vries B.F., Sloof P., Tromp M.C., Van Boom J.H., Benne R.. The sequence of the gene for cytochrome c oxidase subunit I, a frameshift containing gene for cytochrome c oxidase subunit II and seven unassigned reading frames in Trypanosoma brucei mitochrondrial maxi-circle DNA. Nucleic Acids Res. 1984; 12:7327–7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Simpson L., Douglass S.M., Lake J.A., Pellegrini M., Li F.. Comparison of the mitochondrial genomes and steady state transcriptomes of two strains of the trypanosomatid parasite, Leishmania tarentolae. PLOS Negl. Trop. Dis. 2015; 9:e0003841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chung W.-Y., Wadhawan S., Szklarczyk R., Pond S.K., Nekrutenko A.. A first look at ARFome: dual-coding genes in mammalian genomes. PLOS Comput. Biol. 2007; 3:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Missel A., Souza A.E., Nörskau G., Göringer H.U.. Disruption of a gene encoding a novel mitochondrial DEAD-box protein in Trypanosoma brucei affects edited mRNAs. Mol. Cell Biol. 1997; 17:4895–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pelletier M., Read L.K.. RBP16 is a multifunctional gene regulatory protein involved in editing and stabilization of specific mitochondrial mRNAs in Trypanosoma brucei. RNA. 2003; 9:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vondrušková E., van den Burg J., Zíková A., Ernst N.L., Stuart K., Benne R., Lukeš J.. RNA interference analyses suggest a Transcript-specific regulatory role for mitochondrial RNA-binding proteins MRP1 and MRP2 in RNA editing and other RNA processing in Trypanosoma brucei. J. Biol. Chem. 2005; 280:2429–2438. [DOI] [PubMed] [Google Scholar]
- 58. Goulah C.C., Pelletier M., Read L.K.. Arginine methylation regulates mitochondrial gene expression in Trypanosoma brucei through multiple effector proteins. RNA. 2006; 12:1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zíková A., Horáková E., Jirků M., Dunajčíková P., Lukeš J.. The effect of down-regulation of mitochondrial RNA-binding proteins MRP1 and MRP2 on respiratory complexes in procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 2006; 149:65–73. [DOI] [PubMed] [Google Scholar]
- 60. Acestor N., Panigrahi A.K., Carnes J., Zíková A., Stuart K.D.. The MRB1 complex functions in kinetoplastid RNA processing. RNA. 2009; 15:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hernandez A., Madina B.R., Ro K., Wohlschlegel J.A., Willard B., Kinter M.T., Cruz-Reyes J.. REH2 RNA helicase in kinetoplastid mitochondria ribonucleoprotein complexes and essential motifs for unwinding and guide RNA (gRNA) binding. J. Biol. Chem. 2010; 285:1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kafková L., Ammerman M.L., Faktorová D., Fisk J.C., Zimmer S.L., Sobotka R., Read L.K., Lukeš J., Hashimi H.. Functional characterization of two paralogs that are novel RNA binding proteins influencing mitochondrial transcripts of Trypanosoma brucei. RNA. 2012; 18:1846–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zimmer S.L., McEvoy S.M., Menon S., Read L.K.. Additive and Transcript-Specific effects of KPAP1 and TbRND activities on 3′ Non-Encoded tail characteristics and mRNA stability in Trypanosoma brucei. PLoS One. 2012; 7:e37639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carnes J., Ernst N.L., Wickham C., Panicucci B., Stuart K.. KREX2 is not essential for either procyclic or bloodstream form Trypanosoma brucei. PLoS One. 2012; 7:e33405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aphasizheva I., Zhang L., Wang X., Kaake R.M., Huang L., Monti S., Aphasizhev R.. RNA binding and core complexes constitute the U-insertion/deletion editosome. Mol. Cell Biol. 2014; 34:4329–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Simpson R.M., Bruno A.E., Chen R., Lott K., Tylec B.L., Bard J.E., Sun Y., Buck M.J., Read L.K.. Trypanosome RNA editing mediator complex proteins have distinct functions in gRNA utilization. Nucleic Acids Res. 2017; 45:7965–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dixit S., Lukeš J.. Combinatorial interplay of RNA-binding proteins tunes levels of mitochondrial mRNA in trypanosomes. RNA. 2018; 24:1594–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Speijer D. Constructive neutral evolution cannot explain current kinetoplastid panediting patterns. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:E25–E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Speijer D. Is kinetoplastid pan-editing the result of an evolutionary balancing act?. IUBMB Life. 2006; 58:91–96. [DOI] [PubMed] [Google Scholar]
- 70. Speijer D. Göringer HU. Evolutionary aspects of RNA editing. RNA Editing, Nucleic Acids and Molecular Biology. 2008; Berlin, Heidelberg: Springer; 199–227. [Google Scholar]
- 71. Ochsenreiter T., Anderson S., Wood Z.A., Hajduk S.L.. Alternative RNA editing produces a novel protein involved in mitochondrial DNA maintenance in trypanosomes. Mol. Cell Biol. 2008; 28:5595–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Otaka E., Hashimoto T., Mizuta K.. The ribosomal proteins. I: an introduction to a compilation of the protein species equivalents from various organisms by a universal code system. Protein Seq. Data Anal. 1993; 5:285–300. [Google Scholar]
- 73. Käll L., Krogh A., Sonnhammer E.L.L.. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 2007; 35:W429–W432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., Xu J.. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012; 7:1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Peng J., Xu J.. A multiple-template approach to protein threading. Proteins Struct. Funct. Bioinforma. 2011; 79:1930–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Peng J., Xu J.. Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins Struct. Funct. Bioinforma. 2011; 79:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J. et al.. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2014; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SKETCH is available for download on GitHub at https://github.com/laurakirby4/SKETCH. The gRNA search program (26) and gRNA transcriptome dataset is available on GitHub at https://github.com/laurakirby4/gRNASearchProgramAndFiles.
Transcriptome data is available through the NCBI’s Sequence Read Archive under sample numbers: SAMN11233338, SAMN11233339, and SAMN11233340.