

Abstract
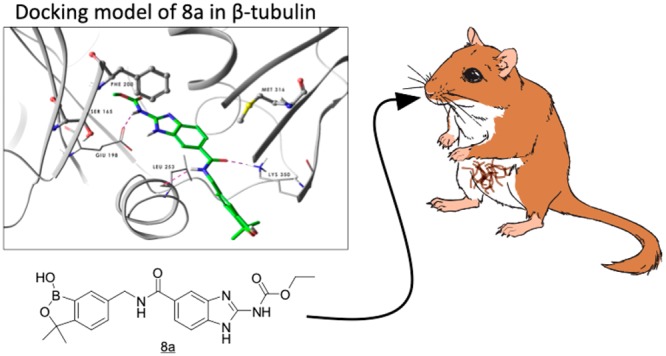
A series of benzimidazole–benzoxaborole hybrid molecules linked via an amide linker are described that exhibit good in vitro activity against Onchocerca volvulus, a filarial nematode responsible for the disease onchocerciasis, also known as river blindness. The lead identified in this series, 8a (AN8799), was found to have acceptable pharmacokinetic properties to enable evaluation in animal models of human filariasis. Compound 8a was effective in killing Brugia malayi, B. pahangi, and Litomosoides sigmodontis worms present in Mongolian gerbils when dosed subcutaneously as a suspension at 100 mg/kg/day for 14 days but not when dosed orally at 100 mg/kg/day for 28 days. The measurement of plasma levels of 8a at the end of the dosing period and at the time of sacrifice revealed an interesting dependence of activity on the extended exposure for both 8a and the positive control, flubendazole.
Keywords: onchocerciasis, lymphatic filariasis, flubendazole, tubulin, organoboron
Diseases caused by infection of an individual with filarial worms are widespread and of particular concern in the endemic countries of the developing world. Two such diseases are onchocerciasis (river blindness), caused by the parasite Onchocerca volvulus, and elephantiasis (lymphatic filariasis, LF), caused by parasites Wuchereria bancroftii, Brugia timori, and B. malayi.1 These diseases are endemic across Asia (LF) and sub-Saharan Africa (LF and onchocherciasis), with the parasites transmitted via black flies (onchocerciasis) or mosquitoes (lymphatic filariasis). Despite significant and long-term efforts to limit the impact of these parasitic infections on the population through mass drug administration (MDA) programs with microfilaricidal drugs (ivermectin for onchocerciasis; albendazole, ivermectin, and/or diethylcarbamazine for LF),2−5 there remains an opportunity to discover, develop, and deliver new drugs that overcome limitations of existing therapies. For example, the current strategy for the treatment of onchocerciasis requires that an infected individual take ivermectin 1–3 times per year for 2–3 years over the lifetime of the adult worms (10–14 years for O. volvulus and 6–8 years for Wuchereria and Brugia spp.), which is logistically challenging in disease endemic areas.2 Long-term treatment is required because the microfilaricidal drugs kill only the microfilariae of O. volvulus or LF; they have little effect on the adult macrofilariae.4,4−7 In addition, coinfection of onchocerciasis or LF patients with the eye worm Loa loa can limit the utility of treatment with ivermectin due to significant side effects resulting from rapidly killing the Loa loa microfilariae.8,9
Flubendazole (1), an inhibitor of tubulin polymerization, has been shown to have the ability to kill adult filarial worms, providing promise that this molecule could have utility in the treatment of onchocerciasis and LF (Figure 1).10−13 Despite this promise, flubendazole has several limitations that complicate its potential as a drug for these human infections. First, flubendazole has limited oral bioavailability, primarily a consequence of its poor aqueous solubility.14,15 Second, though more selective than other members of the benzimidazole class, flubendazole also exhibits affinity for the host’s mammalian tubulin and, consequently, has been demonstrated to be potentially embryotoxic in both a rat whole embryo culture experiment and when dosed to pregnant female rats.16,17 Third, flubendazole has been found to be an aneugen in both in vitro and in vivo micronucleus tests, although it has been argued that the lack of clastogenicity of flubendazole in these tests will limit the risk of carcinogenicity to patients.18 However, the metabolism of flubendazole by the reduction of the ketone leads to short-lived clastogenic metabolites at low levels that may pose a minimal risk.18
Figure 1.
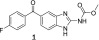
Structure of flubendazole.
Our strategy was to prepare benzoxaborole–benzimidazole analogs of flubendazole that would overcome these limitations. We have observed in other discovery projects that the benzoxaborole core can improve aqueous solubility and oral bioavailability of otherwise poorly soluble molecular frameworks due to the ability of the boron atom to equilibrate between a three coordinate, neutral species (2a) and a four coordinate, negatively charged species (2b) under physiological conditions (Figure 2).19−30
Figure 2.

Equilibrium between three-coordinate, neutral benzoxaborole and four-coordinate, negatively charged benzoxaborole.
A second potential advantage of the incorporation of the benzoxaborole moiety into a molecule was that the benzoxaborole could engage in unique interactions with the subunits of tubulin in a manner that could impart improved selectivity for inhibition of worm tubulin polymerization relative to mammalian host tubulin. Initial efforts to prepare benzoxaborole–benzimidazole hybrids related to flubendazole focused on simple amides. These compounds were easily prepared from an array of previously described amino-(3), aminoalkyl-(4,5), and carboxy-(10) substituted benzoxaboroles and the corresponding carboxy (6)31−33 or amino (11)31 benzimidazole as depicted in Figure 3.
Figure 3.

Strategy for the preparation of the initial benzoxaborole–benzimidazole hybrids.
The first three analogs from the benzimidazole 5-carboxylic acid, namely 7a (from 6-aminobenzoxaborole), 8a (from 6-aminomethyl-3,3-dimethylbenzoxaborole), and 8b (from 3-aminomethylbenzoxaborole), were prepared and tested in an Onchocerca volvulus L3 larval molting assay34,35 and a mammalian G2/M arrest assay36 to evaluate antifilarial activity and selectivity, respectively (Table 1). O. volvulus is the causative agent of river blindness, and the L3 molting assay is the only widely available and reproducible assay of this filariid, the target of novel macrofilaricidal drugs.34,35 Other animal models (e.g., Brugia in gerbils) recapitulate aspects of lymphatic filariasis and are also used as surrogate screens for O. volvulus infections. Of the three compounds tested, 8a emerged as the most attractive compound on the basis of potency and selectivity.
Table 1. Initial Benzoxaborole–Benzimidazole Amide Leadsa.

| ID | link atom | n | R | O. volvulus IC50 (μM)31,32 | G2/M arrest IC50 (μM)33 | MDCK-MDR1 Papp (A-B, ×106 cm/s)34−36 |
|---|---|---|---|---|---|---|
| 1 | NA | NA | NA | 0.004 | 0.67 | 15.6 |
| 7a | 6 | 0 | 4.55 | >100 | NT | |
| 7b | 6 | 0 | 3,3-Me2 | >10 | NT | NT |
| 8a | 6 | 1 | 3,3-Me2 | 0.300 | 13 | 0.8 |
| 8b | 3 | 1 | 0.426 | 10 | NT | |
| 8c | 6 | 1 | 0.421 | >100 | NT | |
| 8d | 5 | 1 | 3,3-Me2 | 0.042 | 2.54 | 0.5 |
| 9a | 6 | 2 | 3,3-Me2 | 0.004 | 0.555 | 0.7 |
| 12a | 6 | 0 | 3,3-Me2 | >10 | >100 | NT |
NT = not tested.
Addition of the 3,3-dimethyl substituents to the 6-aminobenzoxaborole core (7b) resulted in a loss of O. volvulus activity, as did removal of the 3,3-dimethyl substituents from the 6-aminomethyl benzoxaborole (8c). As previous work in the benzoxaborole class had demonstrated that pharmacokinetic properties of 3,3-dimethyl analogs were generally superior to the 3,3-unsubstituted analogs, we focused our attention on this substitution pattern.25 Increasing the length of the linker between the benzoxaborole and benzimidazole cores as in 9a resulted in a significant increase in O. volvulus potency, but also in the G2/M arrest assay we were using as a functional indicator of the interaction with mammalian tubulin. Similarly, changing the point of attachment on the benzoxaborole core from 6- to 5- to afford 8d was also accompanied by increased potency in both the O. volvulus and G2/M arrest assays. Lastly, preparation of a reverse amide from the 5-amino benzimidazole and 6-carboxybenzoxaborole (12a) resulted in loss of activity.
We characterized the in vitro ADME properties of 8a in preparation for evaluation of this compound in our primary in vivo model in gerbils. Metabolic stability of 8a in gerbil microsomes was good (Clint < 4 μL/min/mg), but it was found to be poorly permeable in an MDR1-MDCK monolayer assay (Papp (A-B) = 0.8 × 10–6 cm/s).37−39 The Papp in this assay when the P-glycoprotein (Pgp) efflux inhibitor GF-120918 was added increased to 2.7 × 10–6 cm/s, suggesting that the compound was potentially a substrate for this efflux mechanism.40,41 Taken together, these data prompted us to explore the activity of 8a following subcutaneous administration to gerbils infected by implantation of adult Brugia malayi or Brugia pahangi parasites in the peritoneal cavity.42 We were pleased to find that 8a was able to kill 100% of both male and female worms in the gerbil peritoneum when dosed subcutaneously at 100 mg/kg/day for 14 days as a suspension in a nonsolubilizing HEC/Tween vehicle. Interestingly, when 8a was dosed subcutaneously at 150 mg/kg/day as a solution in a DMSO/water vehicle, in vivo efficacy was substantially reduced. The subsequent assessment of the pharmacokinetics of 8a from these two dosing paradigms provided an interesting observation that we believe to be important in understanding the PK–PD requirements for achieving efficacy in this animal model. In the suspension dose group, plasma levels of 8a were maintained above the in vitro IC50 in the O. volvulus assay (300 nM) for over 42 days after the last dose, whereas in the solution dose group, plasma levels fell below this IC50 within a few days after the last dose. We had made the same observation in a positive control group using flubendazole at a subcutaneous dose of 10 mg/kg/day for 5 days in the nonsolubilizing HEC/Tween vehicle, namely, that plasma levels of flubendazole were maintained above the in vitro IC50 (4 nM) for over 42 days, consistent with data reported in the literature.43 As anticipated from our in vitro ADME data, 8a was not efficacious when dosed by the oral route (at 100 mg/kg/day for 28 days), and plasma levels of the drug were found to be well below the in vitro IC50 at all time points. These observations suggest that the efficacy observed in the in vivo model was dependent upon long-term exposure of worms to the drug, perhaps a consequence of the “depot-like” properties of the subcutaneous suspension.44 We next examined 8a in two additional in vivo models, the first where L3 B. pahangi larvae were injected into the peritoneum of gerbils and allowed to develop into adult worms45 and a second where gerbils were naturally infected by the filarial nematode Litomosoides sigmodontis.46,47 In both of these models, 8a was administered subcutaneously at 100 mg/kg for 14 days or orally at 100 mg/kg for 28 days as a suspension. As with the adult worm infection model, 8a was quite effective via the subcutaneous route in these additional models but essentially inactive when dosed orally as summarized in Table 2. Additionally, when plasma obtained from treated animals at necropsy was analyzed for 8a, we observed an outcome similar to that observed in the adult implantation model, e.g., that measurable levels were present at this time point in the subcutaneous-dosed groups but not in the orally dosed groups, weeks after administration of the drug. In a final L. sigmodontis experiment with 8a, we dosed the compound subcutaneously at 300 mg/kg for 7 days as a suspension. As anticipated, this study demonstrated good activity of 8a, as plasma concentrations of the drug were in excess of the in vitro IC50 for at least 42 days.
Table 2. In Vivo Efficacy and Terminal Plasma Concentrations of 8a and Flubendazolea.
| drug
concentration (μM) |
|||||||
|---|---|---|---|---|---|---|---|
| in vivo model | drug treatment | worm count median ± SEM (range) | adult worm reduction/animals with no worms | P ≤ | 24 h after last dose | interim | necropsy |
| B. malayi; necropsy on day 42 | vehicle (SC study), n = 5 | 12 ± 0.73 (11–15) | N/A | N/A | N/A | ||
| flubendazole, 10 mg/kg × 5 days, QD, SC, n = 10 | 0 ± 0 (0–0) | 100%/100% | 0.0001 | 0.173 | NM | 0.043 | |
| 8a, 150 mg/kg × 14 days, QD, SC (solution), n = 6 | 2 ± 2.14 (0–12) | 83.3%/50% | 0.0622 | 2.47 | 0.07 (day 28) | 0.01 | |
| 8a, 100 mg/kg × 14 days, QD, SC (suspension), n = 16 | 0 ± 0.19 (0–3) | 100%/87.5% | 0.0001 | 4.10 | 5.36 (day 28) | 3.38 | |
| vehicle (PO study), n = 5 | 8 ± 0.81 (5–10) | N/A | N/A | N/A | |||
| 8a, 100 mg/kg × 28 days, QD, PO (suspension), n = 10 | 7 ± 1.38 (4–16) | 12.5%/0% | 0.9999 | 0.128 | NM | <LOQ | |
| B. pahangi; necropsy on day 63; n = 5 per group | vehicle | 89 ± 12.79 (64–146) | N/A | N/A | N/A | ||
| flubendazole, 10 mg/kg × 5 days, QD, SC | 0 ± 0 (0–0) | 100%/100% | 0.0009 | 0.178 | NM | 0.018 | |
| 8a, 100 mg/kg × 14 days, QD, SC (suspension) | 0 ± 0.333 (0–2) | 100%/83% | 0.0024 | 5.87 | NM | 1.15 | |
| 8a, 100 mg/kg × 28 days, QD, PO (suspension) | 67.5 ± 14.86 (2–114) | 24%/0% | 0.9999 | 0.13 | NM | <LOQ | |
| L. sigmodontis; necropsy on day 63; n = 4 vehicle; n = 6 other groups | vehicle | 8.5 ± 3.28 (5–20) | N/A | N/A | |||
| flubendazole, 10 mg/kg × 5 days, QD, SC | 0 ± 0 (0–0) | 100%/100% | 0.0208 | 0.283 | NM | 0.040 | |
| 8a, 100 mg/kg × 14 days, QD, SC (suspension) | 0 ± 0.34 (0–2) | 100%/33.3% | 0.1207 | 9.30 | NM | 0.069 | |
| 8a, 300 mg/kg × 7 days, QD, SC (suspension) | 0 ± 0 (0–0) | 100%/100% | 0.0208 | 7.85 | 10.6 (day 21) | 0.48 | |
| 4.64 (day 42) | |||||||
| 8a, 100 mg/kg × 28 days, QD, PO (suspension) | 20 ± 6.92 (5–50) | –135.3%/0% | 0.999 | 0.210 | 0.006 (day 42) | 0.003 | |
NM = not measured. N/A = not applicable. Statistical significance was tested by Kruskal–Wallis followed by Dunn’s multiple comparisons test.
These observations of the dependence of activity on the extended exposure of B. malayi, B. pahangi, and L. sigmodontis worms to the drug (either flubendazole or 8a) were consistent with observations made in an ex vivo B. malayi/pahangi assay,48 namely, that a short (<7 days) exposure of worms to these drugs was not effective in killing the worms. Taken together, these results suggest that the mechanism of action of these benzimidazole drugs (inhibition of tubulin polymerization) requires a long (>28 day) exposure to the drug to be effective.
While we were encouraged by the proof of concept demonstrated by 8a in these in vivo models, it was clear that this molecule would not meet our target candidate profile that required an orally active drug candidate.
It has been suggested in the literature that the propensity for Pgp efflux is much greater in compounds containing more that 2–3 hydrogen bond donors (HBDs).49,50 Our lead compound (8a) has four potential donors (B–OH, amide NH, benzimidazole NH, and carbamate NH). In order to ameliorate this potential Pgp liability, we prepared and evaluated compounds with fewer HBDs (Table 3). As anticipated on the basis of the flubendazole literature,31 alkylation of the benzimidazole NH (8e, 8f) resulted in loss of activity and also did not improve permeability. Alkylation of the amide nitrogen (8g, 8h) also did not improve permeability. The incorporation of a fluorine substituent on the benzimidazole ring adjacent to the amide (8i, 8j), a strategy that has been shown to “mask” an amide hydrogen bond donor,50,51 did improve the permeability but also affected the potency in both the O. volvulus and G2/M arrest assays. Interestingly, 4-F analog 8i lost activity in the O. volvulus assay, whereas the 7-F analog 8j exhibited greater potency (and hence, poorer selectivity) in both assays.
Table 3. Analogs Designed To Overcome the Permeability Challenge.

| ID | R1 | R2 | R3 | R4 | R5 | O. volvulus IC50 (μM) or % inhibition of molting at 1 μM | G2/M IC50 (μM) | MDCK-MDR1 Papp (A-B, ×106 cm/s) |
|---|---|---|---|---|---|---|---|---|
| 8a | H | H | H | H | H | 0.300 | 13 | 0.8 |
| 8e | CH3 | H | H | H | H | 24%a | >100 | 1.2 |
| 8f | H | H | CH3 | H | H | 29%a | >100 | 1.8 |
| 8g | H | CH3 | H | H | H | NT | 4.1 | 0.6 |
| 8h | H | CH2CH2NMe2 | H | H | H | NT | 35 | 0.14 |
| 8i | H | H | H | F | H | 0%a | 16 | 2.35 |
| 8j | H | H | H | H | F | 0.13 | 1.26 | 2.83 |
% of O. volvulus larvae that completed molting from L3 to L4 compared to control worms.
On the basis of these results, it was clear that more substantial changes needed to be made to the benzoxaborole–benzimidazole hybrids to achieve our objective. The exploration of an additional series of hybrid molecules, most specifically those containing a ketone linker analogous to that found in flubendazole, will be reported in due course.
Acknowledgments
The authors would like to thank the Bill & Melinda Gates Foundation for funding of this program through awards to Anacor (Contract Number 23629), University of Bonn (Grant Number OPP1134310), and the University of California San Francisco (Grant number OPP1017584). We thank Richard Elliott and Ken Duncan of the Bill & Melinda Gates Foundation for their support and guidance, Jason Zhang and his team at Acme Bioscience, Inc. for their contributions to the medicinal chemistry, Mona Luo for the illustration, and Chris Franklin for his help with the graphics.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.9b00396.
Synthesis methods for benzimidazole–benzoxiborole hybrids and methods for testing compounds in larval molt assays and in vivo studies (PDF)
Author Present Address
¶ R.T.J.: Jacobs Scientific Consulting, LLC, 1849 Old College Circle, Wake Forest, NC 27587.
Author Contributions
R.T.J., D.S.C., Y.R.F., and JAS wrote the manuscript and contributed equally to the design and execution of the project. T.A., R.T.J., D.S.C., and J.J.P. designed and coordinated the synthesis of the compounds. R.T.J., K.J., Y.R.F., C.S.L., E.E.E., F.R., J.J.P., R.S., J.M., A.H., J.A.S., and S.L. provided scientific leadership and management of the project. P.W.B. designed, coordinated, and interpreted the in vitro and in vivo pharmacokinetics studies. C.F., C.A.B., K.C.L., B.M.S., N.T., A.M., U.D., S.M., B.C., and S.S. conducted the in vitro and in vivo biological assays, which were designed and coordinated by Y.R.F., C.S.L., F.R., K.J., M.P.H., S.S., A.H., S.L., J.A.S., and J.W.M. The manuscript was edited by R.T.J., D.S.C., Y.R.F., P.W.B., A.H., M.P.H., SL, C.A.B., and J.A.S.
The authors declare no competing financial interest.
Supplementary Material
References
- Simonsen P. E. F., Fischer P. U., Hoerauf A., and Weil G. J. (2014) The Filariases. In Manson’s Tropical Diseases (Farrar J. H., Hotez P., Junghanss T., Kang G., Lalloo D., and White N. J., Eds.) 23rd ed., pp 737–765, Elsevier Saunders. [Google Scholar]
- Ichimori K.; King J. D.; Engels D.; Yajima A.; Mikhailov A.; Lammie P.; Ottesen E. A. (2014) Global programme to eliminate lymphatic filariasis: the processes underlying programme success. PLoS Neglected Trop. Dis. 8 (12), e0003328 10.1371/journal.pntd.0003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper P. J.; Bradley M. H.; Biswas G.; Ottesen E. A. (2009) The Global Programme to Eliminate Lymphatic Filariasis: health impact during its first 8 years (2000–2007). Ann. Trop. Med. Parasitol. 103 (Suppl 1), 17–21. 10.1179/000349809X12502035776513. [DOI] [PubMed] [Google Scholar]
- Thomsen E. K.; Sanuku N.; Baea M.; Satofan S.; Maki E.; Lombore B.; Schmidt M. S.; Siba P. M.; Weil G. J.; Kazura J. W.; Fleckenstein L. L.; King C. L. (2016) Efficacy, safety, and pharmacokinetics of coadministered diethylcarbamazine, albendazole, and ivermectin for treatment of Bancroftian filariasis. Clin. Infect. Dis. 62, 334–341. 10.1093/cid/civ882. [DOI] [PubMed] [Google Scholar]
- Fischer P. U.; King C. l.; Jacobson J. A.; Weil G. J. (2017) Potential value of triple drug therapy with ivermectin, diethylcarbamazine, and albendazole (IDA) to accelerate elimination of lymphatic filariasis and onchocerciasis in Africa. PLoS Neglected Trop. Dis. 11 (1), e0005163 10.1371/journal.pntd.0005163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary T. G. (2005) Ivermectin 20 years on: maturation of a wonder drug. Trends Parasitol. 21 (11), 530–532. 10.1016/j.pt.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Goa K. L.; McTavish D.; Clissold S. P. (1991) Ivermectin. A review of its antifilarial activity, pharmacokinetic properties and clinical efficacy in onchocerciasis. Drugs 42 (4), 640–658. 10.2165/00003495-199142040-00007. [DOI] [PubMed] [Google Scholar]
- Kamgno J.; Pion S. D.; Tejiokem M. C.; Twum-Danso N. A.; Thylefors B.; Boussinesq M. (2007) Randomized, controlled, double-blind trial with ivermectin on Loa loa microfilaraemia: efficacy of a low dose (approximately 25 microg/kg) versus current standard dose (150 microg/kg). Trans. R. Soc. Trop. Med. Hyg. 101 (8), 777–785. 10.1016/j.trstmh.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Gardon J.; Gardon-Wendel N.; Demanga N.; Kamgno J.; Chippaux J. P.; Boussinesq M. (1997) Serious reactions after mass treatment of onchocerciasis with ivermectin in an area endemic for Loa loa infection. Lancet 350 (9070), 18–22. 10.1016/S0140-6736(96)11094-1. [DOI] [PubMed] [Google Scholar]
- Mackenzie C. D.; Geary T. G. (2011) Flubendazole: a candidate macrofilaricide for lymphatic filariasis and onchocerciasis field programs. Expert Rev. Anti-Infect. Ther. 9 (5), 497–501. 10.1586/eri.11.30. [DOI] [PubMed] [Google Scholar]
- Dominguez-Vazquez A.; Taylor H. R.; Greene B. M.; Ruvalcaba-Macias A. M.; Rivas-Alcala A. R.; Murphy R. P.; Beltran-Hernandez F. (1983) Comparison of flubendazole and diethylcarbamazine in treatment of onchocerciasis. Lancet 321 (8317), 139–143. 10.1016/S0140-6736(83)92753-8. [DOI] [PubMed] [Google Scholar]
- Denham D. A.; Brandt E. (1980) Chemoprophylactic activity of flubendazole against adult Brugia pahangi transplanted into the peritoneal cavity of jirds. J. Parasitol. 66 (6), 933–934. 10.2307/3280393. [DOI] [PubMed] [Google Scholar]
- Denham D. A.; Samad R.; Cho S. Y.; Suswillo R. R.; Skippins S. C. (1979) The anthelmintic effects of flubendazole on Brugia pahangi. Trans. R. Soc. Trop. Med. Hyg. 73 (6), 673–676. 10.1016/0035-9203(79)90018-X. [DOI] [PubMed] [Google Scholar]
- Lanusse C. E.; Prichard R. K. (1993) Clinical pharmacokinetics and metabolism of benzimidazole anthelmintics in ruminants. Drug Metab. Rev. 25 (3), 235–279. 10.3109/03602539308993977. [DOI] [PubMed] [Google Scholar]
- Ceballos L.; Moreno L.; Torrado J. J.; Lanusse C.; Alvarez L. (2012) Exploring flubendazole formulations for use in sheep. Pharmacokinetic evaluation of a cyclodextrin-based solution. BMC Vet. Res. 8, 71. 10.1186/1746-6148-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo M.; Zanoncelli S.; Colombo P. A.; Harhay M. O.; Scandale I.; Mackenzie C.; Geary T.; Madrill N.; Mazue G. (2013) Effects of the benzimidazole anthelmintic drug flubendazole on rat embryos in vitro. Reprod. Toxicol. 36, 78–87. 10.1016/j.reprotox.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Longo M.; Zanoncelli S.; Messina M.; Scandale I.; Mackenzie C.; Geary T.; Marsh K.; Lindley D.; Mazue G. (2014) In vivo preliminary investigations of the effects of the benzimidazole anthelmintic drug flubendazole on rat embryos and fetuses. Reprod. Toxicol. 49, 33–42. 10.1016/j.reprotox.2014.06.009. [DOI] [PubMed] [Google Scholar]
- Tweats D. J.; Johnson G. E.; Scandale I.; Whitwell J.; Evans D. B. (2016) Genotoxicity of flubendazole and the impact of a new formulation on in vivo aneugenicity. Mutagenesis 31, 309–321. 10.1093/mutage/gev070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bocxlaer K.; Gaukel E.; Hauser D.; Park S. H.; Schock S.; Yardley V.; Randolph R.; Plattner J. J.; Merchant T.; Croft S. L.; Jacobs R. T.; Wring S. A. (2018) Topical Treatment for Cutaneous Leishmaniasis: Dermato-Pharmacokinetic Lead Optimization of Benzoxaboroles. Antimicrob. Agents Chemother. 62 (5), e02419-17. 10.1128/AAC.02419-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akama T.; Zhang Y. K.; Freund Y. R.; Berry P.; Lee J.; Easom E. E.; Jacobs R. T.; Plattner J. J.; Witty M. J.; Peter R.; Rowan T. G.; Gillingwater K.; Brun R.; Nare B.; Mercer L.; Xu M.; Wang J.; Liang H. (2018) Identification of a 4-fluorobenzyl l-valinate amide benzoxaborole (AN11736) as a potential development candidate for the treatment of Animal African Trypanosomiasis (AAT). Bioorg. Med. Chem. Lett. 28 (1), 6–10. 10.1016/j.bmcl.2017.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. K.; Plattner J. J.; Easom E. E.; Jacobs R. T.; Guo D.; Freund Y. R.; Berry P.; Ciaravino V.; Erve J. C. L.; Rosenthal P. J.; Campo B.; Gamo F. J.; Sanz L. M.; Cao J. (2017) Benzoxaborole Antimalarial Agents. Part 5. Lead Optimization of Novel Amide Pyrazinyloxy Benzoxaboroles and Identification of a Preclinical Candidate. J. Med. Chem. 60 (13), 5889–5908. 10.1021/acs.jmedchem.7b00621. [DOI] [PubMed] [Google Scholar]
- Zhang Y. K.; Plattner J. J.; Easom E. E.; Zhou Y.; Akama T.; Bu W.; White W. H.; Defauw J. M.; Winkle J. R.; Balko T. W.; Guo S.; Xue J.; Cao J.; Zou W. (2015) Discovery of an orally bioavailable isoxazoline benzoxaborole (AN8030) as a long acting animal ectoparasiticide. Bioorg. Med. Chem. Lett. 25 (23), 5589–5593. 10.1016/j.bmcl.2015.10.044. [DOI] [PubMed] [Google Scholar]
- Adamczyk-Wozniak A.; Borys K. M.; Sporzynski A. (2015) Recent Developments in the Chemistry and Biological Applications of Benzoxaboroles. Chem. Rev. 115 (11), 5224–5247. 10.1021/cr500642d. [DOI] [PubMed] [Google Scholar]
- Jacobs R. T.; Plattner J. J.; Keenan M. (2011) Boron-based drugs as antiprotozoals. Curr. Opin. Infect. Dis. 24 (6), 586–592. 10.1097/QCO.0b013e32834c630e. [DOI] [PubMed] [Google Scholar]
- Jacobs R. T.; Nare B.; Wring S. A.; Orr M. D.; Chen D.; Sligar J. M.; Jenks M. X.; Noe R. A.; Bowling T. S.; Mercer L. T.; Rewerts C.; Gaukel E.; Owens J.; Parham R.; Randolph R.; Beaudet B.; Bacchi C. J.; Yarlett N.; Plattner J. J.; Freund Y.; Ding C.; Akama T.; Zhang Y. K.; Brun R.; Kaiser M.; Scandale I.; Don R. (2011) SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Neglected Trop. Dis. 5 (6), e0001151 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nare B.; Wring S.; Bacchi C.; Beaudet B.; Bowling T.; Brun R.; Chen D.; Ding C.; Freund Y.; Gaukel E.; Hussain A.; Jarnagin K.; Jenks M.; Kaiser M.; Mercer L.; Mejia E.; Noe A.; Orr M.; Parham R.; Plattner J.; Randolph R.; Rattendi D.; Rewerts C.; Sligar J.; Yarlett N.; Don R.; Jacobs R. (2010) Discovery of novel orally bioavailable oxaborole 6-carboxamides that demonstrate cure in a murine model of late-stage central nervous system african trypanosomiasis. Antimicrob. Agents Chemother. 54 (10), 4379–4388. 10.1128/AAC.00498-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczyk-Wozniak A.; Cyranski M. K.; Jakubczyk M.; Klimentowska P.; Koll A.; Kolodziejczak J.; Pojmaj G.; Zubrowska A.; Zukowska G. Z.; Sporzynski A. (2010) Influence of the substituents on the structure and properties of benzoxaboroles. J. Phys. Chem. A 114 (6), 2324–2330. 10.1021/jp9086283. [DOI] [PubMed] [Google Scholar]
- Hui X.; Baker S. J.; Wester R. C.; Barbadillo S.; Cashmore A. K.; Sanders V.; Hold K. M.; Akama T.; Zhang Y. K.; Plattner J. J.; Maibach H. I. (2007) In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J. Pharm. Sci. 96 (10), 2622–2631. 10.1002/jps.20901. [DOI] [PubMed] [Google Scholar]
- Baker S. J.; Zhang Y. K.; Akama T.; Lau A.; Zhou H.; Hernandez V.; Mao W.; Alley M. R.; Sanders V.; Plattner J. J. (2006) Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. J. Med. Chem. 49 (15), 4447–4450. 10.1021/jm0603724. [DOI] [PubMed] [Google Scholar]
- Baker S. J.; Akama T.; Zhang Y. K.; Sauro V.; Pandit C.; Singh R.; Kully M.; Khan J.; Plattner J. J.; Benkovic S. J.; Lee V.; Maples K. R. (2006) Identification of a novel boron-containing antibacterial agent (AN0128) with anti-inflammatory activity, for the potential treatment of cutaneous diseases. Bioorg. Med. Chem. Lett. 16 (23), 5963–5967. 10.1016/j.bmcl.2006.08.130. [DOI] [PubMed] [Google Scholar]
- Ram S.; Wise D. S.; Wotring L. L.; McCall J. W.; Townsend L. B. (1992) Synthesis and biological activity of certain alkyl 5-(alkoxycarbonyl)-1H-benzimidazole-2-carbamates and related derivatives: a new class of potential antineoplastic and antifilarial agents. J. Med. Chem. 35 (3), 539–547. 10.1021/jm00081a016. [DOI] [PubMed] [Google Scholar]
- Synthesis of compound 8a. Step 1. A mixture of 3-amino-4-(methylamino)benzoic acid (750 mg, 4.51 mmol) and bis(methoxycarbonyl)-2-methylisothiourea (1.86 g, 9.00 mmol) in AcOH (10 mL) was stirred at 80 °C for 20 min and filtered, and the filter cake washed with methanol (20 mL) and ethyl acetate (20 mL), dried in vacuo to give 2-((methoxycarbonyl)amino)-1-methyl-1H-benzo[d]imidazole-5-carboxylic acid 6 (850 mg, 76%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ 7.98 (s, 1H), 7.80 (dd, J = 8.6, 1.4, 1H), 7.43 (d, J = 8.8 Hz, 1H), 3.62 (s, 3H), 3.50 (s, 3H). Step 2. A mixture of 6 (80 mg, 0.32 mmol), 6-(aminomethyl)-3,3-dimethylbenzo[c]-[1,2]oxaborol-1(3H)-ol (61 mg, 0.32 mmol), HATU (182 mg, 0.48 mmol), and DIPEA (124 mg, 0.96 mmol) in DMF (10 mL) was stirred at room temperature for 16 h under N2. The mixture was concentrated, and the residue was purified by Prep-HPLC to give 8a (40 mg, 30%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ 9.06 (t, J = 5.6 Hz, 1H), 8.00 (s, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.60 (s, 1H), 7.56 (d, J = 8.8 Hz, 1H), 7.41 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 4.49 (d, J = 4.8 Hz, 2H), 3.70 (s, 3H), 3.60 (s, 3H), 1.40 (s, 6H).
- Ding C. Z.; Zhang Y. K.; Li X.; Liu Y.; Zhang S.; Zhou Y.; Plattner J. J.; Baker S. J.; Liu L.; Duan M.; Jarvest R. L.; Ji J.; Kazmierski W. M.; Tallant M. D.; Wright L. L.; Smith G. K.; Crosby R. M.; Wang A. A.; Ni Z. J.; Zou W.; Wright J. (2010) Synthesis and biological evaluations of P4-benzoxaborole-substituted macrocyclic inhibitors of HCV NS3 protease. Bioorg. Med. Chem. Lett. 20 (24), 7317–7322. 10.1016/j.bmcl.2010.10.071. [DOI] [PubMed] [Google Scholar]
- Gloeckner C.; Garner A. L.; Mersha F.; Oksov Y.; Tricoche N.; Eubanks L. M.; Lustigman S.; Kaufmann G. F.; Janda K. D. (2010) Repositioning of an existing drug for the neglected tropical disease Onchocerciasis. Proc. Natl. Acad. Sci. U. S. A. 107 (8), 3424–3429. 10.1073/pnas.0915125107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooyit M.; Harris T. L.; Tricoche N.; Javor S.; Lustigman S.; Janda K. D. (2015) Onchocerca volvulus Molting Inhibitors Identified through Scaffold Hopping. ACS Infect. Dis. 1 (5), 198–202. 10.1021/acsinfecdis.5b00017. [DOI] [PubMed] [Google Scholar]
- Pozarowski P.; Darzynkiewicz Z. (2004) Analysis of cell cycle by flow cytometry. Methods Mol. Biol. 281, 301–11. 10.1385/1-59259-811-0:301. [DOI] [PubMed] [Google Scholar]
- Evers R.; Cnubben N. H.; Wijnholds J.; van Deemter L.; van Bladeren P. J.; Borst P. (1997) Transport of glutathione prostaglandin A conjugates by the multidrug resistance protein 1. FEBS Lett. 419 (1), 112–116. 10.1016/S0014-5793(97)01442-7. [DOI] [PubMed] [Google Scholar]
- Irvine J. D.; Takahashi L.; Lockhart K.; Cheong J.; Tolan J. W.; Selick H. E.; Grove J. R. (1999) MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 88 (1), 28–33. 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- Tang F.; Horie K.; Borchardt R. T. (2002) Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa?. Pharm. Res. 19 (6), 765–772. 10.1023/A:1016140429238. [DOI] [PubMed] [Google Scholar]
- Witherspoon S. M.; Emerson D. L.; Kerr B. M.; Lloyd T. L.; Dalton W. S.; Wissel P. S. (1996) Flow cytometric assay of modulation of P-glycoprotein function in whole blood by the multidrug resistance inhibitor GG918. Clin. Cancer Res. 2 (1), 7–12. [PubMed] [Google Scholar]
- Hyafil F.; Vergely C.; Du Vignaud P.; Grand-Perret T. (1993) In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 53 (19), 4595–4602. [PubMed] [Google Scholar]
- Kinnamon K. E.; Klayman D. L.; Poon B. T.; McCall J. W.; Dzimianski M. T.; Rowan S. J. (1994) Filariasis testing in a jird model: new drug leads from some old standbys. Am. J. Trop. Med. Hyg. 51 (6), 791–6. 10.4269/ajtmh.1994.51.791. [DOI] [PubMed] [Google Scholar]
- Michiels M.; Hendriks R.; Heykants J.; van den Bossche H. (1982) The pharmacokinetics of mebendazole and flubendazole in animals and man. Arch. Int. Pharmacodyn. Ther. 256, 180–191. [PubMed] [Google Scholar]
- Geary T. G.; Mackenzie C. D.; Silber S. A. (2019) Flubendazole as a macrofilaricide: History and background. PLoS Neglected Trop. Dis. 13 (1), e0006436. 10.1371/journal.pntd.0006436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulman C. A.; Bidlow C. M.; Lustigman S.; Cho-Ngwa F.; Williams D.; Rascon A. A. Jr.; Tricoche N.; Samje M.; Bell A.; Suzuki B.; Lim K. C.; Supakorndej N.; Supakorndej P.; Wolfe A. R.; Knudsen G. M.; Chen S.; Wilson C.; Ang K. H.; Arkin M.; Gut J.; Franklin C.; Marcellino C.; McKerrow J. H.; Debnath A.; Sakanari J. A. (2015) Repurposing auranofin as a lead candidate for treatment of lymphatic filariasis and onchocerciasis. PLoS Neglected Trop. Dis. 9 (2), e0003534 10.1371/journal.pntd.0003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner M. P.; Ehrens A.; Koschel M.; Dubben B.; Lenz F.; Frohberger S.J.; Specht S.; Quirynen L.; Lachau-Durand S.; Tekle F.; Baeten B.; Engelen M.; Mackenzie C.D.; Hoerauf A. (2019) Macrofilaricidal efficacy of single and repeated oral and subcutaneous doses of flubendazole in Litomosoides sigmodontis infected jirds. PLoS Neglected Trop. Dis. 13, e0006320. 10.1371/journal.pntd.0006320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris C. P.; Evans H.; Larsen S. E.; Mitre E. (2013) A comprehensive, model-based review of vaccine and repeat infection trials for filariasis. Clin Microbiol Rev. 26 (3), 381–421. 10.1128/CMR.00002-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellino C.; Gut J.; Lim K. C.; Singh R.; McKerrow J.; Sakanari J. (2012) WormAssay: a novel computer application for whole-plate motion-based screening of macroscopic parasites. PLoS Neglected Trop. Dis. 6 (1), e0001494 10.1371/journal.pntd.0001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P. V.; Raub T. J.; Blanco M. J. (2012) How hydrogen bonds impact P-glycoprotein transport and permeability. Bioorg. Med. Chem. Lett. 22 (21), 6540–6548. 10.1016/j.bmcl.2012.08.059. [DOI] [PubMed] [Google Scholar]
- Raub T. J. (2006) P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol. Pharmaceutics 3 (1), 3–25. 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. (2018) Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 61 (14), 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.