

Abstract
Triheptanoin is anticonvulsant in several seizure models. Here, we investigated changes in glucose metabolism by triheptanoin interictally in the chronic stage of the pilocarpine mouse epilepsy model. After injection of [U-13C6]-glucose (i.p.), enrichments of 13C in intermediates of glycolysis and the tricarboxylic acid (TCA) cycle were quantified in hippocampal extracts and maximal activities of enzymes in each pathway were measured. The enrichment of 13C glucose in plasma was similar across all groups. Despite this, we observed reductions in incorporation of 13C in several glycolytic intermediates compared to control mice suggesting glucose utilization may be impaired and/or glycogenolysis increased in the untreated interictal hippocampus. Triheptanoin prevented the interictal reductions of 13C incorporation in most glycolytic intermediates, suggesting it increased glucose utilization or – as an additional astrocytic fuel – it decreased glycogen breakdown. In the TCA cycle metabolites, the incorporation of 13C was reduced in the interictal state. Triheptanoin restored the correlation between 13C enrichments of pyruvate relative to most of the TCA cycle intermediates in “epileptic” mice. Triheptanoin also prevented the reductions of hippocampal pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase activities. Decreased glycogen breakdown and increased glucose utilization and metabolism via the TCA cycle in epileptogenic brain areas may contribute to triheptanoin's anticonvulsant effects.
Keywords: Glucose metabolism, pilocarpine, temporal lobe epilepsy, medium chain fatty acid, anaplerosis
Introduction
Triheptanoin, the triglyceride of the odd-chain fatty acid heptanoate, has been shown to be anticonvulsant in several rodent models of epilepsy when provided as 35% of the caloric intake (35E%). This includes the second hit pentylenetetrazole seizure test during the chronic epileptic stage of our mouse pilocarpine model,1 a rat pilocarpine model,2 a 6 Hz electroshock threshold test,3 delaying the development of corneal kindled seizures1 and a genetic mouse model of generalized seizures.4 Heptanoate produces both acetyl-CoA and propionyl-CoA, the latter being anaplerotic.5,6 Propionyl-CoA can refill the TCA cycle intermediate levels via the propionyl-CoA carboxylation pathway, which produces succinyl-CoA.7 This anaplerotic action may in part be responsible for the anticonvulsant effects of triheptanoin.8–10
In epilepsy there is a high metabolic demand to fuel seizures, to recover from the depolarization of large groups of neurons, as well as to take up and metabolize neurotransmitters. Conversely, 18fluorodeoxyglucose positron emission tomography and 14C-deoxyglucose radiography showed decreased signals in people with epilepsy and animal models, respectively.10–12 In the chronic pilocarpine model – a model for temporal lobe epilepsy, we found reductions in hippocampal % 13C enrichments of glucose 6-phospate after [U-13C6]-glucose administration, while there were no changes in hexokinase activity, indicating that glucose utilization in epileptogenic tissue is reduced.13 Together with an excess release of neurotransmitters such as glutamate, this is thought to lead to a loss of TCA cycle intermediate concentrations in the brain.14 This, along with reductions of the activities of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase, can compromise the oxidative metabolism of glucose, as we recently showed in the chronic epileptic stage of the mouse pilocarpine model.13 Taken together, this can lead to a metabolic deficit and increase the vulnerability of neurons to death or damage in epileptogenic brain areas.9,15,16 Thus, a compound that does not rely on the activity of pyruvate dehydrogenase and can potentially increase TCA cycling by anaplerosis, such as triheptanoin, may be a new successful option to treat temporal lobe epilepsy, which often does not respond to available anti-seizure drugs.17 Please note that in a pilot clinical trial in children with drug-resistant epilepsy, most children tolerated the treatment and five out of eight children showed reduced seizures.18
In the pilocarpine epilepsy model, mice that undergo SE after pilocarpine injection develop spontaneous recurrent seizures in the chronic stage.19,20 Those mice that do not suffer from SE do not show any spontaneous seizures or neuropathological changes. In the chronic stage of this model, triheptanoin prevented the decrease of hippocampal [M+2] enrichment in the TCA cycle intermediates, citrate and malate, produced from the metabolism of [1,2-13C]-glucose.21 Furthermore, infusion of [5,6,7-13C]-heptanoate in wild type mice and the transgenic antisense mouse model of GLUT-1 deficiency that commonly exhibits an epileptic phenotype, indicated that heptanoate enters the TCA cycle in the brain both directly and also indirectly via hepatic metabolism to glucose.8 Together, this suggests that triheptanoin is a good candidate to improve glucose energy metabolism in the epileptogenic brain.
Here, we aimed to identify which pathways of glucose metabolism are altered by 35E% triheptanoin treatment interictally in the chronic phase of the pilocarpine model. The enrichments of the 13C in the glycolytic and TCA cycle intermediates in the hippocampus from the metabolism of [U-13C6]-glucose (Figure 1) were quantified using liquid chromatography-tandem mass spectrometry. Furthermore, the maximal activities of enzymes involved in both glycolysis and TCA cycling, that we have previously shown to be reduced in this model, namely pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase,13 were measured in hippocampal extracts using continuous spectrophotometric assays, to identify if triheptanoin treatment had any effect on their activities.
Figure 1.

Schematic of [U-13C6]-glucose metabolism in the brain. Simplified schematic of 13C-labelling patterns following the metabolism of [U-13C6]-glucose via glycolysis, pentose phosphate pathway (PPP) and the TCA cycle. Empty circles represent 12C and black-filled circles represent 13C. Grey coloured intermediates represent the metabolites that were not measured in this study. Glucose (GLC), glucose 1-phosphate (G1P); glucose 6-phosphate (G6P); fructose 6-phosphate (F6P); fructose 1,6-bisphosphate (F16BP); glyceraldehyde 3-phosphate (GA3P); dihydroxyacetone phosphate (DHAP); 1,3-bisphosphoglycerate (13BPG); 3-phosphoglycerate (3PG); 2-phosphoglycerate (2PG); phosphoenolpyruvate (PEP); pyruvate (PYR); Acetyl CoA (Ac-CoA); citrate (CIT); aconitate (ACO); 2-oxoglutarate (2OG); succinate (SUC); fumarate (FUM); malate (MAL); oxaloacetate (OAA), ribulose 5-phosphate (RL5P). Please note that G6P can be produced from glycogen and lactate can leave the brain.
Methods
Animals
Eight-week-old male CD1 mice (Australian Research Council, Western Australia, Australia) were used for all experiments. The weights of mice prior to experimentation ranged from 26 to 38 g. All animals were individually caged under a 12-h light dark cycle, and given at least one week to adapt to conditions before being used. Mice were housed in a conventional animal facility that has minimal restrictions on entry and exit of researchers and laboratory rodents. Fur mites, pinworm, murine norovirus, murine hepatitis virus, and Pasteurella pneumotropica were all detected in mouse colonies during the time the animals were housed within the facility. All mice were individually housed in opaque cages that contained purachip bedding. All experiments were carried out during the light cycle. All efforts were made to minimize the suffering and number of animals used. All experiments were approved by the University of Queensland's Animal Ethics Committee and followed the guidelines of the Queensland Animal Care and Protection Act 2001. This work was reported according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (https://www.nc3rs.org.uk/arrive-guidelines).
Preregistration of study, randomization, blinding and power analysis
The study was not preregistered. All mice were randomly numbered by animal house staff and then randomly assigned to treatment groups. All experimenters remained blinded throughout the experiments until all analyses were completed. Using power analyses (Statmate, GraphPad Prism 2.0) and previously measured variabilities, experiments were designed to achieve sample sizes enabling us to detect 20–30% of difference to the mean with Student's t-tests at the 0.05 significance level with 80% power.
Pilocarpine-status epilepticus mouse model
The pilocarpine-status epilepticus model was performed as previously described.22 Briefly, mice were transferred to our experimental room 24 h before commencement to allow them to acclimatize to the environment. Mice were transferred to new cages containing only paper towels and no bedding and were injected with methylscopolamine (2 mg/kg intraperitoneally in 0.9% saline; Sigma Aldrich, St Louis, MO, USA), and 15 min later injected with a single dose of pilocarpine (345 mg/kg subcutaneously; Sigma Aldrich). Following pilocarpine injection, their seizure behaviour was assessed for a 90-min period, and then all mice were injected with pentobarbital (22.5 mg/kg intraperitoneally; Provet, Northgate, QLD, Australia) to terminate SE. Mice were defined as developing SE if they were observed to have continuous seizure activity mainly consisting of whole-body clonic seizures. Those that did not display this behavior were classified as No SE. No brain pathology nor electrographic seizures have been found in No SE mice so far.19,23 After a seven-day recovery period, mice were fed either a standard diet (SF11-027) or a diet containing triheptanoin (SF11-079), in which simple sugars and some of the complex carbohydrates and long-chain fats were replaced with triheptanoin, to provide 35% of calories (Specialty Feeds, Western Australia, Australia; triheptanoin was gifted from Ultragenyx Pharmaceuticals Inc., Novato, CA, USA). Diets were assigned to the SE and No SE groups at random. Please note that the data from the untreated No SE and SE mice have been previously analyzed and published.13 Here, the results from the untreated mice are shown again relative to the effects of triheptanoin treatment.
[U-13C6]-glucose injections and tissue collection
For all experiments, mice were transferred to the laboratory in their home cages at least 3 h prior to commencement. The order of mice was selected at random. Between 5 and 8 p.m., mice were injected with [U-13C6]-glucose (0.3 mol/L intraperitoneally, 558 mg/kg; 99% 13C; Cambridge Isotope Laboratories, Woburn, MA, USA) three weeks after the development of SE. Fifteen minutes later, mice were sacrificed by focal microwave fixation (Model MMW-05, Muromachi, Tokyo, Japan) at 5 kW for 0.80 to 0.83 s. This process immediately denatures all enzymes and proteins in the brain. Mice were decapitated and hippocampi dissected out and stored at −80℃ until extracted. To extract the polar metabolites, samples were sonicated in 1 mL of methanol using a Vibra Cell sonicator (Model VCX 750, Sonics and Materials, Newton, CT, USA) with 4 microL of a 1 mM azidothymidine (AZT) solution added as an internal standard, followed by the Bligh-Dyer water/methanol/chloroform extraction procedure at a 2/2/3 ratio as previously described.24 Samples were lyophilized, reconstituted and stored at −80℃ until analyzed.
Liquid chromatography tandem mass spectrometry
Intermediates of [U-13C6]-glucose metabolism (Figure 1) were analyzed following the method described in McDonald et al..13 The scheduled multiple reaction monitoring (sMRM) transitions for all the unlabeled metabolites and their associated instrument parameters are detailed in Table 1 of our previous paper.13 Also as previously described,13 glycolytic intermediates were measured using LC-MS/MS by first isolating the precursor ion (Q1 mass, Da) that is uniformly labelled with 13C. The masses isolated are glucose 6-phosphate (G6P), 265; fructose 6-phosphate (F6P), 265; fructose 1,6-phosphate (F16BP), 345; dihydroxyacetone phosphate (DHAP), 172; 2 and 3 phosphoglycerate (2 + 3PG), 188; phosphoenolpyruvate (PEP), 170; and pyruvate (PYR), 90. Following collision-induced dissociation (Q2), the product ion detected (Q3 mass) for most glycolytic metabolites was dihydrogen phosphate ion (97 Da). For phosphoenolpyruvate, the product ion detected was a phosphite ion (79 Da) and pyruvate loses a carboxyl group resulting in a detectable mass of 45 Da.
Table 1.
Heptanoate and ketone body concentrations in plasma and cortex.
| No SE |
SE |
||||
|---|---|---|---|---|---|
| Metabolites (µM or nmol/g) | Untreated n = 5 | Triheptanoin n = 5 | Untreated n = 4 | Triheptanoin n = 7 | |
| Plasma | Heptanoic acid | 7.5 ± 8.3 | 16 ± 12 | 2.6 ± 1.2 | 28 ± 16** |
| β-hydroxybutyrate | 542 ± 340 | 578 ± 421 | 495 ± 172 | 398 ± 167 | |
| β-hydroxypentanoate | 12 ± 17 | 57 ± 55 | 3.3 ± 1 | 66 ± 42** | |
| Heptanoic acid | 7.0 ± 4.0 | 7.3 ± 9.0 | 4.3 ± 1.1 | 4.2 ± 1.0 | |
| Cortex | β-hydroxybutyrate | 148 ± 72 | 184 ± 81 | 176 ± 51 | 187 ± 59 |
| β-hydroxypentanoate | 15 ± 6.5 | 16 ± 6.0 | 8.8 ± 2.0 | 33 ± 16 | |
**p < 0.01 Triheptanoin-treated SE vs. untreated SE.
[U-13C]-pyruvate resulting from glycolysis can produce [U-13C]-lactate or alternatively enter the TCA cycle via pyruvate dehydrogenase (PDH) to [1,2-13C]-acetyl CoA. This entry of 13C labelled acetyl-CoA results in two 13C carbons in all TCA cycle metabolites (Figure 1). Thus, M+2 isomers are isolated as the precursor ions (Q1, Da), for citrate (CIT), 193; aconitate (ACO), 175; 2-oxoglutarate (2OG), 147; succinate (SUC), 119; fumarate (FUM), 117; and malate (MAL), 135. In the collision cell, all TCA cycle intermediates lose the carboxyl group. As the 13C is within one of the carboxyl groups of all metabolites after the collision, either one or two 13C-carbons remain on the product ion (Q3). Thus, molecular weight (Da) of the product ions are 112 (lost a 13C in the collision, M+1) and 113 (both 13C remain, M+2) for citrate; 85 and 86 aconitate; 102 and 103, 2-oxoglutarate; 72 and 73, fumarate; 72 and 73, malate are produced. The sum of both product ions' percentage enrichment is representative of the first turn of the TCA cycle.
Measurement of [U-13C6]-glucose in plasma
Plasma could not be obtained from the mice that were sacrificed via microwave fixation. Therefore, a separate cohort of mice was injected with [U-13C6]-glucose (0.3 mol/L intraperitoneally, 558 mg/kg; 99% 13C; Cambridge Isotope Laboratories, Woburn, MA, USA), three weeks after the development of SE. Fifteen minutes later, the mice were injected with an overdose of pentobarbitone (60 mg/kg i.p.) and blood was collected from the inferior vena cava in a heparin-coated syringe. The blood was centrifuged at 1000g for 10 min at 4℃, and the resultant plasma was collected and stored at −80℃ until used; 50 µL of plasma was spiked with 20 µL of a 2 mM solution of [1-13C]-glucose. Samples were incubated with acetone for 30 min on ice, and then centrifuged at 25,000g for 10 min at 4℃. The supernatant was transferred to a glass vial and acetone was evaporated at RT under a stream of air. Glucose was converted to its pentaacetate derivative by reacting lyophilized sample with 150 µl of pyridine:acetic anhydride mixture (1:2) at 60℃ for 40 min. Samples were evaporated and reconstituted in 100 µl of ethyl acetate and transferred to limited volume insert. Analyses of glucose pentaacetate derivatives were carried out on an Agilent 5973 mass spectrometer equipped with 6890 Gas Chromatograph. An HP-5MS capillary column (60 m × 0.25 mm × 0.25 μm) was used in all assays with a helium flow of 1 ml/min. Samples were analyzed in Selected Ion Monitoring (SIM) mode using electron impact ionization (EI). Ion dwell time was set to 10 ms. Natural glucose pentaacetate ion (M0, 331) as well as isotopically enriched ions (332, M+1, representing [1-13C]glucose and 337, M+6, representing [U-13C6]glucose). M+6 enrichment was calculated as ratio of peak abundances or 337/(331 + 337) × 100%.
Measurement of heptanoate and ketone bodies in cortex and plasma
Mice were decapitated under light isoflurane anesthesia. Trunk blood was collected in EDTA-coated tubes and centrifuged at 1000g for 10 minutes to collect plasma and cerebral cortex was extracted and frozen on dry ice immediately. Plasma and tissue were stored at −80℃ until extracted. The extraction protocols and analysis methods were previously described in Tan et al.25
Mitochondrial isolation and enzyme activities
Mice were decapitated under light isoflurane anesthesia. Hippocampi were dissected out and stored at −80℃ until extracted. Mitochondria were isolated as previously described.25 Aliquots were stored at −80℃ for up to one month, and used to determine enzyme activities. All enzyme activities were measured on the SpectroMax 190 Microplate reader (Molecular Devices, Sunnyvale, CA, USA) using continuous spectrophotometric assays. All enzyme activities were normalized to protein content measured via a Pierce Bicinchoninic acid (BCA) assay (ThermoFisher Scientific, Scoresby, Victoria, Australia) and activity was expressed as U/mg protein, which was calculated as nmol turnover/min relative to mg protein. The activities of phosphoglucose isomerase (PGI, EC 5.3.1.9), phosphofructokinase (PFK, EC 2.7.1.11), pyruvate kinase (PK, EC 2.7.1.40) and 2-oxoglutarate dehydrogenase (2-OGDH, 1.2.4.2) were measured as previously described.13,25 Pyruvate dehydrogenase (PDH, EC 1.2.4.1) activity was measured using the MTT-PMS method.26
Statistical methods
All statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Software, La Jolla, CA, USA). Two-way ANOVAs followed by Fisher's least significant differences post-tests were used to compare the total metabolite concentrations and percent 13C enrichment for each metabolite individually. Correlation analyses were performed to assess the correlations of % 13C enrichments of glucose 6-phosphate to those of downstream glycolytic intermediates and the % 13C enrichment of pyruvate relative to TCA cycle intermediates' 13C enrichments. Maximal enzyme activities were analyzed using unpaired, two-sided Student's t-tests. P < 0.05 was regarded as significant. Values were excluded if higher or lower than two standard deviations from the mean. All data are represented as individual values and mean ± standard deviation.
Results
Pilocarpine-SE model, body weights and plasma metabolites
Of the 52 animals used in the 13C glucose experiment, 26 animals developed SE, while 24 did not and were classified as “No SE.” Two animals out of the 52 injected with pilocarpine died from seizures during the development of SE. Of the 26 animals that developed SE, three mice were culled within the first three days following SE as per the ethical guidelines, as the mice did not recover well. All other surviving mice were used in the experiments. At the time of [U-13C6]-glucose injections, the body weights of mice were similar in both No SE and SE mice fed both the standard (39.9 ± 0.8g, No SE; 39.9 ± 1.3 g, SE) and 35% triheptanoin diet (39.2 ± 0.7g, No SE; 38.8 ± 1.2g, SE). Also, the percent enrichment of plasma [U-13C6]-glucose was similar in both No SE and SE mice treated with control or triheptanoin, with averages ranging between 11.8% and 14.4% (Figure 2, two-way ANOVA, n = 6–8 mice per group). Therefore, any changes in the total concentrations or percent of 13C enrichment in brain metabolites were not due to differing amounts of [U-13C6]-glucose in plasma. No behavioral seizures were observed before and during the [U-13C6]-glucose injection until sacrifice, indicating that this study investigated interictal metabolism. As shown in Table 1, triheptanoin affected the concentrations of heptanoate and β-hydroxypentanoate in plasma, with two-way ANOVAs indicating it accounted for 32.4% and 37.3% of the found variation, respectively (p = 0.005 for both). A Fisher's LSD post-test indicated a 10.9-fold increase in heptanoate and a 19.8-fold increase in β-hydroxypentanoate (p = 0.016) in the plasma of triheptanoin-treated SE mice compared to untreated SE mice. No changes were found in the β-hydroxybutyrate plasma concentrations nor in the cortical concentrations of heptanoate, β-hydroxybutyrate or β-hydroxypentanoate.
Figure 2.
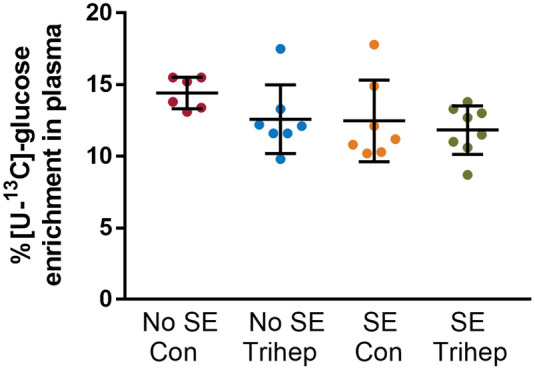
Similar % [U-13C6]-glucose enrichment in plasma from no SE and SE mice in the chronic stage of the pilocarpine model. Plasma enrichment of [U-13C6]-glucose as percentage of total glucose levels after i.p. injection of [U-13C6]-glucose was compared between (“epileptic”) SE and (“non-epileptic') No SE mice, either untreated (Con) or treated with 35E% triheptanoin (trihep). There were no statistically significant differences between the groups (two-way ANOVA; p = 0.47 interaction, p = 0.14 treatment, p = 0.10 SE vs. No SE, n = 6–8 mice per group).
The incorporation of 13C into glycolytic intermediates was reduced interictally in SE mice and improved with triheptanoin treatment
The total concentrations of metabolites in the glycolytic pathway and TCA cycle were similar among mice that had developed SE compared to those that did not, as shown in Table 2. As shown in Figure 3(a), during the chronic stage of the pilocarpine-SE model, mice that had experienced SE have reduced 13C enrichment in glycolytic intermediates. Two-way ANOVAs run on each metabolite independently indicated that the “SE status” of mice, which determines whether mice were in the chronic epileptic stage or not, accounted for between 9.1 and 21.4% (p = 0.002-0.044) of the variation observed in all intermediates of the glycolytic pathway, apart from 2 and 3-phosphoglycerate (p = 0.200). The two-way ANOVAs indicated that triheptanoin treatment also affected the 13C enrichments in glycolytic intermediates, with significance found in glucose 6-phosphate (17.1%, p = 0.004), dihydroxyacetone phosphate (8.9%, p = 0.042), 2- and 3-phosphoglycerate (13.3%, p = 0.018) and phosphoenolpyruvate (10.7%, p = 0.027), indicating that triheptanoin slightly improved the incorporation of 13C from [U-13C6]-glucose. A Fisher's LSD post-test indicated few significant changes in any of the metabolites between specific mouse groups. A significant decrease in 13C incorporation into fructose 6-phosphate was observed in SE mice compared to No SE mice, regardless of treatment (21% untreated, p = 0.039; 23% triheptanoin-treated, p = 0.011). We also analyzed the percent enrichment in the PPP intermediate ribulose 5-phosphate. Treatment equated to 12.2% (p = 0.028), and seizure status accounted for 12.8% of the differences observed, with the only significant difference observed between the No SE and SE mice fed a standard diet (34% decrease, p = 0.0468).
Table 2.
Total metabolite levels in the hippocampal formation.
| No SE |
SE |
|||
|---|---|---|---|---|
| Metabolites (nmol/g weight) | Std diet n = 11 | Trihep diet n = 13 | Std diet n = 10 | Trihep diet n = 9 |
| Ribulose 5-phosphate | 1.3 ± 0.5 | 1.1 ± 0.5 | 1.2 ± 0.5 | 1.1 ± 0.5 |
| Glucose 1-phosphate | 3.2 ± 0.9 | 2.9 ± 1.3 | 3.5 ± 1.6 | 3.2 ± 0.6 |
| Glucose 6-phosphate | 20.1 ± 1.9 | 21.6 ± 1.7 | 24.2 ± 3.4 | 21.2 ± 2.2 |
| Fructose 6-phosphate | 33.0 ± 2.2 | 28.3 ± 4.0 | 36.2 ± 6.0 | 28.0 ± 5.1 |
| Fructose 1,6-bisphosphate | 16.5 ± 1.0 | 18.9 ± 1.2 | 17.8 ± 1.4 | 16.4 ± 0.8 |
| Dihydroxyacetone phosphate | 0.7 ± 0.1 | 0.8 ± 0.1 | 0.7 ± 0.1 | 0.6 ± 0.1 |
| 2/3-phosphoglycerate | 11.2 ± 1.0 | 11.4 ± 1.0 | 10.9 ± 1.2 | 10.6 ± 0.8 |
| Phosphoenolpyruvate | 8.9 ± 1.4 | 9.4 ± 1.3 | 7.5 ± 1.3 | 7.7 ± 1.2 |
| Pyruvate | 38.2 ± 2.7 | 34.2 ± 3.2 | 34.2 ± 6.0 | 35.1 ± 2.8 |
| Citrate | 109 ± 5 | 96 ± 10 | 110 ± 17 | 103 ± 8 |
| Aconitate | 1.8 ± 0.1 | 1.9 ± 0.1 | 2.2 ± 0.3 | 2.0 ± 0.1 |
| 2-oxoglutarate | 90.8 ± 6.5 | 77.2 ± 9.9 | 84.0 ± 17.4 | 91.2 ± 7.9 |
| Succinate | 10.1 ± 0.8 | 10.3 ± 0.6 | 8.2 ± 1.6 | 8.2 ± 1.3 |
| Fumarate | 12.5 ± 1.0 | 12.3 ± 1.1 | 13.2 ± 2.7 | 12.8 ± 0.8 |
| Malate | 45.9 ± 3.9 | 44.6 ± 4.1 | 46.1 ± 6.8 | 44.8 ± 5.2 |
Figure 3.

The effect of triheptanoin on the interictal metabolism of [U-13C6]-glucose via glycolysis in SE mice in the chronic stage of the pilocarpine model. (a) Hippocampal 13C enrichment of glycolytic metabolites after i.p. injection of [U-13C6]-glucose was compared between (“epileptic”) SE and (“non-epileptic') No SE mice, either untreated or treated with 35E% triheptanoin. The top row indicates outcomes of the two-way ANOVAs regarding effects of treatment or SE, if found significant. The horizontal lines with stars indicate significances found in post hoc Fisher's LSD comparisons between specific groups. *p = 0.05 and **p < 0.01. Having undergone SE and thus being in the chronic epileptic stage affected the % 13C enrichment of glucose 6-phosphate (G6P, p = 0.016), fructose 6-phosphate (F6P, p = 0.002), fructose 1,6-bisphosphate (F16BP, p = 0.044), dihydroxyacetone phosphate (DHAP, p = 0.028), phosphoenolpyruvate (PEP, p = 0.040), pyruvate (PYR, p = 0.040) and ribulose 5-phosphate (RL5P, p = 0.024 all two-way ANOVAs). Triheptanoin treatment affected the 13C enrichment of G6P (p = 0.004), DHAP (p = 0.042), 3- and 2-phosphoglycerate (3PG+2PG, p = 0.018), PEP (p = 0.027) and RL5P (p = 0.028). A Fisher's LSD post-test showed a significant increase in the % 13C incorporation in glucose 6-phosphate in triheptanoin-treated mice compared to controls (1.2-fold higher, p = 0.044, No SE mice; 1.3-fold higher, p = 0.032, SE mice; p < 0.05). A significant loss in the % 13C incorporation in fructose 6-phosphate was observed in SE mice compared to No SE mice in both the untreated (21%, p = 0.039) and triheptanoin treated (23%, p = 0.011) groups. There was a 1.3-fold increase in the % 13C incorporation of 3PG+2PG in triheptanoin-treated No SE mice compared to untreated No SE mice (p = 0.031). Finally, there was a 34% decrease in the 13C enrichment of RL5P between the SE and No SE mice fed a standard diet. No significant differences were seen when the SE triheptanoin-treated mice were compared to the No SE untreated group. n = 9–13 mice per group. (b–g) Comparisons between % 13C enrichments of G6P vs. its downstream metabolites with solid lines using the same colours as in panel A showing linear correlations calculated by GraphPad Prism. Significant correlations were observed across all four treatment groups (r = 0.89–0.91, p = 0.003–0.0001) when correlated with the % enrichment of (b) F6P, (c) F16BP, (d) DHAP, (f) PEP and (g) PYR, whereas, (e) 2 + 3PG was only significantly correlated in the No SE group that was not treated (r = 0.76, p = 0.01).
Comparing triheptanoin-treated vs. untreated mice, the enrichment of 13C in glucose 6-phosphate was 1.2-fold higher in the No SE group (p = 0.044), and 1.3-fold higher in the chronic stage of the SE group (p = 0.032). Similarly, there was a 1.3-fold increase in the incorporation of 13C into 2- and 3-phosphoglycerate in the No SE mice treated with triheptanoin compared to untreated No SE mice (p = 0.031). Furthermore, no significant differences were observed when comparing the SE triheptanoin-treated group with the untreated No SE mice, indicating that triheptanoin treatment can at least partially restore alterations in glycolytic glucose metabolism in chronically epileptic mice at an interictal stage (p > 0.05, for all metabolites).
Correlation analyses were performed on the 13C enrichments in the glycolytic intermediates to determine whether the percentage of 13C enrichment in downstream intermediates was related to the percent 13C enrichment of glucose 6-phosphate in individual mice. Most 13C enrichments in downstream metabolites were highly correlated with glucose 6-phosphate 13C enrichment in all four groups (Figure 3(b) to (g), r = 0.64–0.97, p = 0.040-<0.001). Only 2- and 3-phosphoglycerate did not correlate to glucose 6-phosphate in all groups apart from untreated No SE mice (r = 0.76, p = 0.012, untreated No SE mice, Figure 3(e)). No changes in the maximal activities of the glycolytic pathway enzymes phosphoglucose isomerase, phosphofructokinase and pyruvate kinase were found (Table 3), which is consistent with the 13C enrichment data.
Table 3.
Maximal enzyme activities in the hippocampal formation in the chronic stage of the pilocarpine model (nmol turnover/min per mg protein).
| No SE |
SE |
|||
|---|---|---|---|---|
| Enzyme | Std diet n = 7 | Trihep diet n = 8 | Std diet n = 7 | Trihep diet n = 6 |
| Phosphoglucose isomerase | 373 ± 44.3 | 356 ± 24.7 | 332 ± 38.8 | 287 ± 28.4 |
| Phosphofructokinase | 237 ± 52 | 226 ± 52 | 234 ± 54 | 164 ± 32 |
| Pyruvate kinase | 146 ± 12.8 | 156 ± 15.5 | 156 ± 15.9 | 138 ± 27.2 |
| Pyruvate dehydrogenase | 1.3 ± 0.3 | 1.0 ± 0.1 | 0.4 ± 0.1c | 1.0 ± 0.2a |
| 2-oxoglutarate Dehydrogenase | 5.1 ± 0.8 | 3.8 ± 0.5 | 2.3 ± 0.7b,c | 3.1 ± 1.0 |
Note: The activities of the cytosolic enzymes, namely phosphoglucose isomerase (PGI, p = 0.122), phosphofructokinase (PFK, p = 0.525) and pyruvate kinase (PK, p = 0.816) were unaltered between No SE mice and SE the chronic stage of the model, and triheptanoin treatment also had no effect (PGI, p = 0.377; PFK, p = 0.431; PK, p = 0.815 all two-way ANOVAs).
Numbers indicate the following:
An interaction between SE status and treatment was found regarding the activity of PDH (17.3%, p = 0.035).
A two-way ANOVAs indicated having undergone SE and being in the chronic epileptic stage (SE mice) affected the activity of 2-oxoglutarate dehydrogenase (17.2%, p = 0.032).
Untreated SE mice had lower activity of both pyruvate dehydrogenase (PDH, 72%, p = 0.007) and 2-oxoglutarate dehydrogenase (OGDH, 55%, p = 0.019) compared to untreated No SE mice. n = 7–9 mice for all enzymes.
Triheptanoin prevented the decreases of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase activities and the reductions in the % 13C enrichments in the TCA cycle metabolites in the chronic epileptic interictal stage
Similar to the glycolytic intermediates, the percent enrichments of 13C from the metabolism of [U-13C6]-glucose were determined in the TCA cycle intermediates. Please note that oxaloacetate levels were below our detection limit. Two-way ANOVAs were used to analyze each metabolite individually. The chronic epileptic (SE) status was shown to significantly affect the percent 13C enrichments in all of the TCA cycle intermediates (17–30.4% of the variation, p = 0.008–<0.001, Figure 4(a)), whereas triheptanoin only affected the percent 13C enrichment of citrate (7.1% of variation, p = 0.038). A Fisher's Least Significant Differences post-test revealed that the 13C enrichments were reduced in citrate (17%, p = 0.003), aconitate (16%, p = 0.019), succinate (34%, p = 0.002), fumarate (23%, p = 0.004), and malate (17%, p = 0.021) in the untreated SE vs. No SE mice. Similarly, there was a reduction in the 13C enrichment in citrate (15%, p = 0.003), aconitate (18%, p = 0.005), and malate (14%, p = 0.010) between the triheptanoin-treated SE and No SE mouse groups, along with 2-oxoglutarate (18%, p = 0.043).
Figure 4.

The effect of triheptanoin on the interictal metabolism of [U-13C6]-glucose in the TCA cycle in SE mice in the chronic stage of pilocarpine model. (a) Percent 13C enrichments in the TCA cycle metabolites from the first turn of the TCA cycle were compared between SE and No SE mice treated with or without 35E% triheptanoin. The top row shows outcomes of the Two-way ANOVAs regarding effects of treatment or SE when found significant. The horizontal lines with stars indicate significances found in post hoc Fisher's LSD comparisons between specific groups. *p = 0.05 and **p < 0.01. Being in the chronic epileptic stage (SE mice) affected the % 13C enrichment of citrate (CIT, 30.4%, p < 0.001), aconitate (ACO, 26.2%, p < 0.001), 2-oxoglutarate (2OG, 17%, p = 0.08), succinate (SUC, 19.4%, p = 0.003), fumarate (FUM, 24.3%, p = 0.001) and malate (MAL, 21.2%, p = 0.003). However, triheptanoin treatment only affected the % 13C enrichment of CIT (7.1%, p = 0.038). A Fisher's LSD post-test showed that 13C enrichment was reduced in CIT (17%, p = 0.003), ACO (16%, p = 0.019), SUC (34%, p = 0.002), FUM (23%, p = 0.004), and MAL (17%, p = 0.021) in the untreated SE mice compared to No SE mice. The percent 13C enrichments of CIT (15%, p = 0.003), ACO (18%, p = 0.005), 2OG (18%, p = 0.043) and MAL (14%, p = 0.010) were also reduced between the triheptanoin-treated SE and No SE mouse groups. In the SE mice treated with triheptanoin compared to the untreated SE mice, the % 13C enrichment of malate was reduced (15%, p=0.030). Compared to the untreated SE group, the SE mice treated with triheptanoin had a 30% increase in the % 13C enrichment of succinate (p = 0.048) n = 9–13 per group. (b) Comparisons between the 13C enrichment of pyruvate and citrate with solid lines using the same colours as in panel A to depict linear correlations for each of the four treatment groups. Strong positive correlations were found in the No SE untreated, and both No SE and SE groups treated with triheptanoin (r = 0.8–0.86, p = 0.05–0.001), but not in the untreated SE group (yellow, r = 0.34; p = 0.33).
When the triheptanoin-treated SE mice were compared to the No SE untreated group, the percent 13C enrichment in malate remained decreased by 15% (p = 0.030), while it increased in succinate by 30% (p = 0.048) compared to the SE untreated group. The significant decreases in 13C-incorporations in TCA cycle metabolites found in untreated SE vs. no SE mice, namely in citrate, aconitate and fumarate, were prevented with triheptanoin treatment of the SE mice when compared to the untreated No SE mice. This indicates that triheptanoin improved the oxidative metabolism of glucose in the epileptogenic hippocampus.
Correlation analyses were performed between the 13C enrichment of pyruvate to the 13C enrichments of all TCA cycle intermediates, to determine if the enrichment of pyruvate regulates the enrichments observed in all downstream metabolites. A strong positive correlation was observed between the 13C enrichments of pyruvate and most of the downstream metabolites in both the untreated and triheptanoin-treated No SE, and the triheptanoin-treated SE groups. We show the correlation with citrate (Figure 4(b); r = 0.80–0.86, p = 0.005–0.001), and data for the other metabolites except malate are similar (ACO, r = 0.67–0.78, p = 0.04–0.008, 2OG; r = 0.69–0.86, p = 0.02–0.001, SUC; r = 0.58–0.72, p = 0.05–0.02, FUM). In the untreated SE group, no significant correlations were found between any of the metabolites compared to pyruvate (r = 0.34–0.54, p = 0.334–0.104), suggesting that another factor determines entry of pyruvate into the TCA cycle in the chronic epileptic stage (Figure 4(b)).
Consistent with these findings, the maximal activities of the TCA cycle enzymes pyruvate dehydrogenase and 2-oxoglurate dehydrogenase were reduced in untreated SE mice compared to No SE mice by 72% (p = 0.007) and 55% (p = 0.019), respectively (Table 3). These reductions in both of these enzyme activities were not seen between the SE and No SE mice given triheptanoin (p = 0.898, pyruvate dehydrogenase; p = 0.501, 2-oxoglutarate dehydrogenase, Table 3). With triheptanoin treatment, the significance of the reduction in both pyruvate dehydrogenase and 2-oxoglurate dehydrogenase activity in the untreated SE mice was lost.
Discussion
The main findings of this study relating to the effects of triheptanoin treatment in the chronic epileptic stage of the pilocarpine model are as follows: (1) Triheptanoin increased the plasma concentrations of both heptanoate and β-hydroxypentanoate, but not the percentage of [U-13C6]-glucose in the plasma; (2) Based on the increases in the % 13C enrichment of glucose-6-phosphate and most other glycolytic intermediates, triheptanoin alters aspects of interictal hippocampal glucose metabolism in mice. These alterations reestablished most of the reduced hippocampal incorporation of 13C in glycolytic intermediates observed in untreated interictal SE mice and are difficult to interpret as 13C enrichment can be affected by various pathways. We discuss the possibilities that triheptanoin increases glucose uptake, utilization and/or decreases dilution of 13C in glycolytic metabolites by decreasing glycogen breakdown; (3) Triheptanoin treatment in the chronic epileptic stage largely improved the previously reported problems in TCA cycling in the interictal hippocampus.13 The correlations between the 13C enrichments of pyruvate and all TCA cycle metabolites were restored in the treated SE mice when compared to the untreated SE mice. Furthermore, triheptanoin treatment prevented the loss in the activities of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase in the SE mice, and it partially corrected the reduced incorporation of 13C derived from 13C-glucose in most of the TCA cycle intermediates. In summary, triheptanoin effectively improves several important impairments found in hippocampal glucose energy metabolism in the chronic stage of epilepsy in the pilocarpine model.
A limitation of this study was the inability to measure both the total concentrations and the enrichments of 13C in glucose in the brain. Thus, we do not have any direct indications for alterations of glucose uptake in epileptogenic brain areas. On the other hand, we studied functional glucose metabolism in detail in awake animals, which is not possible with other methods. Also, in a separate cohort of mice, we showed that the percent enrichment of [U-13C6]-glucose in plasma was similar between both No SE and SE mice on both treatments. Please note that we refer to glucose uptake, as defined by the methods using arterio-venous differences in glucose levels. In a previous study using the same mouse pilocarpine model in the chronic stage, we were able to measure total glucose and the % enrichment of [1,2-13C2]-glucose and found no changes in % enrichments of glucose in hippocampal tissue.22 Thus, taken together, the observed reductions in 13C incorporation in the hippocampal glycolytic intermediates are not due to a change in the proportion of peripheral [U-13C6]-glucose to unlabeled glucose.
Another challenge for the interpretation of our results is that % 13C enrichments are influenced by dilution from unlabeled carbon sources, which are difficult to estimate. We will discuss effects of glycogen breakdown, lactate efflux and dilution via the PPP. Glycogen breakdown produces unlabeled G1P, which is metabolized to G6P and all downstream metabolites of glycolysis and the PPP. Glycogen is thought to be mostly turned into lactate, which can be released by astrocytes and potentially used as fuel by neurons, but appears to mostly leave the brain. Lactate efflux has been estimated to be 20% from the brain27 and may change in “epileptic” tissue and/or after triheptanoin treatment, but lactate was not measured by our LC-MS/MS method. Our previous data21 showed that overall % enrichment in lactate was low-consistent with being derived mostly from glycogen, namely 6–9%, and there were no changes between healthy and interictal hippocampus. In other studies, extracellular lactate concentrations increased by up to 5-fold in the brain following pilocarpine injections.28,29 However, these levels returned to normal following the termination of behavioral seizures. In our hands, mice generally have one seizure per day during the chronic phase of this model, and it was noted that the mice had no behavioral seizures in the hour before, or during the injection of [U-13C6]-glucose. Therefore, we believe that alterations in lactate efflux are unlikely to be a major cause for observed changes in % 13C enrichment in the TCA cycle intermediates in the SE mice. In addition, another source of dilution of RL5P and glycolytic metabolites, except for G6P, are unlabeled metabolites from the PPP pool, which can dilute RL5P, F6P, GA3P, and downstream metabolites. This can explain the overall low enrichments of below 10% found in RL5P and 3PG+2PG, while most other glycolytic metabolites show higher enrichments around 10–20%. Please note that in this study there were no alterations in total levels of metabolites among the mouse groups, which otherwise could have affected interpretation of % 13C enrichments.
Following the injection of [U-13C6]-glucose, whether mice were in the chronic epileptic stage (SE mice) or not (No SE mice) significantly affected the incorporation of 13C into the glycolytic intermediates, apart from 2 and 3-phosphoglycerate. In the rat lithium-pilocarpine model, it was shown that during the chronic “epileptic” phase glucose utilization was reduced as measured through 14C-2-deoxyglucose analysis,12 which is similar to what occurs interictally in patients investigated with 18FDG-PET.30,31 In our mouse pilocarpine model compared to healthy mice,22 we found trends for reductions in the levels of total glucose by 24% and 13C-glucose by 39% in the hippocampal formation, which can be interpreted as decreased glucose transport and utilization in the epileptic hippocampus, although the % 13C glucose enrichment did not change significantly. In addition, in most of the glycolytic metabolites, the % 13C enrichment decreased in the chronic epileptic hippocampus, while total amounts did not change, which also is consistent with a reduction in glucose utilization in epilepsy.10,13
However, the latter observations are difficult to reconcile with the lack of changes previously seen in the % 13C-glucose enrichments in the epileptic hippocampus.22 Therefore, the carbons entering glycolysis are most likely diluted from a certain compartment largely containing unlabeled carbons, which allows the original small differences in % 13C enrichments in glucose to become larger in the glycolytic intermediates. An obvious option for this dilution of 13C in the glycolytic intermediates is unlabeled G6P derived from G1P and glycogen. Please note that here we were able to show that G1P levels in the hippocampal formation were similar across all groups (Table 1), the first and last intermediate of glycogen production and breakdown, respectively. However, we were unable to detect the 13C enrichment of G1P in this study. The animals in this study were sacrificed between 5 and 8 p.m. at the beginning of their activity period starting around 4:30 p.m. Glycogen breakdown is thought to occur in astrocytes during the awake period of rodents.32 Increases in glycogen metabolism in interictal hippocampus relative to healthy tissue were not considered by us in our previous paper,13 but are consistent with astrogliosis found in this model19,33 and glycogenolysis can at least partially explain the results of reduced interictal % enrichment in the glycolytic intermediates found. In the future, it would be of interest to measure glycogen levels in the hippocampal formation of these animals.
Here, triheptanoin increased the incorporation of 13C into most of the glycolytic intermediates, namely glucose 6-phosphate, dihydroxyacetone phosphate, 2- and 3-phosphoglycerate as well as phosphoenolpyruvate. In addition, triheptanoin prevented the decreases found in the incorporation of 13C into the glycolytic intermediates glucose 6-phosphate and fructose 6-phosphate, dihydroxyacetone phosphate and phosphoenolpyruvate, which were described in previous work.13 Correlation analyses between the percent 13C enrichment in glucose 6-phosphate compared to the enrichment in all downstream glycolytic intermediates indicated the enrichments of most metabolites were strongly correlated to the glucose 6-phosphate enrichments in both mouse groups given triheptanoin. This suggests that triheptanoin has no effect on the glycolytic pathway itself, which is further corroborated by the lack of change in the activities of phosphoglucose isomerase, phosphofructokinase and pyruvate kinase. Interestingly, triheptanoin treatment resulted in increases in the percent 13C enrichment in glucose 6-phosphate in both No SE and SE animals. Taken together, these results can be interpreted in several ways. One possibility is that triheptanoin may increase 13C-glucose utilization in the interictal hippocampal formation, potentially due to increased TCA cycle flux, encouraging glucose breakdown.5,10 Another possibility is that dilution of the unlabeled glycolytic intermediates is decreased with triheptanoin. Due to the increases in % 13C G6P enrichment, the dilution is most likely from unlabeled glycogen-derived carbons. Therefore, we propose that triheptanoin may reduce glycogen breakdown interictally in astrocytes. This fits well with the notion that heptanoate is largely metabolized in astrocytes8 and by providing additional fuel it can spare glycogen breakdown, such as was seen for ketones reducing the use of glucose.34 The use of triheptanoin as a fuel for oxidative metabolism may support neuronal survival, by sparing glucose for use as a neuronal fuel. Taken further, this supports the idea that triheptanoin is useful to treat disorders of glycogen metabolism, such as Adult Polyglucosan Body Disorder35 or potentially Lafora Disease.
The second major finding of this study was that triheptanoin prevented the activity decreases in both pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase in the SE mice (Table 3). We have previously shown that these activities were reduced in the chronic phase of the pilocarpine-SE model,13 and that similar findings have been reported in post mortem tissue from patients with other neurological disorders such as Alzheimer's disease.36 Furthermore, unlike the untreated SE group, triheptanoin-treated SE mice retained the strong correlations between the percent 13C enrichment of pyruvate and those of most of the TCA cycle intermediates. This provides further evidence that the activity reductions of both pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase are important factors mediating the reduced TCA cycle activity in the epileptogenic brain areas. An additional factor that can influence entry of pyruvate into the TCA cycle are changes in the extent of lactate efflux from tissue; however, there are no data from interictal chronic epileptic mice, to our knowledge. In our earlier study, we found no significant changes in the levels of total and 13C-labeled lactate, but a non-significant near doubling of 13C-lactate levels and higher % 13C enrichment in lactate in triheptanoin-treated interictal hippocampus (9%) compared to control-treated No SE tissue (6%), consistent with decreased glycogenolysis with triheptanoin treatment.21 We speculate that there is still slightly reduced entry of pyruvate into the TCA cycle in SE mice when treated with triheptanoin that serves as anaplerotic substrate and oxidative fuel mainly for astrocytes (see below), but there are also other possibilities, including potential changes in lactate efflux.
It is currently unknown how 35E% triheptanoin prevents the reduction in pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase activities in the chronically epileptogenic hippocampus. It is possible that the recently discovered protective and antioxidant effects of triheptanoin preventing SE-induced alterations play a role.37 Triheptanoin pretreatment before SE did not alter SE-severity or duration, but one day later we found reduced lipid peroxidation and prevention of the reductions in the activities of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase complexes.37 Also, triheptanoin pretreatment was shown to prevent ischemic damage in the middle cerebral artery occlusion mouse stroke model, and prevented the loss of mitochondrial respiratory activity, measured via oxygen consumption in isolated mitochondria.38 In the skeletal muscle of MeCP2-KO mice, triheptanoin improved mitochondrial morphology, by reducing swelling.39 Thus, it appears that one of triheptanoin's effects is to restore mitochondrial function in these disorders. On the other hand, we have previously shown that oxidative stress is not increased, nor are there any impairments in mitochondrial respiratory activity during the chronic period of the mouse pilocarpine model of epilepsy.13 Another possibility is that triheptanoin prevents energy deficits through anaplerosis, the refilling of the TCA cycle intermediates.40 This anaplerotic effect may restore TCA cycling and address metabolic impairments in neurological disorders where mitochondria are unable to provide cells with enough energy to maintain normal function. Energy is needed to provide precursors for biosynthesis, neurotransmitters and structural turnover as well as defense mechanisms. The notion that anaplerosis is protective is further supported by several studies that have found that supplementation of anaplerotic molecules, such as pyruvate (anaplerotic via pyruvate carboxylase),41,42 a mixture of 2-oxoglutarate and oxaloacetate,43,44 and a mixture of pyruvate with the anti-oxidants ascorbic acid and alpha-tocopherol,45 reduced seizures and seizure-induced damage in rodent models. Further research needs to be conducted to identify the mechanisms by which triheptanoin alters the activities of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase.
In SE mice, triheptanoin partially restored (significance was lost between No SE and SE groups) the reduced incorporation of 13C derived from 13C-glucose in most of the TCA cycle intermediates and significantly increased it in succinate (Figure 4(a)). Moreover, triheptanoin reinstated the lost correlations between the 13C enrichments in pyruvate vs. the other measured TCA cycle intermediates, indicating that glucose oxidation via the TCA cycle is at least partially improved. The full extent of this improvement is difficult to quantify, as heptanoate metabolism is expected to produce acetyl-CoA, which may dilute entry of 13C-acetyl-CoA derived from 13C-glucose into the TCA cycle. However, we also expected to see this dilution in the triheptanoin-treated No SE mice. As the extent to which heptanoate metabolism contributes to the production of acetyl-CoA in the epileptogenic and normal brain is so far unknown, future studies assessing the metabolism of [1,2,3,4]-13C4-labelled heptanoate are needed.
Overall, we have provided evidence that 35E% triheptanoin treatment may have increased glucose utilization and/ or reduced glycogen breakdown and improved glucose oxidative metabolism in the hippocampal formation of mice in the chronic epileptic stage. Furthermore, the activities of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase were restored with triheptanoin treatment, indicating that triheptanoin can increase the capacity for glucose metabolism in the mitochondria. In conclusion, the partial improvement in glucose utilization together with improved glucose entry into the TCA cycle and TCA cycling in epileptogenic brain areas may contribute to triheptanoin' s anticonvulsant effects.
Authors' contributions
TSM and KB designed the study. TSM performed most the experiments, final data analysis and wrote the first draft of the manuscript. Quantifications and data analysis were performed by MPH for all LCMS-MS data, by IB regarding plasma glucose levels and by MP regarding ketone body levels. All authors contributed to the writing. KB finalized and submitted the manuscript and obtained most of the funding.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We are grateful to funding by the Australian National Health and Medical Research Council (NHMRC) 1044007 and the Brain Foundation (Australia) to K.B and the APA Scholarship to T.S.M as well as Ultragenyx Pharmaceuticals Inc. for providing triheptanoin, and the National Collaborative Research Infrastructure Strategy (NCRIS) through BioPlatforms Australia for funding of Metabolomics Australia.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. KB has received research support from Ultragenyx Pharmaceuticals Inc.. She has filed for a complete US patent regarding triheptanoin as a treatment for seizures, which has been has been licensed to Ultragenyx Pharmaceuticals Inc. The remaining authors have no conflicts of interest.
References
- 1.Willis S, Stoll J, Sweetman L, et al. Anticonvulsant effects of a triheptanoin diet in two mouse chronic seizure models. Neurobiol Dis 2010; 40: 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gama IR, Trindade-Filho EM, Oliveira SL, et al. Effects of ketogenic diets on the occurrence of pilocarpine-induced status epilepticus of rats. Metab Brain Dis 2015; 30: 93–98. [DOI] [PubMed] [Google Scholar]
- 3.McDonald TS, Tan KN, Hodson MP, et al. Alterations of hippocampal glucose metabolism by even versus uneven medium chain triglycerides. J Cereb Blood Flow Metab 2014; 3: 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim TH, Borges K, Petrou S, et al. Triheptanoin reduces seizure susceptibility in a syndrome-specific mouse model of generalized epilepsy. Epilepsy Res 2013; 103: 101–105. [DOI] [PubMed] [Google Scholar]
- 5.Brunengraber H, Roe CR. Anaplerotic molecules: current and future. J Inherit Metab Dis 2006; 29: 327–331. [DOI] [PubMed] [Google Scholar]
- 6.Mochel F, DeLonlay P, Touati G, et al. Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy. Mol Genet Metab 2005; 84: 305–312. [DOI] [PubMed] [Google Scholar]
- 7.Kinman RP, Kasumov T, Jobbins KA, et al. Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. Am J Physiol Endocrinol Metab 2006; 291: E860–866. [DOI] [PubMed] [Google Scholar]
- 8.Marin-Valencia I, Good LB, Ma Q, et al. Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J Cereb Blood Flow Metab 2013; 33: 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borges K, Sonnewald U. Triheptanoin – a medium chain triglyceride with odd chain fatty acids: a new anaplerotic anticonvulsant treatment?. Epilepsy Res 2012; 100: 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald T, Puchowicz M, Borges K. Impairments in oxidative glucose metabolism in epilepsy and metabolic treatments thereof. Front Cell Neurosci 2018; 12: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guedj E, Bonini F, Gavaret M, et al. 18FDG-PET in different subtypes of temporal lobe epilepsy: SEEG validation and predictive value. Epilepsia 2015; 56: 414–421. [DOI] [PubMed] [Google Scholar]
- 12.Dube C, Boyet S, Marescaux C, et al. Relationship between neuronal loss and interictal glucose metabolism during the chronic phase of the lithium-pilocarpine model of epilepsy in the immature and adult rat. Exp Neurol 2001; 167: 227–241. [DOI] [PubMed] [Google Scholar]
- 13.McDonald TS, Carrasco-Pozo C, Hodson M, et al. Alterations in cytosolic and mitochondrial [U-13C]-glucose metabolism in a chronic epilepsy mouse model. eNeuro 2017; 4: e0341–16.2017. doi: 10.1523/ENEURO.0341-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eid T, Williamson A, Lee T-SW, et al. Glutamate and astrocytes – key players in human mesial temporal lobe epilepsy?. Epilepsia 2008; 49: 42–52. [DOI] [PubMed] [Google Scholar]
- 15.Tan KN, McDonald TS, Borges K.Metabolic dysfunctions in epilepsy and novel metabolic treatment approaches. In: Preedy WaV. (ed). Bioactive nutraceuticals and dietary supplements in neurological and brain disease: Prevention and therapy, New York NY, USA: Oxford University Press, 2015, pp. 461–470. [Google Scholar]
- 16.Tefera TW, Tan KN, McDonald TS, et al. Alternative fuels in epilepsy and amyotrophic lateral sclerosis. Neurochem Res 2017; 42: 1610–1620. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt D, Schachter SC. Drug treatment of epilepsy in adults. BMJ 2014; 348: g254. [DOI] [PubMed] [Google Scholar]
- 18.Calvert S, Barwick K, Par M, et al. A pilot study of add-on oral triheptanoin treatment for children with medically refractory epilepsy. Eur J Paediatr Neurol 2018; 22: 1074–1080. [DOI] [PubMed] [Google Scholar]
- 19.Borges K, Gearing M, McDermott DL, et al. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol 2003; 182: 21–34. [DOI] [PubMed] [Google Scholar]
- 20.Benson MJ, Thomas NK, Talwar S, et al. A novel anticonvulsant mechanism via inhibition of complement receptor C5ar1 in murine epilepsy models. Neurobiol Dis 2015; 76: 87–97. [DOI] [PubMed] [Google Scholar]
- 21.Hadera MG, Smeland OB, McDonald TS, et al. Triheptanoin partially restores levels of tricarboxylic acid cycle intermediates in the mouse pilocarpine model of epilepsy. J Neurochem 2014; 129: 107–119. [DOI] [PubMed] [Google Scholar]
- 22.Smeland OB, Hadera MG, McDonald TS, et al. Brain mitochondrial metabolic dysfunction and glutamate level reduction in the pilocarpine model of temporal lobe epilepsy in mice. J Cereb Blood Flow Metab 2013; 3: 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kharatishvili I, Shan ZY, She DT, et al. MRI changes and complement activation correlate with epileptogenicity in a mouse model of temporal lobe epilepsy. Brain Struct Funct 2014; 219: 683–706. [DOI] [PubMed] [Google Scholar]
- 24.Le Belle J, Harris N, Williams S, et al. A comparison of cell and tissue extraction techniques using high-resolution 1H-NMR spectroscopy. NMR Biomed 2002; 15: 37–44. [DOI] [PubMed] [Google Scholar]
- 25.Tan KN, Carrasco-Pozo C, McDonald TS, et al. Tridecanoin is anticonvulsant, antioxidant, and improves mitochondrial function. J Cereb Blood Flow Metab 2017; 37: 2035–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ke C-J, He Y-H, He H-W, et al. A new spectrophotometric assay for measuring pyruvate dehydrogenase complex activity: a comparative evaluation. Analyt Meth 2014; 6: 6381–6388. [Google Scholar]
- 27.Dienel GA, Cruz NF. Exchange-mediated dilution of brain lactate specific activity: implications for the origin of glutamate dilution and the contributions of glutamine dilution and other pathways. J Neurochem 2009; 109(Suppl 1): 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fornai F, Bassi L, Gesi M, et al. Similar increases in extracellular lactic acid in the limbic system during epileptic and/or olfactory stimulation. Neuroscience 2000; 97: 447–458. [DOI] [PubMed] [Google Scholar]
- 29.Imran I, Hillert MH, Klein J. Early metabolic responses to lithium/pilocarpine-induced status epilepticus in rat brain. J Neurochem 2015; 135: 1007–1018. [DOI] [PubMed] [Google Scholar]
- 30.Arnold S, Schlaug G, Niemann H, et al. Topography of interictal glucose hypometabolism in unilateral mesiotemporal epilepsy. Neurology 1996; 46: 1422–1422. [DOI] [PubMed] [Google Scholar]
- 31.Henry TR, Mazziotta JC, Engel J. Interictal metabolic anatomy of mesial temporal lobe epilepsy. Arch Neurol 1993; 50: 582–589. [DOI] [PubMed] [Google Scholar]
- 32.Bellesi M, de Vivo L, Koebe S, et al. Sleep and wake affect glycogen content and turnover at perisynaptic astrocytic processes. Front Cell Neurosci 2018; 12: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borges K, McDermott D, Irier H, et al. Degeneration and proliferation of astrocytes in the mouse dentate gyrus after pilocarpine-induced status epilepticus. Exp Neurol 2006; 201: 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Kuang Y, Xu K, et al. Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab 2013; 33: 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiffmann R, Wallace ME, Rinaldi D, et al. A double-blind, placebo-controlled trial of triheptanoin in adult polyglucosan body disease and open-label, long-term outcome. J Inherited Metab Dis 2018; 41: 877–883. [DOI] [PubMed] [Google Scholar]
- 36.Bubber P, Haroutunian V, Fisch G, et al. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 2005; 57: 695–703. [DOI] [PubMed] [Google Scholar]
- 37.Tan KN, Simmons D, Carrasco-Pozo C, et al. Triheptanoin protects against status epilepticus-induced hippocampal mitochondrial dysfunctions, oxidative stress and neuronal degeneration. J Neurochem 2018; 144: 431–442. [DOI] [PubMed] [Google Scholar]
- 38.Schwarzkopf TM, Koch K, Klein J. Reduced severity of ischemic stroke and improvement of mitochondrial function after dietary treatment with the anaplerotic substance triheptanoin. Neuroscience 2015; 300: 201–209. [DOI] [PubMed] [Google Scholar]
- 39.Park MJ, Aja S, Li Q, et al. Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS One 2014; 9: e109527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu L, Zhang GF, Kombu RS, et al. Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. II. Effects on lipolysis, glucose production, and liver acyl-CoA profile. Am J Physiol Endocrinol Metab 2010; 298: E362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim TY, Yi JS, Chung SJ, et al. Pyruvate protects against kainate-induced epileptic brain damage in rats. Exp Neurol 2007; 208: 159–167. [DOI] [PubMed] [Google Scholar]
- 42.Carvalho AS, Torres LB, Persike DS, et al. Neuroprotective effect of pyruvate and oxaloacetate during pilocarpine induced status epilepticus in rats. Neurochem Int 2011; 58: 385–390. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto H. Protection against cyanide-induced convulsions with alpha-ketoglutarate. Toxicology 1990; 61: 221–228. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto HA, Mohanan PV. Effect of alpha-ketoglutarate and oxaloacetate on brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Toxicol Lett 2003; 143: 115–122. [DOI] [PubMed] [Google Scholar]
- 45.Simeone KA, Matthews SA, Samson KK, et al. Targeting deficiencies in mitochondrial respiratory complex I and functional uncoupling exerts anti-seizure effects in a genetic model of temporal lobe epilepsy and in a model of acute temporal lobe seizures. Exp Neurol 2014; 251: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]