

Abstract
G-protein-coupled receptors (GPCRs) are the largest family of human membrane proteins and serve as primary targets of ~1/3 of currently marketed drugs. In particular, adenosine A1 receptor (A1AR) is an important therapeutic target for treating cardiac ischemia-reperfusion injuries, neuropathic pain and renal diseases. As a prototypical GPCR, the A1AR is located within a phospholipid membrane bilayer and transmits cellular signals by changing between different conformational states. It is important to elucidate the lipid-protein interactions in order to understand the functional mechanism of GPCRs. Here, all-atom simulations using a robust Gaussian accelerated molecular dynamics (GaMD) method were performed on both the inactive (antagonist bound) and active (agonist and G protein bound) A1AR, which was embedded in a 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) lipid bilayer. In the GaMD simulations, the membrane lipids played a key role in stabilizing different conformational states of the A1AR. Our simulations further identified important regions of the receptor that interacted distinctly with the lipids in highly correlated manner. Activation of the A1AR led to differential dynamics in the upper and lower leaflets of the lipid bilayer. In summary, GaMD enhanced simulations have revealed strongly coupled dynamics of the GPCR and lipids that depend on the receptor activation state.
Keywords: G-protein-coupled receptors, Adenosine A1 receptor, Enhanced sampling, Gaussian accelerated molecular dynamics, Protein-lipid interactions
Graphic Abstract:
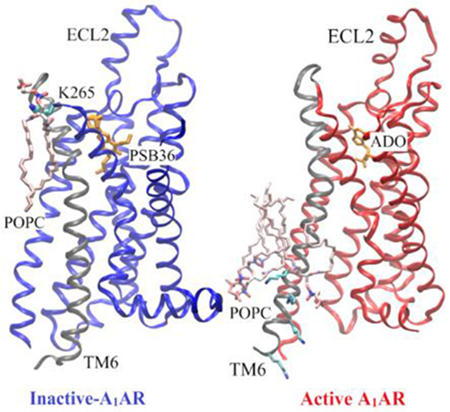
Using adenosine A1 receptor (A1AR) as a model G-protein-coupled receptor (GPCR), Gaussian accelerated molecular dynamics (GaMD) was applied to explore GPCR-membrane interactions. Membrane lipids played a key role in stabilizing different conformational states of the A1AR. Activation of the A1AR led to differential dynamics in the upper and lower leaflets of the lipid bilayer. The GaMD simulations revealed strongly coupled dynamics of the GPCR and lipids that depend on the receptor activation state.
Introduction
G-protein-coupled receptors (GPCRs) are primary cell surface receptors that account for vital physiological and pathological functions in the human body. About 1/3 of currently marketed drugs approved by the Food and Drug Administration (FDA) target GPCRs. Four subtypes of adenosine receptors (ARs), the A1AR, A2AAR, A2BAR and A3AR, mediate a broad range of physiological functions. Particularly, the Adenosine A1 Receptor (A1AR) has emerged as an important therapeutic target for treating cardiac ischemia-reperfusion injuries, neuropathic pain and renal diseases.(1) Being a GPCR, the A1AR is embedded in cell membrane, maintaining close contact with lipid molecules. Lipids have been suggested to affect the receptor conformation and dynamics, which play an important role in transmitting cellular signals from extracellular environment to the cytoplasm. Similarly, lipid metabolites are also known to bind proteins and act as messengers(2). These include lysolipids, sphingo-1-phosphate (S1P), diacylglycerol and fatty acyl derivates. In addition, lipids help in the partitioning of membrane and receptors. Membrane proteins are affected by lipid compositions and function differently in healthy and diseased individuals.(3) Therefore, it is important to study GPCR-membrane interactions in order to elucidate functional mechanism of the membrane proteins.
Experimental techniques including fluorescence resonance energy transfer (FRET), fluorescence correlation spectroscopy (FCS), fluorescence recovery after photobleaching (FRAP) and fluorescence-based monitoring of solvent relaxations rates have been utilized to study protein-membrane biology(4–7). Experiments showed that cholesterol could affect the stability, oligomerization, and ligand binding properties of membrane proteins(8–21). X-ray crystal structures identified allosteric sites for cholesterol binding to GPCRs(13, 15, 22). Phospholipids were found to modulate dynamic processes of GPCRs such as G protein association and ligand binding(23, 24). Recently, Dawaliby et. al. showed experimentally that lipids with different head groups favor different activation states of the β2-adrenergic receptors (β2AR)(25). Lipids with phosphatidylglycerol (PG) headgroups preferred agonist binding and receptor activation, whereas lipids with phosphatidylethanolamine (PE) headgroups preferred antagonist binding and inactive state of the β2AR. Despite these advances, there remains a knowledge gap in the understanding of protein-membrane interactions. From atomic motions of lipid molecules to curvature change across the cell membrane, protein-membrane interactions span a wide range of time scales(26). It is often difficult to directly examine protein-membrane interactions in experiments due to limited time resolution.
Molecular dynamics (MD) simulation has emerged as a powerful computational technique to bridge the gap of knowledge for studying membrane-protein interactions. Both atomistic and coarse-grained MD simulations have been applied to study the effects of lipids in protein dynamics and cellular signaling.(3) Bruzzese et al. confirmed the above mentioned experimental results obtained by Dawaliby et. al. that different charges of PG and PE lipid headgroups affected the GPCR activation and deactivation in MD simulations(27). The net negative charge in PG molecules favored interaction with positively-charged residues in the intracellular loop 3 (ICL3) and intracellular end of transmembrane helix 6 (TM6). This stabilized the outward movement of TM6 and hence the active state of the β2AR. Neale et. al.(28) showed that the PG lipid blocked formation of the R1313.50-E2686.30 ionic lock by interacting with R1313.50 in the β2AR. Residue superscripts denote Ballesteros and Weinstein (BW) numbering of GPCRs(29). The R3.50 and E6.30 are highly conserved residues in GPCRs and often form an ionic lock in inactive receptors. In contrast, net-neutral zwitterionic lipids such as PE with strongly favored the inactive structure of β2AR and destabilized the active structure(27). Salas-Estrada et. al(30) showed that activation in rhodopsin induced changes in the membrane structure, including increase in the local order and effective length of lipid acyl chains in the vicinity of the protein. Dror et. al. showed that gradual inactivation of the β2AR occurred in the neutral lipid 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC)(31, 32). This was consistent with another study favored partial deactivation of the β2AR was found in the 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipids(27). In coarse-grained MD simulations, Song et. al. showed that PIP2 (Phosphatidylinositol 4,5-bisphosphate) stabilized the outward movement of TM6 by binding in the crevice between TM6 and TM7 of adenosine A2A receptor (A2AAR)(33).
In addition, enhanced sampling techniques have been applied to investigate protein membrane interaction. Using steered MD(34) and umbrella sampling(35) techniques, Song et. al. suggested that the PIP2 facilitated recruitment of the G protein by forming bridging interactions with basic residues of the Gα subunit and hence stabilizing the active A2AAR(33). However, these enhanced sampling techniques require predefined collective variables, which are often difficult to identify in the context of protein-membrane interactions. In this context, Gaussian accelerated MD (GaMD) is a robust technique that provides unconstrained enhanced sampling without the need to set predefined collective variables(36, 37). GaMD simulations have been successfully applied to investigate GPCR activation(36, 38, 39), protein folding(36, 39), ligand binding and unbinding(36, 38, 39), protein-protein interactions(40–42) and protein-nucleic acid interactions(43, 44).
Here, we have applied GaMD to investigate lipid interactions with the A1AR in two different conformational states, the cryo-EM structure of the active adenosine ADO-bound A1AR coupled with the Gi protein (referred to as ADO-A1AR-Gi)(45) and the X-ray structure(46, 47) of the inactive antagonist PSB36-bound A1AR (referred to as PSB36-A1AR). Our simulations showed that the protein-membrane interactions depended on different conformational states of the A1AR. The membrane lipids played an important role in stabilizing different conformations of the A1AR. GaMD simulations further identified important regions of the receptor that interacted distinctly with the lipids. Activation of the A1AR led to differential dynamics in the upper and lower leaflets of the lipid bilayer.
Materials and Methods
Gaussian Accelerated Molecular Dynamics (GaMD)
GaMD is an enhanced sampling technique, in which a harmonic boost potential is added to reduce the system energy barriers(36). GaMD is able to accelerate biomolecular simulations by orders of magnitude(39, 48). GaMD does not need predefined collective variables. Moreover, because GaMD boost potential follows a gaussian distribution, biomolecular free energy profiles can be properly recovered through cumulant expansion to the second order(36). GaMD has successfully overcome the energetic reweighting problem in free energy calculations that was encountered in the previous aMD (accelerated molecular dynamics) method(49, 50) for free energy calculations. GaMD has been implemented in widely used software packages including AMBER (36, 51) and NAMD(52). A brief summary of GaMD is provided here.
Consider a system with N atoms at positions . When the system potential is lower than a reference energy E, the modified potential of the system is calculated as:
| (1) |
where k is the harmonic force constant. The two adjustable parameters E and k are automatically determined based on three enhanced sampling principles(36). The reference energy needs to be set in the following range:
| (2) |
where Vmax and Vmin are the system minimum and maximum potential energies. To ensure that Eqn. (2) is valid, k has to satisfy: Let us define , then 0 < k0 ≤ 1. The standard deviation of ΔV needs to be small enough (i.e., narrow distribution) to ensure proper energetic reweighting(53): σΔV = k(E − Vavg)σV ≤ σ0 where Vavg and σV are the average and standard deviation of the system potential energies, σΔV is the standard deviation of ΔV with σ0 as a user-specified upper limit (e.g., 10kBT) for proper reweighting. When E is set to the lower bound E=Vmax, k0 can be calculated as:
| (3) |
Alternatively, when the threshold energy E is set to its upper bound , k0 is set to:
| (4) |
if is found to be between 0 and 1. Otherwise, k0 is calculated using Eqn. (3).
Similar to aMD, GaMD provides schemes to add only the total potential boost ΔVP, only dihedral potential boost ΔVD, or the dual potential boost (both ΔVP and ΔVD). The dual-boost simulation generally provides higher acceleration than the other two types of simulations(54). The simulation parameters comprise of the threshold energy E for applying boost potential and the effective harmonic force constants, k0P and k0D for the total and dihedral potential boost, respectively.
Energetic Reweighting of GaMD Simulations
To calculate potential of mean force (PMF)(55) from GaMD simulations, the probability distribution along a reaction coordinate is written as p*(A). Given the boost potential of each frame, p*(A) can be reweighted to recover the canonical ensemble distribution, p(A), as:
| (5) |
where M is the number of bins, β = kBT and is the ensemble-averaged Boltzmann factor of for simulation frames found in the jth bin. The ensemble-averaged reweighting factor can be approximated using cumulant expansion:
| (6) |
where the first two cumulants are given by
| (7) |
The boost potential obtained from GaMD simulations usually follows near-Gaussian distribution. Cumulant expansion to the second order thus provides a good approximation for computing the reweighting factor(36, 53). The reweighted free energy F(A) = −kBT ln p(A) is calculated as:
| (8) |
where F*(A) = −kBT ln p*(A) is the modified free energy obtained from GaMD simulation and Fc is a constant.
Lipid -SCD Order Parameter
The -SCD order parameter measures orientational anisotropy of the C-H bond in sn-2 acyl chains of lipids that are usually obtained from NMR experiments. It is a function of the angle between the C-H bond and lipid bilayer normal. It is defined by following equation:
| (9) |
where θ is the angle between the bilayer normal and C-H bond and 〈…〉 denotes an ensemble average. Here, the -SCD order parameter is averaged over all the lipids in the system and all the frames in the simulation trajectory. The -SCD order parameter calculated from GaMD simulations is not reweighted due to complexity of the function. However, since GaMD maintains the overall shape of the original potential energy surface(36), the resulting order parameter is found to be close to the experimental values(56) (see Results). The -SCD value usually ranges from −0.25 to 0.5, with 0.5 for the C-H bond being fully ordered along the bilayer normal and −0.25 being parallel to the bilayer plane. The -SCD approximates the mobility of each C-H bond and hence estimates the membrane fluidity.
System Setup
The cryo-EM structure of the ADO-A1AR-Gi complex (PDB: 6D9H(45)) and X-ray structure of PSB36-A1AR complex (PDB: 5N2S(46)) were used to prepare the simulation systems. As helix 8 region was missing in the crystal structure of PSB36-A1AR, atomic coordinates were added using another X-ray structure of the inactive A1AR (PDB: 5UEN(47)) after aligning the receptor TM domain. All chain termini were capped with neutral groups, i.e. the acetyl group (ACE) for the N-terminus and methyl amide group (CT3) for C terminus. Protein residues were set to the standard CHARMM protonation states at neutral pH with the psfgen plugin in VMD(57). Then the receptor was inserted into a POPC bilayer with all overlapping lipid molecules removed using the Membrane plugin in VMD(57). The system charges were then neutralized at 0.15 M NaCl using the Solvate plugin in VMD(57). Periodic boundary conditions were applied on the simulation systems. The simulation systems of the active and inactive A1AR systems are summarized in Table 1.
Table 1:
Summary of GaMD simulations performed on the adenosine A1 receptor (A1AR).
| System | aNatoms | Dimension (Å3) | bMethod | Simulation | cΔVavg (kcal/mol) | dσΔv (kcal/mol) |
|---|---|---|---|---|---|---|
| PSB36-A1AR | 77,809 | 93x100x101 | GaMD_Dih | 300 ns x 2 | 4.47 | 1.81 |
| GaMD_Dual | 300 ns x 2 | 8.45 | 3.33 | |||
| ADO-A1AR-Gi | 180,394 | 93x111x167 | GaMD_Dih | 300 ns x 2 | 5.04 | 2.22 |
| GaMD_Dual | 150 ns x 2 | 9.94 | 2.57 |
Natoms is number of atoms in the simulation systems.
GaMD_Dih and GaMD_Dual represent the dihedral and dual boost GaMD simulations respectively.
ΔVavg and dσΔV are the average and standard deviation of the GaMD boost potential.
Simulation Protocol
The CHARMM36 parameter set(58) was used for the protein and POPC lipids. For agonist ADO and antagonist PSB36, the force field parameters were obtained from the CHARMM ParamChem web server(59, 60). Initial energy minimization and thermalization of the A1AR system follow the same protocol as used in the previous GPCR simulations(61). The simulation proceeded with equilibration of lipid tails. With all the other atom fixed, the lipid tails were energy minimized for 1000 steps using the conjugate gradient algorithm and melted with constant number, volume, and temperature (NVT) run for 0.5ns at 310 K. Each system was further equilibrated using constant number, pressure, and temperature (NPT) run at 1 atm and 310 K for 10 ns with 5 kcal (mol Å2)−1 harmonic position restraints applied to the protein. Further equilibration of the systems was performed using an NPT run at 1 atm and 310 K for 0.5ns with all atoms unrestrained. Conventional MD simulation was performed on each system for 10 ns at 1atm pressure and 310 K with a constant ratio constraint applied on the lipid bilayer in the X-Y plane. The GaMD simulations were carried out using NAMD2.13(52, 62). Both dihedral and dual-boost GaMD simulations were then performed to study the protein-membrane interactions in the inactive and active A1AR systems (Table 1). In the GaMD simulations, the threshold energy E for adding boost potential is set to the lower bound, i.e. E = Vmax(36, 52). The simulations included 50ns equilibration after adding the boost potential and then multiple independent production runs lasting 150 – 300 ns with randomized initial atomic velocities. GaMD production simulation frames were saved every 0.2ps for analysis.
Simulation analysis
The VMD(57) and CPPTRAJ(63) tools were used for trajectory analysis. In particular, distance was calculated between the Cα atoms residues Arg3.50 and Glu6.30. Root-mean-square fluctuations (RMSFs) were calculated for the protein residues and ligands, averaged over two independent GaMD simulations and color coded for schematic representation of each complex system. MEMBPLUGIN, a plugin for the VMD package was used to calculate the -SCD order parameter for POPC lipid tails(64). The -SCD order parameters were averaged over all lipids and frames of the two independent GaMD simulations for each system. The CPPTRAJ tool was used to calculate the correlation matrices. The Cα atoms of the receptor and phosphorous atoms in the POPC lipid head groups were used for the calculations. In addition to the phosphorous atom, the C8 and C18 atoms representing different regions of the lipids were also used to calculate dynamic correlations with the receptor. The PyReweighting toolkit(53) was applied to reweight GaMD simulations for free energy calculations by combining independent trajectories for each system. A bin size of 1 Å was used for the Arg3.50-Glu6.30 distance and 1 for the number of lipids. The cutoff was set to 500 for calculating the 2D PMF profiles.
Results
Structural flexibility of the A1AR depended on the receptor conformational state
All-atom GaMD simulations were performed on two different conformational states of the A1AR, active (ADO-A1AR-Gi) and inactive (PSB36-A1AR) states (Table 1). For the inactive A1AR system, the boost potential was 4.47±1.81 kcal/mol and 8.45±3.33 kcal/mol in dihedral and dual-boost GaMD simulations, respectively. For the active A1AR system, the boost potential was 5.04±2.22 kcal/mol and 9.94±2.57 kcal/mol in dihedral and dual-boost GaMD simulations, respectively (Table 1). Thus, dual-boost GaMD provided higher acceleration in the simulations with greater boost potential. In the dihedral GaMD simulations of the inactive A1AR, the TM helices of the receptor were rather rigid. Only the intracellular end of TM6, the terminus of helix 8 (H8), extracellular end of TM1 and extracellular loop 2 (ECL2) regions were flexible (Figure 1A). Similar results were obtained for the active A1AR in the dihedral GaMD simulations (Figure 1B). However, the intracellular ends of TM6 and TM5 of the A1AR exhibited more fluctuations in the active state compared to the inactive state. The ECL2 region was relatively more flexible in the active A1AR than in the inactive A1AR. In both systems, the ligands remained stably bound at the orthosteric site throughout the simulations. In comparison, the G protein coupled to the active A1AR exhibited higher fluctuations. In particular, C terminus of the α5 helix, α4-β5 loop and α4-β6 loop of the Gα subunit and terminal ends of the Gβγ subunits exhibited high fluctuations up to 3 Å. Similar results were also found in the dual-boost GaMD simulations of the inactive and active A1AR systems (Figure S1).
Figure 1:
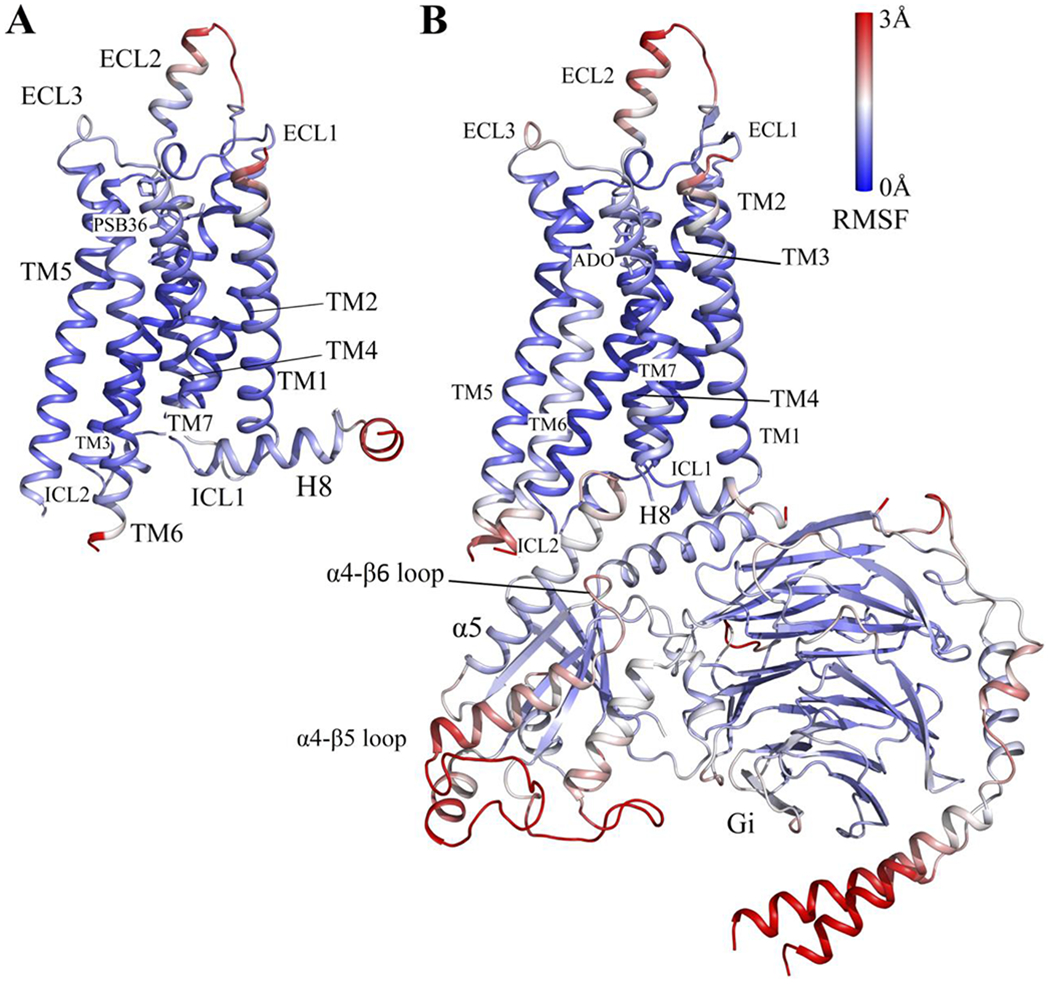
Comparison of structural flexibility of the inactive and active A1AR systems obtained from dihedral GaMD simulations: (A) Root-mean-square fluctuations (RMSFs) of the inactive PSB36-A1AR complex. (B) RMSFs of the active ADO-A1AR-Gi protein complex. A color scale of 0 Å (blue) to 3 Å (red) was used.
Lipids in the lower leaflet of the active A1AR system showed higher fluidity than in the inactive-A1AR system
The lipid -SCD order parameters were calculated for the upper (extracellular) and lower (cytoplasmic) leaflets from GaMD simulations of the inactive and active A1AR systems (Figure 2). In both inactive and active A1AR systems, lower leaflet was more fluid than the upper leaflet with smaller -SCD order parameters. The lower leaflet exhibited significant differences between the inactive and active A1AR systems. In particular, the -SCD order parameter of the fifth carbon atom in POPC was ~0.20 in the lower leaflet in the inactive A1AR system (Figure 2A, 2C), but decreased to ~0.17 in the active A1AR system (Figure 2B, 2D). This indicated higher inclination of C-H bonds being ordered along the bilayer normal in the lower leaflet of the active A1AR system. This appeared to correlate with the outward movement of TM6 as the A1AR changed from the inactive to active state. In the inactive A1AR, the R1313.50-E2686.30 distance at the free energy minimum was ~7 Å (Figure 3A, 3C). In comparison, this distance at the free energy minimum increased to ~17 Å in the active A1AR (Figure 3B, 3D). The lateral movement of the TM6 could push the surrounding lipids. Higher flexibility of the receptor TM6 intracellular end was accompanied by increased fluidity of lipids in the lower leaflet of membrane. Similar results were found from the dihedral and dual-boost GaMD simulations (Figure 3). Therefore, the structural conformation and flexibility of the GPCR are strongly coupled with the surrounding membrane lipids.
Figure 2:

The -SCD order parameters calculated for sn-2 acyl chains of POPC lipids in different simulation systems: (A) Inactive A1AR using dihedral-boost GaMD, (B) Active A1AR using dihedral-boost GaMD, (C) Inactive A1AR using dual-boost GaMD and (D) Active A1AR using dual-boost GaMD. Red diamond lines represent the average -SCD order parameters for the cytoplasmic lower leaflet and blue diamond lines for the extracellular upper leaflet.
Figure 3:
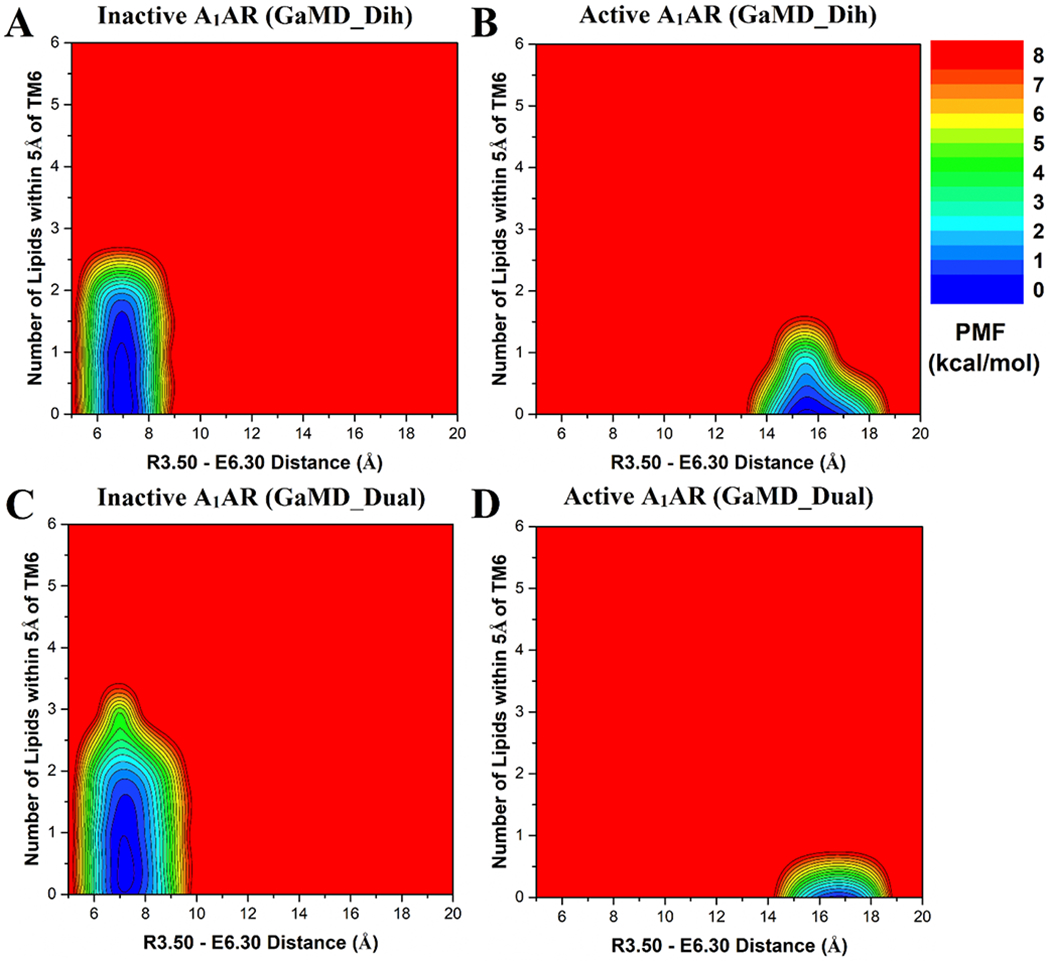
Free energy profiles of the extracellular upper leaflet of membrane in different simulation systems regarding the number of lipids within 5 Å of the receptor TM6 and the receptor R3.50 – E6.30 distance: (A) Inactive A1AR using dihedral-boost GaMD, (B) Active A1AR using dihedral-boost GaMD, (C) Inactive A1AR using dual-boost GaMD and (D) Active A1AR using dual-boost GaMD. The R3.50 – E6.30 distance is ~7 Å in the inactive A1AR and increases to ~17 Å in the active A1AR due to outward movement of TM6.
The inactive A1AR attracted more lipids in the upper leaflet to the TM6 than the active A1AR
Considering significant conformational changes (especially in the TM6) during receptor activation, we hypothesized that the number of lipids interacting with the active and inactive receptor are different. In order to identify the low-energy states of the membrane-receptor interactions, potential of mean force (PMF) profiles were calculated by reweighting the GaMD simulations (Figure 3). The R1313.50-E2686.30 distance was chosen as one reaction coordinate to characterize activation of the GPCR. The number of POPC phosphate head groups within 5 Å of TM6 was calculated as the other reaction coordinate (Figures S2 and S3). In the upper leaflet, approximately one lipid molecule was found interacting with TM6 in the inactive A1AR (Figure 3A, 3C). But no lipid in the upper leaflet was found within 5 Å of TM6 in the active A1AR (Figure 3B, 3D). Further analysis revealed that one positively-charged residue was located in the receptor ECL3 (K265ECL3), being close to the extracellular end of the TM6 (Figure 5A). In the inactive A1AR, this lysine pointed towards the lipid membrane and thus attracted the negatively-charged phosphate head group of a POPC molecule. Instead, the positively-charged side chain of K265ECL3 formed a stable salt-bridge with negatively-charged glutamate (E172ECL2) of ECL2 in the active A1AR (Figure S4). Residue K265ECL3 did not interact with the lipid in the active A1AR. Therefore, the inactive A1AR interacted with more phospholipids in the upper leaflet compared to the active A1AR.
Figure 5:
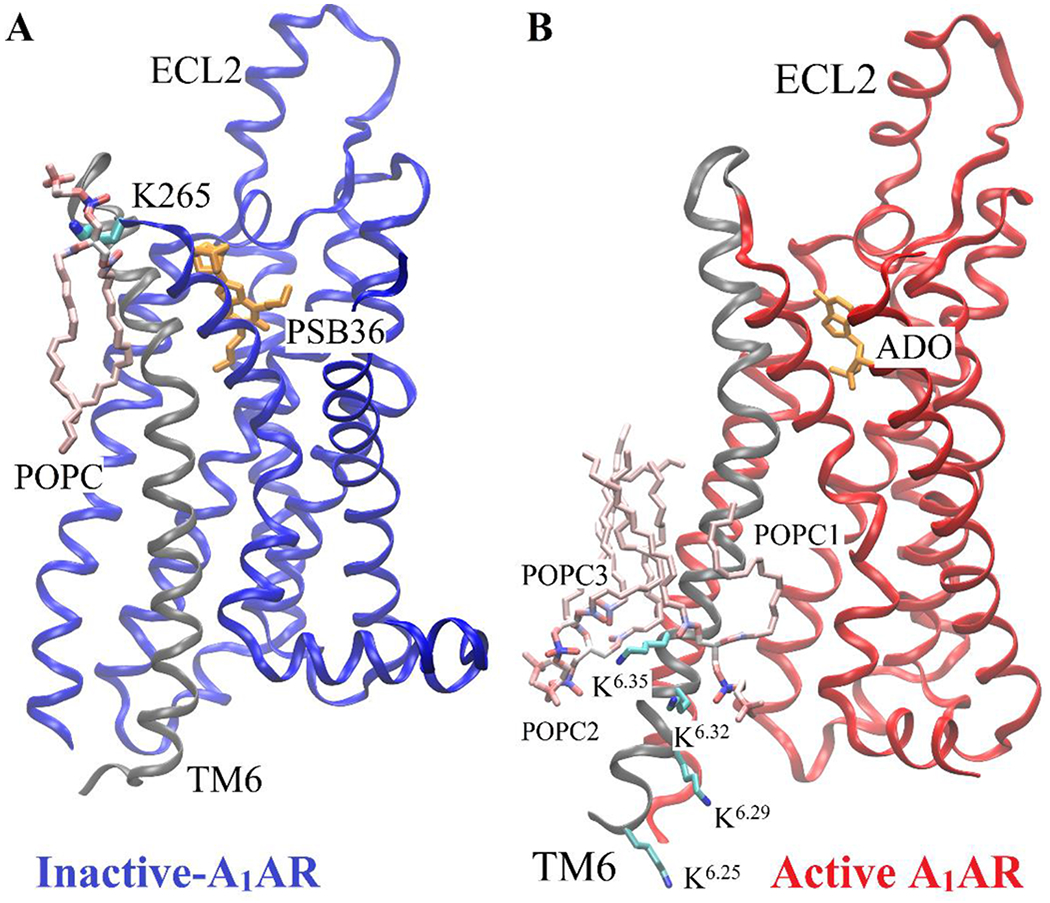
Minimum energy states of POPC lipid interacting with the positively-charged lysine residues in TM6 of the receptor obtained from dihedral GaMD simulations. (A) One POPC molecule in the upper leaflet interacts with one lysine residue (K265ECL3) of the inactive A1AR. (B) Three POPC molecules (POPC1, POPC2, POPC3) in the lower leaflet interact with four Lysine residues (K2636.25, K2676.29, K2706.32 and K2736.35) of the active A1AR. The receptor TM6 is colored in gray.
The active A1AR attracted more lipids in the lower leaflet to the TM6 than the inactive A1AR
In contrast to the upper leaflet, the lower leaflet had more lipids within 5 Å of TM6 in the active A1AR than in the inactive system (Figure 4). In the lowest energy state, the inactive A1AR interacted with approximately two lipids within 5 Å of TM6 (Figure 4A, 4C). In comparison, the active A1AR exhibited a relatively broader energy well. The TM6 intracellular domain interacted with ~2-4 lipid molecules (Figure 4B, 4D). Upon activation of the A1AR, the TM6 moved outwards by ~10 Å and exposed its positively-charged residues to the membrane. Therefore, the lipids that diffused in the lower leaflet of the membrane interacted more frequently with the receptor. Fewer lipids interacted with the inactive A1AR because the receptor has a narrower curvature in the TM6 intracellular region. Moreover, the lower leaflet had a larger number of lipids interacting with receptor TM6 than the upper leaflet. Out of five positively-charged residues in TM6, four were located in the intracellular region (K2246.25, K2286.29, K2316.32 and K2346.35) (Figure 5B). The negatively-charged phosphate head groups of POPC tended to interact with these positively-charged lysine residues to stabilize the active receptor conformation. Therefore, the lipid-GPCR interaction should play an important role in the conformational changes during activation of the A1AR.
Figure 4:
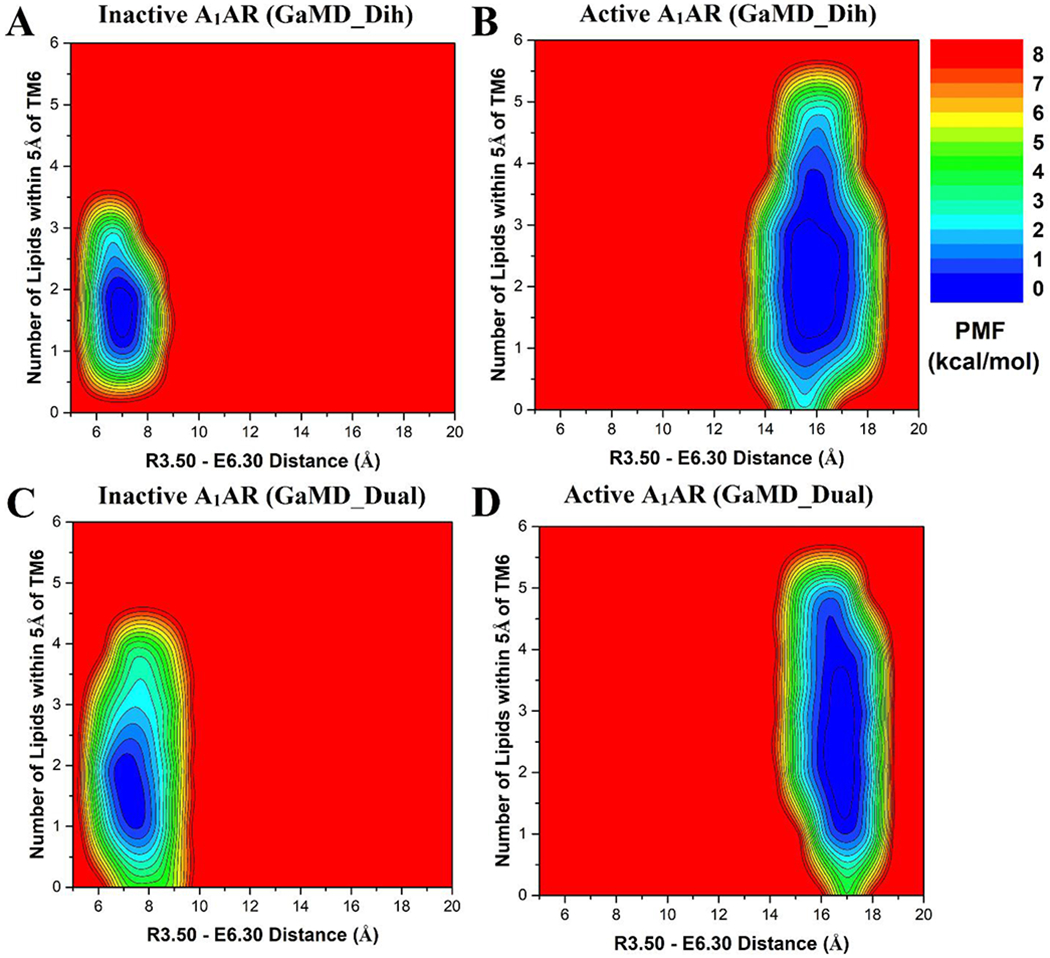
Free energy profiles of the cytoplasmic lower leaflet of membrane in different simulation systems regarding the number of lipids within 5 Å of the receptor TM6 and the receptor R3.50 – E6.30 distance: (A) Inactive A1AR using dihedral-boost GaMD, (B) Active A1AR using dihedral-boost GaMD, (C) Inactive A1AR using dual-boost GaMD and (D) Active A1AR using dual-boost GaMD. The R3.50 – E6.30 distance is ~7 Å in the inactive A1AR and increases to ~17 Å in the active A1AR due to outward movement of TM6.
GaMD simulations revealed strongly coupled dynamics between the GPCR and membrane lipids
Dynamic correlations were identified between residues of the A1AR and lipids in both the upper and lower leaflets. The Cα atoms in the receptor residues and the phosphorous atoms in the lipid head groups were used to calculate the correlation matrices (see details in Methods). Similar results were obtained using the C8 and C18 atoms in the lipid hydrophobic tails to calculate the dynamic correlation matrices (Figure S5). In all the simulation systems, motions of the receptor N-terminus, ECL1, ECL2 and ECL3 regions were positively correlated to those of lipids in upper leaflet (Figure 6). Similarly, motions of the receptor ICL1, ICL2 and ICL3 were positively correlated to those of lipids in the lower leaflet (Figure 7). For the inactive A1AR, most TM helix residues exhibited negatively correlated motions with the lipids. In this regard, the TM helices appeared to move in the opposite direction relative to the lipids in the inactive A1AR simulation system (Figure 6A, 6C, 7A and 7A). For the active A1AR, correlations between TM helix residues and the lipids in both the upper and lower leaflets were very weak, being close to zero (Figure 6B, 6D, 7B and 7D). However, marked positive correlations were identified between the intracellular region of the receptor TM6 and lipids in the lower leaflet (Figures 7B and 7D). Therefore, the TM6 intracellular region of the active A1AR appeared to move in the same direction with the surrounding lipids. This was highly consistent with the simulation finding that significantly more lipids were found within 5 Å of the TM6 intracellular domain in the active A1AR system (Figure 4) and they formed remarkably stronger electrostatic interactions (Figure 5) compared with the inactive A1AR system. In summary, the GaMD simulations revealed strongly coupled dynamics between the GPCR and membrane lipids.
Figure 6:
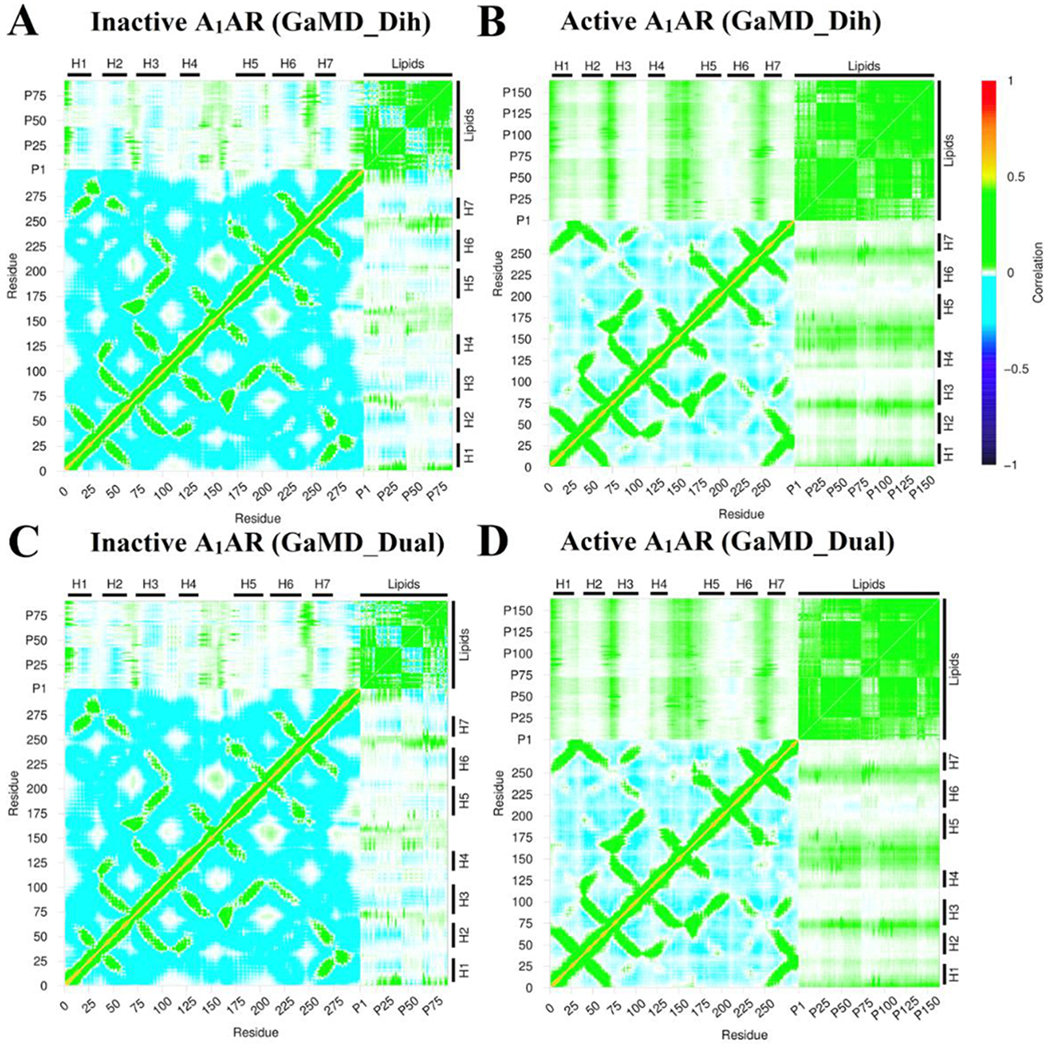
Dynamic correlation matrices calculated for lipids in the extracellular upper leaflet with residues in the A1AR in different simulation systems: (A) Inactive A1AR using dihedral-boost GaMD, (B) Active A1AR using dihedral-boost GaMD, (C) Inactive A1AR using dual-boost GaMD and (D) Active A1AR using dual-boost GaMD. The Cα atoms of the receptor and phosphorous atoms in the lipid head groups were used for calculating the correlation matrices here. Similar results were obtained using the C8 and C18 atoms in the lipid hydrophobic tails as shown in Figure S5. The receptor ICL1, ICL2 and ICL3 represent intracellular loops between TM helices 1-2, 3-4, and 5-6 respectively. Similarly, the receptor ECL1, ECL2 and ECL3 represent extracellular loops between TM helices 2-3, 4-5, and 6-7 respectively.
Figure 7:
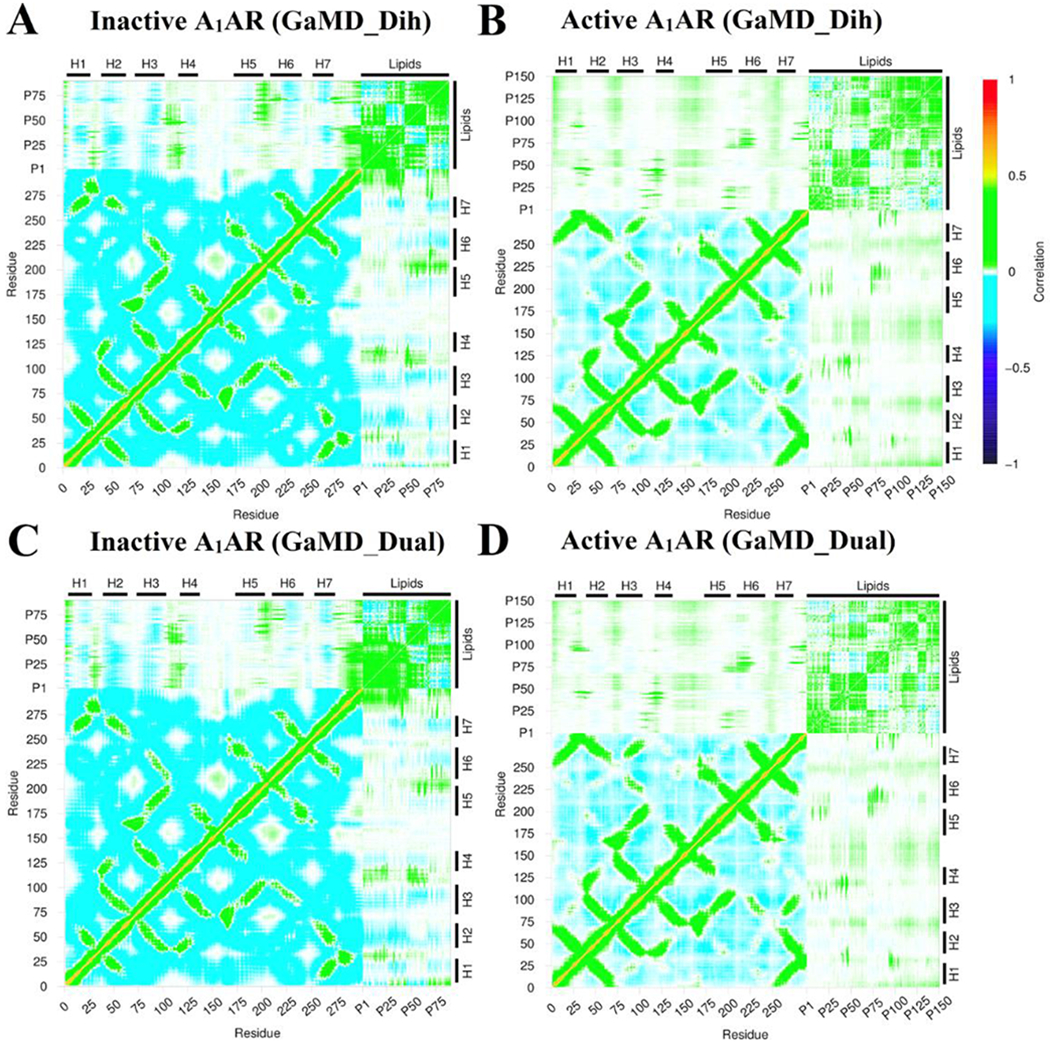
Dynamic correlation matrices calculated for lipids in the intracellular lower leaflet with residues in the A1AR in different simulation systems: (A) Inactive A1AR using dihedral-boost GaMD, (B) Active A1AR using dihedral-boost GaMD, (C) Inactive A1AR using dual-boost GaMD and (D) Active A1AR using dual-boost GaMD. The Cα atoms of the receptor and phosphorous atoms in the lipid head groups were used for calculating the correlation matrices here. Similar results were obtained using the C8 and C18 atoms in the lipid hydrophobic tails as shown in Figure S5. The receptor ICL1, ICL2 and ICL3 represent intracellular loops between TM helices 1-2, 3-4, and 5-6 respectively. Similarly, the receptor ECL1, ECL2 and ECL3 represent extracellular loops between TM helices 2-3, 4-5, and 6-7 respectively.
Discussion
In this study, we have applied all-atom GaMD simulations to investigate GPCR-membrane interactions, using the A1AR as a model receptor. In the GaMD simulations, the inactive and active A1AR showed different structural flexibility profiles. The ECL2 region, intracellular ends of TM6 and TM5 exhibited higher fluctuations in the active A1AR compared to the inactive A1AR. The receptor TM domain was rigid and the ligands remained tightly bound at the orthosteric site. However, the G protein coupled to the active A1AR exhibited high flexibility during the simulations, especially in the α5 helix, α4-β5 loop and α4-β6 loop of the Gα subunit and terminal ends of the Gβγ subunits. These results were consistent to our earlier simulation findings of AR-G protein complexes(65).
The -SCD order parameter values obtained from GaMD simulations were consistent with experimental data. In NMR experiments, the -SCD order parameter for the fifth carbon C-H bond of POPC was observed to be at ~0.18-0.20(56). The -SCD order parameter of POPC’s fifth carbon atom was ~0.20±0.02 in the lower leaflet in the inactive A1AR system. It decreased to ~0.17±0.02 in the active A1AR system. The -SCD order parameter of the ninth carbon C-H bond in POPC was measured as ~0.10 in NMR experiments(56), for which the same value was obtained from GaMD simulations. Furthermore, the GaMD simulations showed that POPC lipids in the lower leaflet of the active A1AR system were more fluid than in the inactive A1AR system. The -SCD order parameters for the lower leaflet in the active A1AR system were smaller than those in the inactive A1AR system. This finding correlated with the outward movement of TM6 in the active A1AR, which caused higher inclination of the C-H bonds to be aligned along the bilayer normal. The smaller -SCD order parameters suggested higher membrane fluidity in the lower leaflet of the active A1AR system.
In the GaMD simulations, the inactive A1AR attracted more lipids in the upper leaflet than the active A1AR. The membrane facing positively-charged lysine residue (K265ECL3) interacted with the negatively-charged phosphate head group of POPC. In contrast, this lysine pointed towards ECL2 in the active A1AR. This was consistent with our previous study(66), in which the positive allosteric modulator (PAM) enhanced the agonist binding at the orthosteric site by forming a salt-bridge between E172ECL2-K265ECL3. Moreover, the active A1AR attracted more lipids in the lower leaflet compared with the inactive A1AR. When exposed to the membrane, the positively-charged residues in intracellular region of TM6 of the A1AR interacted with negatively-charged head groups. This was further verified by the correlation matrix. The only positive correlation between the transmembrane helices and the lipids was observed between the intracellular region of TM6 and lipids in the lower leaflet of active A1AR. Four lysine residues (K2636.25, K2676.29, K2706.32 and K2736.35) present in the intracellular end in the β2AR of TM6 were also known to interact with negatively-charged headgroups of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) lipid molecules and thus stabilize the GPCR active state(27).
In summary, all-atom GaMD simulations have revealed strongly coupled dynamics between a GPCR and the membrane lipids that depend on the receptor activation state. The GaMD method has greatly enhanced sampling of the lipid-protein interactions, which would take significantly longer simulation time using cMD. Nonetheless, the activation or deactivation conformational transitions of the GPCR were not observed in the presented GaMD simulations. Longer GaMD simulations (e.g., microseconds) are expected to capture such conformational transitions (67) and the related effects of lipid-receptor interactions will be investigated in the future. Furthermore, the effects different lipid types (e.g., cholesterol, PIP2, etc.) on GPCR-membrane interactions are subject to future studies. It is important to study specific lipid interactions with GPCRs during the receptor activation. Developments of enhanced sampling methodologies and computing power would aid to further address these challenges.
Supplementary Material
Acknowledgements
We dedicate this manuscript to Prof. Benoit Roux’s 60th birthday for his contributions in particularly membrane protein simulations and free energy calculations. This work used supercomputing resources with allocation award TG-MCB180049 through the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562, and project M2874 through the National Energy Research Scientific Computing Center (NERSC), which is a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231. This work was supported in part by the American Heart Association (Award 17SDG33370094), the National Institutes of Health (R01GM132572) and the startup funding in the College of Liberal Arts and Sciences at the University of Kansas.
Footnotes
Competing Interests Statement
There is no competing interest.
References
- 1.Jacobson KA & Gao Z-G (2006) Adenosine receptors as therapeutic targets. Nature Reviews Drug Discovery 5:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandis AZ & Wenk MR (2007) Membrane lipids as signaling molecules. Current opinion in lipidology 18(2):121–128. [DOI] [PubMed] [Google Scholar]
- 3.Manna M, Nieminen T, & Vattulainen I (2019) Understanding the Role of Lipids in Signaling Through Atomistic and Multiscale Simulations of Cell Membranes. Annual review of biophysics 48. [DOI] [PubMed] [Google Scholar]
- 4.Chattopadhyay A & Raghuraman H (2004) Application of fluorescence spectroscopy to membrane protein structure and dynamics. Curr Sci 87(2):175–180. [Google Scholar]
- 5.CHATTOPADHYAY A (2016) Experimental and Computational Approaches to Study Membranes and Lipid–-Protein Interactions. Computational Biophysics of Membrane Proteins:137. [Google Scholar]
- 6.Chattopadhyay A & Haldar S (2013) Dynamic insight into protein structure utilizing red edge excitation shift. Accounts of chemical research 47(1):12–19. [DOI] [PubMed] [Google Scholar]
- 7.Chattopadhyay A & Jafurulla M (2015) Novel insights in membrane biology utilizing fluorescence recovery after photobleaching Biochemical Roles of Eukaryotic Cell Surface Macromolecules, (Springer; ), pp 27–40. [DOI] [PubMed] [Google Scholar]
- 8.Pucadyil TJ & Chattopadhyay A (2004) Cholesterol modulates ligand binding and G-protein coupling to serotonin1A receptors from bovine hippocampus. Biochimica et Biophysica Acta (BBA)-Biomembranes 1663(1-2):188–200. [DOI] [PubMed] [Google Scholar]
- 9.Saxena R & Chattopadhyay A (2012) Membrane cholesterol stabilizes the human serotonin1A receptor. Biochimica et Biophysica Acta (BBA)-Biomembranes 1818(12):2936–2942. [DOI] [PubMed] [Google Scholar]
- 10.Casiraghi M, et al. (2016) Functional modulation of a G protein-coupled receptor conformational landscape in a lipid bilayer. Journal of the American Chemical Society 138(35):11170–11175. [DOI] [PubMed] [Google Scholar]
- 11.Manna M & Mukhopadhyay C (2011) Cholesterol driven alteration of the conformation and dynamics of phospholamban in model membranes. Physical Chemistry Chemical Physics 13(45):20188–20198. [DOI] [PubMed] [Google Scholar]
- 12.Muth S, Fries A, & Gimpl G (2011) Cholesterol-induced conformational changes in the oxytocin receptor. Biochemical Journal 437(3):541–553. [DOI] [PubMed] [Google Scholar]
- 13.Hanson MA, et al. (2008) A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 16(6):897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zocher M, Zhang C, Rasmussen SG, Kobilka BK, & Müller DJ (2012) Cholesterol increases kinetic, energetic, and mechanical stability of the human β2-adrenergic receptor. Proceedings of the National Academy of Sciences 109(50):E3463–E3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gimpl G (2016) Interaction of G protein coupled receptors and cholesterol. Chemistry and physics of lipids 199:61–73. [DOI] [PubMed] [Google Scholar]
- 16.Gimpl G, Burger K, & Fahrenholz F (1997) Cholesterol as modulator of receptor function. Biochemistry 36(36):10959–10974. [DOI] [PubMed] [Google Scholar]
- 17.Oates J & Watts A (2011) Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Current opinion in structural biology 21(6):802–807. [DOI] [PubMed] [Google Scholar]
- 18.Paila YD & Chattopadhyay A (2009) The function of G-protein coupled receptors and membrane cholesterol: specific or general interaction? Glycoconjugate journal 26(6):711. [DOI] [PubMed] [Google Scholar]
- 19.Paila YD, Jindal E, Goswami SK, & Chattopadhyay A (2011) Cholesterol depletion enhances adrenergic signaling in cardiac myocytes. Biochimica et Biophysica Acta (BBA)-Biomembranes 1808(1):461–465. [DOI] [PubMed] [Google Scholar]
- 20.Pontier SM, et al. (2008) Cholesterol-dependent Separation of the β2-Adrenergic Receptor from Its Partners Determines Signaling Efficacy INSIGHT INTO NANOSCALE ORGANIZATION OF SIGNAL TRANSDUCTION. Journal of biological chemistry 283(36):24659–24672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pucadyil TJ & Chattopadhyay A (2006) Role of cholesterol in the function and organization of G-protein coupled receptors. Progress in lipid research 45(4):295–333. [DOI] [PubMed] [Google Scholar]
- 22.Hedger G & Sansom MS (2016) Lipid interaction sites on channels, transporters and receptors: recent insights from molecular dynamics simulations. Biochimica et Biophysica Acta (BBA)-Biomembranes 1858(10):2390–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inagaki S, et al. (2012) Modulation of the interaction between neurotensin receptor NTS1 and Gq protein by lipid. Journal of molecular biology 417(1-2):95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oates J, et al. (2012) The role of cholesterol on the activity and stability of neurotensin receptor 1. Biochimica et Biophysica Acta (BBA)-Biomembranes 1818(9):2228–2233. [DOI] [PubMed] [Google Scholar]
- 25.Dawaliby R, et al. (2016) Allosteric regulation of G protein–coupled receptor activity by phospholipids. Nature chemical biology 12(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sengupta D, Kumar GA, Prasanna X, & Chattopadhyay A (2016) Experimental and Computational Approaches to Study Membranes and Lipid–Protein Interactions. Computational Biophysics of Membrane Proteins), pp 137–160. [Google Scholar]
- 27.Bruzzese A, Gil C, Dalton JA, & Giraldo J (2018) Structural insights into positive and negative allosteric regulation of a G protein-coupled receptor through protein-lipid interactions. Scientific reports 8(1):4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neale C, Herce HD, Pomes R, & García AE (2015) Can specific protein-lipid interactions stabilize an active state of the beta 2 adrenergic receptor? Biophysical journal 109(8):1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballesteros JA & Weinstein H (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors Methods in Neurosciences, ed Sealfon SC (Academic Press; ), Vol 25, pp 366–428. [Google Scholar]
- 30.Salas-Estrada LA, Leioatts N, Romo TD, & Grossfield A (2018) Lipids alter rhodopsin function via ligand-like and solvent-like interactions. Biophysical journal 114(2):355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dror RO, et al. (2011) Activation mechanism of the β2-adrenergic receptor. Proceedings of the National Academy of Sciences 108(46):18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Latorraca NR, Venkatakrishnan A, & Dror RO (2016) GPCR dynamics: structures in motion. Chemical reviews 117(1):139–155. [DOI] [PubMed] [Google Scholar]
- 33.Song W, Yen H-Y, Robinson CV, & Sansom MS (2019) State-dependent lipid interactions with the A2a receptor revealed by MD simulations using in vivo-mimetic membranes. Structure 27(2):392–403. e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isralewitz B, Baudry J, Gullingsrud J, Kosztin D, & Schulten K (2001) Steered molecular dynamics investigations of protein function. Journal of Molecular Graphics and Modelling 19(1):13–25. [DOI] [PubMed] [Google Scholar]
- 35.Torrie GM & Valleau JP (1977) Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. Journal of Computational Physics 23(2):187–199. [Google Scholar]
- 36.Miao Y, Feher VA, & McCammon JA (2015) Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J Chem Theory Comput 11(8):3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhattarai A & Miao Y (2018) Gaussian accelerated molecular dynamics for elucidation of drug pathways. Expert opinion on drug discovery 13(11):1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miao Y & McCammon JA (2016) Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proceedings of the National Academy of Sciences 113(43):12162–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao Y & McCammon JA (2017) Gaussian accelerated molecular dynamics: Theory, implementation, and applications Annual reports in computational chemistry, (Elsevier; ), Vol 13, pp 231–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sibener LV, et al. (2018) Isolation of a structural mechanism for uncoupling T cell receptor signaling from peptide-MHC binding. Cell 174(3):672–687. e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miao Y & McCammon JA (2018) Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor. Proceedings of the National Academy of Sciences 115(12):3036–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salawu EO (2018) The Impairment of TorsinA’s Binding to and Interactions With Its Activator: An Atomistic Molecular Dynamics Study of Primary Dystonia. Frontiers in molecular biosciences 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palermo G, Miao Y, Walker RC, Jinek M, & McCammon JA (2017) CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proceedings of the National Academy of Sciences 114(28):7260–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ricci CG, et al. (2019) Deciphering Off-Target Effects in CRISPR-Cas9 through Accelerated Molecular Dynamics. ACS Central Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Draper-Joyce CJ, et al. (2018) Structure of the adenosine-bound human adenosine A 1 receptor–G i complex. Nature 558(7711):559. [DOI] [PubMed] [Google Scholar]
- 46.Cheng RK, et al. (2017) Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 25(8):1275–1285. e1274. [DOI] [PubMed] [Google Scholar]
- 47.Glukhova A, et al. (2017) Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 168(5):867–877. e813. [DOI] [PubMed] [Google Scholar]
- 48.Miao Y (2018) Acceleration of biomolecular kinetics in Gaussian accelerated molecular dynamics. The Journal of chemical physics 149(7):072308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamelberg D, Mongan J, & McCammon JA (2004) Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J Chem Phys 120(24):11919–11929. [DOI] [PubMed] [Google Scholar]
- 50.Shen TY & Hamelberg D (2008) A statistical analysis of the precision of reweighting-based simulations. J Chem Phys 129(3):034103. [DOI] [PubMed] [Google Scholar]
- 51.Case D, et al. (2014) Amber 14, University of California, San Francisco. [Google Scholar]
- 52.Pang YT, Miao Y, Wang Y, & McCammon JA (2017) Gaussian Accelerated Molecular Dynamics in NAMD. J Chem Theory Comput 13(1):9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miao Y, Sinko W, Pierce L, Bucher D, & McCammon JA (2014) Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J Chem Theory Comput 10(7):2677–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamelberg D, de Oliveira CAF, & McCammon JA (2007) Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J Chem Phys 127(15):155102. [DOI] [PubMed] [Google Scholar]
- 55.Roux B (1995) The Calculation of the Potential of Mean Force Using Computer-Simulations. Comput Phys Commun 91(1-3):275–282. [Google Scholar]
- 56.Perly B, Smith IC, & Jarrell HC (1985) Acyl chain dynamics of phosphatidylethanolamines containing oleic acid and dihydrosterculic acid: deuteron NMR relaxation studies. Biochemistry 24(17):4659–4665. [DOI] [PubMed] [Google Scholar]
- 57.Humphrey W, Dalke A, & Schulten K (1996) VMD: Visual molecular dynamics. Journal of Molecular Graphics & Modelling 14(1):33–38. [DOI] [PubMed] [Google Scholar]
- 58.Vanommeslaeghe K & MacKerell AD Jr (2014) CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochimica et biophysica acta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanommeslaeghe K, et al. (2010) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31(4):671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nguyen AT, et al. (2016) Role of the Second Extracellular Loop of the Adenosine A1 Receptor on Allosteric Modulator Binding, Signaling, and Cooperativity. Molecular pharmacology 90(6):715–725. [DOI] [PubMed] [Google Scholar]
- 61.Kappel K, Miao Y, & McCammon JA (2015) Accelerated Molecular Dynamics Simulations of Ligand Binding to a Muscarinic G-protein Coupled Receptor. Quarterly Reviews of Biophysics 48(04):479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phillips JC, et al. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26(16):1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roe DR & Cheatham TE (2013) PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput 9(7):3084–3095. [DOI] [PubMed] [Google Scholar]
- 64.Guixa-González R, et al. (2014) MEMBPLUGIN: studying membrane complexity in VMD. Bioinformatics 30(10):1478–1480. [DOI] [PubMed] [Google Scholar]
- 65.Wang J & Miao Y (2019) Mechanistic Insights into Specific G Protein Interactions with Adenosine Receptors Revealed by Accelerated Molecular Simulations. bioRxiv:541250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miao Y, Bhattarai A, Nguyen AT, Christopoulos A, & May LT (2018) Structural Basis for Binding of Allosteric Drug Leads in the Adenosine A 1 Receptor. Scientific reports 8(1):16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miao Y, Nichols SE, Gasper PM, Metzger VT, & McCammon JA (2013) Activation and dynamic network of the M2 muscarinic receptor. Proceedings of the National Academy of Sciences 110(27):10982–10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.