


Abstract
Multiplexed RNA in situ hybridization for the analysis of gene expression patterns plays an important role in investigating development and disease. Here, we present a method for multiplexed RNA-ISH to detect spatial tumor heterogeneity in tissue sections. We made use of a microfluidic chip to deliver ISH-probes locally to regions of a few hundred micrometers over time periods of tens of minutes. This spatial multiplexing method can be combined with ISH-approaches based on signal amplification, with bright field detection and with the commonly used format of formalin-fixed paraffin-embedded tissue sections. By using this method, we analyzed the expression of HER2 with internal positive and negative controls (ActB, dapB) as well as predictive biomarker panels (ER, PgR, HER2) in a spatially multiplexed manner on single mammary carcinoma sections. We further demonstrated the applicability of the technique for subtype differentiation in breast cancer. Local analysis of HER2 revealed medium to high spatial heterogeneity of gene expression (Cohen effect size r = 0.4) in equivocally tested tumor tissues. Thereby, we exemplify the importance of using such a complementary approach for the analysis of spatial heterogeneity, in particular for equivocally tested tumor samples. As the method is compatible with a range of ISH approaches and tissue samples, it has the potential to find broad applicability in the context of molecular analysis of human diseases.
INTRODUCTION
Personalized medicine relies on the molecular analysis of individual tumors. To understand differences in progression and survival probabilities between patients affected by cancer, it is necessary to differentiate tumors into subtypes. For example, the current molecular classification schemes for breast cancer describe five molecular subtypes: luminal A, luminal B, human epidermal growth factor receptor 2 (HER2)-positive, triple-negative, and normal-like (1). For predictive and prognostic reasons, breast tumors are classified into these subtypes according to their molecular characteristics. Standard clinical methods for subtype differentiation include immunohistochemistry (IHC) to detect the biomarker expression status at the protein level and fluorescence in situ hybridization (FISH) to analyze gene amplification. While most of the tumors can be classified by using these methods, equivocal cases do exist. For HER2 status determination, for example, 32% of tumors are described as equivocal when using IHC and 5% when using FISH (2). Repeated testing using both methods can reduce the total number of equivocal cases to 5%. Ohlschlegel et al. showed that equivocal testing is largely influenced by the molecular heterogeneity of the tumor tissue (3). While genomic heterogeneity was described for 14.7% of the cases in a general cohort, the number increased to 41% for equivocal cases. The analysis of molecular heterogeneity is therefore highly valuable for the subtype differentiation of breast tumors and thus also for prognostic and predictive purposes.
Cellular heterogeneity is a driving factor in development and disease. It arises from modulations at the gene, transcript, and protein level and can be detected molecularly and phenotypically. Patient-by-patient or intertumor molecular heterogeneity is commonly analyzed in the clinical setting (subtype differentiation) and can be distinguished and addressed by using existing methods recommended in clinical guidelines (4). However, the impact of the prognostic and predictive power of spatial intratumor heterogeneity is less known. Retaining spatial information is highly important to link the molecular state to function, development, and disease. This is of special significance in tumors, as ∼60–70% of all somatic mutations are not detectable in all tumor areas (5) and cell variants with an abundance of as little as 0.1% of the total cell population can affect the treatment response (6). Recently, the coexistence of multiple breast cancer subtypes and their plastic conversion within a single tumor has been proposed (7) and several clinical trials are currently analyzing the impact of intratumor heterogeneity spatially and temporally (8–11). Interestingly, intermediate levels of genetic intratumor heterogeneity have been linked to a poor prognosis (12,13), while, for high levels of heterogeneity, the prognosis improved in certain cases (12). To personalize tumor treatment based on heterogeneity profiles, it is therefore important to capture the molecular tumor profile to the full extent in the spatial and temporal dimension. In situ techniques intrinsically provide spatial information and in situ protein detection has been extended to multiplexed approaches, thus allowing the spatial detection of multiple proteins of interest. In tumor analysis, multiplexing is commonly used for the detection of protein biomarkers, e.g. using the Opal multiplex assay (14), and tumor associated immune cells (15–17). Through using automated microfluidic systems, the duration of four-plex experiments has been reduced from two days to few hours (14,18). Quantitation of the signal is facilitated by immunofluorescence approaches and spectral unmixing or sequential detection (19–21) and can be performed using automated quantitative analysis (AQUA) (22). Nonetheless, when using current standard immunohistochemistry approaches (23), the quantification of protein expression levels is difficult (24). Therefore, the analysis and quantification of transcripts is a promising alternative, as it provides information about the genetic content as well as gene expression levels (25). Recent novel diagnostic developments for breast cancer thus place a focus on the detection of gene expression on the RNA level, e.g. 21-gene Oncotype DX Recurrence Score (26), 70-gene MammaPrint Assay (27) and a multitude of other signatures (28,29), as mRNA is readily quantifiable in quantitative real-time polymerase chain reactions (qRT-PCR) or microarray-based approaches. Clinical guidelines approve of the use of gene expression profiles for additional prognostic and predictive purposes under certain conditions (29,30) and ongoing studies of tumors in the central nervous system show great promise in using RNA sequencing as a predictive measure (31). Translating such methods for quantifying gene expression from the research to the clinical setting therefore plays an important role in developing novel diagnostic approaches for addressing intratumor heterogeneity.
In research settings, several techniques for the detection and quantification of transcripts exist. While many of the early quantitative methods required the extraction of RNA from the sample (such as Northern blotting, qRT-PCR, digital PCR, microarrays or high-throughput sequencing), new developments allow for in situ detection and quantification of transcripts. These methods can be divided into four categories: (I) FISH (32–36), (II) FISH with barcoding (37–40), (III) in situ sequencing (41–44) and (IV) patterned barcoded microarrays (45,46). These methods have in common that they provide information about transcript localization in the sample or even on a subcellular level. They thus enable the analysis of subpopulations of cells, whose signals would be averaged in extraction-based approaches. A methodology combining the advantages of both, localization-retaining methods and high-throughput sequencing, uses laser capture microdissection (47–49). While spatial transcriptomic methods, such as in situ sequencing or combined laser capture microdissection and RNA sequencing, provide quantitative spatial information about a large number of transcripts (up to 6000), they require the use of specialized instruments and techniques, which are currently not used in the clinical environment. As DNA-FISH is routinely performed in the clinical setting, the equipment required for in situ hybridization experiments is available and can be readily used for transcript analysis. Wang et al. described the use of RNA in situ hybridization as a quantitative companion diagnostic application for HER2 testing, with special importance for cases with intratumoral heterogeneity or equivocal FISH results (2). Previous studies have shown the applicability of RNA-ISH for formalin-fixed paraffin-embedded (FFPE) tissue sections (50–52). Compared to other spatial transcriptomic techniques, it thus poses an important alternative for clinical applications because the majority of tumor tissues stored in biobanks are FFPE tissues. Furthermore, nearly all mRNA molecules can be detected using single molecule FISH, while single-cell mRNA sequencing captures 5–40% and microarray or in situ barcode sequencing only around 5% of mRNA molecules (34). This makes RNA-ISH a wide-spread technique for quantitative or semi-quantitative analyses of transcripts, although limited in the number of species that can be detected simultaneously. The detection of single RNA transcripts and their quantification can either be performed directly, by using hybridization probes containing fluorophores and highly sensitive detection optics (53), or by amplification of the reaction using branched FISH (54–56), enzymatic processes (57) and antibodies (58). The use of amplification-based systems obliviates the need for expensive optical setups and detection can even be performed using brightfield microscopy. Its application is currently limited, however, to one or two detection channels depending on the enzyme-substrate pairs used. We therefore use a microfluidic system for the local delivery of probes, which allows for spatial multiplexing. We make use of a variant of a microfluidic system called the vertical microfluidic probe (MFP) (59,60) in this work, which enables the localized delivery of reagents on the nanoliter scale. To avoid confusion with the usage of FISH probes, we term the MFP as microfluidic chip in this manuscript. A basic implementation of the chip consists of two channels with a size in the range of 50–100 μm, whose openings are positioned in parallel to and tens of micrometers above a surface of interest (59,60). Simultaneous injection and aspiration of liquid (at certain ratios of flow rates) from the channels confines the injected reagent to a small region between the chip and a surface immersed in aqueous solution (hydrodynamic flow confinement). Owing to its compatibility with biological substrates in a liquid environment, variants of the microfluidic chip (60) have found use in life science and biomedical applications (24,61,62).
Here, we established a microfluidic method for the detection of multiple transcripts in FFPE tissues through using spatial multiplexing in branched probe-based or enzymatic amplification RNA-ISH. Even though current clinical approaches are based on the semi-quantitative analysis of proteins and DNA, the possibility of quantification on the transcript level will likely provide advantages in the description of the spatial heterogeneity of cancers, where gene amplification, RNA and protein levels correlate (2,63). By using this multiplexed RNA-ISH, transcripts of interest along with tissue internal controls, complex cancer biomarker panels and spatial tumor heterogeneity can be evaluated on a single tissue section.
MATERIALS AND METHODS
FFPE cell blocks and tissues
FFPE cell blocks of the cell lines MCF7, BT474 and SKBR3 (AMS Biotechnology, Abingdon, UK) with HER2 expression levels of 1+, 2+ and 3+, respectively, were used as references. Breast carcinoma tissue sections were either obtained from BioChain (T2235086) or were kindly provided and anonymized by the tissue biobank of the Department of Pathology and Molecular Pathology of the Universitätsspital Zürich.
Hematoxylin and eosin staining of FFPE sections
Tissue sections were stained by incubation in hematoxylin for 5:30 min. A subsequent washing process in tap water for 5 min, incubation in 1% HCl for 10–20 s, and another wash in tap water for 1 min were used for colorimetric changes and removal of excess hematoxylin. Eosin was applied for 1 min, followed by a wash in tap water for 10 s and posterior dehydration of the tissue section using 95% and 100% ethanol for 1 min each.
Microfluidic system for probe delivery
The microfluidic chip, consisting of one layer of silicon and one of glass and containing four channels (channel size: 200, 100, 100, 200 μm) was fabricated in clean-room facilities in-house (60) and is reusable under stringent cleaning conditions. It was positioned on the microscopic system on a stage adjustable along the z-axis, using a connector system consisting of a screw, a gasket ring and a circular connector (Dolomite Microfluidics) (Supplementary Figure S1A–C). Tubings emerging from four syringes serving as reservoirs and a flow system were attached to the microfluidic chip via the connector. Liquid flows were generated using NeMESYS low pressure module syringe pumps (Cetoni, Korbussen, Germany) controlled by the software QMix Elements (Supplementary Figure S1D). The microfluidic chip was mounted on a Nikon Ti Eclipse microscope body placed in an incubating chamber (Life Imaging Services) and equipped with a Lumencor Sola 6-LCR-SB illumination (Lumencor, Beaverton, USA) and a Hamamatsu Orca Flash 4.0 camera (Hamamatsu, Nakaku, Japan). The position of the sample was adjusted in the x- and y-direction using high-precision stages (Phytron, Gröbenzell, Germany) controlled by the software WinCommander5.
RNA in situ hybridization
RNA-ISH experiments were performed following the instructions of the manufacturer (ViewRNA, Thermo-Fisher Scientific). Solutions for hybridization and washing were provided as part of the in situ hybridization kit. ISH probes targeted against dapB (dapB ViewRNA Probe Type 6, Bacillus subtilis), HER2 (HER2 ViewRNA Probe Type 1), PgR (PGR ViewRNA Probe Type 1 and Type 6), ERα (ESR ViewRNA Probe Type 1 and Type 6) and Actinβ (ActB ViewRNA Probe Type 1) RNA were used.
The FFPE tumor sections were incubated at 60°C for 1 h prior to the removal of paraffin using a 10 min incubation in xylene, two 5 min incubations in ethanol and washing steps in 70% ethanol and H2O. The pretreatment of the sections was a 20 min incubation of the slides in a 1× preincubation solution at 95°C. While incubating the sections in PBS, a hydrophobic pen was applied (ImmEdge Pen, Vector Laboratories) to limit the spread of the immersion liquid to a defined area around the tumor section. The sections were then digested using protease at 40°C for 5 min (cell pellet sections) or 10 min (mammary carcinoma sections). After washing in PBS, the sections were fixed using 10% neutral buffered formalin solution for 5 min. Sections were washed in PBS before the primary probe was delivered either through using the microfluidic chip (see Delivery of primary ISH-probes using the microfluidic chip) or by incubation of the whole slide in probe solution at 40°C. Varying hybridization times of the primary probe between 10 min and 3 h were used as indicated. After washing the sections in wash buffer three times, they were incubated with preamplifier solution (for dual color experiments: preamplifier mixture) at 40°C for 25 min. Following three washes in wash buffer at room temperature, the sections were incubated in amplifier solution (for dual color experiments: amplifier mixture) at 40°C for 15 min. Residual probes were removed by washing the sections three times using wash buffer. When dual-color experiments were performed, the sections were incubated with a label-6 alkaline phosphatase (AP) probe at 40°C for 15 min, washed three times, and incubated at room temperature for 30 min using a blue reagent as enzymatic substrate. After three washes using wash buffer, residual alkaline phosphatase was deactivated using AP stop solution at room temperature for 30 min, and the sections were washed using PBS and wash buffer. The sections were then incubated with a label-1 alkaline phosphatase probe at 40°C for 15 min, residual probes were removed using wash buffer, and the reaction was enhanced by an incubation in AP enhancer solution at room temperature for 5 min. The sections were incubated with Fast Red substrate at 40°C for 30 min, before being stained using hematoxylin and 3 μg/ml 4′-6-diamidino-2-phenylindole (DAPI). Sections were mounted using aqueous ImmunoHistoMount (Sigma-Aldrich) and imaged using brightfield and fluorescence detection (see Signal Visualization). Using CellProfiler (64), nuclei and cytoplasm of cells were detected and the ISH signal intensity was determined and allocated to each of the cells.
Delivery of the primary ISH-probes using the microfluidic chip
We performed hematoxylin and eosin staining on sections consecutive to the ones used for local RNA-ISH. Regions for delivery of the ISH probes were defined by microscopic inspection of the hematoxylin and eosin stained tissue sections. Primary probes were delivered using a microfluidic chip positioned ∼50 μm above the tissue section immersed in PBS. The inner channels of the chip with channel widths of 100 μm were used for the delivery of the primary probe, while the outer channels (channel width 200 μm) were used for the visualization of the hydrodynamic flow confinement using a red food dye (Trawosa). The flow rates of the inner and outer injections were 1 μl/min and 0.5 μl/min and the ones of the inner and outer aspirations were 1 μl/min and 5 μl/min, respectively. When performing the experiments using the microfluidic chip, primary probe incubation times ranged from 10 min to 1 h. The positions of local delivery of the probes were recorded using the software of the x–y–z stages (WinCommander5).
Whenever multiplexed RNA-ISH experiments of a single detection color and involving the use of primary probes of type 1 and type 6 were performed using the microfluidic chip, preamplifier and amplifier mixtures were used and the label probes of type 1 and type 6 were combined in a single hybridization reaction.
Global and microfluidic chip-based immunohistochemistry
ER, PgR and HER2 detection and quantification through immunohistochemistry was performed following the guidelines of the manufacturer (Herceptest, Dako Denmark A/S) and primary antibodies were delivered either globally or locally using the microfluidic chip as described (24,59). Briefly, after deparaffinization, epitope retrieval was performed at 95–99°C for 40 min (+20 min cool-down). After washing in wash buffer, the sections were incubated in a peroxidase-blocking solution for 5 min. For global immunohistochemistry, the sections were incubated in 2 μg/ml anti-HER2 antibody (ErbB2 Monoclonal Mouse Antibody, Clone e2-4001, ThermoFisher) for 1 h, in a 1:40 dilution of anti-ER antibody (Monoclonal Rabbit Anti-Human Estrogen Receptor α, Clone EP1, Dako) for 20 min, or in a 1:50 dilution of anti-PgR antibody (Monoclonal Mouse Anti-Human Progesterone Receptor, Clone PgR 1294, Dako) for 20 min. For local immunohistochemistry, 20 μg/ml anti-HER2 antibody (Polyclonal Rabbit Anti-Human c-erbB-2, Dako) was delivered to the tissue sections using the microfluidic chip for incubation times ranging from 28 to 600 s. The sections were then washed in wash buffer and incubated with a visualization reagent containing secondary antibodies against mouse and rabbit (EnVision Dual Link System-HRP, Dako) for 30 min. After washing the sections, the signal was visualized using 3,3′-diaminobenzidine for 10 min. A hematoxylin counterstain was performed to visualize the cells.
Signal visualization and analysis
Brightfield and fluorescence imaging was performed on a Nikon Eclipse 90i microscope using 4 × 0.2 NA, 10 × 0.45 NA, 20 × 0.75 NA, and 40 × 0.6 NA objectives. Brightfield signals were detected on a DXM 1200C camera.
For fluorescence imaging, the sample was excited using a Lumencor Sola SE 5-LCR-SB with the excitation filters 531/40 nm or 630/38 nm for visualization of the Fast Red or the Fast Blue signal, respectively. Fluorescence was collected by 4 × 0.2 NA, 10 × 0.45 NA, 20 × 0.75 NA and 40 × 0.6 NA objectives and separated according to wavelength by a 562 nm dichroic combined with a 593/40 nm emission filter or a 757 nm dichroic and a 775/59 nm emission filter. Stained nuclei were visualized using a DAPI filter set (377/50, 409, 447/60 nm). The signal was detected on a Digital Sight DS-1QM camera. Signal intensities were extracted from the images as integrated intensities of single cells using CellProfiler (64).
Heterogeneity analysis
The extracted signal intensities were compared between different regions of the tumor tissue section and a Mann–Whitney U-test was used to determine differences in the expression levels between regions. The test statistic U was calculated as
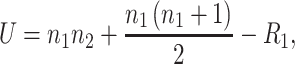 |
where R1 is the larger of the two rank sums, n1 the number of samples in the group with the larger rank sum and n2 the number of samples in the group with the smaller rank sum. The significance z was standardized using
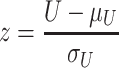 |
with the mean μ and the standard error σ of the distribution U. The Cohen effect size r, describing the importance of the significance in the difference of the means, was determined as
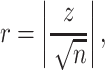 |
where n is the sum of the number of samples in both groups. A Cohen effect size r = 0.1 typically corresponds to a small effect, r = 0.3 to a moderate effect, and r = 0.5 to a strong effect (65). For visualization, the effect size was displayed as a color-coded matrix.
RESULTS
Localized RNA-ISH by microfluidic implementation
The molecular heterogeneity of tumor tissues is reflected in spatial differences in gene expression. To investigate these differences at the transcript level, we developed a microfluidic implementation of the RNA-ISH assay (Figure 1A, Supplementary Figure S1). It makes it possible to increase the number of transcripts that can be investigated on a single tissue section by using amplification-based transcript detection. Theoretically, up to 3000 hybridizations could be performed on a tissue area of 10 mm × 10 mm. However, the number of reactions that one can practically perform depends on multiple factors, ranging from the morphology of the tissue to availability of probes and design of the microfluidic chips.
Figure 1.

Local RNA in situ hybridization. (A) Schematic overview of a microfluidic implementation of chromogenic RNA in situ hybridization. The initial preparation of the tissue section including paraffin removal (1), heat (2) and protease (3) pretreatment, as well as fixation was performed globally. The primary probe was then hybridized locally to the RNA of interest (4). In this case the detection of HER2 is shown with internal negative (dapB) and positive (ActB) controls. Finally, the amplification of the signal using preamplifier and amplifier strands (5), the binding of enzyme (6), and the enzymatic reactions (7) were performed globally on the whole slide. (B) Fluorescence images of RNA-ISH experiments detecting the gene expression of HER2 in cell pellet sections of the breast cancer cell lines MCF7 (left), SKBR3 (center) and BT474 (right). Probes directed against bacterial dapB and ActB were used as negative and positive controls, respectively. Scale bar: 100 μm.
The initial pretreatment of the tumor tissue section, including paraffin removal, heat-induced pretreatment and protease digestion, was performed on the whole tissue slide. Precise control of the pretreatment steps, especially the protease digestion times, specific to the tissue of origin was required (Supplementary Note SN1, Figure S2). For the subsequent delivery of the primary probe, we used an open space microfluidic chip (59,60,66). Through the simultaneous injection and aspiration of the primary probe solution, it was delivered to a spatially defined region of ∼50–100 cell sections (∼150 μm in diameter). Probes for multiple transcripts of interest can thus be used in neighboring regions within the same tumor area. Additionally, through using hierarchical flow confinements (66), inner and outer regions of the confinement can be targeted using different probes and thus patterns of local delivery can be generated. The RNA-ISH signal increased for primary probe delivery times from 10 to 60 minutes, even under conditions with a constant flow (Supplementary Figure S3). Further increasing the hybridization time of the primary probe beyond 60 min, however, did not increase the signal intensity. Therefore, an incubation time of 60 min was chosen, when using the microfluidic chip. Constant delivery of probes under flow leads to the convection-enhanced reduction in hybridization time, which facilitates the sequential delivery of multiple probes without affecting the time required for the complete workflow (11–14 h depending on the spatial and spectral multiplexing conditions). Subsequent signal amplification and visualization was performed on the whole tumor tissue section, which enables direct comparisons of the signals within each tumor tissue section without having to take additional properties of the fluorophores and detection optics into consideration.
The use of the microfluidic chip system thus allows for the spatial multiplexing of a number of different RNA targets. The global delivery of oligonucleotides for amplification of the signal and enzymatic substrates permits different targets to be simultaneously amplified and visualized in a single reaction, thereby facilitating quantitative comparisons that are independent of the amplification process.
Biomarker analysis with intrinsic positive and negative controls
The use of the microfluidic chip for spatially defined RNA-ISH experiments enables the sequential or even simultaneous deposition of primary probes targeted at several RNA sequences of interest on a single tissue section. We leveraged this ability to introduce probes for the biomarker of interest, HER2, as well as 4-hydroxy-tetrahydrodipicolinate reductase (dapB) from Bacillus subtilis and Actinβ (ActB) as negative and positive controls on the same slide. The positive control serves as an indicator for RNA integrity, while the negative control, which is not expressed in human tissue, provides information about background enzymatic reactions, unspecific binding of amplification probes, and staining. The absence of dapB signal in the area of local delivery and the absence of ISH signal in cells surrounding the spatially defined regions serve as negative controls. Figure 1B shows FFPE cell pellet sections of the cell lines MCF7, SKBR3 and BT474 with RNA-ISH signals of dapB, HER2 and ActB in spatially defined regions of the section (see also Supplementary Figures S4). The relative quantification of the signal intensity was based on the mean integrated intensity of the FISH signal per cell section (Supplementary Figure S5). The relative comparison of expression levels within a tissue section is independent of the amplification, enzymatic visualization reaction, and imaging conditions, as these are equal to all transcripts of interest in the same section. The expression levels based on ISH signal intensities were therefore directly compared within a single tissue section. The signal of the positive control ActB was detected for all three cell lines with a mean integrated intensity per cell between 40 and 100, while the dapB signal was low in all cases (mean integrated intensity of 3–10) (Figures 1B, 2). The signal intensity of HER2 showed a strong variation between the three cell lines with a high mean integrated intensity per cell of 438 for SKBR3 and 208 for BT474 and a weak signal (int = 7) for MCF7. This is consistent with the expression levels detected for these cell lines when using immunohistochemistry, where SKBR3 and BT474 are described as HER2 3+ and MCF7 as HER2 0–1+ (Supplementary Figure S6) (67). The method thus enables the estimation of HER2 overexpression in known breast cancer cell lines. Therefore, we applied the same method to investigate the HER2 expression in breast carcinoma sections and found HER2 to be strongly expressed when compared to the positive control ActB in the tumor region analyzed (Figure 2A). The analyzed tumor is thus HER2 positive.
Figure 2.

Detection of HER2 expression status by RNA in situ hybridization using internal positive and negative controls. (A) Fluorescence images of the signals detected for HER2, dapB, and ActB in a mammary carcinoma section. Scale bar: 100 μm. (B) Quantitative analysis of the fluorescence intensity of FISH signals detected for HER2, dapB, and ActB integrated per sectioned cell for FFPE sections of the breast cancer cell lines MCF7, SKBR3, and BT474 (see Figure 1) as well as the mammary carcinoma from (A). 100 ms excitation.
Using this approach, the comparison to a house-keeping gene, such as ActB, can be used to quantify differences in the expression level of the gene of interest (68). We calculated the ratios of HER2 and ActB expression levels based on the mean integrated intensities per cell section. For MCF7 cells, a ratio of 0.07 was calculated. BT474 and SKBR3 expressed HER2 and ActB in ratios of 5.65 and 6.64, respectively. The investigated mammary carcinoma section showed an expression ratio of 23. However, as shown by Aerts et al. (69), we observed that the expression level of ActB at the transcriptomic level, based on the integrated intensity of the ISH signal, differs between the breast cancer cell lines MCF7, SKBR3 and BT474 (Figure 2B, Supplementary Note SN2). A direct comparison between the different cell lines therefore has to be considered carefully when basing the quantification on the house-keeping gene ActB. While different cell cycle states within a certain cell line may indeed be compared, additional house-keeping genes should be considered for the comparison between cell lines or tumor tissues.
We demonstrated here that transcripts of interest can be analyzed in a multiplexed fashion using the microfluidic chip-based delivery of ISH probes. Nonetheless, house-keeping genes used for the relative quantification of transcript levels must be evaluated carefully. Integrating negative and positive controls into the experiments provides measures for unspecific signal and ensures that pre-analytical and analytical conditions (e.g. fixation, pretreatment of the tissue, assay parameters) do not impact RNA integrity. Our approach thus provides a robust basis for a controlled gene expression analysis.
Multiplexed RNA-ISH analysis of a predictive breast cancer biomarker panel
Spatially multiplexed RNA-ISH allows the analysis of biomarker panels using single color detection. We focused the analysis on the predictive breast cancer biomarker panel estrogen receptor (ER), progesterone receptor (PgR), and HER2. By using microfluidic chip-based local delivery, we determined their expression levels in cell pellet sections of breast cancer cell lines and mammary carcinoma sections (Figure 3, Supplementary Figures S7). The imaging conditions were kept constant for the analysis of the different cell lines and the carcinoma tissue in Figure 3. We observed an elevated expression of ER in MCF7 (int = 29) and PgR in MCF7 and BT474 cells (int = 16 and int = 535, respectively). HER2 was strongly overexpressed in SKBR3 (int = 1057) and BT474 (int = 836) and mildly expressed in MCF7 (int = 20) (Supplementary Figure S7). The mammary carcinoma section showed HER2 overexpression, while ER and PgR signals were not elevated (Figure 3). These results correspond to the ones of the molecular analysis of the cell lines by immunohistochemistry. In MCF7, the Allred scores for ER and PgR are 6, whereas HER2 expression is quantified as 0–1+ (67). SKBR3, on the other hand, shows a high expression of HER2 (3+) and no overexpression of ER and PgR (Allred score = 0) and BT474 highly expresses HER2 (3+) and PgR (Allred score = 8) (67).
Figure 3.

Single color multiplexed RNA-ISH for the detection of breast cancer biomarkers. (A) The primary probes for ER, PgR, and HER2 were delivered to spatially distinct regions of a mammary carcinoma section using a microfluidic chip (schematic in upper left corner). Fluorescence images of the signal detected for the breast cancer biomarkers ER, PgR and HER2 in a mammary carcinoma section. Scale bar: 100 μm. (B) Fluorescence intensity of FISH signals detected for ER, PgR and HER2 integrated per sectioned cell for FFPE sections of the breast cancer cell lines MCF7, SKBR3 and BT474 (see Supplementary Figure S7) as well as the mammary carcinoma from (A). 500 ms excitation.
These results show that the multiplexed analysis of multiple transcripts, e.g. a biomarker panel, is possible through using single-color RNA-ISH in fluorescence or brightfield detection mode. We believe that this facilitates the quantification of multiple transcripts at the same time and allows for the detection of a multitude of transcripts requiring only a single tissue section or small amounts of valuable tissue.
We further increased the multiplexing modalities by combining spatial multiplexing with dual color detection. By using only two spatial positions, we could thus visualize gene expression patterns of the biomarker panel ER/PgR/HER2 and the positive control ActB as well as negative controls in forms of the surrounding tissue (Supplementary Figure S8). ActB and HER2 were visualized by using Fast Red, and ER and PgR using Fast Blue. The ActB signal and the area surrounding the local delivery of probes show the integrity of the RNA in the sections as well as a negligible signal owing to unspecific reactions. As expected, MCF7 showed an overexpression of ER and PgR, while the HER2 signal was significantly lower than for the other cell lines. On the other hand, SKBR3 and BT474 as well as the carcinoma sections, showed a strong HER2 signal. PgR was dominantly overexpressed in BT474 cell pellet sections. This agrees with the subtype differentiation of these cell lines into luminal A, luminal B, and HER2-overexpressing with an Allred score of 8 for PgR in BT474 and one of 6 in MCF7 cells.
Although the sensitivity of detection is higher when using fluorescence excitation and detection, the spatial multiplexing approach allows the detection of multiple different types of transcripts when using brightfield microscopy on a single tissue section (Supplementary Figures S4, S9). By using micrometer sized areas of hybridization, thousands of transcripts could theoretically be detected on a single tissue section through spatial multiplexing. By additionally employing dual-color detection schemes, the number of different types of detected transcripts can be doubled.
Spatially multiplexed RNA-ISH for detecting molecular tumor heterogeneity
Having shown that multiplexed RNA-ISH detection methods allow for the spatial analysis of several biomarkers simultaneously, including internal positive and negative controls (Figure 2) or panels of biomarkers (Figure 3), we applied the technique to investigate spatial heterogeneity in the expression levels of the detected transcripts. By using globally or locally applied RNA-ISH, we analyzed the spatial heterogeneity of HER2 expression in mammary carcinoma sections (Figure 4, Supplementary Figure S10). Each tumor region (distance between regions 5 mm to 1 cm, separated by fibrous tissue) was represented by multiple positions within the region (areas with a distance of a few 100 μm). Differences within each of the regions (intraregional differences) and between the different regions (interregional differences) were analyzed. Regions are shown in hematoxylin-stained tissue sections in Supplementary Figure S11. Intraregional and interregional differences in expression level were analyzed by using a Mann–Whitney test and the Cohen effect size (65). For a carcinoma strongly expressing HER2 (35 HER2 copies, HER2 IHC 3+), the interregional heterogeneity was weak with Cohen effect sizes ranging from r = 0.09 to r = 0.11 (Figure 4B, left, Supplementary Table S1). The intraregional differences within regions 2 and 3 were low, while region 1 showed a slightly increased heterogeneity in HER2 expression levels (Figure 4Bright). The carcinoma section with a lower HER2 protein expression status (five HER2 copies, HER2 IHC 2+) showed weak interregional heterogeneity between regions 2 and 3 (r = 0.04) and moderate to strong differences in HER2 expression levels when comparing them to region 1 (r = 0.36 and r = 0.34 for regions 1 and 2, and 1 and 3, respectively) (Figure 4C). Additionally, intraregional heterogeneity was moderate for region 1 with effect sizes ranging from r = 0.05 to r = 0.4 between different areas within the region. These results show that the molecular heterogeneity of the HER2 status differs between tumors and patients. While some tumors display a homogeneous distribution of HER2 expression levels, others show moderate to large variations. Histological and pathological evaluations of the tissue sections corresponded to the HER2 expression patterns observed using RNA-ISH. The highly HER2-overexpressed tissue section (Figure 4B) contained 35 copies of the HER2 gene and was classified as HER2 3+ when using immunohistochemistry, while the low HER2 expressing carcinoma carried 5 HER2 gene copies and had a HER2 score of 2+. The evaluation of this tumor tissue was equivocal. Interestingly, these equivocal results corresponded to a tissue with moderate to high spatial heterogeneity in HER2 transcript levels, suggesting the importance of molecular tumor heterogeneity for the differentiation of breast carcinomas into subtypes.
Figure 4.

Spatially multiplexed RNA-ISH to detect spatial tumor heterogeneity in mammary carcinoma sections. (A) Fluorescence images of RNA-ISH signals detected for HER2 and ActB by global ISH or local ISH experiments on mammary carcinoma sections. Nuclei visualized using DAPI. The images display the inter- and intratumor heterogeneity of the HER2 expression. Scale bar: 100 μm. (B, C) Integrated fluorescence intensity of the HER2 signal per sectioned cell in three different regions of a strongly (B) (35 HER2 copies, HER2 IHC 3+) and low (C) (five HER2 copies, HER2 IHC 2+) HER2-expressing carcinoma tissue section (left). Using a Mann–Whitney test, the Cohen effect size (r) for interregional heterogeneity was calculated from the combined areas (I–IV) within a region and displayed by the values given in the graph on the left. A matrix of Cohen effect sizes calculated for the individual areas within or between regions (intraregional and interregional heterogeneity) is shown on the right. Corresponding regions are shown on hematoxylin and immunohistochemically stained tissue sections in Supplementary Figure S11A and B.
Multiplexed analysis revealed heterogeneity in HER2 gene expression in relation to the expression levels of ActB (Figure 5A). While only a low variation in the expression of ActB was detected between three different regions of the tissue (r = 0.08–0.21), the expression level of HER2 was significantly lower in region 3 compared to the other two regions (r = 0.74 for both, comparison between regions 1 and 3, and between regions 2 and 3). This suggests a differential regulation of HER2, while the overall RNA content between the regions is constant. A comparison of the HER2/ActB expression ratios showed an expression of HER2 in region 3 that was 15 times lower than in both other regions analyzed (HER2/ActB (region 1) = 0.7, HER2/ActB (region 2) = 0.6, HER2/ActB (region 3) = 0.04).
Figure 5.

Differential HER2 gene expression in mammary carcinoma sections. (A) Integrated RNA-ISH fluorescence intensity per cell section for ActB and HER2 in a mammary carcinoma section (lower image). Cohen effect sizes describing interregional differences in expression levels calculated using a Mann–Whitney test are shown as matrices for ActB and HER2, respectively (upper images). (B) Histological overview of the tumor tissue section stained with hematoxylin and eosin (upper image) and HER2 immunohistochemistry (lower image) with the regions 1–4 used for local RNA-ISH detection in (C). Scale bar: 1 mm. (C) Integrated RNA-ISH fluorescence intensity (HER2) per cell section for four areas in each of the four different tumor regions in a mammary carcinoma section with a HER2 IHC status of 3+. The entire tissue sections including the regions of interest are shown as hematoxylin and IHC stained tissues in Supplementary Figure S11C. The Cohen effect size (r) for interregional heterogeneity was calculated from all areas (1–4) within each region combined by using a Mann-Whitney test and its values are displayed above the graph. (D) Quantitative immunohistochemistry using a gradient of primary antibody incubation times by local antibody delivery (24). The graph shows the relative mean intensity of the diaminobenzidine (DAB) signal after the amplification of the secondary antibody signal through a horseradish peroxidase reaction versus the incubation time of the primary antibody. It provides a quantitative measure of protein expression levels.
Correlation between transcript and protein expression
We further investigated the correlation between differences in expression at the transcript and protein level. We determined HER2 expression levels and their intraregional and interregional differences of a carcinoma tissue section with HER2 status of 3+ in immunohistochemistry results (Figure 5B and C, Supplementary Table S1). HER2 protein levels were analyzed using standard immunohistochemistry and quantitative immunohistochemistry (Figure 5B and D) (24). Between the four regions analyzed, the heterogeneity of HER2 gene expression showed a small effect size between regions 1, 2, and 4. It was moderate when comparing these regions to region 3 (r = 0.26 for regions 1 and 3 as well as 4 and 3, and r = 0.19 for regions 2 and 3). Correspondingly, HER2 protein showed a higher expression in region 3 when compared to the other regions (Figure 5C). Evidently, we observed a correlation in the spatial heterogeneity of HER2 between the transcript and protein levels.
In summary, spatially multiplexed RNA-ISH can be used to detect spatial molecular tumor heterogeneity of a panel of biomarkers or in combination with house-keeping genes and internal controls. The transcript level can thereby provide additional quantitative information about the gene expression status and may be used in a complementary fashion to existing approaches for biomarker analysis, being especially advantageous for equivocal tumor cases.
DISCUSSION
Spatial tumor heterogeneity is an important driver in tumor progression and resistance development and is currently rarely considered in clinical decision making. Therefore, a feasible approach describing molecular tumor heterogeneity is needed. A variety of research methods to study spatial cellular heterogeneity at the protein and transcript expression level exists. Multiplexed immunohistochemistry, which is commercially available as Opal and UltiMapper detection schemes for automated platforms, allows to spatially detect up to 16 different proteins (14,70–72). Recent developments additionally explore the label-free detection of biomarkers using infrared imaging (73–76) or deep learning approaches on histological images (77–80). Furthermore, a multitude of new techniques for the spatial analysis of transcripts have recently been developed. They range from location-inference for single-cell sequencing data (81–85) or next-generation sequencing of laser capture microdissected sections (47–49) to in situ sequencing (41–44) or patterned microarrays (45,46). Depending on the application, each of the methods has its own advantages, such as the number of different transcripts detectable, the efficiency of detection, or the quantification of gene expression. However, many of these methods require the use of live cells or fresh frozen tissue to ensure a stable quality of RNA. As FFPE samples are one of the most common sample types in pathology laboratories and tissue biobanks (86), a method to detect gene expression levels despite formalin-fixation is highly advantageous and may lead to translation of the technique to other laboratories. To limit the technical complexity and make spatial transcript analysis potentially accessible for clinical use, we therefore developed a novel method for multiplexed RNA-ISH, which has been shown to be compatible with FFPE tissues (50–52). RNA-ISH provides the additional advantage of detecting transcripts with low expression levels, such as non-coding RNAs. The method is based on existing RNA detection schemes that use amplification and brightfield or single-color fluorescence detection. We leveraged the microfluidic chip's ability to precisely and locally deliver chemicals (59,60) to spatially confine primary ISH-probes on FFPE sections and thus achieve spatial multiplexing with simple detection schemes.
By using spatial multiplexing, the number of different targets for amplification-based RNA-ISH techniques can be increased. Current non-fluorescent and enzyme-based amplification approaches for ISH have the advantage of abolishing the need for highly sensitive detection optics (56). However, they are limited to two different target samples. By applying spatial multiplexing, the number of different targets can easily be scaled and is only limited by the number of positions available in a homogeneous region of the tissue section. The established method provides the additional advantage that signal intensities within a section can be directly compared without the need to control for variations in pretreatment conditions or saturation of the amplification reaction. As multiplexing using a single color is possible, differences in quantum yield, excitation power, and excitation and emission filter bandwidth do not have to be corrected for. Furthermore, the spectral multiplexing of transcripts may be advantageous when assessing highly expressed genes, where the dense distribution hinders hybridization to multiple targets.
We demonstrated the utility of the technique by the detection of HER2 transcripts with tissue internal positive and negative controls and the analysis of the predictive breast cancer biomarker panel ER/PgR/HER2 in a single tumor tissue section. In clinical decision making, control reactions are extremely important as they provide information about tissue processing conditions, integrity of the molecular content, and performance of the analytical assay. Internal controls are the most precise measure and are therefore preferred over external controls on consecutive tissue sections or other samples (87). Including internal controls in RNA-ISH facilitates the evaluation of RNA integrity and stability of the experimental conditions and is thus an important measure for assay performance. The established assay can thereby aid in implementing these internal controls.
The information from the multiplexed RNA-ISH analysis of panels of biomarkers can be used to allocate breast cancer cell pellet sections to their molecular subtype (MCF7: luminal A, BT474: luminal B, SKBR3: HER2-overexpressed (67)). Complementary to protein expression analysis, the same information derived from transcript expression levels can therefore be used to classify tumor tissue sections into their molecular subtypes. This is in accordance with the results by Alba et al. (2012), who have shown a 95.2% correlation of gene expression in RNA-ISH compared to immunohistochemistry and a 96.5% concordance of RNA-ISH with gene amplification by DNA-FISH (63). Baehner et al. (2010) additionally investigated the correlation between the RNA expression levels of HER2 detected by qRT-PCR and DNA-FISH. The observed concordance of 97% between the two methods shows that RNA expression levels are a reliable indicator for patient stratification (88), a finding further confirmed by Vassilakopoulou et al. (89), Wang et al. (2), and Wu et al. (90). Although multiplexing with spectral unmixing and AQUA analysis enable a certain degree of quantitation of immunohistochemistry and immunofluorescence results (14,18,22,23), the quantification of immunohistochemistry results is still not precise and leads to 32% equivocal results, 84% of which can be resolved by subsequent DNA-FISH (2,63). The analysis of expression at the RNA level presents an alternative approach with the additional advantage of using oligonucleotide probes over antibodies, many of which are not suitable for obtaining reproducible IHC results on FFPE tissue (91,92). For PD-L1 testing, for example, different antibodies, assays, and evaluation criteria are used to predict PD-1/PD-L1 inhibitor therapy (93). Through using RNA-ISH, the standardization of PD-L1 testing may be facilitated. The clinical application of approaches such as Oncotype DX, 70-gene signature, or PAM-50 have illustrated the usefulness of analyzing transcript expression levels for breast cancer prognosis and prediction (26–29,94,95). Further studies showed the advantages of using mRNA expression profiles for the therapeutic stratification of patients with breast cancer (TAILORx (96), MINDACT (27), EndoPredict (97,98)). When using these techniques, the relative quantification of gene expression levels is achieved by using PCR and microarrays, while the spatial information is not preserved. Therefore, combinations of quantitative gene expression analysis at the transcript level with spatially resolved DNA-FISH or immunohistochemistry can reveal additional important information on molecular heterogeneity. Alternatively, RNA-ISH offers the possibility of the relative quantification of transcripts directly in a spatially defined manner on a single tissue section. Annaratone et al. quantified ER and HER2 transcripts in FFPE tissue sections and successfully stratified the tissues into molecular subtypes (99). Using the method described in this paper, spatial differences of multiple transcripts of interest can be assessed on a single tissue section. We used the approach to evaluate the spatial heterogeneity of HER2 transcripts and observed varying interregional and intraregional differences in the gene expression level for the carcinoma sections investigated. Interestingly, we observed heterogeneity in HER2 transcript levels with moderate to strong effect size in carcinoma tissue with equivocal results from HER2 gene copy number analysis and immunohistochemistry. This suggests that spatial heterogeneity contributes to the significance of molecular subtype differentiation, making it an even more important parameter to consider in clinical decisions. Especially in equivocal cases, spatially defined transcript levels can provide a complementary approach and additional indicator for predictive and prognostic decisions. We observed a correlation of interregional differences in the HER2 expression between the transcript and protein level. This is in accordance with previous results revealing spatial heterogeneity of HER2 breast cancer on the transcript or protein level (24,99,100).
We believe that the local delivery of probes and antibodies will facilitate the multimodal detection of RNA and proteins on single tissue sections by combining RNA-ISH and immunohistochemistry. Previous studies have shown the simultaneous detection of transcripts and proteins on a single monolayer of cells or tissue section (101–103) and recent elaborate techniques combine RNA-ISH and immunofluorescence with metal-based detection using mass cytometry (104). Furthermore, owing to its immense flexibility, the microfluidic chip may be used for the simultaneous local delivery of in situ detection moieties (e.g. hybridization probes, antibodies) and local removal of cells for genetic or transcriptomic analysis (61,105). We can thus imagine its capabilities to provide spatial information about whole genomes, transcriptomes, or even proteomes. The quantitation of transcripts and proteins in a spatially defined manner holds great promise for the elucidation of tumor development and progression. Future studies on cohorts of tumor biopsies can provide information on the applicability of the technique for clinical interpretation.
To summarize, our work provides a versatile approach for multiplexed in situ detection of transcripts using single-color or brightfield detection. As methods that recover spatial information provide the possibility to investigate dynamic processes, development of lineages, connectivity, and alterations in tissue architecture in development and disease, we believe that the developed technique will find application in a wide variety of biological and medical contexts.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Susanne Dettwiler for generating and providing tumor tissue sections. We are grateful to Daniel Schulz and Bernd Bodenmiller for their advice in the initial phase of our experiments and to Szymon Stoma and Simon F. Noerrelykke for the discussion on image data analysis. Thanks are due to Robert D. Lovchik for his advice on imaging. We thank Natascha Brinskelle-Schmal for providing feedback on initial experiments. L.V. wishes to thank Alexander Borodavka and Waldemar Schrimpf for in-depth scientific discussions. We are grateful to Linda Rudin for valuable comments on the manuscript. We thank Emmanuel Delamarche, Walter Riess, and Heike E. Riel for their continuous support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council [ERC 727761 (BioProbe-PIT) to G.V.K.]. Funding for open access charge: European Research Council.
Conflict of interest statement. L.V., A.F.K., D.H. and G.V.K. were employed by IBM Research GmbH and P.S. was employed by Universitätsspital Zürich while working on this project. The authors have no conflict of interest.
REFERENCES
- 1. Dai X., Li T., Bai Z., Yang Y., Liu X., Zhan J., Shi B.. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015; 5:2929–2943. [PMC free article] [PubMed] [Google Scholar]
- 2. Wang Z., Portier B.P., Gruver A.M., Bui S., Wang H., Su N., Vo H.-T., Ma X.-J., Luo Y., Budd G.T. et al.. Automated quantitative RNA in situ hybridization for resolution of equivocal and heterogeneous ERBB2 (HER2) status in invasive breast carcinoma. J. Mol. Diagn. 2013; 15:210–219. [DOI] [PubMed] [Google Scholar]
- 3. Ohlschlegel C., Zahel K., Kradolfer D., Hell M., Jochum W.. HER2 genetic heterogeneity in breast carcinoma. J. Clin. Pathol. 2011; 64:1112–1116. [DOI] [PubMed] [Google Scholar]
- 4. Duffy M.J.J., Harbeck N., Nap M., Molina R., Nicolini A., Senkus E., Cardoso F.. Clinical use of biomarkers in breast cancer: updated guidelines from the European Group on Tumor Markers (EGTM). Eur. J. Cancer. 2017; 75:284–298. [DOI] [PubMed] [Google Scholar]
- 5. Farhangfar C.J., Meric-Bernstam F., Mendelsohn J., Mills G.B., Lucio-Eterovic A.K.. The impact of tumor heterogeneity on patient treatment decisions. Clin. Chem. 2013; 59:38–40. [DOI] [PubMed] [Google Scholar]
- 6. Fedele C., Tothill R.W., McArthur G.A.. Navigating the challenge of tumor heterogeneity in cancer therapy. Cancer Discov. 2014; 4:146–148. [DOI] [PubMed] [Google Scholar]
- 7. Yeo S.K., Guan J.-L.. Breast cancer: multiple subtypes within a tumor. Trends Cancer. 2017; 3:753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerlinger M., Rowan A.J., Horswell S., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P. et al.. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012; 366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Albiges L., Gerlinger M.. Abstract 1746: the PREDICT (Personalised RNA Interference to Enhance the Delivery of Individualised Cytotoxic and Targeted Therapeutics) approach to biomarker discovery in renal cell carcinoma. Cancer Res. 2012; 72:1746–1746. [Google Scholar]
- 10. Jamal-Hanjani M., Hackshaw A., Ngai Y., Shaw J., Dive C., Quezada S., Middleton G., de Bruin E., Le Quesne J., Shafi S. et al.. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol. 2014; 12:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bancroft H., Langman G., Kerr A., Naidu B.. Lessons learnt from the initiation of PEACE (Posthumous Evaluation of Advanced Cancer Environment) at a regional thoracic centre. Lung Cancer. 2018; 115:S85. [Google Scholar]
- 12. Andor N., Graham T.A., Jansen M., Xia L.C., Aktipis C.A., Petritsch C., Ji H.P., Maley C.C.. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016; 22:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morris L.G.T., Riaz N., Desrichard A., Şenbabaoğlu Y., Hakimi A.A., Makarov V., Reis-Filho J.S., Chan T.A.. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget. 2016; 7:2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stack E.C., Wang C., Roman K.A., Hoyt C.C.. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014; 70:46–58. [DOI] [PubMed] [Google Scholar]
- 15. Tsujikawa T., Kumar S., Borkar R.N., Azimi V., Thibault G., Chang Y.H., Balter A., Kawashima R., Choe G., Sauer D. et al.. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 2017; 19:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parra E.R., Uraoka N., Jiang M., Cook P., Gibbons D., Forget M.-A., Bernatchez C., Haymaker C., Wistuba I.I., Rodriguez-Canales J.. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci. Rep. 2017; 7:13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang W., Hubbard A., Jones T., Racolta A., Bhaumik S., Cummins N., Zhang L., Garsha K., Ventura F., Lefever M.R. et al.. Fully automated 5-plex fluorescent immunohistochemistry with tyramide signal amplification and same species antibodies. Lab. Investig. 2017; 97:873–885. [DOI] [PubMed] [Google Scholar]
- 18. Cappi G., Dupouy D.G., Comino M.A., Ciftlik A.T.. Ultra-fast and automated immunohistofluorescent multistaining using a microfluidic tissue processor. Sci. Rep. 2019; 9:4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bolognesi M.M., Manzoni M., Scalia C.R., Zannella S., Bosisio F.M., Faretta M., Cattoretti G.. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. J. Histochem. Cytochem. 2017; 65:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin J.-R., Fallahi-Sichani M., Sorger P.K.. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 2015; 6:8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gerdes M.J., Sevinsky C.J., Sood A., Adak S., Bello M.O., Bordwell A., Can A., Corwin A., Dinn S., Filkins R.J. et al.. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:11982–11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harigopal M., Barlow W.E., Tedeschi G., Porter P.L., Yeh I.-T., Haskell C., Livingston R., Hortobagyi G.N., Sledge G., Shapiro C. et al.. Multiplexed Assessment of the southwest oncology group-directed intergroup breast cancer trial S9313 by AQUA shows that both high and low levels of HER2 are associated with poor outcome. Am. J. Pathol. 2010; 176:1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carvajal-Hausdorf D.E., Schalper K.A., Neumeister V.M., Rimm D.L.. Quantitative measurement of cancer tissue biomarkers in the lab and in the clinic. Lab. Investig. 2015; 95:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kashyap A., Fomitcheva Khartchenko A., Pati P., Gabrani M., Schraml P., Kaigala G.V.. Quantitative microimmunohistochemistry for the grading of immunostains on tumour tissues. Nat. Biomed. Eng. 2019; 3:478–490. [DOI] [PubMed] [Google Scholar]
- 25. Xi X., Li T., Huang Y., Sun J., Zhu Y., Yang Y., Lu Z.. RNA biomarkers: frontier of precision medicine for cancer. Non-Coding RNA. 2017; 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paik S., Shak S., Tang G., Kim C., Baker J., Cronin M., Baehner F.L., Walker M.G., Watson D., Park T. et al.. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004; 351:2817–2826. [DOI] [PubMed] [Google Scholar]
- 27. Cardoso F., van’t Veer L.J., Bogaerts J., Slaets L., Viale G., Delaloge S., Pierga J.-Y., Brain E., Causeret S., DeLorenzi M. et al.. 70-Gene signature as an aid to treatment decisions in early-stage breast cancer. N. Engl. J. Med. 2016; 375:717–729. [DOI] [PubMed] [Google Scholar]
- 28. Huang S., Murphy L., Xu W.. Genes and functions from breast cancer signatures. BMC Cancer. 2018; 18:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Senkus E., Kyriakides S., Ohno S., Penault-Llorca F., Poortmans P., Rutgers E., Zackrisson S., Cardoso F.. Primary breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015; 26:v8–v30. [DOI] [PubMed] [Google Scholar]
- 30. Garcia-Saenz J.A., Bermejo B., Estevez L.G., Palomo A.G., Gonzalez-Farre X., Margeli M., Pernas S., Servitja S., Rodriguez C.A., Ciruelos E.. SEOM clinical guidelines in early-stage breast cancer 2015. Clin. Transl. Oncol. 2015; 17:939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller C.A., Dahiya S., Li T., Fulton R.S., Smyth M.D., Dunn G.P., Rubin J.B., Mardis E.R.. Resistance-promoting effects of ependymoma treatment revealed through genomic analysis of multiple recurrences in a single patient. Mol. Case Stud. 2018; 4:a002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Femino A.M., Fay F.S., Fogarty K., Singer R.H.. Visualization of single RNA transcripts in situ. Science. 1998; 280:585–590. [DOI] [PubMed] [Google Scholar]
- 33. Raj A., Peskin C.S., Tranchina D., Vargas D.Y., Tyagi S.. Stochastic mRNA Synthesis in Mammalian Cells. PLoS Biol. 2006; 4:e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lein E., Borm L.E., Linnarsson S.. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 2017; 358:64–69. [DOI] [PubMed] [Google Scholar]
- 35. La Manno G., Gyllborg D., Codeluppi S., Nishimura K., Salto C., Zeisel A., Borm L.E., Stott S.R.W., Toledo E.M., Villaescusa J.C. et al.. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell. 2016; 167:566–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shah S., Lubeck E., Zhou W., Cai L.. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron. 2016; 92:342–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lubeck E., Cai L.. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods. 2012; 9:743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lubeck E., Coskun A.F., Zhiyentayev T., Ahmad M., Cai L.. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods. 2014; 11:360–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen K.H., Boettiger A.N., Moffitt J.R., Wang S., Zhuang X.. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015; 348:aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moffitt J.R., Hao J., Wang G., Chen K.H., Babcock H.P., Zhuang X.. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:11046–11051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Larsson C., Koch J., Nygren A., Janssen G., Raap A.K., Landegren U., Nilsson M.. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nat. Methods. 2004; 1:227–232. [DOI] [PubMed] [Google Scholar]
- 42. Larsson C., Grundberg I., Söderberg O., Nilsson M.. In situ detection and genotyping of individual mRNA molecules. Nat. Methods. 2010; 7:395–397. [DOI] [PubMed] [Google Scholar]
- 43. Ke R., Mignardi M., Pacureanu A., Svedlund J., Botling J., Wählby C., Nilsson M.. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods. 2013; 10:857–860. [DOI] [PubMed] [Google Scholar]
- 44. Lee J.H., Daugharthy E.R., Scheiman J., Kalhor R., Yang J.L., Ferrante T.C., Terry R., Jeanty S.S.F., Li C., Amamoto R. et al.. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014; 343:1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stahl P.L., Salmen F., Vickovic S., Lundmark A., Navarro J.F., Magnusson J., Giacomello S., Asp M., Westholm J.O., Huss M. et al.. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016; 353:78–82. [DOI] [PubMed] [Google Scholar]
- 46. Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R., Welch J., Chen L.M., Chen F., Macosko E.Z.. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019; 363:1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen J., Suo S., Tam P.P., Han J.-D.J., Peng G., Jing N.. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat. Protoc. 2017; 12:566–580. [DOI] [PubMed] [Google Scholar]
- 48. Moor A.E., Golan M., Massasa E.E., Lemze D., Weizman T., Shenhav R., Baydatch S., Mizrahi O., Winkler R., Golani O. et al.. Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science. 2017; 357:1299–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nichterwitz S., Chen G., Aguila Benitez J., Yilmaz M., Storvall H., Cao M., Sandberg R., Deng Q., Hedlund E.. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat. Commun. 2016; 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kurobe M., Kojima T., Nishimura K., Kandori S., Kawahara T., Yoshino T., Ueno S., Iizumi Y., Mitsuzuka K., Arai Y. et al.. Development of RNA-FISH assay for detection of oncogenic FGFR3-TACC3 fusion genes in FFPE samples. PLoS One. 2016; 11:e0165109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Henke R.T., Kim E.S., Maitra A., Paik S., Wellstein A.. Expression analysis of mRNA in formalin-fixed, paraffin-embedded archival tissues by mRNA in situ hybridization. Methods. 2006; 38:253–262. [DOI] [PubMed] [Google Scholar]
- 52. Bingham V., Mcilreavey L., Greene C., Doherty E.O., Clarke R., Craig S., Salto-tellez M., Mcquaid S., Lewis C., James J.. RNAscope in situ hybridization confirms mRNA integrity in formalin-fixed, paraffin-embedded cancer tissue samples. Oncotarget. 2017; 8:93392–93403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S.. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008; 5:877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Battich N., Stoeger T., Pelkmans L.. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat. Methods. 2013; 10:1127–1136. [DOI] [PubMed] [Google Scholar]
- 55. Player A.N., Shen L.-P., Kenny D., Antao V.P., Kolberg J.A.. Single-copy gene detection using branched DNA (bDNA) in situ hybridization. J. Histochem. Cytochem. 2001; 49:603–611. [DOI] [PubMed] [Google Scholar]
- 56. Wang F., Flanagan J., Su N., Wang L.C., Bui S., Nielson A., Wu X., Vo H.T., Ma X.J., Luo Y.. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012; 14:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ramsköld D., Luo S., Wang Y.-C., Li R., Deng Q., Faridani O.R., Daniels G.A., Khrebtukova I., Loring J.F., Laurent L.C. et al.. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012; 30:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Paré A., Lemons D., Kosman D., Beaver W., Freund Y., McGinnis W.. Visualization of Individual Scr mRNAs during Drosophila Embryogenesis Yields Evidence for Transcriptional Bursting. Curr. Biol. 2009; 19:2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Juncker D., Schmid H., Delamarche E.. Multipurpose microfluidic probe. Nat. Mater. 2005; 4:622–628. [DOI] [PubMed] [Google Scholar]
- 60. Kaigala G.V., Lovchik R.D., Drechsler U., Delamarche E.. A vertical microfluidic probe. Langmuir. 2011; 27:5686–5693. [DOI] [PubMed] [Google Scholar]
- 61. Kashyap A., Autebert J., Delamarche E., Kaigala G.V. Selective local lysis and sampling of live cells for nucleic acid analysis using a microfluidic probe. Sci. Rep. 2016; 6:29579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huber D., Kaigala G.V.. Rapid micro fluorescence in situ hybridization in tissue sections. Biomicrofluidics. 2018; 12:042212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alba J., Gutierrez J., Coupe V.M., Fernández B., Vázquez-Boquete Á., Alba J., Forteza J., García-Caballero T.. HER2 status determination using RNA-ISH–a rapid and simple technique showing high correlation with FISH and IHC in 141 cases of breast cancer. Histol. Histopathol. 2012; 27:1021–1027. [DOI] [PubMed] [Google Scholar]
- 64. Lamprecht M.R., Sabatini D.M., Carpenter A.E.. CellProfilerTM: free, versatile software for automated biological image analysis. BioTechniques. 2007; 42:71–75. [DOI] [PubMed] [Google Scholar]
- 65. Cohen J. A power primer. Psychol. Bull. 1992; 112:155–159. [DOI] [PubMed] [Google Scholar]
- 66. Autebert J., Kashyap A., Lovchik R.D., Delamarche E., Kaigala G.V.. Hierarchical hydrodynamic flow confinement: efficient use and retrieval of chemicals for microscale chemistry on surfaces. Langmuir. 2014; 30:3640–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Subik K., Lee J.-F., Baxter L., Strzepek T., Costello D., Crowley P., Xing L., Hung M., Bonfiglio T., Hicks D.G. et al.. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer (Auckl). 2010; 4:35–41. [PMC free article] [PubMed] [Google Scholar]
- 68. Liu L.-L., Zhao H., Ma T.-F., Ge F., Chen C.-S., Zhang Y.-P.. Identification of valid reference genes for the normalization of RT-qPCR expression studies in human breast cancer cell lines treated with and without transient transfection. PLoS One. 2015; 10:e0117058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aerts J.L., Gonzales M.I., Topalian S.L.. Selection of appropriate control genes to assess expression of tumor antigens using real-time RT-PCR. BioTechniques. 2004; 36:84–91. [DOI] [PubMed] [Google Scholar]
- 70. Tóth Z.E., Mezey É. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. J. Histochem. Cytochem. 2007; 55:545–554. [DOI] [PubMed] [Google Scholar]
- 71. Liu Y., Zugazagoitia J., Ahmed F.S., Henick B.S., Gettinger S., Herbst R.S., Schalper K.A., Rimm D.L.. Immune cell PD-L1 co-localizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin. Cancer Res. 2019; doi:10.1158/1078-0432.CCR-19-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Manesse M., Patel K.K., Bobrow M., Downing S.R.. The InSituPlex® staining method for multiplexed immunofluorescence cell phenotyping and spatial profiling of tumor FFPE samples. Methods Mol. Biol. 2020; 2055:585–592. [DOI] [PubMed] [Google Scholar]
- 73. Eaton S.L., Cumyn E., King D., Kline R.A., Carpanini S.M., Del-Pozo J., Barron R., Wishart T.M.. Quantitative imaging of tissue sections using infrared scanning technology. J. Anat. 2016; 228:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Witzke K.E., Großerueschkamp F., Jütte H., Horn M., Roghmann F., von Landenberg N., Bracht T., Kallenbach-Thieltges A., Käfferlein H., Brüning T. et al.. Integrated fourier transform infrared imaging and proteomics for identification of a candidate histochemical biomarker in bladder cancer. Am. J. Pathol. 2019; 189:619–631. [DOI] [PubMed] [Google Scholar]
- 75. Pilling M.J., Henderson A., Bird B., Brown M.D., Clarke N.W., Gardner P.. High-throughput quantum cascade laser (QCL) spectral histopathology: a practical approach towards clinical translation. Faraday Discuss. 2016; 187:135–154. [DOI] [PubMed] [Google Scholar]
- 76. Kuepper C., Kallenbach-Thieltges A., Juette H., Tannapfel A., Großerueschkamp F., Gerwert K.. Quantum cascade laser-based infrared microscopy for label-free and automated cancer classification in tissue sections. Sci. Rep. 2018; 8:7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shamai G., Binenbaum Y., Slossberg R., Duek I., Gil Z., Kimmel R.. Artificial intelligence algorithms to assess hormonal status from tissue microarrays in patients with breast cancer. JAMA Netw. Open. 2019; 2:e197700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Couture H.D., Williams L.A., Geradts J., Nyante S.J., Butler E.N., Marron J.S., Perou C.M., Troester M.A., Niethammer M.. Image analysis with deep learning to predict breast cancer grade, ER status, histologic subtype, and intrinsic subtype. npj Breast Cancer. 2018; 4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Djuric U., Zadeh G., Aldape K., Diamandis P.. Precision histology: how deep learning is poised to revitalize histomorphology for personalized cancer care. npj Precis. Oncol. 2017; 1:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bera K., Schalper K.A., Rimm D.L., Velcheti V., Madabhushi A.. Artificial intelligence in digital pathology — new tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 2019; 16:703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Treutlein B., Brownfield D.G., Wu A.R., Neff N.F., Mantalas G.L., Espinoza F.H., Desai T.J., Krasnow M.A., Quake S.R.. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014; 509:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A.. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015; 33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Durruthy-Durruthy R., Gottlieb A., Hartman B.H., Waldhaus J., Laske R.D., Altman R., Heller S.. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell. 2014; 157:964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Halpern K.B., Shenhav R., Matcovitch-Natan O., Tóth B., Lemze D., Golan M., Massasa E.E., Baydatch S., Landen S., Moor A.E. et al.. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 2017; 542:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Achim K., Pettit J.B., Saraiva L.R., Gavriouchkina D., Larsson T., Arendt D., Marioni J.C.. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol. 2015; 33:503–509. [DOI] [PubMed] [Google Scholar]
- 86. Wang Y. Development of cancer diagnostics-from biomarkers to clinical tests. Transl. Cancer Res. 2015; 4:270–279. [Google Scholar]
- 87. Torlakovic E.E., Nielsen S., Vyberg M., Taylor C.R.. Getting controls under control: the time is now for immunohistochemistry. J. Clin. Pathol. 2015; 68:879–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baehner F.L., Achacoso N., Maddala T., Shak S., Quesenberry C.P., Goldstein L.C., Gown A.M., Habel L.A.. Human epidermal growth factor receptor 2 assessment in a case-control study: comparison of fluorescence in situ hybridization and quantitative reverse transcription polymerase chain reaction performed by central laboratories. J. Clin. Oncol. 2010; 28:4300–4306. [DOI] [PubMed] [Google Scholar]
- 89. Vassilakopoulou M., Togun T., Dafni U., Cheng H., Bordeaux J., Neumeister V.M., Bobos M., Pentheroudakis G., Skarlos D.V., Pectasides D. et al.. In situ quantitative measurement of HER2mRNA predicts benefit from trastuzumab-containing chemotherapy in a cohort of metastatic breast cancer patients. PLoS One. 2014; 9:e99131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wu N.C., Wong W., Ho K.E., Chu V.C., Rizo A., Davenport S., Kelly D., Makar R., Jassem J., Duchnowska R. et al.. Comparison of central laboratory assessments of ER, PR, HER2, and Ki67 by IHC/FISH and the corresponding mRNAs (ESR1, PGR, ERBB2, and MKi67) by RT-qPCR on an automated, broadly deployed diagnostic platform. Breast Cancer Res. Treat. 2018; 172:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. O’Hurley G., Sjöstedt E., Rahman A., Li B., Kampf C., Pontén F., Gallagher W.M., Lindskog C.. Garbage in, garbage out: a critical evaluation of strategies used for validation of immunohistochemical biomarkers. Mol. Oncol. 2014; 8:783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Uhlén M., Björling E., Agaton C., Szigyarto C.A.-K., Amini B., Andersen E., Andersson A.-C., Angelidou P., Asplund A., Asplund C. et al.. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell Proteomics. 2005; 4:1920–1932. [DOI] [PubMed] [Google Scholar]
- 93. Udall M., Rizzo M., Kenny J., Doherty J., Dahm S., Robbins P., Faulkner E.. PD-L1 diagnostic tests: a systematic literature review of scoring algorithms and test-validation metrics. Diagn. Pathol. 2018; 13:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bastien R.R.L., Rodríguez-Lescure Á., Ebbert M.T.W., Prat A., Munárriz B., Rowe L., Miller P., Ruiz-Borrego M., Anderson D., Lyons B. et al.. PAM50 breast cancer subtyping by RT-qPCR and concordance with standard clinical molecular markers. BMC Med. Genomics. 2012; 5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ohara A.M., Naoi Y., Shimazu K., Kagara N., Shimoda M., Tanei T., Miyake T., Kim S.J., Noguchi S.. PAM50 for prediction of response to neoadjuvant chemotherapy for ER-positive breast cancer. Breast Cancer Res. Treat. 2019; 173:533–543. [DOI] [PubMed] [Google Scholar]
- 96. Sparano J.A., Gray R.J., Makower D.F., Pritchard K.I., Albain K.S., Hayes D.F., Geyer C.E., Dees E.C., Goetz M.P., Olson J.A. et al.. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N. Engl. J. Med. 2018; 379:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Müller B.M., Kronenwett R., Hennig G., Euting H., Weber K., Bohmann K., Weichert W., Altmann G., Roth C., Winzer K.-J. et al.. Quantitative determination of estrogen receptor, progesterone receptor, and HER2 mRNA in formalin-fixed paraffin-embedded tissue–a new option for predictive biomarker assessment in breast cancer. Diagn. Mol. Pathol. 2011; 20:1–10. [DOI] [PubMed] [Google Scholar]
- 98. Denkert C., Kronenwett R., Schlake W., Bohmann K., Penzel R., Weber K.E., Höfler H., Lehmann U., Schirmacher P., Specht K. et al.. Decentral gene expression analysis for ER+/Her2-breast cancer: Results of a proficiency testing program for the EndoPredict assay. Virchows. Arch. 2012; 460:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Annaratone L., Simonetti M., Wernersson E., Marchiò C., Garnerone S., Scalzo M.S., Bienko M., Chiarle R., Sapino A., Crosetto N.. Quantification of HER2 and estrogen receptor heterogeneity in breast cancer by single-molecule RNA fluorescence in situ hybridization. Oncotarget. 2017; 8:18680–18698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Raphael B.J., Hruban R.H., Aguirre A.J., Moffitt R.A., Yeh J.J., Stewart C., Robertson A.G., Cherniack A.D., Gupta M., Getz G. et al.. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017; 32:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kochan J., Wawro M., Kasza A.. Simultaneous detection of mRNA and protein in single cells using immunofluorescence-combined single-molecule RNA FISH. BioTechniques. 2015; 59:209–221. [DOI] [PubMed] [Google Scholar]
- 102. Kwon S., Chin K., Nederlof M., Gray J.W.. Quantitative, in situ analysis of mRNAs and proteins with subcellular resolution. Sci. Rep. 2017; 7:16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chaumeil J., Augui S., Chow J.C., Heard E.. Combined immunofluorescence, RNA fluorescent in situ hybridization, and DNA fluorescent in situ hybridization to study chromatin changes, transcriptional activity, nuclear organization, and X-chromosome inactivation. Methods Mol. Biol. 2008; 463:297–308. [DOI] [PubMed] [Google Scholar]
- 104. Schulz D., Zanotelli V.R.T., Fischer J.R., Schapiro D., Engler S., Lun X.K., Jackson H.W., Bodenmiller B.. Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst. 2018; 6:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. van Kooten X.F., Petrini L.F., Kashyap A., Voith von Voithenberg L., Bercovici M., Kaigala G.V. Spatially resolved genetic analysis of tissue sections enabled by microscale flow confinement retrieval and isotachophoretic purification. Angew. Chemie. 2019; 58:15259–15262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.