

Abstract

For realization of a wearable artificial kidney based on regeneration of a small volume of dialysate, efficient urea removal from dialysate is a major challenge. Here a potentially suitable polymeric sorbent based on phenylglyoxaldehyde (PGA), able to covalently bind urea under physiological conditions, is described. Sorbent beads containing PGA groups were obtained by suspension polymerization of either styrene or vinylphenylethan-1-one (VPE), followed by modification of the aromatic groups of poly(styrene) and poly(VPE) into PGA. It was found that PGA-functionalized sorbent beads had maximum urea binding capacities of 1.4–2.2 mmol/g and removed ∼0.6 mmol urea/g in 8 h at 37 °C under static conditions from urea-enriched phosphate-buffered saline, conditions representative of dialysate regeneration. This means that the daily urea production of a dialysis patient can be removed with a few hundred grams of this sorbent which, is an important step forward in the development of a wearable artificial kidney.
Keywords: chemisorption, dialysis, kinetics, phenylglyoxaldehyde, sorbent, urea
Introduction
Uremic toxins such as creatinine and urea accumulate in the body of patients suffering from end-stage kidney disease (ESKD).1 In order to remove these waste compounds, ESKD patients undergo hemodialysis (HD) 3–6 times a week for 2–8 h, usually performed in a dialysis center or hospital. To improve the flexibility and quality of life of dialysis patients, a wearable HD device is urgently needed that uses a small volume of dialysate (preferably <0.5 L compared to 120 L presently used for a conventional HD session) that is continuously regenerated by a purification unit so that it can be reused in a closed-loop system.2−4 Urea removal from dialysate is crucial in realizing such a wearable HD device. Urea is the uremic toxin with the highest daily production (∼400 mmol per day5), and currently, there is no adequate urea removal technology available that enables reduction of the dialysate volume to <0.5 L.6
Ideally, a suitable urea sorbent has a high density of functional groups that are able to form covalent or noncovalent bonds with urea at 37 °C, resulting in its removal from dialysate,7 without forming toxic side-products that can be released into the patient. As noncovalent bonds between urea and physisorbents are reversible and generally weaker than covalent bonds, these sorbents have low binding capacities (BCs). Chemisorption by the formation of covalent bonds between urea and the electrophilic groups in a sorbent is therefore preferred to remove high amounts of urea from dialysate at 37 °C. A few urea chemisorbents have been reported, in which urea removal was based on the reaction with an electrophilic carbonyl group, such as an aldehyde (BC = 0.2 mmol/g),8,9 an indanetrione (BC = 2.0 mmol/g),10,11 or a phenylglyoxaldehyde (PGA, BC = 0.3–1.5 mmol/g).12−14
PGA is a very electrophilic functional group (2a) and forms a (reversible) hydrate in contact with water, i.e., phenylglyoxaldehyde hydrate (1a, PGAH). In our previous work, we systematically studied the reactivity of urea with a variety of carbonyl compounds in phosphate-buffered saline (PBS) spiked with urea (30 mM) and found that PGAH was one of the compounds with the highest reactivity. Importantly, PGAH is synthetically easily accessible, and a urea sorbent based on PGAH would therefore be a logical choice for application in dialysate regeneration. In addition, we found that PGAH and urea can irreversibly react in a 1:1 ratio (3a) and a 2:1 ratio (3′a) (Scheme 1) and proposed a mechanism for both reactions.15
Scheme 1. Reaction of PGAH with Urea15.

PGAH-type urea sorbent beads based on polystyrene (PS) were claimed in a few patents. These beads were obtained via acetylation of PS, followed by halogenation of the acetyl group and Kornblum oxidation of the primary halide.12−14 Poss et al. synthesized macroporous particles based on this procedure to allow easier diffusion of urea into the polymeric matrix, and thereby, faster and higher urea removal was achieved than with nonporous beads (BC of 1.5 vs 0.29 mmol/g, respectively).12,13 However, the experimental procedures to obtain these materials and their characteristics were not described in detail. In theory, a polymeric sorbent based on vinyl-substituted PGAH has a maximum urea BC of 2.8 or 5.6 mmol/g, depending on whether PGAH and urea react in a 2:1 ratio or a 1:1 ratio, which is much higher than those reported by Poss et al., which suggests a low extent of conversion of the phenyl groups into PGAH groups. In the present work, we aim to develop PGAH-type sorbents for urea removal from dialysate by studying and optimizing the modification of the aromatic groups of PS into PGAH groups. In addition, we explored a new route to obtain PGAH-functionalized sorbents that circumvents the acetylation step by polymerization of vinylphenylethanone (VPE), an acetylated styrene-like monomer, and potentially results in a higher amount of PGAH groups per gram of sorbent. The kinetics of the urea uptake by PGAH-type sorbents under physiological conditions were studied. Finally, we determined whether the PGAH groups in the sorbents react with urea in a 1:1 or a 2:1 molar ratio.
Materials and Methods
Materials
4-Nitrophenylglyoxaldehyde and 4-methylphenylglyoxaldehyde were purchased from CombiBlocks (CA, USA). 4-Ethynylacetophenone was purchased from Acros Organics (NJ, USA). PBS (pH = 7.4, ion composition: Na+ 163.9 mM, Cl– 140.3 mM, HPO42– 8.7 mM, H2PO4– 1.8 mM) was obtained from B. Braun (Melsungen AG, Germany). Anhydrous dicalcium phosphate (CaHPO4) was obtained from Chemtrade International (Bussum, The Netherlands). Polymethacrylic acid (Degalan RG S mv) was obtained from Evonik Industries (Darmstadt, Germany). ShellSolTD, a mixture of alkenes, was a kind gift from Shell (Amsterdam, The Netherlands). All other chemicals were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands) and used as received, unless stated otherwise. Nickel filters with a cutoff of 200 μm were obtained from Veco B.V. (Eerbeek, The Netherlands).
NMR, UV, and IR Spectroscopy
NMR spectra were recorded on a Bruker 600 MHz with a BBI probe at room temperature (RT). Residual solvent signals were used as internal standard (1H: δ 7.26 ppm, 13C (1H): δ 77.16 ppm for CDCl3). Chemical shifts (δ) are given in ppm, and coupling constants (J) are given in hertz (Hz). Resonances are reported as s (singlet), d (doublet), t (triplet), q (quartet), bs (broad singlet), and m (multiplet) or combinations thereof. UV absorption spectra were recorded in triplicate with a BMG LABTECH SpectroStar Nano plate reader using a UV-Star Microplate 96 well obtained from Greiner Bio-One (Alphen aan de Rijn, The Netherlands). Infrared (IR) spectra were recorded neat using a PerkinElmer ATRU Spectrum 2.
Determination of the Pseudo-First-Order Rate Constants
PGAH (1a) and two PGAH derivatives (1b and 1c) (0.3 mmol, 1.0 equiv) were individually dissolved in a 1:1 v/v mixture of PBS:dimethyl sulfoxide (DMSO) (10 mL). Urea (901 mg, 15 mmol, 50 equiv) was dissolved in the PGAH solution, which was subsequently magnetically stirred at 50 °C. Samples (50 μL) from the reaction mixture were taken at different time points, diluted 10 (1a) or 15 (1b and 1c) times with 1:1 v/v DMSO:PBS (500 or 700 μL), and subsequently diluted another 10× using the same solvent mixture (thus resulting in a final 100 or 150 times dilution, respectively). The concentrations of the PGAH (derivatives) 1a–c in the 100 or 150 times diluted samples were determined by UV spectroscopy (260, 263, and 270 nm, for 1a, 1b, and 1c, respectively). A calibration curve was prepared using a dilution series in a 1:1 v/v mixture of PBS:DMSO (final concentrations varied from 0.030 to 0.360 mM) from a stock solution of the PGAH (derivatives) (30 mM) in 1:1 (v/v) DMSO:PBS. The kPFO values for the PGAH analogues were determined from the slopes of the plots of ln[PGAH] versus time.
Calculation of Gibbs Free Energy (G)
Density functional theory (DFT) calculations were performed using the Gaussian 09 software package, using the B3LYP (Becke, three-parameter, Lee–Yang–Parr) functional with 6-31g(d,p) as the basis set on all atoms.16 Structure optimizations were carried out with water as solvent without any symmetry restraints. Frequency analyses were performed on all optimized geometries to check that they are in fact minima. Standard temperature (298.15 K) and pressure (1 atm) were used for thermochemical calculations. Input: #B3LYP/6-31G(d,p) opt=tight freq scf=tight int=ultrafine pop=regular SCRF=(Solvent=Water).
Preparation of 10% Polymethacrylic Acid Sodium Salt Solution in Water
In a glass reactor equipped with a mechanical stirrer, polymethacrylic acid (10 g) was dissolved in water (84 mL) by heating to 80 °C and stirring for 30 min. Next, an aqueous 50% NaOH (2.67 mL; 68 mmol NaOH) solution was added, and stirring was continued for 60 min at the same temperature. The obtained viscous solution was transferred into a Falcon tube and stored at 4 °C for later use as a thickening agent of the aqueous phase in suspension polymerization.
Suspension Polymerization of Styrene
For the suspension polymerization of styrene, we essentially used a method as described by Jong et al.17 However, ShellSolTD and a poly(methacrylic acid) sodium salt solution were used instead of hexane and poly(acrylic acid) sodium salt. The detailed procedure was as follows.
The aqueous phase was prepared by addition of NaCl (340 mg), poly(methacrylic acid) sodium salt solution (8.32 g of a 10% solution in water), and CaHPO4 (3.06 g) to water (540 mL) in a glass reactor equipped with a Teflon blade stirrer (see Supporting Information section 3 for a picture). The aqueous phase was stirred for 30 min at RT, and the pH was 6.9. The organic phase was prepared by mixing styrene (229 mL, 2.0 mol), ShellSolTD (276 mL), and toluene (27 mL) in a beaker. Next, 55% technical grade divinylbenzene (DVB) (13 mL, 50 mmol, 2.5 mol %) and a 50% benzoylperoxide blend with dicyclohexyl phthalate (6.0 g, 12.4 mmol, 0.6 mol %) was added to the organic phase and stirred until the initiator was dissolved and a homogeneous solution was formed at RT. The organic phase was subsequently added to the aqueous phase in the glass reactor under continuous mechanical stirring at 180 rpm, by which an o/w emulsion was formed, and oxygen was removed by flushing with nitrogen gas for 20 min. Next, the emulsion was heated at 73 °C in an oil bath for 16 h under mechanical stirring. The resulting suspension was allowed to cool to RT and was poured over a sieve (cutoff 200 μm, Veco B.V.) and washed with acetone and water. The white beads were collected and dried over P2O5 under vacuum, resulting in 216 g of PS beads. Thermographic analysis (TGA) showed ∼14% volatiles present, indicating a yield of solid material of ∼186 g (yield 86%).
Friedel–Crafts Acetylation of Polystyrene12
In a glass reactor equipped with a Teflon blade stirrer, PS beads (80.9 g, 0.77 mol aromatic groups, 1.0 equiv) were swollen in 1,2-dichloroethane (DCE, 750 mL) for 30 min under mechanical stirring. Anhydrous AlCl3 (156 g, 1.17 mol, 1.5 equiv) was added portion-wise (3–5 g) to the suspension over the course of 15 min. After all AlCl3 was added, acetyl chloride (66 mL, 0.94 mol, 1.2 equiv) was added slowly, and the suspension was heated to 50 °C in an oil bath for 5 h, after which the formation of HCl gas (caused by the reaction of an aromatic group and acetyl chloride) stopped. The suspension was allowed to cool to RT and filtered (cutoff, 200 μm). The residue was suspended in 500 mL of 6 M HCl solution at 0 °C in an ice bath and stirred for 30 min to remove aluminum salts; this step was repeated twice. The suspension was filtered (cutoff 200 μm, Veco B.V.) and washed with acetone and water until the pH of the filtrate was >5. The residue was dried over P2O5 under vacuum, resulting in acetylated polystyrene (PS-Ac, 71.6 g).
Halogenation and Kurnblum Oxidation of Acetylated Polystyrene12
In a glass reactor equipped with a Teflon blade stirrer, PS-Ac beads (60.0 g) were swollen in DMSO (600 mL, 8.45 mol) for 30 min under continuous stirring, after which an aqueous solution of 48% HBr (175 mL, 1.55 mol) was slowly added. One of the outlets of the reactor was capped with a septum containing a needle allowing escape of the formed Me2S. The suspension was stirred at 80 °C for 8 h, after which the reaction mixture was filtered (cutoff 200 μm, Veco B.V.). The residue was washed with water until the pH of the filtrate was >5. The residue was dried over P2O5 under vacuum, resulting in PS-Ac-Ox (55.2 g).
Synthesis of p-Vinylphenylethenone (VPE)
In a three-neck round-bottom flask p-ethynylphenylethenone (6, 10.0 g, 69.4 mmol) was suspended in EtOH (350 mL), and Lindlar’s catalyst (300 mg, 3 wt %) was added. Air was replaced by H2 (balloon), and the suspension was stirred at RT for 2–16 h. To monitor the conversion (and thus preventing over-reduction of VPE (7) into the alkane (8)), samples were frequently taken from the reaction mixture, and after evaporation of EtOH under reduced pressure, the conversion was determined by 1H NMR (CDCl3). After the conversion was >90%, the H2-filled balloon was removed, and the reaction mixture was concentrated under reduced pressure. The crude product was redissolved in CH2Cl2 and purified by filtration over Hyflo. The filtrate was concentrated under reduced pressure, giving crude VPE (7) as a yellow liquid in a 99% yield (10.1 g, 69.0 mmol). Melting point 29 °C, melting enthalpy 90.6 J/g (Supporting Information Figure S11). 1H NMR (CDCl3, 600 MHz) δ 7.92 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 8.2 Hz, 2H), 6.75 (dd, J = 17.6 Hz, 10.9 Hz, 1H), 5.87 (d, J = 17.6 Hz, 1H), 5.39 (d, J = 10.9 Hz, 1H), 2.59 (s, 3H). The melting point and the NMR spectrum correspond with the ones reported in the literature.18−20
Suspension Polymerization of VPE
The same procedure as that for the preparation of PS beads was employed, with some minor modifications. In brief, the aqueous phase was prepared by addition of NaCl (11 mg), polymethacrylic acid sodium salt solution (452 mg of a 10% gel in water), and CaHPO4 (84 mg) to water (15 mL). The organic phase was composed of VPE (2.1 g, 14.4 mmol, 2 mL), porogen (2.9 mL), 80% technical grade DVB (3–6 mol %), and a 50% benzoylperoxide blend with dicyclohexyl phthalate (174 mg, 0.36 mmol, 2.5 mol %). After mixing and polymerization (same procedure followed as for “Suspension Polymerization of Styrene”), the resulting suspension was allowed to cool to RT and poured over a filter (cutoff 200 μm, Veco B.V.). The residue was washed with acetone and water and finally dried over P2O5 under vacuum, resulting in pVPE (1.1–1.9 g, yield 52–90%).
Halogenation and Kurnblum Oxidation of pVPE
The same procedure as that for the halogenation and Kurnblum oxidation of PS-Ac beads was employed for 12 h, downscaled to 600 mg of pVPE. After washing, 606 mg of yellow beads (pVPE-Ox) was obtained.
Scanning Electron Microscopy Analysis of Sorbent Particles
The morphology of the beads was analyzed by scanning electron microscopy (SEM, Phenom, FEI Company, The Netherlands). Dried beads were transferred onto 12 mm diameter aluminum specimen stubs (Agar Scientific Ltd., England) using double-sided adhesive tape. Prior to analysis, the beads were coated with platinum using an ion coater under vacuum. The samples were imaged using a 5 kV electron beam.
Determination of the Size of the Beads by Light Microscopy
The diameters of the beads were measured using optical microscopy, utilizing a size-calibrated Nikon eclipse TE2000-U microscope equipped with a digital camera (Nikon DS-2Mv camera and Nikon DS-U1 digital adapter, with a 4× magnification) and the NIS-elements basic research software package. Images of the beads were taken in the dry state, and for 30 arbitrary beads, 3 points on the perimeter of the beads were identified to allow calculation of circular diameter by the program.21 The average diameters and standard deviations are reported.
Quantitative 13C Solid-State NMR Analysis of the Different Beads
For solid-state 13C NMR measurements, beads were crushed and transferred into a 3.2 mm rotor for the magic-angle spinning (MAS) solid-state NMR analysis. The analysis of the samples was performed either on a Bruker 700 MHz wide-bore magnet with an AVANCE-III console or on a Bruker 400 MHz spectrometer. The spectra were recorded at RT (298 K) and using a MAS frequency between 10 and 14 kHz, chosen to minimize the overlap of the signal with spinning sidebands. For the 13C direct excitation spectra, 30° pulses were applied with field strengths of 55 kHz and 80 kHz SPINAL64.221H decoupling was applied during acquisition. The 13C T1 relaxation time for each sample was determined using inverse recovery and used to establish the repetition time for the different samples, set to 2*T1. Except for the pVPE-Ox-(4) sample, which showed a very short relaxation time of 1 s, for the other samples, the T1 varied from 40 to 80 s. Details for the specific experiments are given in the Supporting Information section 7. The NMR spectra were processed with 200 Hz line-broadening and analyzed with Bruker Topspin3.5.
Determination of the Surface Area of the Beads Using Nitrogen Physisorption
N2 physisorption isotherms were measured at −196 °C using a Micromeritics TriStar 3000 and TriStar II Plus apparatus. Prior to analysis, the samples were dried under vacuum for 16 h at RT. Surface areas of the beads were determined using the Brunauer–Emmett–Teller (BET) method, and the total pore volumes were derived from the amount of N2 adsorbed at p/p0 = 0.995.23 A Barrett–Joyner–Halenda (BJH) analysis was employed to determine pore size/volume distributions of the samples with the use of a Harkins–Jura thickness curve.24,25 Due to the shrinking of the porous polymeric beads and collapsing of the pores with increasing pressure and subsequent expansion with decreasing pressure, the correction of the dead volume is incorrect as by default it assumes that the solid fraction of the sample does not change in volume with pressure. As the dead volume was determined at p/p0 ≈ 0 and assumed constant during the measurement, the default dead volume-corrected isotherms decreased slightly with increasing pressures, which is physically meaningless. The relative deviation is largest for materials with low surface areas (<5 m2/g) and high materials volume fractions in the measurement tubes, such as for pVPE-Ox. A correction for this deformation, i.e., a change in dead volume with pressure was applied to these isotherms by a linear swelling function (Vadjusted = a · (p/p0) + Voriginal), in which a represents the swelling factor relative to the material’s volume at p/p0 ≈ 0, until dV/d(p/p0) > 0 is achieved for all pressures. Values for a were between 1.2 and 7.2, indicating a significant deformation of these materials. The SBET surface areas of the pVPE-Ox beads were calculated from the isotherms that were corrected for these volume changes as a function of pressure.
Determination of Urea Binding
The sorbent beads (15 mg) were dispersed with urea solution (1.5 mL, 30 mM) in PBS in Eppendorf tubes. The samples were placed in an oven at 37 °C on a rotating device. After 1, 2, 4, 8, 16, and 24 h, two Eppendorf tubes per time point were taken, the beads were allowed to settle, and the supernatant was removed. To determine the maximum BC (control experiment shown in Supporting Information section 8), the sorbent beads (50 mg per vial) were incubated for 24 h at 70 °C with a urea solution (5 mL, 30 or 50 mM) in PBS in two glass vials, after which the beads were allowed to settle, and the urea concentrations in the supernatants were determined with an AU 5800 routine chemistry analyzer (Beckman Coulter, Brea, CA) using a coupled enzyme reaction, which resulted in a colorimetric (570 nm) product proportional to the urea concentration.26
Thermal Analysis of Monomer and Beads
TGA was done as follows. In a platinum pan, the beads were heated at a rate of 10 °C/min. The weight loss during the ramp heating (and thereby the decomposition temperature) was determined on a TA Instruments TGA Q50.
Differential scanning calorimetry (DSC) analysis of the different samples was done as follows. In an open aluminum pan, the monomer or beads was heated from −50 to 250 °C at a rate of 10 °C/min, and the heat flow was monitored. Next, the sample was quickly cooled from 250 to −50 °C and subsequently heated again to 250 °C at a rate of 10 °C/min. The Tg or melting point was determined with a TA Instruments Discovery DSC. For the beads, residual solvent evaporated during the first run, and therefore, the results of the second run are reported. For the monomer (VPE), events of the first run are reported.
Synthesis of 5-(1-Hydroxy-2-oxo-2-phenylethyl)-5-phenylimidazolidine-2,4-dione
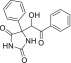 5-(1-Hydroxy-2-oxo-2-phenylethyl)-5-phenylimidazolidine-2,4-dione
was synthesized as described earlier15 and
used as a reference compound for the IR analysis of the sorbent beads
before and after urea saturation.
5-(1-Hydroxy-2-oxo-2-phenylethyl)-5-phenylimidazolidine-2,4-dione
was synthesized as described earlier15 and
used as a reference compound for the IR analysis of the sorbent beads
before and after urea saturation.
Results and Discussion
First, we investigated whether the reactivity of a PGAH-based urea sorbent could be increased by appropriate substituents because in our previous publication about the reactivity of ninhydrin with urea we found that substituents on the aromatic ring have a substantial impact on the reaction rate due to their effect on the indanetrione–ninhydrin equilibrium in water.27 Thus, the kinetics of the reaction of urea with para-methyl-PGAH (1b), a PGAH derivative with an electron-donating group (EDG), and with para-nitro-PGAH (1c), a PGAH derivative with an electron withdrawing group (EWG) (Figure 1A), were analyzed and compared to the kinetics of unsubstituted PGAH (1a) with urea. Substituents on the meta position were not investigated because in our previous study on substituent effects on the reaction of ninhydrin analogues with urea we found that the position of the EDG has a marginal effect on the overall reactivity of ninhydrin derivatives with urea.27 Moreover, the Friedel–Crafts acetylation of the aromatic group in PS (as the first step toward the PGAH sorbent) lead to ortho/para substitution because the aliphatic polymeric backbone is considered as an activating group28 and therefore acetylation will not take place at the meta position.
Figure 1.

(A) Structures of PGAH (1a), para-methyl PGAH (1b), and para-nitro PGAH (1c). (B) Reaction scheme of the PGAH (derivatives) with urea. (C) Plot of the logarithm of the concentration of PGAH (derivatives) versus time. Reaction conditions: PGAH (derivatives) (compound 1, 0.3 mmol, 1.0 equiv, 30 mM) and urea (15 mmol, 50 equiv, 1.5 M) in a 1:1 (v/v) mixture of PBS and DMSO (10 mL) at 50 °C.
As urea and PGAH can react with each other in aqueous solution both in a 1:1 and 1:2 ratio (Scheme 1),15 a large excess (50 equiv) of urea was used to limit the formation of the 1:2 urea–PGA adduct (Figure 1B). The rate of reaction of PGAH with urea is expressed by eq 1.15 Because the urea concentration is much higher than the PGAH concentration, its concentration stays almost constant, and thus, pseudo-first-order conditions are valid (eq 2), thereby making the reaction rate (−d[PGAH]/dt) dependent on the PGAH concentration only (eq 3). The pseudo-first-order kinetics of the reaction of PGAH (and its derivatives) with urea were analyzed by determining the concentrations of 1a–c in time using UV spectroscopy (see Supporting Information section 1 for the raw data and calculations). The solvent for this reaction was a 1:1 (v/v) PBS/DMSO mixture due to the very low solubility of 1b and 1c in PBS only. The pseudo-first-order rate constants (kPFO) correspond with the negative slopes in the plot of the logarithm of the PGAH (derivatives) concentration divided by log(e) versus time (Figure 1C) and are reported in Table S4.
| 1 |
| 2 |
| 3 |
The results presented in Figure 1 and Table S4 show that methyl-substituted PGAH (1b) reacted slightly slower with urea than unsubstituted PGAH (1a) (kPFO = 0.061 versus 0.085 min–1); however, nitro-substituted PGAH (1c) reacted more than a factor of 10 slower than unsubstituted PGAH (1a) (kPFO = 0.006 min–1). In our previous work, we found that EDGs and EWGs decrease the reaction rate of urea with ninhydrin derivatives due to the changes in the rate of dehydration of these derivatives, as suggested by calculations of the Gibbs free energies (G) of the starting material and intermediates.27 Similarly, we calculated G values of PGAH (1), PGA (2), and the first urea–PGA intermediate (4, Supporting Information Scheme S1) at the B3LYP/6-31G(d,p) DFT level (Supporting Information sections 2 and 12) and calculated the change in energy for dehydration of PGAH (ΔGdehydration) and the reaction of PGA with urea (ΔGintermediate) (Scheme S1). The influence of the substituent (Me and NO2) on both reaction steps was quantified by subtraction of the ΔG values of unsubstituted species from the ΔG of substituted species, thereby yielding a ΔΔG value for each substituent (eqs 4 and 5).
| 4 |
| 5 |
On the basis of the ΔΔG values of the substituents, we found that an EDG (i.e., compound 1b) results in a more favorable and hence presumably faster dehydration into 2b as compared to the dehydration of PGAH (1a) into PGA (2a) (ΔΔGMe,dehydration = −0.27 kcal/mol). On the other hand, the subsequent reaction of 2b with urea is less favorable compared to reaction of 2a with urea (ΔΔGMe,intermediate = 0.35 kcal/mol). In other words, the reaction of 2b with urea is enhanced to a greater extent than the dehydration of 1b, thus explaining the slightly lower kPFO value of 1b as compared to that of 1a. In contrast, the influence of the EWG (i.e., compound 1c) results in an increase of Gibbs free energy for the dehydration of 1c into 2c (ΔΔGNO2,dehydration = 0.94 kcal/mol) and in a more favorable reaction of 2c with urea (ΔΔGNO2,intermediate = −0.41 kcal/mol), as compared to unsubstituted PGAH. For PGAH bearing an EWG (1c), the less favorable dehydration affects the overall rate to a greater extent than the reaction with urea, resulting in substantially slower reaction kinetics as well. On the basis of this analysis, it is concluded that a PGAH-containing sorbent in which the aromatic ring is directly connected to an electron-donating polycarbon backbone (such as in PS) has probably a somewhat reduced reactivity as compared to unsubstituted PGAH but that this cannot readily be compensated by other or additional (EDG or EWG) substituents.
An overview of the synthesis route toward PGAH-type sorbents based on PS is shown in Scheme 2A. For the preparation of PS, we essentially used a method as described by Jong et al.17 Macroporous PS beads were synthesized by suspension copolymerization of styrene and a low content of divinylbenzene (DVB, 2.5%) in a cylindrical reactor with a mechanical stirrer (Supporting Information section 3). A mixture of toluene and ShellSolTD (9:91 v/v) was used as a nonsolvating porogen,29 and spherical beads (Figure 2A) were obtained in 97% yield. The average diameter of the beads as determined by light microscopy was 0.49 ± 0.18 mm. SEM analysis (Figure 2B) showed that pores are clearly visible on the surface of the beads. The surface area (SBET) and pore volume of the beads as determined by nitrogen physisorption were 36.3 m2/g and 0.32 mL/g, respectively. The plot of the pore volume versus the pore diameter (Figure 2C) shows that the pores present in the material were mainly in the range of 50–100 nm, demonstrating that the obtained beads are indeed macroporous.30
Scheme 2. Synthesis of PGAH-Type Sorbents from (A) Styrene and (B) VPE.

R1 = crosslinker, R2 = unmodified styrene, R3 = side product; see Figure 3C and Scheme 3.
Figure 2.

Characteristics of the styrene-based PGAH sorbent (PS-Ac-Ox: G-I) and its precursors (PS beads: A–C; PS-Ac beads: D-F): photographs (left images), typical SEM images (middle), and pore size/volume distributions (right figures).
The aromatic groups of the PS beads suspended in DCE were subsequently acetylated in a Friedel–Crafts reaction using acetyl chloride as the reactant and AlCl3 as the catalyst. The obtained acetylated PS beads (71.6 g, PS-Ac, Figure 2D) were characterized using SEM (Figure 2E), light microscopy, and nitrogen physisorption (Figure 2F). The PS-Ac beads showed similar characteristics as PS; the SBET surface area was similar (43.4 m2/g) as were the size (0.61 ± 0.17 mm) and the pore size/volume distribution (Figure 2F), demonstrating that the Friedel–Crafts reaction did not adversely affect the macroporosity or degrade the beads, possibly because the reaction temperature (50 °C) was far below the glass transition temperatures of both the PS and PS-Ac beads (Tg’s were 119 and 184 °C, respectively, Supporting Information section 4.1). Infrared (IR) spectroscopic analysis of PS-Ac showed the presence of a new peak at 1675 cm–1, assigned to the C=O stretching vibration of the acetyl group,31 demonstrating that acetylation indeed had taken place (Supporting Information section 6). To quantify the degree of acetylation of the aromatic groups, the PS-Ac beads were analyzed by quantitative 13C solid-state NMR spectroscopy (Supporting Information section 7). Comparison of the integral from the carbonyl carbons (180–200 ppm) with that from the aromatic peaks (110–160 ppm) and the aliphatic peaks (10–50 ppm) shows that approximately 60% of the styrene units have been acetylated.
The acetyl aromatic groups in PS-Ac beads were halogenated and subsequently converted into PGAH groups by a Kornblum oxidation in a one-pot procedure using a mixture of concentrated aqueous HBr and DMSO, thereby yielding PS-Ac-Ox beads.32,33 To establish the optimal reaction time for these oxidizing conditions to obtain the highest PGAH density, beads were taken from the reaction mixture at different time points, and their urea BC was determined (Supporting Information sections 8 and 10 and Figure 3A).
Figure 3.

(A) Plot of the urea BC of PS-Ac-Ox beads as a function of oxidation time of Ps-Ac. Conditions for oxidation: Ps-Ac (3.5 g) in DMSO (35 mL) and 48% aqueous HBr (10 mL) stirred mechanically at 80 °C. (B) IR spectra of PS-Ac-Ox-8h (lower spectrum) and PS-Ac-Ox-48h (upper spectrum). (C) Reaction scheme of the oxidation of PS-Ac using HBr and DMSO. PGA = phenylglyoxaldehyde, PGAH = phenylglyoxaldehyde hydrate, PGOA = phenylglyoxilic acid.
Figure 3A shows that the urea BC of the beads increased with oxidation time during the first 8 h to 1.8 mmol/g, demonstrating successful oxidation of the acetyl group into PGAH/PGA (Figure 3A, blue line). However, at longer reaction times, the BC decreased (Figure 3A, red line). IR analysis of the sorbent obtained after 8 h of oxidation (Ps-Ac-Ox-8h; which had the highest BC) showed a single carbonyl peak at 1675 cm–1 with a minor shoulder peak at 1740 cm–1, whereas the sorbent obtained after 48 h of oxidation (Ps-Ac-Ox-48h) showed two carbonyl peaks at 1675 and 1740 cm–1 (Figure 3B). Floyd et al. reported that oxidation of 2-bromoacetophenone with DMSO resulted in a mixture of PGAH and phenylglyoxilic acid (PGOA, structure shown in Figure 3C). It is therefore concluded that overoxidation occurred when the beads were exposed to the oxidizing mixture for more than ∼8 h, resulting in the formation of carboxylic acids groups (Figure 3C). The shoulder peak at 1740 cm–1 in the PS-Ac-Ox-8h sample (Figure 3B) indicates that the overoxidation of PGA/PGAH into PGOA already occurred during the first 8 h of reaction, but it is slower than the oxidation of the acetyl group into the PGA/PGAH group.
The sorbent was analyzed by quantitative 13C solid-state NMR to quantify the amount of PGAH groups in PS-Ac-Ox (Supporting Information Figure S20). The CH3 peak of the acetyl group detected in the 13C NMR spectrum of PS-Ac (Supporting Information Figure S19) had disappeared, indicating that all acetyl groups had been converted. Comparison of the area under the hydrate carbon peak (80–100 ppm) with that of the aromatic peaks (110–160 ppm) and the aliphatic peaks (10–50 ppm) shows that ∼40% of the aromatic groups (thus ∼67% of the acetyl groups) had been converted into PGAH groups. In addition, a minor peak at around 165 ppm was detected, which is assigned to the carboxylic acid carbonyl peak from PGOA (Supporting Information Figure S20).
PS-Ac beads were also oxidized for 8 h on a larger scale (i.e., 60 instead of 3.5 g), and the obtained beads (PS-Ac-Ox) were characterized by SEM, light microscopy, and nitrogen physisorption (Figure 2G–I). PS-Ac-Ox beads had a similar size (0.54 ± 0.11 mm), surface area (37.0 m2/g), and pore volume and pore size/volume distribution (Figure 2I) as PS and PS-Ac. This confirms that also the oxidation reaction had neither affected the macroporosity nor degraded the beads, likely because the reaction temperature (80 °C) was below the glass transition temperatures (Tg) of both the PS-Ac and PS-Ac-Ox beads (Tg’s of dry beads were 184 and >230 °C, respectively (Supporting Information section 4.1)). The PGAH content of the sorbent according to 13C NMR (Supporting Information Figure S21) was similar to the sorbent prepared at small scale, while the urea BC of PS-Ac-Ox was lower than that of the batch prepared at small scale (i.e., 1.4 versus 1.8 mmol/g at the 3.5 g scale) (Table 1 and Supporting Information section 10). This difference can be explained by the relatively large error (∼10%) in quantification of the PGAH groups by 13C NMR.
Table 1. Characteristics of PS, PS-Ac, and PS-Ac-Ox Beads (after 8 h of Oxidation of PS-Ac).
| beads | diameter (mm)a | surface area (m2/g)b | total pore volume (mL/g)b | functionalizationc | urea BC (mmol/g) |
|---|---|---|---|---|---|
| PS | 0.49 ± 0.18 | 36.3 | 0.32 | ||
| PS-Ac | 0.61 ± 0.17 | 43.4 | 0.31 | ∼60% acetylation | |
| PS-Ac-Ox (small scale) | n.d. | n.d. | n.d. | ∼40% PGAH groups | 1.8 |
| Ps-Ac-Ox (large scale) | 0.54 ± 0.11 | 37.0 | 0.31 | ∼40% PGAH groups | 1.4 |
Determined by light microscopy.
Determined by N2 physisorption.
Determined by 13C solid-state NMR.
One option to obtain a sorbent with higher BC is to increase the density of reactive PGAH units. We hypothesized that this can be accomplished by bypassing the incomplete Friedel–Crafts acetylation of PS (∼60%). Therefore, a novel route was explored in which VPE was used as the monomer, which can be polymerized, yielding a polymer with 100% acetylated aromatic groups, which requires only one postpolymerization step, i.e., the oxidation of the acetyl group (Scheme 2B). To allow proper comparison with the route based on styrene (Scheme 2A), we selected VPE with the vinyl group at the para position with respect to the acetyl group (Scheme 2B). The VPE monomer was synthesized on a 10 g scale from ethynylphenylethanone, in which the triple bond was reduced to a double bond using Lindlar’s catalyst and hydrogen. The reaction time was carefully monitored to prevent over-reduction of the double bond into the single bond (Supporting Information section 5).
Next, the conditions needed to obtain macroporous sorbent beads based on the monomer VPE (Scheme 2B) were determined. Crucial to obtain macroporous sorbent beads is the identification of the optimal porogen.30,34,35 The suspension polymerization conditions of styrene were selected as a starting point, i.e., using toluene with ShellSolTD as the nonsolvating porogen. Importantly, the nonsolvating porogen should dissolve VPE and precipitate pVPE. When the cross-linked polymer does not swell in the porogen, this will result in phase separation and yield porosity.29 However, ShellSolTD and VPE are not miscible, and therefore, ShellSolTD/toluene mixtures with high volumes of ShellSolTD are nonsolvents for VPE, and no polymeric beads were obtained by suspension polymerizations at various volumetric ratios of ShellSolTD as the porogen.
The Hildebrand solubility parameter (δ) of a molecule or polymer, which is defined as the square root of the cohesive energy density, is commonly used to predict the miscibility of solvents, monomers, and polymers because compounds with a similar δ value are likely miscible. This solubility parameter can be determined experimentally or calculated based on the molecular structure, i.e., by the method reported by Fedors.36 ShellSolTD is a mixture of alkanes, and therefore, ShellSolTD was replaced by heptane, for which δ = 15.2 J1/2 m–3/2,37 and the heptane/toluene ratio was adjusted to vary the δ value between 15.2 and 18.2 J1/2 m–3/2 (i.e., the δ value of toluene).37,38 The calculated δ values of VPE (δ = 19.0 J1/2 m–3/2)36 and of pVPE (δ = 21.1 J1/2 m–3/2)36 are higher than those of styrene (17.8 J1/2 m–3/2)39 and PS (δ = 17.4–19.0 J1/2 m–3/2);37,40 thus, the δ of a suitable porogen mixture (δmix) is also expected to be higher than the δmix of the porogen used for the suspension polymerization of PS. Volumetric mixtures of 75:25 and 50:50 heptane and toluene with δmix values of 15.6 and 16.9 J1/2 m–3/2, respectively, did dissolve VPE and resulted in the formation polymeric beads (Table 2, entries 1 and 2). However, the SBET surface areas of the beads as determined with nitrogen physisorption were low (SBET < 0.05 m2/g), indicating that the δmix value of these porogen mixtures is too low, resulting in early precipitation of pVPE without the formation of macropores.
Table 2. Effect of the Porogen in the Suspension Copolymerization of VPE with DVB on the Surface Area of Polyvinylphenylethanone.
| entry | porogen | porogen ratio | δmix porogen (J1/2 m–3/2)b | % DVB | bead diameter (mm) | yield (%) | SBET surface area (m2/g) |
|---|---|---|---|---|---|---|---|
| 1a | heptane/toluene | 75:25 | 15.6 | 3 | 0.62 ± 0.22 | 93 | <0.05 |
| 2 | heptane/toluene | 50:50 | 16.9 | 3 | 0.40 ± 0.28 | 99 | <0.05 |
| 3 | heptane/toluene | 40:60 | 17.5 | 3 | 0.48 ± 0.14 | 65 | 0.1 |
| 4 | heptane/toluene | 30:70 | 17.8 | 3 | 0.61 ± 0.23 | 75 | 2.0 (1.9c) |
| 5 | heptane/toluene | 20:80 | 18.1 | 3 | 0.66 ± 0.21 | 66 | 0.2 |
| 6 | heptane/toluene | 10:90 | 18.2 | 3 | 0.47 ± 0.10 | 70 | <0.05 |
| 7 | toluene | - | 18.2 | 6 | 0.71 ± 0.23 | 69 | 0.2 |
| 8a | toluene/nitrobenzene | 90:10 | 18.2 | 3 | 0.57 ± 0.34 | 52 | <0.05 |
| 9 | toluene/nitrobenzene | 80:20 | 18.4 | 6 | 0.55 ± 0.19 | 82 | <0.05 |
| 6 |
To further tune the porogen mixture to induce precipitation in a later stage of the polymerization, the δ value of the porogen mixture (δmix) was stepwise increased and therefore closer to the δ value of pVPE, from 16.9 to 18.2 J1/2 m–3/2 using mixtures of heptane and toluene (Table 2, entries 2–7). To further increase the δmix, mixtures of nitrobenzene (δ = 21.7 J1/2 m–3/2)41 and toluene (δ = 18.2 J1/2 m–3/2)37 were used in 10:90 and 20:80 ratios, respectively (Table 2, entries 8 and 9). The 30:70 mixture of heptane and toluene (entry 4), for which the calculated δmix value of the porogen mixture is 17.8 J1/2 m–3/2, yielded the sorbent with the largest SBET surface area of 2.0 m2/g. Interestingly, the difference in the calculated δ value of pVPE and the 30:70 heptane/toluene mixture (21.1–17.8 = 3.3 J1/2 m–3/2) is similar to the difference between the δ value of PS and that of the 91:9 ShellSolTD/heptane mixture (18.2 ± 0.8–15.2 = 3.0 ± 0.8 J1/2 m–3/2). However, the surface area of the obtained pVPE beads was substantially smaller than that of the PS beads (2.0 and 36.3 m2/g, respectively). It is important to note that at the start of the polymerization the δmix of the porogen is higher than at the end of the polymerization, due to VPE dissolved in the porogen (δmix = 18.3 J1/2 m–3/2; the δmix gradually decreases during the polymerization reaction as a result of VPE depletion). Therefore, the initial difference between the calculated δ values of pVPE and the porogen–VPE mixture (21.1–18.3 = 2.8 J1/2 m–3/2) is actually bigger than the initial difference between the δ value of PS and that of the porogen–styrene mixture (18.2 ± 0.8–16.5 = 1.7 ± 0.8 J1/2 m–3/2), and therefore, precipitation of pVPE probably occurred at an earlier stage of the polymerization reaction than PS did.
To assess the influence of porosity on the urea BC, the pVPE beads of entries 2 (SBET < 0.05 m2/g) and 4 (SBET = 2.0 m2/g) were selected for oxidation. The beads of low surface area (entry 2) were oxidized for 4–12 h under the same conditions as those applied for PS-Ac. The urea BCs of the resulting oxidized pVPE beads (pVPE-Ox-(2) were 1.8–2.2 mmol/g, of which the highest BC (2.2 mmol/g) was obtained after 12 h of oxidation (Supporting Information sections 9 and 10). The pVPE beads with the highest surface area (entry 4) were therefore oxidized for 12 h, and the urea BC of these beads (pVPE-Ox-(4)) was 1.8 mmol/g (Supporting Information section 10). The surface area of the pVPE-Ox-(4) determined by nitrogen physisorption was similar to that of the corresponding pVPE beads (1.9 vs 2.0 m2/g), most likely because the reaction temperature of the oxidation reaction (80 °C) is much lower than the Tg of pVPE beads (147 °C, Supporting Information Figure S12), and the beads therefore remain dimensionally stable under these oxidizing reaction conditions.
As hypothesized, the VPE-based materials showed a higher urea BC than the styrene-based materials because of the increase in the density of acetyl groups and therefore a higher PGAH content after oxidation (1.4–1.8 vs. 1.8–2.2 mmol/g). Surprisingly, the surface area of pVPE beads had no influence on the urea BC (1.8–2.2 and 1.8 mmol/g for pVPE-Ox-(2) and pVPE-Ox-(4), respectively). This shows that PGAH groups are accessible for urea also in materials without macroporosity, possibly because the sorbents swell to a minor but sufficient extent in water due to the polar and hydrophilic carbonyl groups and the carboxylic acid groups of PGOA due to overoxidation of PGA/PGAH (Figure 3C). In addition, upon urea binding, the beads become more hydrophilic, further enhancing accessibility for water and urea, thereby further improving urea binding kinetics.
The average size of pVPE-Ox-(4) beads determined by light microscopy was slightly larger than that of the pVPE beads (0.77 ± 0.20 and 0.61 ± 0.23 mm, respectively). Due to the swelling/deswelling of the beads during nitrogen physisorption experiments, the pore/volume distribution for these materials could not be determined.
The pVPE-Ox-(4) and corresponding pVPE beads were analyzed by SEM (Figure 4B,C,E,F). Unlike the beads obtained in the suspension polymerization of styrene, the obtained pVPE beads were hollow as they were deflated after drying under vacuum, which suggests core–shell phase separation during the polymerization reaction. In general, core–shell structures are thermodynamically favorable in water–oil–polymer mixtures when the surface tension between the water and the oil phase (γWO) is greater than the surface tension between the water and the polymer phase (γWP) and the polymer and oil phase (γPO) combined (γWO > γWP + γPO).42,43 Apparently, due to the polar carbonyl groups present in pVPE, γWP decreased as compared to the γWP between water and PS.
Figure 4.

Photographs of the VPE-based PGAH sorbent beads (pVPE-Ox-(4): D–F) and their precursor (pVPE beads: A–C): photographs (left images) and typical SEM images (middle and right).
To determine the density of PGAH groups in pVPE-Ox-(2) and pVPE-Ox-(4), these materials were analyzed by 13C solid-state NMR spectroscopy (Supporting Information Figures S22 and S23). Comparison of the hydrate peak integral (80–100 ppm) with the backbone peak integral (10–50 ppm) demonstrates a PGAH content of ∼50% for both pVPE-Ox-(2) and pVPE-Ox-(4), which confirms that higher PGAH contents are obtained using the VPE instead of the styrene route (∼50 and ∼40%, respectively).
Like PS-Ac-Ox beads, pVPE-Ox beads also clearly show the peak at around 165 ppm (assigned to the carboxylic acid group of PGOA) due to overoxidation of PGAH, resulting in the formation of PGOA (Figure 3C). Moreover, an additional peak at around 70 ppm was detected in the 13C NMR spectrum of pVPE-Ox. Presumably, due to the higher density of acetyl groups in pVPE as compared to PS-Ac, an aldol condensation between neighboring PGA and remaining acetyl groups had occurred during the oxidation reaction, as was described for the oxidation of acetophenone into PGA/PGAH (Scheme 3).44,45 Because PS was only acetylated for 60%, this side reaction likely also occurred to a smaller extent in this material, as indicated by a very minor peak at 70 ppm in the spectrum of PS-Ac-Ox (Supporting Information Figures S20 and S21).
Scheme 3. Aldol Reaction between a Dehydrated PGA Group and the Enol Tautomer of the Acetyl Group, Giving Rise to the Signal at 70 ppm in the Quantitative 13C Solid-State NMR Spectrum.

It was found that the pVPE-Ox sorbent beads, which were ∼50% functionalized with PGAH groups, had a urea BC of ∼2 mmol/g. However, a 100% functionalized sorbent contains 5.5 mmol/g PGAH groups (including a 3 mol % cross-linker) based on the molecular weight of the monomer (178 g/mol), which implies that a sorbent with ∼50% PGAH groups would have a urea BC of 2.8 mmol/g at most. There are two reasons why the actual urea BC for a sorbent functionalized with PGAH groups is lower than the theoretical urea BC based on a 1:1 reaction of urea with PGAH. First, some of the PGAH groups might be inaccessible for urea. Second, PGAH can react with urea in both a 1:1 and a 2:1 ratio (Scheme 1), and therefore, one potential binding site is lost when PGAH reacts with urea in a 2:1 ratio.
Quantification of the inaccessible and therefore unreacted PGAH groups in beads with ∼2 mmol urea/g sorbent with 13C NMR spectroscopy is not possible because unreacted PGAH and reacted PGAH give rise to signals in the same region of the spectrum. Therefore, the sorbent beads that had reacted with urea (PS-Ac-Ox@urea and pVPE-Ox-(4)@urea) were analyzed with IR spectroscopy, along with PGAH and the 2:1 adduct of PGAH and urea (3′a) (Figure S18). PGAH shows a clear ketone–carbonyl stretching vibration at 1700 cm–1 and a C–O stretching vibration at 1210 cm–1. However, these peaks have a lower intensity in the IR spectra of PS-Ac-Ox@urea and pVPE-Ox-(4)@urea, and the main carbonyl peak is clearly shifted (from 1700 to 1740 cm–1). On the basis of these observations, it is concluded that the majority of the PGAH groups were indeed accessible for reaction with urea and had reacted. This agrees with the observation that the surface area does not influence the urea BC. Moreover, the IR spectra of PS-AC-Ox@urea and pVPE-Ox-(4)@urea are more similar to the IR spectrum of the isolated 2:1 addition product 3′a (Figure S18). The several peaks arising from the carbonyl stretching vibration in the region of 1650–1800 cm–1 of 3′a are also present in the spectra of PS-AC-Ox@urea and pVPE-Ox-(4)@urea. Therefore, it is concluded that reaction of the 1:1 PGAH:urea adduct with a second PGA group takes place in the sorbent beads at least to some extent, thereby explaining the difference between the urea BC of the sorbents (∼2.0 mmol/g) and the theoretical capacity based on the actual PGAH content (2.8 mmol/g). From this difference, we then calculated that (2.8 – 2.0 =) 0.8 mmol/g PGAH was lost because it reacted with the 1:1 PGAH:urea adduct. Therefore (2 × 0.8 =) 1.6 mmol/g (57%) of PGAH reacted in a 2:1 ratio with urea, and thus, (2.8 – 1.6 =) 1.2 mmol/g (43%) of the PGAH groups present in the sorbent reacted in a 1:1 ratio with urea.
The kinetics of the urea binding of the two different types of PGAH-type sorbents was investigated by incubating them in a 30 mM urea solution in PBS at 37 °C, conditions representative for the regeneration of dialysate.7 The urea binding was determined by measuring the urea concentration in the solution at different time points (Supporting Information section 11 and Figure 5A). The PS-Ac-Ox sorbent showed a binding of 0.5–0.6 mmol/g after 24 h. However, the sorbent pVPE-Ox-(4), which showed a higher maximum BC than PS-Ac-Ox (1.8 vs 1.4 mmol/g), bound 0.5–0.6 mmol per gram already within 8 h, which increased to 0.8–0.9 mmol/g after 24 h. In fact, when the amount of urea bound was normalized to the maximum BC of the respective sorbent (Figure 5B), both materials reached ∼45% of the maximum BC after 24 h, which shows that both PS-Ac-Ox and pVPE-Ox-(4) displayed similar urea removal kinetics.
Figure 5.

Urea binding of PS-Ac-Ox and pVPE-Ox-(4) in time. (A) Binding expressed in mmol urea/g sorbent. (B) Relative urea binding (percentage of the maximum BC). Conditions: sorbent (10 mg/mL) in 30 mM urea solution in PBS at 37 °C (N = 4).
These PGA-type sorbents are not specific for urea and can likely also form covalent bonds with other nucleophilic species present in dialysate such as creatinine and amino acids. However, because only ∼30% of the PGA groups has reacted in 8 h, it is expected that other nucleophilic solutes present in the dialysate in low concentration (such as creatinine and amino acids) do not limit the urea removal in a 8 h dialysis session. In addition, reaction of PGA groups with urea (both in a 1:1 and a 2:1 ratio) is irreversible; therefore, the sorbent cannot be regenerated for reuse and is thus disposable.
Conclusions
Urea sorbent beads containing phenylglyoxaldehyde hydrate (PGAH) groups were successfully prepared via suspension polymerization of either styrene or vinylphenylethanone (VPE) followed by acetylation and oxidation or oxidation only respectively. The VPE route turned out to be the best choice as it saves one postpolymerization modification step and, importantly, resulted in a sorbent with higher PGAH content (∼50 vs ∼40% for PS-based sorbents) and concomitantly higher BC (1.8–2.2 vs 1.4–1.8 mmol/g for PS-based sorbents), which makes them among the best urea sorbents reported in the literature. In addition, the PS-based sorbents are inexpensive and synthetically accessible and therefore potentially suitable for application as urea sorbents in a wearable artificial kidney.
The accessibility of the PGAH groups in the VPE-based sorbents is not dependent on the surface area of the material, possibly because the beads swell to a minor extent.
The kinetics of urea sorption from simulated dialysate showed that ∼30% of the BC is reached after 8 h at 37 °C. The best sorbent developed (pVPE-Ox-(4)) bound ∼0.5–0.6 mmol/g in 8 h, which demonstrates that ∼700 g of this PGAH-type sorbent is needed to remove the daily urea production of 400 mmol of ESKD patients during a dialysis session of 8 h.
Acknowledgments
The authors would like to thank Carl C. L. Schuurmans for his help with determining the size of the sorbent particles and Lies A. L. Fliervoet for making Figures 2 and 4 and Shell Amsterdam for the gift of ShellSolTD. This research was supported by the Dutch Organization for Scientific Research (NWO-TTW, Project 14433) and the Dutch Kidney Foundation. The NMR experiments were supported by a TOP-PUNT grant to M.B. (NWO Grant Number 718.015.001). R.D. acknowledges the European Research Council (ERC) for funding (ERC-2014-CoG 648991). M.E.M. acknowledges the Sectorplan Natuur- en Scheikunde (Tenure-track grant at Utrecht University) for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsapm.9b00948.
Raw data of the experiments for determination of rate constants; calculated G values for all PGAH analogues, PGA analogues and intermediates, and ΔG and ΔΔG values for dehydration and the reaction of PGA analogues with urea; pictures of the reactor used for suspension polymerization; DSC and TGA analyses of the monomers and polymers; reaction scheme of the synthesis of VPE; IR characterization of the sorbents; quantitative 13C solid-state NMR spectra; urea sorption kinetics at 70 °C to determine the maximum BC; optimization of the oxidation time of pVPE; raw data for the determination of the maximum BCs and static urea sorption of PS-Ac-Ox and pVPE-Ox-(4); and coordinates for optimized structures calculated compounds and intermediates (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Vanholder R.; Gryp T.; Glorieux G. Urea and chronic kidney disease: the comeback of the century? (in uraemia research). Nephrol., Dial., Transplant. 2018, 33 (1), 4–12. 10.1093/ndt/gfx039. [DOI] [PubMed] [Google Scholar]
- Gura V.; Rivara M. B.; Bieber S.; Munshi R.; Smith N. C.; Linke L.; Kundzins J.; Beizai M.; Ezon C.; Kessler L.; Himmelfarb J. A wearable artificial kidney for patients with end-stage renal disease. JCI Insight 2016, 1 (8), 15. 10.1172/jci.insight.86397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agar J. W. Review: understanding sorbent dialysis systems. Nephrology 2010, 15 (4), 406–11. 10.1111/j.1440-1797.2010.01321.x. [DOI] [PubMed] [Google Scholar]
- Davenport A.; Gura V.; Ronco C.; Beizai M.; Ezon C.; Rambod E. A wearable haemodialysis device for patients with end-stage renal failure: a pilot study. Lancet 2007, 370 (9604), 2005–10. 10.1016/S0140-6736(07)61864-9. [DOI] [PubMed] [Google Scholar]
- Wester M.; van Gelder M. K.; Joles J. A.; Simonis F.; Hazenbrink D. H. M.; van Berkel T. W. M.; Vaessen K. R. D.; Boer W. H.; Verhaar M. C.; Gerritsen K. G. F. Removal of urea by electro-oxidation in a miniature dialysis device: a study in awake goats. Am. J. Physiol. Renal. Physiol. 2018, 315 (5), F1385–F1397. 10.1152/ajprenal.00094.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann H. D.; Marten R.; Fahrner I.; Gullberg C. A. Urea elimination using a cold activated-carbon artificial tubulus for hemofiltration. Artif. Organs 1981, 5 (4), 351–356. 10.1111/j.1525-1594.1981.tb04016.x. [DOI] [PubMed] [Google Scholar]
- Van Gelder M. K.; Mihaila S. M.; Jansen J.; Wester M.; Verhaar M. C.; Joles J. A.; Stamatialis D.; Masereeuw R.; Gerritsen K. G. F. From portable dialysis to a bioengineered kidney. Expert Rev. Med. Devices 2018, 15 (5), 323–336. 10.1080/17434440.2018.1462697. [DOI] [PubMed] [Google Scholar]
- Shimizu T.; Fujishige S. A newly prepared surface-treated oxystarch for removal of urea. J. Biomed. Mater. Res. 1983, 17 (4), 597–612. 10.1002/jbm.820170405. [DOI] [PubMed] [Google Scholar]
- Deepak D. Evaluation of adsorbents for the removal of metabolic wastes from blood. Med. Biol. Eng. Comput. 1981, 19 (6), 701–6. 10.1007/BF02441331. [DOI] [PubMed] [Google Scholar]
- Smakman R.Macromolecular carbonyl groups containing material suitable for use as sorbent for nitrogen compounds. U.S. Patent 4,897,200, 1990.
- Smakman R.; van Doorn A. W. Urea removal by means of direct binding. Clin. Nephrol. 1986, 26 (Suppl 1), S58–62. [PubMed] [Google Scholar]
- Poss M. J.; Blom H.; Odufu A.; Smakman R.. Macromolecular ketoaldehydes. U.S. Patent 6,861,473 B2, 2005.
- Kuntz E.; Quentin J. P.. Alkenylaromatic polymers with α-ketoalhydic groups. U.S. Patent 3.933.753, 1976.
- Kuntz E.; Quentin J. P.. Process for extracting urea from a solution with alkenylaromatic polymers with a-ketoalhydic groups. U.S. Patent 3.933.753, 1977.
- Jong J. A. W.; Smakman R.; Moret M.-E.; Verhaar M. C.; Hennink W. E.; Gerritsen K. G. F.; Van Nostrum C. F. Reactivity of (vicinal) carbonyl compounds with urea. ACS Omega 2019, 4 (7), 11928–11937. 10.1021/acsomega.9b01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch Micheal J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J.; Fox D.. Gaussian 09, revision A.02; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Jong G. J. D.Ion exchangers from poly(aminostyrene) and ethylene imine. U.S. Patent US3582505A, 1971.
- Bassetti M.; Ciceri S.; Lancia F.; Pasquini C. Hydration of aromatic terminal alkynes catalyzed by iron(III) sulfate hydrate under chlorine-free conditions. Tetrahedron Lett. 2014, 55 (9), 1608–1612. 10.1016/j.tetlet.2014.01.083. [DOI] [Google Scholar]
- Molander G. A.; Brown A. R. Suzuki-Miyaura cross-coupling reactions of potassium vinyltrifluoroborate with aryl and heteroaryl electrophiles. J. Org. Chem. 2006, 71 (26), 9681–6. 10.1021/jo0617013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmark S. E.; Butler C. R. Vinylation of aryl bromides using an inexpensive vinylpolysiloxane. Org. Lett. 2006, 8 (1), 63–66. 10.1021/ol052517r. [DOI] [PubMed] [Google Scholar]
- Schuurmans C. C. L.; Abbadessa A.; Bengtson M. A.; Pletikapic G.; Eral H. B.; Koenderink G.; Masereeuw R.; Hennink W. E.; Vermonden T. Complex coacervation-based loading and tunable release of a cationic protein from monodisperse glycosaminoglycan microgels. Soft Matter 2018, 14 (30), 6327–6341. 10.1039/C8SM00686E. [DOI] [PubMed] [Google Scholar]
- Fung B. M.; Khitrin A. K.; Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142 (1), 97–101. 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- Thommes M.; Kaneko K.; Neimark A. V.; Olivier J. P.; Rodriguez-Reinoso F.; Rouquerol J.; Sing K. S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051. 10.1515/pac-2014-1117. [DOI] [Google Scholar]
- Barrett E. P.; Joyner L. G.; Halenda P. P. The determination of pore volume and area distributions in porous substances. I., Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73 (1), 373–380. 10.1021/ja01145a126. [DOI] [Google Scholar]
- Jura G.; Harkins W. D. Surfaces of Solids. XI. Determination of the decrease (π) of free surface energy of a solid by an adsorbed film. J. Am. Chem. Soc. 1944, 66 (8), 1356–1362. 10.1021/ja01236a046. [DOI] [Google Scholar]
- Talke H.; Schubert G. E. Enzymatic urea determination in the blood and serum in the warburg optical test. Klin. Wochenschr. 1965, 43, 174–5. 10.1007/BF01484513. [DOI] [PubMed] [Google Scholar]
- Jong J. A. W.; Moret M. E.; Verhaar M. C.; Hennink W. E.; Gerritsen K. G. F.; van Nostrum C. F. Effect of substituents on the reactivity of ninhydrin with urea. Chemistry Select 2018, 3 (4), 1224–1229. 10.1002/slct.201800040. [DOI] [Google Scholar]
- Clayden J.; Warren S.; Greeves N.; Wothers P.. Organic Chemistry; Oxford University Press, 2000; p 561. [Google Scholar]
- Gokmen M. T.; Du Prez F. E. Porous polymer particles—A comprehensive guide to synthesis, characterization, functionalization and applications. Prog. Polym. Sci. 2012, 37 (3), 365–405. 10.1016/j.progpolymsci.2011.07.006. [DOI] [Google Scholar]
- Durie S.; Jerabek K.; Mason C.; Sherrington D. C. One-Pot synthesis of branched poly(styrene-divinylbenzene) suspension polymerized resins. Macromolecules 2002, 35 (26), 9665–9672. 10.1021/ma0209794. [DOI] [Google Scholar]
- Cacchi S.; Fabrizi G.; Gavazza F.; Goggiamani A. Palladium-catalyzed reaction of aryl iodides with acetic anhydride. A carbon monoxide-free synthesis of acetophenones. Org. Lett. 2003, 5 (3), 289–291. 10.1021/ol027243b. [DOI] [PubMed] [Google Scholar]
- Floyd M. B.; Du M. T.; Fabio P. F.; Jacob L. A.; Johnson B. D. The oxidation of acetophenones to arylglyoxals with aqueous hydrobromic acid in dimethyl sulfoxide. J. Org. Chem. 1985, 50 (25), 5022–5027. 10.1021/jo00225a004. [DOI] [Google Scholar]
- Kornblum N.; Jones W. J.; Anderson G. J. A New and selective method of oxidation - the conversion of alkyl halides and alkyl tosylates to aldehydes. J. Am. Chem. Soc. 1959, 81 (15), 4113–4114. 10.1021/ja01524a080. [DOI] [Google Scholar]
- Liu Q. Q.; Wang L.; Xiao A. G. Research progress in macroporous styrene-divinylbenzene co-polymer microspheres. Des. Monomers Polym. 2007, 10 (5), 405–423. 10.1163/156855507781833620. [DOI] [Google Scholar]
- Wood C. D.; Cooper A. I. Synthesis of macroporous polymer beads by suspension polymerization using supercritical carbon dioxide as a pressure-adjustable porogen. Macromolecules 2001, 34 (1), 5–8. 10.1021/ma001514l. [DOI] [Google Scholar]
- Fedors R. F. A method for estimating both the solubility parameters and molar volumes of liquids. Polym. Eng. Sci. 1974, 14 (2), 147–154. 10.1002/pen.760140211. [DOI] [Google Scholar]
- Van Krevelen D. M.Properties of Polymers; 1990; pp 198–203. [Google Scholar]
- Cameron N. R.; Barbetta A. The influence of porogen type on the porosity, surface area and morphology of poly(divinylbenzene) PolyHIPE foams. J. Mater. Chem. 2000, 10 (11), 2466–2471. 10.1039/b003596n. [DOI] [Google Scholar]
- Cai Y.; Yan W.; Peng X.; Liang M.; Yu L.; Zou H. Influence of solubility parameter difference between monomer and porogen on structures of poly (acrylonitrile-styrene-divinylbenzene) resins. J. Appl. Polym. Sci. 2019, 136 (3), 46979. 10.1002/app.46979. [DOI] [Google Scholar]
- Matsukawa H.; Yoda S.; Okawa Y.; Otake K. Phase behavior of a carbon dioxide/methyl trimethoxy silane/polystyrene ternary system. Polymers 2019, 11 (2), 246. 10.3390/polym11020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen C. M.The three dimensional solubility parameter and solvent diffusion coefficient, their importance in surface coating formulation; Danish Technical Press: Copenhagen, 1967. [Google Scholar]
- Jonsson M.; Nordin O.; Malmström E.; Hammer C. Suspension polymerization of thermally expandable core/shell particles. Polymer 2006, 47 (10), 3315–3324. 10.1016/j.polymer.2006.03.013. [DOI] [Google Scholar]
- Sundberg D. C.; Casassa A. P.; Pantazopoulos J.; Muscato M. R.; Kronberg B.; Berg J. Morphology development of polymeric microparticles in aqueous dispersions. I., Thermodynamic considerations. J. Appl. Polym. Sci. 1990, 41 (7–8), 1425–1442. 10.1002/app.1990.070410706. [DOI] [Google Scholar]
- Liu Y.; Zhao H.; Tian G.; Du F.; Qi Y.; Wen Y. A novel coupling reaction of α-halo ketones promoted by SmI3/CuI. RSC Adv. 2016, 6 (31), 26317–26322. 10.1039/C5RA26604A. [DOI] [Google Scholar]
- Dudley H. W.; Ochoa S. 157. Benzoylphenacylcarbinol. J. Chem. Soc. 1933, 0, 625. 10.1039/jr9330000625. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.