

Graphical abstract
Keywords: Heterogeneity, Clonal evolution, Breast cancer, Metastasis, Patient derived xenograft models
Abstract
Genetic heterogeneity within a tumor arises by clonal evolution, and patients with highly heterogeneous tumors are more likely to be resistant to therapy and have reduced survival. Clonal evolution also occurs when a subset of cells leave the primary tumor to form metastases, which leads to reduced genetic heterogeneity at the metastatic site. Although this process has been observed in human cancer, experimental models which recapitulate this process are lacking. Patient-derived tumor xenografts (PDX) have been shown to recapitulate the patient’s original tumor’s intra-tumor genetic heterogeneity, as well as its genomics and response to treatment, but whether they can be used to model clonal evolution in the metastatic process is currently unknown. Here, we address this question by following genetic changes in two breast cancer PDX models during metastasis. First, we discovered that mouse stroma can be a confounding factor in assessing intra-tumor heterogeneity by whole exome sequencing, thus we developed a new bioinformatic approach to correct for this. Finally, in a spontaneous, but not experimental (tail-vein) metastasis model we observed a loss of heterogeneity in PDX metastases compared to their orthotopic “primary” tumors, confirming that PDX models can faithfully mimic the clonal evolution process undergone in human patients during metastatic spreading.
1. Introduction
Patient-derived tumor xenograft (PDX) models, in which tumor cells from a human patient are implanted into an immunocompromised mouse, can resemble the original patient tumor in many ways (reviewed in [1], [2]). PDX models’ similarities to patient tumors make them uniquely well-suited for studying phenomena like metastasis and intra-tumor clonal heterogeneity.
Intra-tumor genetic heterogeneity reflects the underlying clonal evolution that occurs within tumors, encompassing competition, cooperation, microenvironment, spatial structure, tumor size, and historical contingency [3], [4], [5], [6], [7], [8], [9], [10]. Genetic heterogeneity within tumors has been widely observed [11], [12], [13], [14] and can lead to negative outcomes like resistance to therapy [15], [16], [17], [18], and progression or reduced patient survival [12], [13], [19], [20]. The heterogeneity in patient tumors and their matched PDX models suggest that the original tumor’s heterogeneity can be recapitulated [21], although it can change during xenoplantation [17], [22], [23], [24], [25], evolve over time [24], [25], [26], or reflect adaptation to the mouse environment [25].
Metastasis is also an evolutionary process in which one or more clones from a primary tumor seed a new tumor at a distant site [27]. How metastasis affects genetic heterogeneity depends on the whether a distant tumor was seeded by one or several clones. When a clone from a heterogeneous primary tumor seeds a distant metastasis, heterogeneity can decrease due to a so-called ‘population bottleneck’. Metastases that are seeded by multiple clones can instead result in unchanged or increased heterogeneity, depending on the clonal composition of the metastasis sample. Heterogeneity can also increase in an initially clonal metastasis given enough time for new mutations to accumulate. Most studies of patients have shown reduced heterogeneity or monoclonality in metastases, for example in breast, renal, and ovarian cancer [28], [29], [30]. Nevertheless, metastases may also be seeded by more than one clone [30], [31], [32] and increased heterogeneity has been observed in rare cases, for example in metastases from small intestine neuroendocrine tumors [33].
Whether PDX models can be used to study how genetic heterogeneity changes during metastasis is an open question. To answer this question, we focused on triple negative breast cancer (TNBC) for two reasons. First, TNBC primary tumors in patients are heterogeneous [34], [35], [36]. Second, PDX models of breast cancer and TNBC are well-studied [37], [38], [39], [40], [41], [42], [43], [44], and can reflect the heterogeneity [23], [24] and important biology of patient TNBC tumors (reviewed in [42], [45], [46], [47]).
Here, the genetic heterogeneity of two PDX models of triple negative breast cancer (TNBC) and their metastases was evaluated. Two commonly used methods to generate PDX metastases in mice were employed [48], [49]: 1) PDX fragments implanted in the mammary fat pad were allowed to develop metastases spontaneously; and 2) suspensions of PDX cells were injected in the tail vein to seed experimental metastases. We found that the level of heterogeneity in PDX tumors depends on whether metastases were generated by the spontaneous or experimental metastases method and is sensitive to the amount of mouse stromal cells in the tumor. After controlling for these factors, we observed a loss of heterogeneity in PDX metastases compared to their orthotopic ‘primary’ tumors, consistent with a population bottleneck.
2. Methods
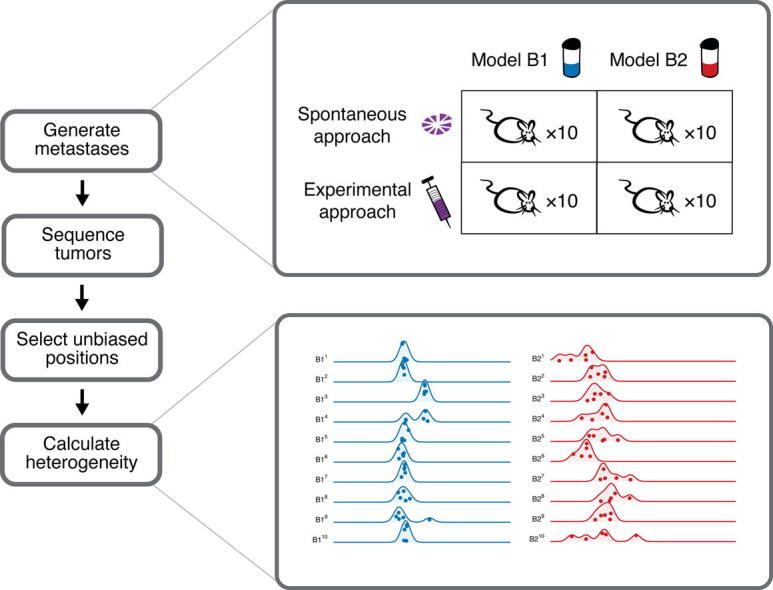
More detailed methods are available in the Supplementary Methods and an overview of the study design is provided in Fig. 1. Two established and characterized TNBC primary tumor models were selected: B1 (1004-HBRX) and B2 (1921-HBRX) [41]. Model B1 was obtained from a Grade III primary TNBC with lymph node metastases and B2 was obtained from a Grade III primary TNBC with no evidence of metastases. We used two common approaches to obtain either ‘spontaneous’ or ‘experimental’ metastases (Fig. 1A, Fig. S1, Supplementary Methods). To collect spontaneous metastases, a PDX tumor was dissected into fragments that were implanted orthotopically into the mammary fat pad of 10 untreated NOG mice (NOD.Cg-Prkdcscid il2rgtm1Sug/JicTac, Taconic). The resulting primary tumors were resected and the mice were monitored for development of spontaneous metastases (Fig. 2A, Fig. S2A). To generate experimental metastases a mixture of dissociated cells from five (B1) or seven (B2) PDX tumors were injected into the tail veins of NOG mice that were monitored for development of experimental metastases. In total, 10 NOG mice were monitored for metastasis per approach and model (Fig. 1B), which resulted in eight mice with spontaneous metastases and 14 mice with experimental metastases (and three mice did not survive the implantation, Fig. 2B, Fig. S2B). The resected orthotopic primary tumors were dissected into five fragments and large metastases into several fragments for whole exome sequencing (Fig. 2C, D, and Supplementary Methods). We have matched primary tumors and metastases from the same mice for the spontaneous approach experiments. However, we do not have matched primary tumor and metastasis samples for the experimental approach because the experimental metastases originated from the cells injected in the tail vein.
Fig. 1.

(A) Two common approaches were used to obtain either spontaneous (top) or experimental (bottom) metastases. For spontaneous metastasis (top), a fragment of the patient-derived tumor xenograft was implanted in the mammary fat pad of a mouse to generate the ‘primary orthotopic’ tumor. This primary tumor was removed, and divided into five fragments for sequencing. Spontaneous metastases are any metastases that subsequently arose in the mouse. Experimental metastases (bottom) are any metastases that arose in a mouse after the tail vein injection of a patient-derived tumor xenograft cell solution. (B) Metastases were obtained from two PDX models of breast cancer (B1 and B2) using the spontaneous and experimental approaches with 10 mice per condition. The primary tumors from the mice in the spontaneous metastasis experiments and all metastases were collected. Large tumors were dissected into several pieces and sequenced.
Fig. 2.

(A, B) The number of tumor samples sequenced for each of the 40 mice (rows) implanted with B1 (blue) or B2 (red) PDX tissue using the spontaneous (panel A) or the experimental (panel B) approaches. An empty box indicates that no metastasis was found in that mouse’s organ. For example in mouse B11, no metastases were found in its lungs or lymph nodes, but five samples from its primary tumor and one spontaneously-arising metastasis from its liver was sequenced. Dashes indicate that a mouse did not survive the implantation procedure (e.g., B118, B119, B120). (C) Primary orthotopic tumors were collected from 20 mice (10 B1 and 10 B2), and each primary tumor was dissected into five pieces for sequencing, as depicted in a representative image from mouse B25. (D) Large metastases were also divided into smaller pieces for sequencing. The lungs often contained multiple metastases that were dissected into multiple pieces for sequencing, as shown here for mouse B219. (E) The mouse content for all sequenced PDX tumor samples for the orthotopic primary tumors (mammary gland) and the metastases for models B1 (blue) and B2 (red) was estimated from the number of sequencing reads that uniquely mapped to the mouse and human genomes. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Results
3.1. Generation of orthotopic primary tumors and metastases from TNBC PDX models
We used two PDX models in this study: B1 and B2. For each model, one tumor was resected and divided into 10 fragments of approximately 5 mm3, which were implanted in the mammary fat pad of 10 immunocompromised NOG mice (one fragment per mouse). Each of these fragments generated a primary tumor, which was further resected and collected for sequencing. Mice were monitored after resection of the primary tumor for the development of spontaneous metastases. Only model B1 generated spontaneous metastases (B1: 80%, 8 of 10; B2: 0%, 0 of 10), which were found most commonly in the liver but also in the lymph nodes (Fig. 2A). Experimental metastases were generated by injecting approximately 1 million cells of a cell suspension into the tail veins of 10 NOG mice per model. The cell suspensions were created by combining several resected tumors (B1: 5 tumors, B2: 7 tumors), dissociating the cells, and filtering for human cells. Most mice injected with cells from the B1 or B2 model developed experimental metastases (B1: 57%, 4 of 7; B2: 100%, 10 of 10), which were primarily found in the lungs (Fig. 2B).
3.2. Assessment of tumor purity
One common confounding factor for studying heterogeneity is tumor purity because differences in tumor purity can cause biased heterogeneity estimates. Tumor purity decreases when the tumor samples also contain non-tumor cells, typically from the patient’s surrounding normal tissue [50]. Bulk samples from PDX models can contain cells that originate from the mouse host that can affect the calculation of heterogeneity. We assessed the purity of our samples bioinformatically by estimating their mouse content from whole exome sequencing data, and validated our approach on a subset of nine samples by comparing our bioinformatic estimates to those obtained from qPCR experiments (Fig. S3). While the bioinformatic approach found less mouse content than we measured experimentally, the estimates were well correlated between the two methods (Spearman’s rank correlation ρ: 0.88, p = 0.003, n = 9) and we thus used the bioinformatic approach to estimate the mouse content in all sequenced samples.
The mouse content in our samples differed depending on the tissue from which the sample was obtained (Fig. 2E). We found high levels of mouse content in the liver and lung metastases (median 39% liver and 27% lung; n = 10 and 30 respectively) and lower levels in the primary orthotopic tumors and lymph node metastases (median 3% mammary gland and 16% lymph nodes; n = 100 and 6, respectively). The difference in mouse content between the primary orthotopic tumors and most metastases is important, because it can lead to incorrect biological conclusions when comparing primary tumors and metastases that could simply be an artifact of the systematic differences in mouse content.
3.3. Estimation of genetic heterogeneity
Care must be taken when estimating the heterogeneity within a tumor because such an estimate can reflect experimental artifacts rather than biological variation [51]. Better heterogeneity estimates can be obtained by using standard variant callers to call somatic mutations and their allele frequencies from matched tumor-normal samples sequenced to high read depth [51]. PDX samples present two main challenges to the standard approach that we addressed in our analyses: matched normal samples are often not available, and PDX samples contain cells from the host (as shown in Fig. 2E). Fortunately, conducting PDX experiments allowed us a unique opportunity to address these challenges with PDX-derived tumor sequence data in two ways. First, we increased our ability to detect genomic positions with heterogeneity by collecting multiple biological replicates for each PDX model; we obtained 10 primary orthotopic tumors per model, and sequenced five samples from each tumor (50 sequenced samples from primary tumors per model). We used these samples to identify heterogeneous genomic positions with at least 100-fold coverage in which an alternative allele is found in at least five samples. Second, we controlled for mouse content in our PDX samples by sequencing four host mouse control samples that we used to select human-specific and eliminate mouse-biased genomic regions. In doing so, we identified 4641 (model B1) and 587 (model B2) human-specific heterogeneous genomic positions distributed across the genome (Fig. 3A and B, Fig. S4). We used these positions to estimate the genetic heterogeneity of each sample as the average minor allele frequency of the heterogeneous genomic positions (Fig. S5, Supplementary Methods). Importantly, our method for estimating genetic heterogeneity is not correlated with mouse content, such that we have minimized the bias that can arise from increased mouse content (Fig. 3C, D, Spearman’s rank correlation; B1: ρ = 0.013, p = 0.93; B2: ρ = 0.25, p = 0.08; n = 50 primary samples per model).
Fig. 3.

(A) Sequenced samples from PDX models contain sequences from the host mouse that can bias heterogeneity estimates. Unbiased genomic positions were selected using the steps outlined in the boxes. Each step is described in full detail in the Supplementary Methods. The number of genomic positions remaining for model B1 and model B2 after each step are indicated in the blue and red bars. (B) The distribution of the unbiased genomic positions across the genome for models B1 (blue) and B2 (red). The heterogeneity for each (C) model B1 and (D) model B2 sample was estimated using the unbiased genomic positions selected for each model (vertical axes) and plotted against the amount of mouse contamination in the sample (horizontal axes). Heterogeneity and mouse contamination are uncorrelated for the primary samples depicted in the inset panels (Spearman’s rank correlation; B1: ρ = 0.013, p = 0.93, n = 50; B2: ρ = 0.25, p = 0.08, n = 50). However, heterogeneity increased with mouse content for samples with more than 55% mouse content; therefore, these samples were removed from downstream analyses. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. Consistency of genetic heterogeneity in primary orthotopic tumors
We were interested in determining the extent to which heterogeneity is consistent between replicated primary tumors of the same PDX model, and regionally within each primary orthotopic tumor. We first compared whether primary tumors from the same model have similar levels of genetic heterogeneity. Heterogeneity was relatively consistent between the 10 different B1 primary tumors, but much more variable between the 10 B2 primary tumors (Fig. 4A top panel). We found only two B1 primary tumors with statistically different heterogeneity from the other B1 primary tumors (resected from mice B13 and B14), but most B2 primary tumors were different from each other (Mann-Whitney test for all primary tumor pairs).
Fig. 4.

The heterogeneity score (horizontal axes) of the samples obtained from each mouse (vertical axes) for PDX model B1 (blue) and B2 (red). Five samples were collected from each primary tumor (one solid circle is shown for each of the five sequenced regions from a primary tumor), in addition to metastases from the liver (triangles), lungs (diamonds), and lymph nodes (squares). (A) The primary tumors collected during the spontaneous metastasis experiments are depicted in the top panels, while the metastases collected during the experimental metastasis experiments are depicted in the bottom panels. Comparing heterogeneity between primary tumors and experimental metastases was inconclusive for model B1 (primary tumors B11 to B110 vs. experimental metastases B111 to B113, Mann-Whitney, p = 0.95) and increased for model B2 (primary tumors B21 to B210 vs. experimental metastases B211 to B220, Mann-Whitney, p = 0.01). (B) Primary tumors and their metastases collected during spontaneous metastasis experiments. Heterogeneity decreased in the mice (B11 to B16) when comparing matched primary tumors and spontaneous metastases (linear mixed effects analysis, ??2(1) = 6.2, p = 0.013, n = 38). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Next, we looked at the regional differences in genetic heterogeneity within each primary orthotopic tumor by measuring the heterogeneity of five spatially-distinct subsamples. We observed a broad range of genetic heterogeneity within most B2 primary tumors, consistent with both regional variation and the high variability between B2 primary tumors. In contrast, we found few regional differences within B1 primary tumors with a notable exception in the tumor resected from mouse B14. Strikingly, there appear to be two subclones within primary tumor B14, and the heterogeneity of those subclones is consistent with the two levels of heterogeneity found across all B1 primary tumors. Specifically, two regional samples within the B14 primary tumor had higher levels of heterogeneity similar to those found in the primary tumor from mouse B13, while the other three regional samples had lower levels similar to those found in the primary tumors of the other B1 mice (Fig. 4A, top left panel). This pattern is consistent with the transfer of regional genetic heterogeneity from the expanded precursor B1 tumor to the mammary fat pads of mice B11 - B110. Indeed, spatial heterogeneity in the originating tissue has been previously observed to have such an effect in one PDX model of colon cancer [52].
3.5. Changes in genetic heterogeneity during metastasis
To understand the effect of metastasis on genetic heterogeneity, we next compared the primary tumor and metastasis samples. Because we found inherent differences in heterogeneity between primary orthotopic tumors (Fig. 4A top panels), we focused on comparing the heterogeneity between pairs of primary tumors and the spontaneous metastases they formed (Fig. 4B, B1: 6 mice with a primary tumor and spontaneous metastases, B2: 0 mice). We found a significant decrease in heterogeneity in the B1 spontaneous metastasis samples (linear mixed effects analysis, Χ2(1) = 6.2, p = 0.013, n = 38; primary: 0.0386 ± 0.0003, metastasis: 0.0379 ± 0.0004, heterogeneity ± standard error, Supplementary Methods) which is consistent with previous observations in patients [28], [29], [30]. Thus, orthotopic implantation of PDX tissue to generate spontaneous mutations can provide a biologically consistent model of changing heterogeneity during metastasis.
As described above, model B2 only forms metastases by tail vein injection, not spontaneous metastases (Fig. 2A, B). We wondered whether experimental metastases could also be used to model heterogeneity. We compared the experimental metastasis samples to the primary tumors using a Mann-Whitney test for unpaired data (Fig. 4A, Supplementary Methods), and found that they either showed no difference (B1, Mann-Whitney, p = 0.95) or that they unexpectedly increased (B2, Mann-Whitney, p = 0.010). Thus, in the models described herein, experimental metastases obtained by tail vein injection do not provide a biologically consistent model of reduced heterogeneity during metastasis.
4. Discussion
We present here the first study of how genetic heterogeneity changes during metastasis in two patient-derived tumor xenograft models of triple-negative breast cancer. To this end, we generated metastases using two approaches. In the first, we obtained spontaneously arising metastases and matching primary tumor samples by orthotopically implanting patient-derived tumor samples into the mammary fat pads of 10 mice. In the second approach, we obtained experimentally generated metastases that arose after injecting cells from patient-derived tumor samples into the tail veins of 10 mice. We isolated the DNA from these samples, sequenced their exomes, and computed the genetic heterogeneity of each sample. From our data, we concluded that spontaneously-arising metastases are a more realistic method to study changes in heterogeneity during metastasis than experimental metastases obtained from tail-vein injections.
Presence of mouse host stroma in human xenograft tumors is required for tumor growth and may mimic some aspects of the human tumor microenvironment, despite its reduced immune context (discussed in [46], [47], [53]. Additionally, different tumor microenvironments can influence the clonal evolution of the tumor by exerting different selective pressures (for example [54], [55]). However, our study focuses on the genetic heterogeneity of the patient-derived tumor itself. In this context, stromal cells can bias genetic heterogeneity estimates because the sequenced samples are a mixture of human tumor cells and mouse stromal cells. Regions of the mouse genome that are homologous with the human genome can falsely overinflate heterogeneity measures from bulk sequenced samples of mixed human tumor and mouse cells. The potential scope of the problem is large as 80% of mouse genes have an orthologue in the human genome [56], and between 0.9% and 97.5% of each of our PDX tumor samples consisted of mouse-specific DNA. A variety of experimental and computational approaches to handle mouse contamination have been developed [50], [57], but can still leave a small fraction of false positives from the host mouse [50]. Additionally, several methods to measure intra-tumor heterogeneity or reconstruct the subclonal phylogeny have been developed that can account for tumor cellularity from the surrounding normal tissue [13], [58], [59], [60], [61], [62], but they do not explicitly address the issues faced with data from PDX models and some measures may be insufficient to describe evolutionary dynamics [20]. We thus developed a bioinformatics approach to mitigate the bias in heterogeneity that can arise from contamination with the mouse by estimating each sample’s heterogeneity using only unbiased genomic positions (Fig. 3A). There are two consequences of our approach to removing mouse bias. First, the remaining unbiased genomic positions are not suitable to use to identify driver mutations – many functionally important regions (like cancer drivers) have high homology with the mouse genome, and so are removed from our analysis. Second, our heterogeneity scores can only be used to compare samples within a given model (e.g., all of the B1 model tumors) because different unbiased and heterogeneous sites are identified for each model. Thus, we cannot compare heterogeneity across models unless we explicitly set out to do so.
Heterogeneity is a reflection of clonal evolution within a tumor, and it can differ within a tumor based on how the tumor evolved and grew [63]. Because of this, we wondered to what extent clonal evolution affected the heterogeneity of replicates from the same model. We used the heterogeneity score for each sample to evaluate to what extent heterogeneity is consistent between replicate tumors of the same model. Model B1 had relatively consistent heterogeneity both within and between primary tumors, though we did find that our samples formed two distinct groups with higher and lower heterogeneity. An intriguing explanation consistent with finding two distinct groups is that regional genetic heterogeneity from the expanded precursor B1 tumor was transferred to the mammary fat pads of mice B11 - B110. Indeed, regional differences in genetic variation is unsurprising [64]; for example, a previous observation showed that tumor cells originating from nearby regions within a colon cancer tumor were more similar to each other than those found further away [65]. On the other hand, model B2 showed less consistency both within and between primary tumors. Model B2’s higher variability in heterogeneity could either be inherent to the patient’s tumor itself, or because its heterogeneity score is derived from fewer genomic positions than that of model B1. Regardless, the variability between tumors for both models suggests that we should strive to evaluate changes in heterogeneity during metastasis relative to the primary tumor from the same mouse.
In addition, we looked at the effect of metastasis on heterogeneity in our PDX models. Metastasis is an evolutionary process [27] and a successful metastasis requires that cells undergo several steps [66], [67], [68], [69] each of which can generate a population bottleneck and reduce heterogeneity. Indeed, current evidence from patients and PDX models suggest that metastases are formed by a single clone or small cluster of cells from the primary tumor [22], [28], [70], [71], [72]. While we therefore expected genetic heterogeneity to decrease during metastasis, we did not know whether this would be the case in PDX models.
The two approaches we used to generate metastases (spontaneous and experimental) present different selective pressures on the primary tumor cells. Spontaneous metastases would need to escape from the primary tumor, and then both survive the trip to the distant organ and successfully colonize it [66], [67], [68], [69]. Because experimental metastases are generated by tail vein injection of tumor cells, they would only need to survive and colonize after injection. However, these cells must also survive the experimental steps necessary for the injection, namely generating a cell solution by tumor cell dissociation and mouse cell depletion. Thus, it is possible that the same model may have a different propensity to metastasize spontaneously and experimentally. Indeed, we observed such differences in our two models – model B1 was relatively poor at forming experimental metastases, while model B2 was incapable of forming spontaneous metastases, consistent with the lack of metastases in the patient from which it was derived. Despite differences in selective pressures, a previous study showed no gene expression differences between metastases obtained by orthotopic implantation and tail vein injection of a breast cancer cell line (but they did show differences in morphology) [73]. The locations of the metastases can also depend on the model. Spontaneous metastases in previous studies were found in the mouse lung in one study [39], or the lymphatics, lung, and peritoneum in another [37], while the B1 model spontaneously metastasized to the liver and lymph nodes.
Finally, we looked at how heterogeneity changed during spontaneous and experimental metastasis and found differences between the two methods. Heterogeneity decreased as expected during a population bottleneck in spontaneous mutations, but either showed no change or an unexpected increase in experimental metastases. The increased heterogeneity could arise from a combination of several factors. First, the sample itself could contain many small experimental metastases as shown in Fig. 1D, where the lung was split into several pieces, each of which contains multiple experimental metastases. Second, there could be more heterogeneity in the cell solution injected in the mouse tail veins because it is comprised of cells from multiple tumors. Third, individual cells injected into the tail-vein could have a higher propensity to aggregate into a multi-clonal metastasis than cells from spontaneous metastases. Indeed, this explanation is consistent with recent observations in PDX breast cancer models of metastasis [32]. Measuring the original heterogeneity from the PDX tumors that were implanted in the mammary fat pad or injected into the tail vein could help to distinguish between these options, however we unfortunately do not have these samples. Regardless, we found that the method for generating metastases is important and that spontaneous metastases show a reduction in heterogeneity consistent with a population bottleneck.
Our study suggests that the method to generate metastases is important when studying heterogeneity using PDX models. For the two PDX models we studied here, the expected changes in heterogeneity during metastasis were obtained by implanting PDX tissue in the mammary fat pads and waiting for spontaneous metastases. However, the spontaneous approach has drawbacks – not every PDX model is capable of generating metastases in this way (only one of our two PDX models worked), and it generates fewer metastases (between 0 and 2 per mouse). When we generated experimental metastases by injecting patient-derived tumor cells in the tail vein of mice, both PDX models generated metastases, but showed unrealistically high levels of heterogeneity. We therefore recommend using spontaneously-generated metastases for studies involving genetic heterogeneity and clonal evolution.
Conflict of interest
All authors were employed by Novartis Institutes for BioMedical Research while conducting research for this paper.
Acknowledgment
We thank Louise Barys, Eusebio Manchado, Joana Pinto Couto, and Luca Tordella for their valuable advice and feedback.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.01.008.
Contributor Information
Kathleen Sprouffske, Email: kathleen.sprouffske@novartis.com.
Audrey Kauffmann, Email: audrey.kauffmann@novartis.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hidalgo M., Amant F., Biankin A.V., Budinska E., Byrne A.T. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrne A.T., Alférez D.G., Amant F., Annibali D., Arribas J. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17:254–268. doi: 10.1038/nrc.2016.140. [DOI] [PubMed] [Google Scholar]
- 3.Nowell P. The clonal evolution of tumor cell populations. Sci N Y NY. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 4.Merlo L.M.F., Pepper J., Reid B., Maley C.C. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 5.Marusyk A., Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merlo L.M.F., Maley C.C. The role of genetic diversity in cancer. J Clin Invest. 2010;120:401–403. doi: 10.1172/JCI42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greaves M., Maley C.C. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sprouffske K., Merlo L.M.F., Gerrish P.J., Maley C.C., Sniegowski P.D. Cancer in light of experimental evolution. Curr Biol. 2012;22:R762–R771. doi: 10.1016/j.cub.2012.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aparicio S., Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842–851. doi: 10.1056/NEJMra1204892. [DOI] [PubMed] [Google Scholar]
- 10.McGranahan N., Swanton C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 11.Navin N., Krasnitz A., Rodgers L., Cook K., Meth J. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andor N., Graham T.A., Jansen M., Xia L.C., Aktipis C.A. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2015;22:105–113. doi: 10.1038/nm.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mroz E.A., Tward A.M., Hammon R.J., Ren Y., Rocco J.W. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from The Cancer Genome Atlas. PLOS Med. 2015;12 doi: 10.1371/journal.pmed.1001786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamal-Hanjani M., Wilson G.A., McGranahan N., Birkbak N.J., Watkins T.B.K. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–2121. doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 15.Landau D.A., Carter S.L., Stojanov P., McKenna A., Stevenson K. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhang H.C., Ruddy D.A., Krishnamurthy Radhakrishna V., Caushi J.X., Zhao R. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. 2015;21:440–448. doi: 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- 17.Kemper K., Krijgsman O., Cornelissen-Steijger P., Shahrabi A., Weeber F. Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts. EMBO Mol Med. 2015;7:1104–1118. doi: 10.15252/emmm.201404914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearson A., Smyth E., Babina I.S., Herrera-Abreu M.T., Tarazona N. High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov. 2016;6:838–851. doi: 10.1158/2159-8290.CD-15-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maley C.C., Galipeau P., Finley J., Wongsurawat V., Li X. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 20.Noorbakhsh J., Kim H., Namburi S., Chuang J.H. Distribution-based measures of tumor heterogeneity are sensitive to mutation calling and lack strong clinical predictive power. Sci Rep. 2018;8:11445. doi: 10.1038/s41598-018-29154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guenot D., Guérin E., Aguillon-Romain S., Pencreach E., Schneider A. Primary tumour genetic alterations and intra-tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J Pathol. 2006;208:643–652. doi: 10.1002/path.1936. [DOI] [PubMed] [Google Scholar]
- 22.Ding L., Ellis M.J., Li S., Larson D.E., Chen K. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eirew P., Steif A., Khattra J., Ha G., Yap D. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2014;518:422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruna A., Rueda O.M., Greenwood W., Batra A.S., Callari M. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;167:260–274.e22. doi: 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben-David U., Ha G., Tseng Y.-Y., Greenwald N.F., Oh C. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49:1567–1575. doi: 10.1038/ng.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen L.V., Cox C.L., Eirew P., Knapp D.J.H.F., Pellacani D. DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat Commun. 2014;5:5871. doi: 10.1038/ncomms6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turajlic S., Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–175. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 28.Navin N., Kendall J., Troge J., Andrews P., Rodgers L. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turajlic S., Xu H., Litchfield K., Rowan A., Chambers T. Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell. 2018;173 doi: 10.1016/j.cell.2018.03.057. 581–594.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McPherson A., Roth A., Laks E., Masud T., Bashashati A. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet. 2016;48:758. doi: 10.1038/ng.3573. [DOI] [PubMed] [Google Scholar]
- 31.Reeves M.Q., Kandyba E., Harris S., Del Rosario R., Balmain A. Multicolour lineage tracing reveals clonal dynamics of squamous carcinoma evolution from initiation to metastasis. Nat Cell Biol. 2018;20:699–709. doi: 10.1038/s41556-018-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X., Taftaf R., Kawaguchi M., Chang Y.-F., Chen W. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discov. 2019;9:96–113. doi: 10.1158/2159-8290.CD-18-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walter D., Harter P.N., Battke F., Winkelmann R., Schneider M. Genetic heterogeneity of primary lesion and metastasis in small intestine neuroendocrine tumors. Sci Rep. 2018;8:3811. doi: 10.1038/s41598-018-22115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah S.P., Roth A., Goya R., Oloumi A., Ha G. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koren S., Bentires-Alj M. Breast tumor heterogeneity: Source of fitness, hurdle for therapy. Mol Cell. 2015;60:537–546. doi: 10.1016/j.molcel.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 36.Pereira B., Chin S.-F., Rueda O.M., Vollan H.-K.M., Provenzano E. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeRose Y.S., Wang G., Lin Y.-C., Bernard P.S., Buys S.S. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17:1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabos P., Finlay-Schultz J., Li C., Kline E., Finlayson C. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res Treat. 2012;135:415–432. doi: 10.1007/s10549-012-2164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X., Claerhout S., Prat A., Dobrolecki L.E., Petrovic I. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–4897. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H., Cohen A.L., Krishnakumar S., Wapnir I.L., Veeriah S. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014;16:R36. doi: 10.1186/bcr3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao H., Korn J.M., Ferretti S., Phane Eacute, Monahan J.E., Wang Y. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- 42.Dobrolecki L.E., Airhart S.D., Alferez D.G., Aparicio S., Behbod F. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016;35:547–573. doi: 10.1007/s10555-016-9653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung J., Jang K., Ju J.M., Lee E., Lee J.W. Novel cancer gene variants and gene fusions of triple-negative breast cancers (TNBCs) reveal their molecular diversity conserved in the patient-derived xenograft (PDX) model. Cancer Lett. 2018;428:127–138. doi: 10.1016/j.canlet.2018.04.020. [DOI] [PubMed] [Google Scholar]
- 44.Coussy F., Koning L., Lavigne M., Bernard V., Ouine B. A large collection of integrated genomically characterized patient-derived xenografts highlighting the heterogeneity of triple-negative breast cancer. Int J Cancer. 2019 doi: 10.1002/ijc.32266. [DOI] [PubMed] [Google Scholar]
- 45.Whittle J.R., Lewis M.T., Lindeman G.J., Visvader J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. doi: 10.1186/s13058-015-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cassidy J.W., Batra A.S., Greenwood W., Bruna A. Patient-derived tumour xenografts for breast cancer drug discovery. Endocr Relat Cancer. 2016;23:T259–T270. doi: 10.1530/ERC-16-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murayama Gotoh. Patient-derived xenograft models of breast cancer and their application. Cells. 2019;8:621. doi: 10.3390/cells8060621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ottewell P.D., Coleman R.E., Holen I. From genetic abnormality to metastases: murine models of breast cancer and their use in the development of anticancer therapies. Breast Cancer Res Treat. 2006;96:101–113. doi: 10.1007/s10549-005-9067-x. [DOI] [PubMed] [Google Scholar]
- 49.Couto J.P., Bentires-Alj M. Springer International Publishing; Breast Cancer; Cham: 2017. Mouse models of breast cancer: deceptions that reveal the truth; pp. 49–60. [Google Scholar]
- 50.Callari M., Batra A.S., Batra R.N., Sammut S.-J., Greenwood W. Computational approach to discriminate human and mouse sequences in patient-derived tumour xenografts. BMC Genomics. 2018;19:19. doi: 10.1186/s12864-017-4414-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi W., Ng C.K.Y., Lim R.S., Jiang T., Kumar S. Reliability of whole-exome sequencing for assessing intratumor genetic heterogeneity. Cell Rep. 2018;25:1446–1457. doi: 10.1016/j.celrep.2018.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pompili L., Porru M., Caruso C., Biroccio A., Leonetti C. Patient-derived xenografts: a relevant preclinical model for drug development. J Exp Clin Cancer Res. 2016;35:189. doi: 10.1186/s13046-016-0462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cassidy J.W., Caldas C., Bruna A. Maintaining tumor heterogeneity in patient-derived tumor xenografts. Cancer Res. 2015;75:2963–2968. doi: 10.1158/0008-5472.CAN-15-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fidler I. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 55.Alzubi M.A., Turner T.H., Olex A.L., Sohal S.S., Tobin N.P. Separation of breast cancer and organ microenvironment transcriptomes in metastases. Breast Cancer Res. 2019;21 doi: 10.1186/s13058-019-1123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 57.Schneeberger V.E., Allaj V., Gardner E.E., Poirier J.T., Rudin C.M. Quantitation of murine stroma and selective purification of the human tumor component of patient-derived xenografts for genomic analysis. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0160587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andor N., Harness J.V., Müller S., Mewes H.W., Petritsch C. EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics. 2014;30:50–60. doi: 10.1093/bioinformatics/btt622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beerenwinkel N., Schwarz R.F., Gerstung M., Markowetz F. Cancer evolution: mathematical models and computational inference. Syst Biol. 2014;64:e1–e25. doi: 10.1093/sysbio/syu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fischer A., Vázquez-García I., Illingworth C.J.R., Mustonen V. High-definition reconstruction of clonal composition in cancer. Cell Rep. 2014;7:1740–1752. doi: 10.1016/j.celrep.2014.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li B., Li J.Z. A general framework for analyzing tumor subclonality using SNP array and DNA sequencing data. Genome Biol. 2014;15:473. doi: 10.1186/s13059-014-0473-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roth A., Khattra J., Yap D., Wan A., Laks E. PyClone: Statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kostadinov R., Maley C.C., Kuhner M.K. Bulk genotyping of biopsies can create spurious evidence for hetereogeneity in mutation content. PLOS Comput Biol. 2016;12 doi: 10.1371/journal.pcbi.1004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martens E., Kostadinov R., Maley C.C., Hallatschek O. Spatial structure increases the waiting time for cancer. New J Phys. 2011;13 doi: 10.1088/1367-2630/13/11/115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roerink S.F., Sasaki N., Lee-Six H., Young M.D., Alexandrov L.B. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature. 2018;556:457–462. doi: 10.1038/s41586-018-0024-3. [DOI] [PubMed] [Google Scholar]
- 66.Nguyen D.X., Bos P.D., Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 67.Chaffer C.L., Weinberg R.A. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 68.Massagué J., Obenauf A.C. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Welch D.R., Hurst D.R. Defining the hallmarks of metastasis. Cancer Res. 2019;79:3011–3027. doi: 10.1158/0008-5472.CAN-19-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yates L.R., Gerstung M., Knappskog S., Desmedt C., Gundem G. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gkountela S., Castro-Giner F., Szczerba B.M., Vetter M., Landin J. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 2019;176:98–112. doi: 10.1016/j.cell.2018.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merino D., Weber T.S., Serrano A., Vaillant F., Liu K. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat Commun. 2019;10:766. doi: 10.1038/s41467-019-08595-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rashid O.M., Nagahashi M., Ramachandran S., Dumur C.I., Schaum J.C. Is tail vein injection a relevant breast cancer lung metastasis model? J Thorac Dis. 2013;5:385–392. doi: 10.3978/j.issn.2072-1439.2013.06.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.