

Abstract
Phosphatidylinositol phosphates (PIPs) function as important second messengers in many cellular events. In the human intestinal protist Entamoeba histolytica, where phagocytosis/trogocytosis plays an indispensable role in proliferation and pathophysiology during infection, various PIPs are involved in multiple steps of phago/trogocytosis. PI3‐phosphate (PI3P) plays a pivotal role in the biogenesis of phagosome/trogosomes via recruitment of PI3P effectors. Because no known PI3P downstream effectors are conserved in E. histolytica, we exploited a unique method to identify the proteins PI3P dependently recruited to phagosomes. We rationalised that overexpression of PI3P‐binding GFP‐HrsFYVE competes for PI3P on phagosomal membranes and results in dissociation of PI3P effectors from phagosomes. EhVps26 and EhVps35, but not sorting nexins (SNXs), of the retromer complex were detected from phagosomes only without GFP‐HrsFYVE overexpression. Two potential SNXs, EhSNX1 and EhSNX2, identified in the genome, possess only phox homology domain and specifically bound to PI3P, but retromer components, EhVps26 and EhVps35, did not bind to PI3P. Live and immunofluorescence imaging showed that EhSNX1 was recruited to the trogocytic cup and tunnel‐like structures, and subsequently, EhSNX2 was recruited to trogosomes. Furthermore, EhSNX1, but not EhSNX2, specifically bound to Arp2/3 and EhVps26, which were localised to the tunnel‐like structures and the trogosomes, respectively. EhSNX2 gene silencing increased trogocytosis, suggesting that EhSNX2 plays an inhibitory role in trogocytosis.
Keywords: pathogenesis, phagocytosis, phosphoinositide, trogocytosis, vesicular traffic
Phosphatidylinositol (PI) phosphates function as important second messengers in many cellular events. PI3‐phosphate plays a pivotal role in phagosome/trogosome biogenesis, which is essential for pathogenesis of Entamoeba histolytica, causing amebiasis. We report novel PI3‐phosphate‐mediated regulation of phagocytosis/trogocytosis via two isotypes of sorting nexins (SNXs) that function as PI3‐phosphate effectors, which play distinct and tempo‐spatial regulatory roles. SNX1 is involved in the recruitment of the retromer complex to the ingestion site, while SNX2 functions as a negative regulator of phagocytosis/trogocytosis.
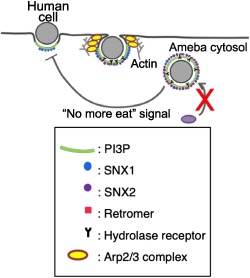
1. INTRODUCTION
Phosphatidylinositol (PI) is one class of glycerophospholipids that is composed of an inositol ring, a phosphate, a glycerol skeleton, and two acyl chains. PI converts to seven types of phosphatidylinositol phosphates (PIPs): three types of PIP, three types of PIP2, and one PIP3. These seven different types of PIPs are localised on different cellular compartments and mediate a variety of the intracellular events by recruiting proteins possessing PIPs‐recognizing domains such as FYVE (Fab1, YOTB1, Vac1, early endosomal antigen 1 [EEA1]), pleckstrin‐homology, and phox homology (PX) domains. In mammalian cells, the forming phagocytic cup is partially decorated with PI(4,5)P2 and PI(3,4,5)P3, and subsequently with PI3P as the phagosome encloses. Each PIP species recruits specific effector proteins to endosomes and phagosomes to promote formation and maturation. For example, PI3P recruits FYVE domain‐containing EEA1, which is recruited to early endosomes and facilitates endosome maturation, whereas Rabenosyn‐5, hepatocyte growth factor‐regulated tyrosine kinase substrate (Hrs), and endosomal sorting complex required for transport (ESCRT) complex are also recruited to PI3P‐rich membranes and play unique roles (Burman & Ktistakis, 2010).
Entamoeba histolytica is the protozoan parasite that displays inherited capacity of ingestion of foreign cells by phagocytosis and trogocytosis (“trogo” means “nibble” or “chew” and thus, the word implies ingestion of live cells by pieces; Ralston, Solga, MacKey‐Lawrence, Bhattacharya, & Petri, 2014) and responsible for human amebiasis causing an estimated 73,800 deaths annually (Lozano et al., 2012). Phagocytosis and trogocytosis have been implicated in self‐defense, proliferation, and pathogenicity of this organism (Ralston, 2015; Ralston et al., 2014). In E. histolytica, it has been suggested that PI3P is involved in the early to late phases of phagocytosis/trogocytosis as it is localised on the phagocytic and trogocytic cup/the phagosome and trogosome (Nakada‐Tsukui, Okada, Mitra, & Nozaki, 2009), whereas PI(3,4,5)P3, which is localised on the preclosed phagocytic and trogocytic cups and dissociated from them after sealing, in a relatively early phase (Byekova, Powell, Welter, & Temesvari, 2010). E. histolytica lacks an EEA1 homolog, but possesses 12 FYVE domain containing proteins (EhFPs, Nakada‐Tsukui et al., 2009), 7 PX domain containing proteins, and 13 pleckstrin‐homology domain containing proteins. However, the roles of these potential PIP‐binding proteins in phagocytosis and trogocytosis remain elusive. We identified and characterised AGC kinases as PI(3,4,5)P3‐binding proteins, which are involved in phagocytosis and trogocytosis. It has been shown that AGC kinase 1 is specifically involved in trogocytosis, whereas AGC kinase 2 is involved in both phagocytosis of dead mammalian cells and trogocytosis of live cells (Somlata, Nakada‐Tsukui & Nozaki, 2017). Although PI(3,4,5)P3 effectors have been in part characterised, the identity of PI3P effectors remains to be investigated because of the promiscuity of lipid‐binding specificities of FYVE domain containing proteins, as shown for EhFP4, for instance.
In this study, we attempted to identify PI3P effectors recruited to phagosomes and trogosomes in a PI3P‐dependent fashion by proteome analysis of purified phagosomes under PI3P‐deprived (by overexpression of human HrsFYVE‐GFP, using a tetracycline inducible system) and normal conditions. Finally, we proposed the model of how these sorting nexins (SNXs), the PI3P effectors, are involved in both positive and negative regulation of phagocytosis and trogocytosis.
2. RESULTS
2.1. Identification of retromer complex components as potential PI3P effectors that were recruited to phagosomes
To identify PI3P‐binding effectors that are involved in the phagosome biogenesis, particularly on the late stage of maturation, in E. histolytica trophozoites, we opted to utilise differential proteomic analysis of the phagosomes under normal versus PI3P‐deprived (competed) conditions. We rationalised that overexpression of a soluble PI3P‐binding protein should compete for PI3P with other PI3P effectors that play a role in phagosome maturation to be recruited (Figure 1a). To this end, the E. histolytica strain that expresses GFP‐fused HrsFYVE, which is the well‐documented human PI3P‐binding domain (Nakada‐Tsukui et al., 2009) in a tetracycline‐inducible fashion, was established. Expression of GFP‐HrsFYVE in E. histolytica peaked at 24 hr of cultivation with 10 μg/ml of tetracycline (Figure S1). At this time point, we confirmed by immunofluorescence assay (IFA) that GFP‐HrsFYVE was localised on the phagosomes containing human serum‐coated Dynabeads (Figure 1b). We verified the purity of the phagosome‐enriched fraction by the significant enrichment of GFP‐HrsFYVE (Figure 1c) and the mature CP‐A5 in the phagosome‐enriched fraction. Altogether, these results suggest that our protocol of phagosome purification using serum‐coated Dynabeads allowed us to successfully enrich phagosomes that were associated with GFP‐HrsFYVE and thus PI3P, on phagosome membranes (Figure 1c). We isolated phagosomes using previously established protocols (Marion, Laurent, & Guillén, 2005) and analysed their proteome under the conditions of GFP‐HrsFYVE being expressed and non‐expressed. After all detected proteins were categorised by their functions, those detected at least twofold higher under the GFP‐HrsFYVE non‐expressed condition compared with those under the GFP‐HrsFYVE expressed condition were considered to be potential candidates of PI3P‐binding proteins (Table S1). Only proteins categorised to lipid metabolism and vesicular trafficking are shown in Figure 1d. Among them, vacuolar protein sorting (Vps) 26 and Vps35, both of which are the components of the retromer complex, were detected, which were mainly investigated in this study, whereas analysis of other components will be described elsewhere.
Figure 1.
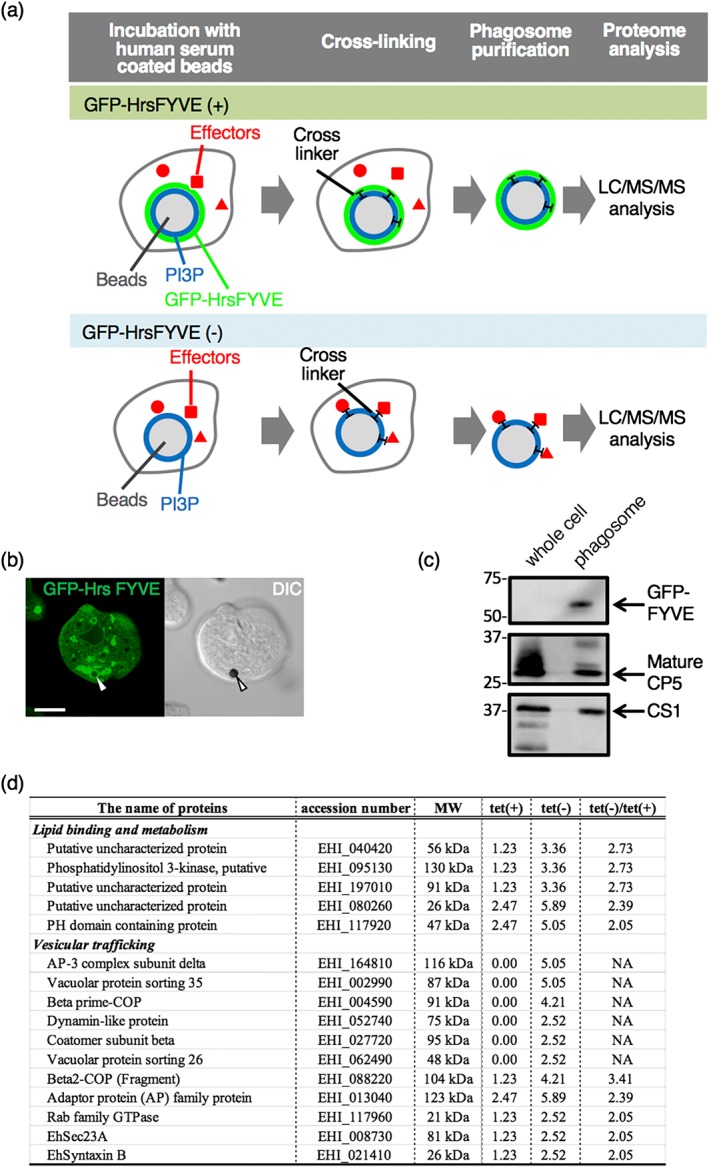
Identification of PI3P‐binding effector proteins involved in phagosome biogenesis. (a) The strategy and rationale used in this study to identify PI3P‐binding effector proteins involved in phagosome biogenesis. See text for details. (b) Verification of the recruitment of GFP‐HrsFYVE to phagosomes by fluorescence microscopy. The Entamoeba histolytica transformant cells that expressed GFP‐HrsFYVE under tetracycline induction were cultured in the presence of 10 μg/ml of tetracycline for 24 hr and then incubated with human serum‐coated beads for 30 min. Arrowheads indicate ingested beads. (c) Validation of the recruitment of GFP‐HrsFYVE to phagosomes by phagosome purification followed by immunoblot analysis. The bead‐containing phagosomes were purified as described in Section 4 and subjected to SDS‐PAGE and immunoblot analyses. GFP‐HrsFYVE and mature CP‐A5 were detected with anti‐myc, anti‐GFP, and anti‐CP‐A5 antibody, respectively. (d) The categorised list of proteins that are differentially excluded from phagosomes in GFP‐HrsFYVE‐dependent fashion. Tet(+) and tet(−) indicate the conditions where GFP‐HrsFYVE expression was induced with 10 μg/ml of tetracycline or not induced. The values indicate the relative frequency of the detected peptides corresponding to each protein. “tet(−)/tet(+)” indicates the division of the value of tet(−) by the value of tet(+). A whole list of the detected proteins from purified phagosomes are found in Table S1
2.2. In silico identification of SNXs in E. histolytica
In mammalian cells, the retromer complex is known to be involved in recycling of transmembrane receptors from endosomes to the trans‐Golgi network (Bonifacino & Hurley, 2008). It has been demonstrated that the retromer complex consists of two subcomplexes: the major subcomplex of Vps26/Vps29/Vps35 and a combination of SNX1 or 2, SNX5 or 6, and SNX3. The retromer is recruited to PI3P‐rich endosomes via PX domain of SNX3 (Seaman, 2012). The components of the major subcomplex (Vps26/Vps29/Vps35) have been identified and investigated in E. histolytica (Nakada‐tsukui, et al., 2005; Picazarri et al., 2015; Srivastava et al., 2017).
However, it remains elusive whether or not SNXs are also conserved in E. histolytica and if they are, what the roles of SNXs in vesicular traffic are, particularly during phagocytosis and trogocytosis. There are several classes of SNXs known in mammals: those with PX and BAR (Bin/Amphiphysin/Rvs) domains, those with PX domain and other domain such as FERM, PDZ, and SH3 domains, and those containing PX domain only (Worby & Dixon, 2002). Human SNX1 or SNX3 belongs to the first or third group, respectively (Cullen, 2008). Human SNX3 is responsible for recruiting the retromer complex to the membrane of late endosomes via binding to PI3P (Seaman, 2012). Human SNX3 is phosphorylated on Ser72 (Lenoir et al., 2018). When phosphorylated, it cannot bind to PI3P and is localised to the cytosol.
We conducted an in silico survey of SNX genes in the E. histolytica genome. We used SNX1 isoform a (NP_003090.2) from Homo sapiens as a query for Blastp. We identified two proteins encoded by EHI_060320 and EHI_004400 as possible SNX homologs (E‐value of 4e‐6 and 1e‐4, respectively). Both of the proteins have a PX domain but lack any other recognisable domains, and thus, based on domain configuration, they are considered to be orthologous to human SNX3. These E. histolytica SNX orthologs were designated as EhSNX1 and EhSNX2. Alignment of EhSNX1, EhSNX2, and human SNX3 protein sequences shows that the residue corresponding to conserved Ser72 (conserved among about the half of human SNXs) is conserved in the PX domain of EhSNX1 (Ser132) and EhSNX2 (Ser53; Figure 2). Two out of four residues shown to be involved in the binding to PI3P are conserved in EhSNX1 and EhSNX2. Three out of ten residues implicated in the binding to Human Vps26 and Vps35 are conserved in EhSNX1, but not in EhSNX2, suggesting a possibility of isotype‐specific interactions with EhVps26 and EhVps35. It is also worth noting that EhSNX1, but not EhSNX2, has an approximately 60 a.a.‐long amino terminal extension and a 10 a.a.‐long carboxyl terminal extension rich in serine and negatively charged amino acids, which may be involved in protein–protein interaction.
Figure 2.

Alignment of protein sequences of EhSNX1, EhSNX2, and HsSNX3. Protein sequences were aligned using clustalw algorithm (http://clustalw.ddbj.nig.ac.jp). Phox homology domain sequences were predicted by HMMER (Meng & Ji, 2013) and are shown with yellow background. The numbers by three amino acid sequences (EhSNX1, EhSNX2, and HsSNX3) correspond to the a.a. positions of each protein, whereas those on top of the sequences (Arg69, Ser72, Lys95, Ile109, and Arg118) correspond to the positions of the residues in HsSNX3 (Lenoir et al., 2018). Ser72 was previously shown to be phosphorylated and marked with a blue rectangle, whereas the other residues implicated for PI3P binding are marked with red rectangles. Important residues for binding to Vps26 and Vps35 are shown with blue and pink background, respectively
2.3. Determination of EhSNXs binding specificity to PI3P
To investigate whether EhSNX1 and EhSNX2 bind to PIPs and, if so, what PIP species they bind, we created E. histolytica lines that expressed EhSNX1 and EhSNX2 with HA tag. A single band corresponding to the expected molecular mass of HA‐EhSNX1 (27.7 + 3 kDa for the HA tag) or HA‐EhSNX2 (17.0 + 3 kDa for the HA tag) was observed in the respective transformants (Figure 3a). The lipid overlay assay using amebic lysates from these transformants showed that both EhSNX1 and EhSNX2 specifically bound to PI3P (Figure 3b). We also verified the lipid‐binding activity of some PI3P‐binding protein candidates in the proteome list (Figure 1d) by lipid overlay assay: β‐adaptin, Vps35, Vps26, and Syntaxin. However, they did not bind to any phospholipids including PI and PIPs (data not shown).
Figure 3.
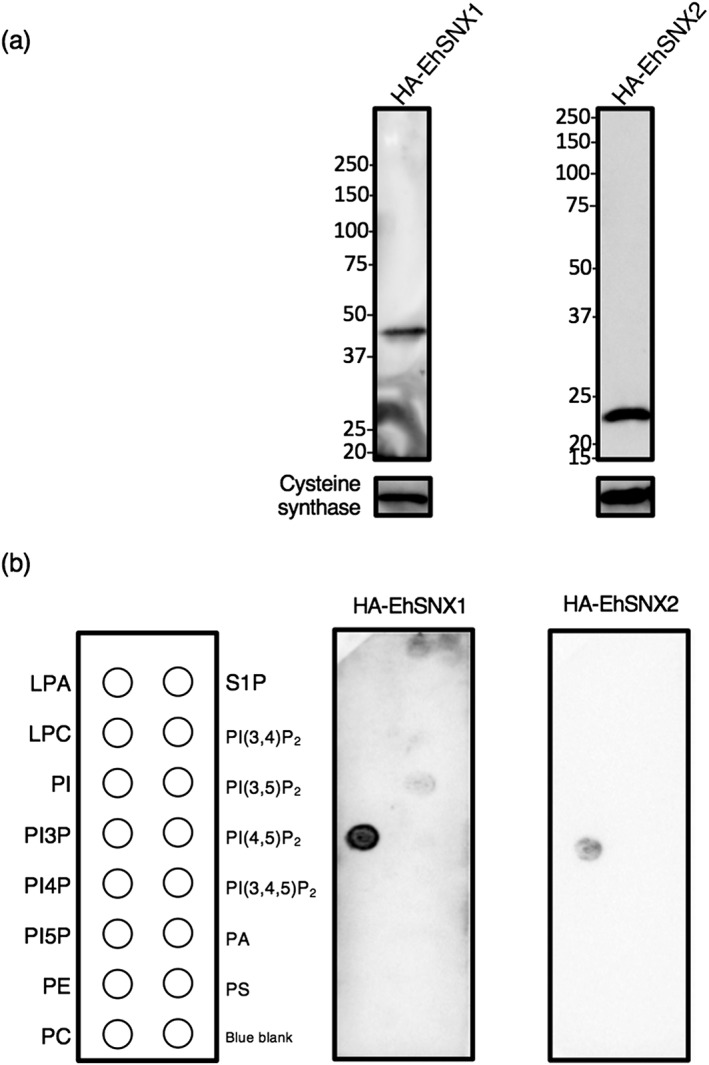
Lipid‐binding specificity of EhSNX1 and EhSNX2. (a) Verification of the expression of EhSNX1 and EhSNX2 in HA‐EhSNX1‐ and HA‐EhSNX2‐expressing transformants by immunoblotting using anti‐HA antibody. Approximately 20 μg of total lysates from these strains were loaded. Cysteine synthase 1 was detected by anti‐CS1 antiserum as a loading control. (b) Lipid‐binding specificity of EhSNX1 and EhSNX2 observed by lipid overlay assay. A panel of PIPs and phospholipids spotted on nitrocellulose membrane was incubated with total lysates from HA‐EhSNX1‐ and HA‐EhSNX2‐expressing transformants. The membranes were reacted with anti‐HA antibody. LPA, lysophosphatidic acid; LPC, lysophosphocholine; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; S1P, sphingosine‐1‐phosphate
2.4. Immunofluorescence imaging showing colocalisation of EhSNXs with EhVps26
As EhSNXs were shown to bind to PI3P and thus, work as PI3P effectors, they were expected to be localised to PI3P‐rich membranes (compartments) during phagocytosis and trogocytosis. Because it was not clear whether EhSNXs make complex with the other major retromer subcomplex, we investigated whether EhSNXs are colocalised with Vps26 during trogocytosis of Chinese hamster ovary (CHO) cells by IFA using HA‐EhSNX1‐ and HA‐EhSNX2‐expressing strains. In steady state, both EhSNX1 and EhSNX2 were localised on some small vesicles associated with EhVps26 (Figure 4a,b). To investigate the localisation and kinetics of EhSNXs during trogocytosis, we performed IFA at 15 min after addition of CellTracker Blue‐stained live CHO cells to E. histolytica trophozoites. EhSNX1 was localised on the bottom of the trogocytic cup associated with EhVps26 (Figure 4a). EhSNX1 appeared to be dissociated from the trogosome after phagocytic cup membrane was closed (data not shown, see below for live imaging quantitation). Unlike EhSNX1, EhSNX2 was seldom present on the preclosure trogocytic cup, but localised on the closed trogosomes associated with EhVps26 (Figure 4b).
Figure 4.
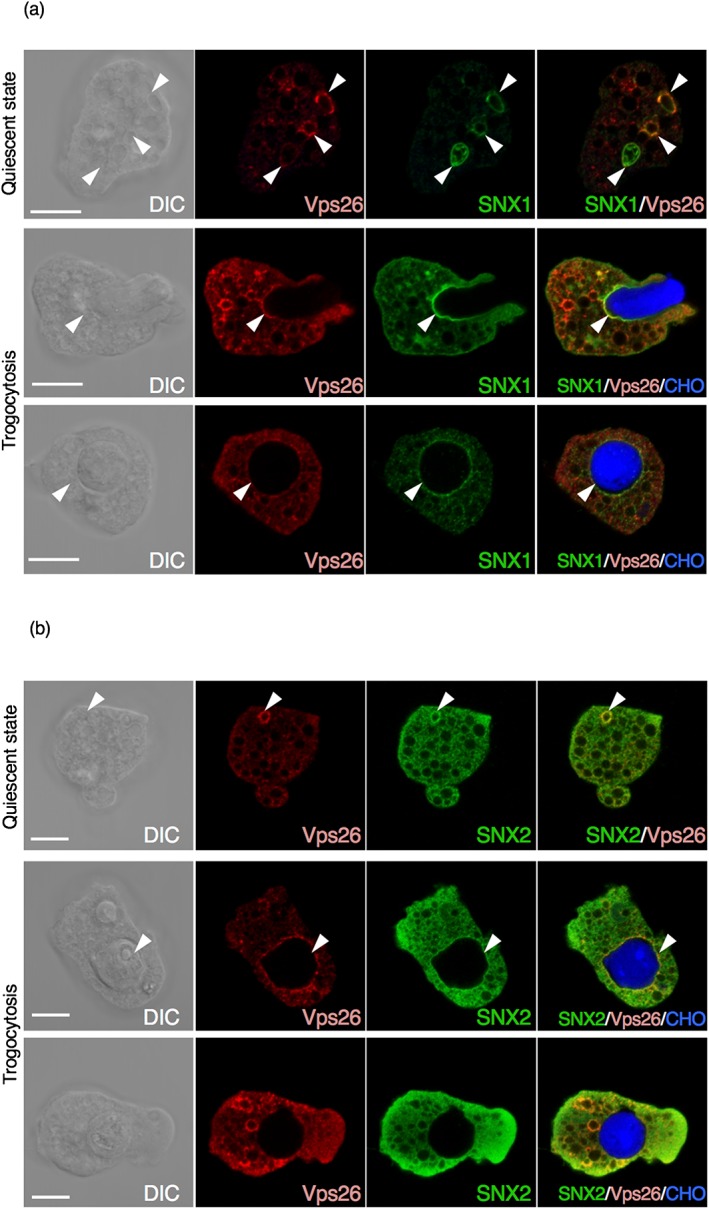
Localisation of HA‐EhSNX1 and HA‐EhSNX2. Localisation of EhSNX1 and EhSNX2 (a, b) was examined by immunofluorescence assay in quiescent state and during phagocytosis. HA‐EhSNX1/2 and EhVps26 were detected by anti‐HA antibody and anti‐EhVps26 antiserum, respectively. HA‐EhSNX1/2 are shown in green, EhVps26 in red, and Chinese hamster ovary (CHO) cells were stained by CellTracker Blue. Differential interference contrast (DIC) and fluorescence images are shown. Scale bars are 10 μm. Upper panels, in quiescent state; middle panels, during internalisation of a CHO cell (a), after closure of the trogosome (b); lower panels, after closure of the trogosome. Arrowheads depict vesicles, vacuoles, phagosomes, and the phagocytic cup where EhSNX1 or 2 is colocalised with EhVps26
2.5. Live imaging and quantification of GFP‐EhSNX1/2 during trogocytosis
We monitored (Movies S1 and S2) and quantified (Figure 5) GFP‐EhSNX1/2 on the trogocytic cup (and tunnel‐like structures) and the trogosome during trogocytosis, according to the protocol described in Section 4. GFP‐EhSNX1 was recruited to the trogocytic cup when the cup was formed and remained associated until 108 s (note that the movie started to be recorded 4–7 s after adherence; Movie S2, Figure 5). The intensity of GFP‐EhSNX1 was detected after the attachment of the trophozoite to the CHO cell and gradually decreased during trogosome maturation. On the contrary, GFP‐EhSNX2 was recruited to the trogosome after the trogocytic cup was enclosed (Movie S2). The signal of GFP‐EhSNX2 remained low until 108 s; it suddenly increased at 124 s (Figure 5). This live imaging suggests sequential processes in which GFP‐EhSNX2 recruitment to the trogosome occurred after GFP‐EhSNX1 dissociation from the trogosome.
Figure 5.
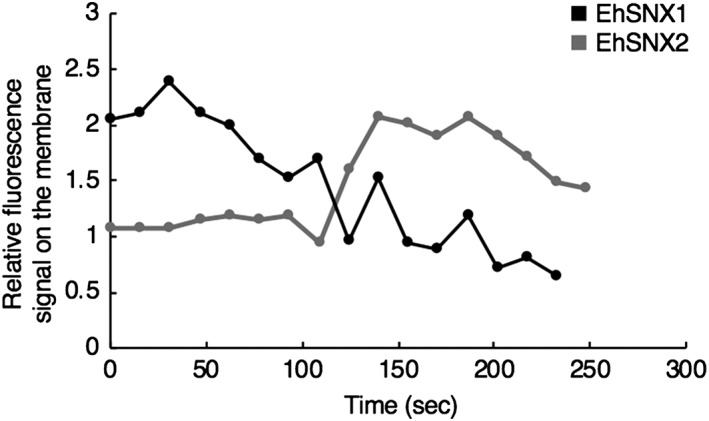
Kinetics of association of GFP‐EhSNX1/2 to the trogosome membrane during Chinese hamster ovary (CHO) cell internalisation. Fluorescence intensity of GFP‐EhSNX1/2 on the trogosome membrane relative to the cytoplasm was monitored during trogocytosis of CHO cells. The images started to be collected 4–7 s after the initiation of the trogocytic cup formation, and this timing was set to zero. Y‐axis represents the mean fluorescence signal per pixel of GFP‐SNX1/2 on the trogosome membrane relative to that in the cytosol. One representative image each of three independent experiments is shown. Only representative movies were analysed, but other movies also showed similar kinetics
2.6. Interaction between EhSNX1 and the retromer complex demonstrated by immunoprecipitation
To demonstrate physical interaction between EhSNX1/2 and EhVps26 containing the retromer complex during trogocytosis, immunoprecipitation was performed with anti‐EhVps26 antiserum using lysates from EhSNX1‐HA expressing cells. Expression of EhSNX1‐HA was confirmed by immunoblot analysis using anti‐HA antibody (Figure 6a). For unknown reasons, EhSNX2‐HA‐expressing transformant could not be established (data not shown), and HA‐EhSNX2 was used as control. Because EhSNX1 was found on the trogocytic cup/trogosome during trogocytosis of CHO cells, EhSNX1‐HA‐expressing cells were coincubated with CHO cells for 30 min and then, subjected to immunoprecipitation with anti‐EhVps26 antiserum, followed by immunoblot analysis using anti‐HA antibody, anti‐EhVps26, and anti‐EhVps35 antisera. EhVps26, EhVps35, and, in a lesser amount, EhSNX1‐HA were immunoprecipitated from EhSNX1‐HA‐expressing cells (Figure 6b). HA‐EhSNX2 was not coprecipitated with EhVps26, suggesting that the interaction with EhVps26 was specific to EhSNX1, but not EhSNX2.
Figure 6.
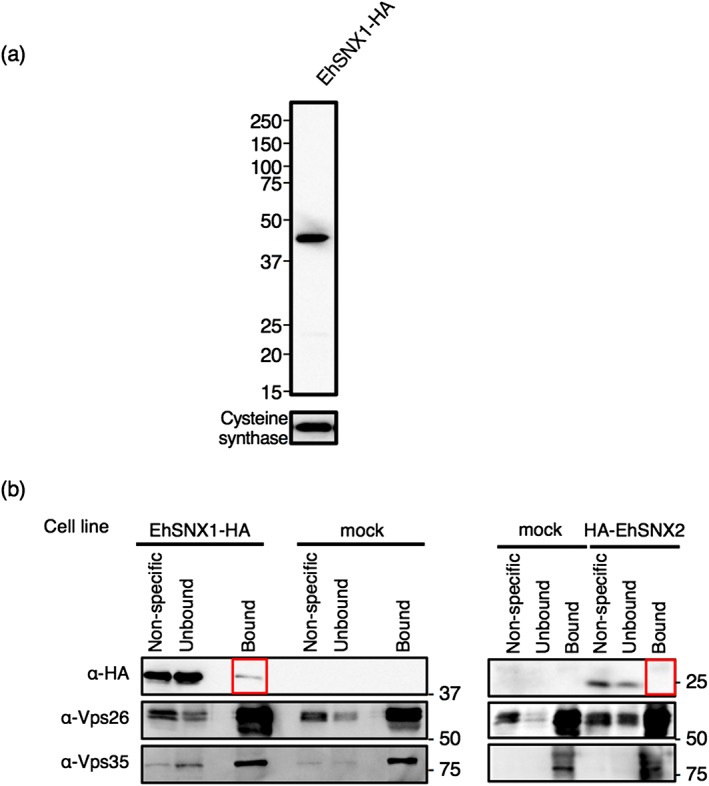
Binding of EhSNX1 with the retromer complex. (a) Verification of the expression of EhSNX1‐HA by immunoblot analysis using anti‐HA antibody. Approximately 20 μg of total lysates from these strains were loaded. Cysteine synthase 1 was detected by anti‐CS1 antiserum as a loading control. (b) Immunoprecipitation of EhVps26 and EhSNX1‐HA followed by immunoblot analysis. After 30 min co‐culture with EhSNX1‐HA, HA‐EhSNX2, or mock transformant cells with CHO cells, the amoebae were collected to produce lysates, which were subjected to immunoprecipitation by anti‐Vps26 antiserum. The immunoprecipitated (“Bound”), unbound supernatant (“Unbound”), and non‐specific (protein G‐Sepharose control beads that were preincubated with lysates, washed, and boiled with SDS loading buffer) fractions were analysed with SDS‐PAGE and immunoblot analyses using anti‐HA antibody, anti‐Vps26, and anti‐Vps35 antisera
2.7. Interaction between Arp 2/3 complex component and EhSNX1 demonstrated by immunoprecipitation
To further understand the role of EhSNX1, immunoprecipitation was performed using the strain expressing EhSNX1 with the amino‐terminal HA. HA‐EhSNX1 was precipitated with anti‐HA agarose after co‐incubation with CHO cells for 30 min. One specific band around 20 kDa was detected after SDS‐PAGE and silver staining (Figure 7a) and subjected to mass spectometry analysis (Figure 7b). Notably, two of eight detected proteins (EHI_174910; EHI_030820) were C3 and C4 components of actin‐related protein (Arp) 2/3 complex. To verify the interaction, the strain expressing FLAG‐tagged 2/3 complex subunit 3 (EhArpC3; FLAG‐EhArpC3) was established. The FLAG‐EhArpC3 localisation was examined by IFA using anti‐FLAG antibody. FLAG‐EhArpC3 was localised on the tunnel‐like structure (corresponding to the neck of a flask) during trogocytosis of CHO cells (Figure 7c). On the other hand, EhVps26 was never found on the tunnel‐like structure, but on the bottom region of the trogocytic cup (before closure) and on the trogosome (after closure).
Figure 7.
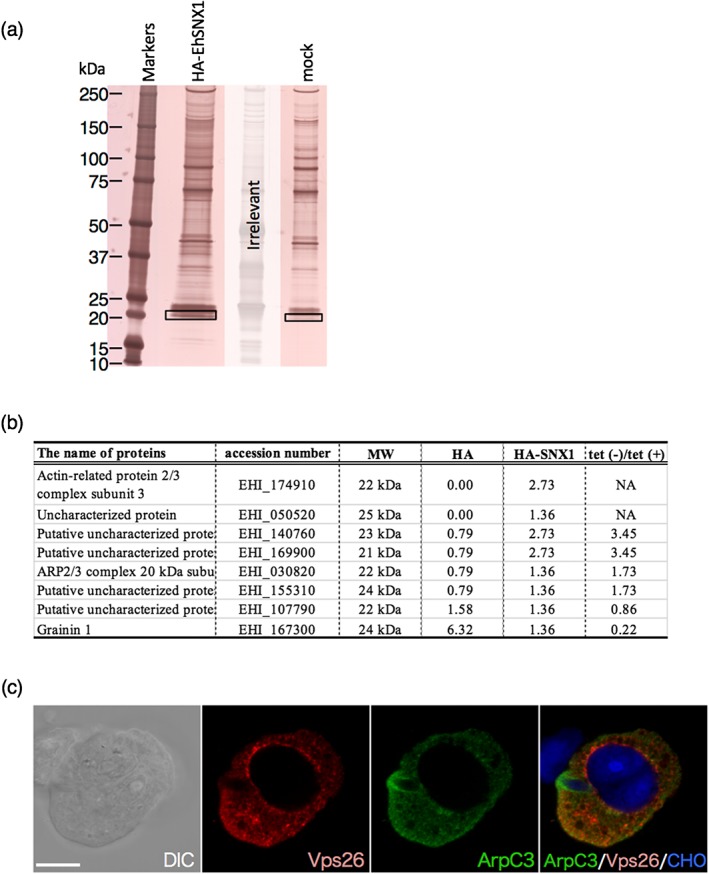
Identification of candidate binding proteins of EhSNX1. (a) Silver‐stained SDS‐PAGE gel of the immunoprecipitated samples from HA‐EhSNX1 and mock transformants using anti‐HA antibody. The rectangles indicate the region of the specific band excised from HA‐EhSNX1 and its corresponding region from control. (b) The proteins that were detected from the unique band specifically recognised from HA‐EhSNX1‐expressing transformants by immunoprecipitation using anti‐HA antibody. Proteins that were not detected from the HA‐EhSNX1 are excluded from the table. The values indicate the relative frequency of the detected peptides corresponding to each protein. “HA‐EhSNX1/HA” indicates the division of the value of HA‐EHhSNX1 by the value of HA control. A whole list of the detected proteins is found in Table S2. (c) The localisation of EhArpC3 and EhVps26 during trogocytosis. FLAG‐EhArpC3 transformant was co‐cultured with CHO cells stained with CellTracker Blue for 15 min. FLAG‐EhArpC3 and EhVps26 were detected using anti‐FLAG antibody and anti‐EhVps26 antiserum, respectively. FLAG‐EhArpC3 is shown in green; EhVps26 is shown in red. Scale bar is 10 μm
2.8. Transcriptional gene silencing of EhSNX1 gene reduced recruitment of EhVps26 to the trogosome, whereas that of EhSNX2 gene upregulated trogocytosis
To better understand how EhSNXs are involved in trogocytosis and phagocytosis, we performed transcriptional gene silencing (gs) of EhSNX1 and EhSNX2 genes. In the parental G3 line transformed with the mock vector, the level of EhSNX2 mRNA is approximately 3.0‐fold higher than that of EhSNX1. The level of EhSNX1 transcript was reduced by 81 % by EhSNX1 gene silencing, whereas that of EhSNX2 was conversely increased by 41 % (Figure 8a). In contrast, the level of EhSNX1 and EhSNX2 transcripts was reduced by 55 % and 94 % by EhSNX2, respectively (Figure 8a).
Figure 8.
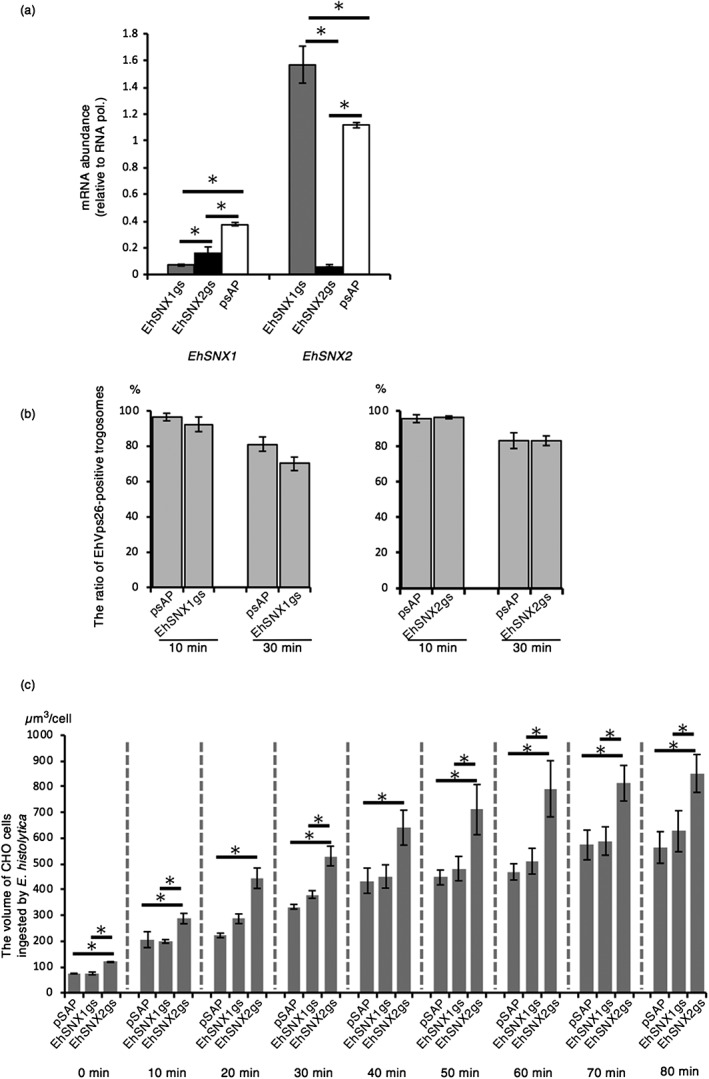
The effect of gene silencing of EhSNX1 and EhSNX2 on the retromer recruitment to trogosomes and trogocytosis. (a) The levels of EhSNX1 and EhSNX2 transcripts in EhSNX1gs, EhSNX2gs, and control strains. The transcript levels are shown relative to that of RNA polymerase II. (b) The effects of gene silencing of EhSNX1 and EhSNX2 on the recruitment of EhVps26 to trogosomes. Trophozoites were allowed to ingest live Chinese hamster ovary (CHO) cells for 10 or 30 min and subjected to immunofluorescence assay. Images of trophozoites that had ingested CHO cells were randomly captured, and the number of all trogosomes and Vps26‐positive trogosomes were counted to estimate the ratio of Vps26‐positive trogosomes. (c) The effects of gene silencing of EhSNX1 and EhSNX2 genes on trogocytosis. Trophozoites of EhSNX1gs and EhSNX2gs strains stained with CellTracker Green were incubated with CHO cells stained with CellTracker Blue as described in Section 4. The images were taken on CQ1 every 10 min for 80 min. The volume of the ingested CHO cells (blue) was calculated using three‐dimensionally reconstituted data. Bars indicate standard errors. Asterisks indicate statistical significance by Tukey test (p < .05). Experiments were conducted three times independently, and a representative data set is shown
Because EhSNX1 physically interacts with the major retromer subcomplex (see above), gene silencing of EhSNXs can potentially affect localisation of the complex during trogocytosis and phagocytosis and also efficiency of ingestion. EhVps26 was swiftly recruited, together with EhSNX1, to the trogocytic cup during trogocytosis of CHO cell (Figure 4a). Almost all (96%) of CHO cell‐containing trogosomes in the control strain were decorated with EhVps26 at 10 min of co‐incubation with CHO cells (Figure 8b). At 30 min, a slightly less proportion (81%) of trogosomes in the control were still decorated with EhVps26. Although statistically insignificant (p value .44 and .11, at 10 and 30 min, respectively), gene silencing of EhSNX1 caused less recruitment of EhVps26 to trogosomes compared with the control (4% and 14% reduction compared with the control at 10 and 30 min, respectively). Gene silencing of EhSNX2 caused no change in the association of EhVps26 with trogosomes at 10 and 30 min. Next, we measured the total volume of ingested CHO cells per trophozoite, in EhSNX1gs, EhSNX2gs, and mock strains, on image cytometer CQ1 to estimate the efficiency of trogocytosis. The amoebic transformants were cultivated with CHO cells, and images were captured every 10 min for 80 min. Interestingly, trogocytosis significantly increased by 1.4‐ to 2.0‐fold in EhSNX2gs strain compared with the control at all time points (p < .05; Figure 8c). On the other hand, the volume of ingested CHO cells was not changed in EhSNX1gs strain at any time point. Phagocytosis was also affected by EhSNX2 gene silencing; the volume of ingested prekilled CHO cells increased by 1.4‐ to 2.0‐fold in EhSNX2gs strain at all time points, similar to the effects on trogocytosis (Figure S4).
3. DISCUSSION
3.1. EhSNX1/2 are downstream effectors of PI3P signals on the trogosome membrane
PIPs‐mediated signalling plays a pivotal role in phagocytosis and trogocytosis in E. histolytica. In this study, we identified for the first time the retromer complex, in particular SNXs, as a PI3P downstream effector on the phagosome and trogosome membrane in E. histolytica. It has been well established that in mammalian cells, PI3P is recruited to phagosomes and trogosomes after complete closure of the phagocytic/trogocytic cup and formation of the phagosome/trogosome, and that PI3P effectors such as EEA1 is recruited to the enclosed phagosomes to facilitate maturation of phagosome/trogosomes. It has been shown in E. histolytica that PI3P is localised on the phagocytic cup before closure as well as on the enclosed phagosomes (Nakada‐Tsukui et al., 2009), and the timing of PI3P recruitment to the phagocytic and trogocytic cup in E. histolytica appears to be different from human cells (Yeung & Sergio, 2007). However, as no known PI3P effector homologs were found in the E. histolytica genome, the role of PI3P on the phagocytic/trogocytic cup and phagosome/trogosome and its downstream effectors remained elusive. To identify the proteins that are recruited to phagosomes in a PI3P‐dependent manner, we took a unique approach using our unique reverse genetic system in which PI3P‐binding Hrs‐FYVE domain fused with GFP, expressed in a tetracycline‐inducible manner, competes with endogenous PI3P binding proteins for PI3P on phagosome and trogosomes. Because such interaction between PI3P and its effectors is transient and unstable, we needed to use a cross‐linker and a phosphatase inhibitor to immobilise such interaction (Figure S2).
SNXs were not identified from E. histolytica in our previous study by biochemical approaches (Nakada‐Tsukui et al., 2005). Furthermore, an in silico gene survey also failed to identify SNX homologs (Nakada‐Tsukui et al., 2005), which was likely due to our using of yeast Vps5p as a query for Blastp search. E. histolytica SNXs were identified when human SNX1 or SNX3 was used as a query, but not by using PX domain from other protein as a query (the E‐values of the Blastp search using HsSNX1 or HsSNX3 as a query were 4e‐6 and 5e‐10 for EHI_060320, respectively; 1e‐04 and 3e‐13 for EHI_004400; whereas that using yeast Vps5p was 0.007 for EHI_060320 and 0.002 for EHI_004400).
3.2. PIP specificity of EhSNX1/2 and human SNXs
The PIP specificity of EhSNX1 and EhSNX2 compared with that of human SNX1‐3 is intriguing. In humans, there are >30 members of SNXs known (Cullen, 2008) containing only several members with known PIP specificity. Human SNX1 and SNX2 showed preference in the order of PI(3,4,5)P3>PI(3,5)P2>PI3P and PI3P>PI4P>PI5P, respectively, whereas human SNX3 almost exclusively binds to PI3P, similar to EhSNX1 and EhSNX2 (Worby & Dixon, 2002). Amino acid alignment shows six amino acid residues that are shared by EhSNX1, EhSNX2, and HsSNX3, but not conserved in both HsSNX1 and HsSNX2, including EhSNX1 T107 of (EhSNX2 T28), EhSNX1 Y131 (EhSNX2 Y53), EhSNX1 L160 (EhSNX2 L82), EhSNX1 G162 (EhSNX2 G84), EhSNX1 F175 (EhSNX2 F96), and EhSNX1 I191 (EhSNX2 I112; Figure S3). The conservation of EhSNX1 G162 (EhSNX2 G84) corresponding to HsSNX3 G94 is of particular interest as these residues are also exclusively present in HsSNX11‐13 and 27 (Lenoir et al., 2018), among which PIP specificity of only HsSNX13 was shown: preferred binding to PI3P and PI5P over PI(3,5)P2 and PI4P (Worby & Dixon, 2002). These data suggest that EhSNX1 G162 (EhSNX2 G84) potentially contributes to PI3P specificity. Although PIP specificity may be affected by other domains than PX for the proteins that contain multiple domains such as HsSNX13 (Cullen, 2008), those of EhSNX1 and EhSNX2 are likely to be attributable to only the PX domain, similar to HsSNX3.
3.3. EhSNX1 and EhSNX2 have different roles during trogocytosis
Two SNXs from E. histolytica have similar domain structure (containing only PX domain, but lacking BAR domain) and lipid‐binding specificities, but showed distinct patterns of localisation and trogosome/phagosome recruitment, and phenotypes by gene silencing. We have shown by biochemical, genetic analyses and live imaging that EhSNX1, but not EhSNX2, is involved in the recruitment of EhVps26 during trogocytosis. We have also shown that EhSNX2 is negatively involved in trogocytosis per se, as demonstrated with partial augmentation of trogocytosis by EhSNX2 gene silencing. Our data indicate, together with our previous finding showing that EhVps26 of the retromer complex is recruited to the trogocytic cup (Nakada‐Tsukui et al., 2005; Picazarri et al., 2015), that EhSNX1 recruits the retromer complex to the trogosome/phagosome in a PI3P‐dependent manner. Unlike EhSNX1, EhSNX2 did not interact with the retromer complex as indicated. This result is consistent with the fact that the amino acid residues implicated for the binding with the retromer complex are not conserved in EhSNX2 (see Figure 2 and Section 2.2).
The fact that gene silencing of EhSNX2, but not EhSNX1, increased the trogocytosis efficiency was counter‐intuitive and suggestive of EhSNX2 being a negative regulator of trogocytosis. It has been shown that in dendritic cells, SNX3 overexpression causes defect in early endosome maturation by competing PI3P on early endosomes and reducing EEA1 recruitment (Chua & Wong, 2013). This observed phenotype may mirror the apparent upregulation of trogocytosis by EhSNX2 gene silencing. However, the loss of competition for PI3P by EhSNX2 gene silencing may not be the reason of the observed upregulation of trogocytosis because no change in trogocytosis was observed by EhSNX1 gene silencing, which in theory also causes reduced competition for PI3P. The lack of phenotype by EhSNX1 gene silencing may be explained at least in part by compensatory upregulation of EhSNX2 gene expression by EhSNX1 gene silencing. One possible scenario to explain these phenotypic changes in both human and E. histolytica is that both HsSNX3 and EhSNX2 employ a possible negative downstream effector/machinery needed for maturation of early endosomes and trogosomes.
3.4. EhSNX1 binds to Arp2/3 complex for downstream signalling to cytoskeletal rearrangement during trogocytosis
We demonstrated that EhSNX1 specifically bound to ArpC3/ArpC4 of the Arp2/3 complex. ArpC3 and ArpC4 were found to interact with EhSNX1 as indicated by LC/MS/MS analysis of the immunoprecipitated samples using HA‐EhSNX1 as a bait. In general, Arp2/3 complex is known to be composed of seven subunits and involved in elongation, branching, and cross‐linking of actin filaments via regulation of actin nucleation and elongation (Goley & Welch, 2006). Because all components of the Arp2/3 complex were found in the previous phagosomal proteome analyses (Boettner et al., 2008; Furukawa, Nakada‐tsukui, & Nozaki, 2013; Marion et al., 2005; Okada et al., 2006), the Arp2/3 complex was suggested to be involved in phagocytosis and trogocytosis. The recruitment of EhSNX1 to the trogocytosis initiation site was specific to EhSNX1 and not observed for EhSNX2. Precise timing of the recruitment of EhSNX1 to the trogocytosis initiation site relative to that of Arp2/3 was not determined as live imaging was unable to be performed. However, the recruitment of EhSNX1 appeared to slightly precede that of Arp2/3. The continuity of the association of EhSNX1 and Arp2/3 clearly indicates close and direct interplay between them, triggered by PI3P‐dependent recruitment of EhSNX1 to the trogocytosis initiation site. It is worth noting that the localisation of EhVps26 and EhArpC3 on the trogocytic cup were overlapping but not identical. Together with the observation that EhSNX1 partially colocalised with EhVps26, these observations indicate that EhSNX1 may play a role in the bridging cytoskeletal rearrangement by EhArpC3 and receptor recycling mediated by the retromer.
3.5. The model proposed by EhSNX1/2 live imaging
Based on the biochemical, live imaging, and immunofluorescence image data, we propose the following model of how key players are engaged during trogocytosis (Figure 9). Upon the adherence of the amoebic trophozoite to the live mammalian cell, PI3P is locally synthesised in situ on the trogocytosis initiation site. EhSNX1 is recruited via PI3P binding mediated by its PX domain to the trogocytosis initiation site, which then becomes the trogocytic cup. EhSNX1 recruits Arp2/3 complex, which subsequently leads to actin cytoskeleton reorganisation needed for the establishment of the tunnel‐like structure of the trogosomes and also the closure of trogosomes. At the same time, EhSNX1 also recruits the retromer complex via interaction with Vps26. The retromer complex then binds to the hydrolase receptors for the retrieval (and recycling) of the receptors. Once the trogocytic cup (or the trogosome) is enclosed, EhSNX2 is recruited to the trogosome by virtue of its PI3P‐binding ability with a concomitant dissociation of EhSNX1. This model depicts a sequential involvement of EhSNX1 and EhSNX2 during the formation and maturation of trogosomes.
Figure 9.
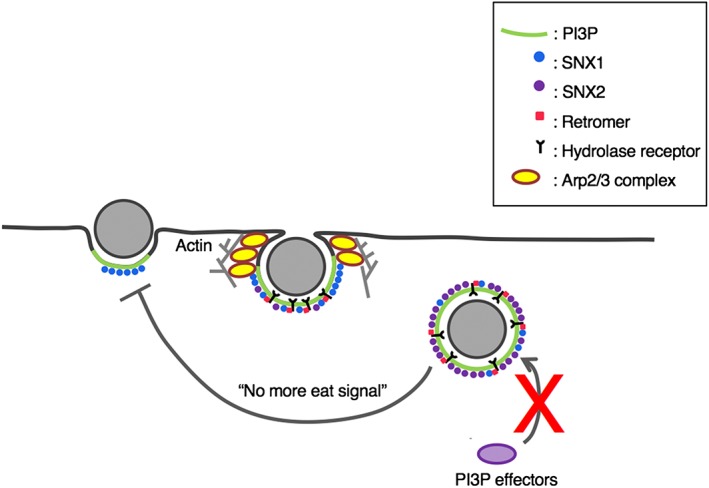
A model of the involvement of PI3P, SNXs, the retromer, the hydrolase receptor, and Arp2/3 complex in phagocytosis/trogocytosis
The apparent enhancement of trogocytosis by EhSNX2 gene silencing is possibly due to the loss of negative feedback regulation toward the initial trogocytosis event(s) as explained above. However, its mechanism and the possible negative effector(s) remain elusive and need further investigation. The recruitment of EhSNX1 and 2 to the trogocytic cup and the trogosome was time‐ and localisation (domain)‐specific. Although the underlying mechanism of the spaciotemporal recruitment of EhSNXs remains unknown, it is plausible that phosphorylation of the conserved Ser72 (of HsSNX3; Figure S3) may regulate the binding of EhSNX1/2 to PI3P and other possible accessory and effector proteins in an isotype‐specific manner.
The retromer complex was previously identified as an effector of GTP‐bound active Rab7A in E. histolytica (Nakada‐Tsukui et al., 2005). EhRab7A was reported to be localised to the preparatory vacuolar organelle that emerges upon erythrophagocytosis (pre‐phagosomal vacuole), where EhRab7A and EhVps26 were colocalised. In this study, the effects of EhSNX1 gene silencing on the inhibition of Vps26 recruitment to the trogosome was not large and rather marginal, suggesting that the recruitment of the retromer may be also regulated via EhRab7A.
4. EXPERIMENTAL PROCEDURES
4.1. Cells and reagents
Trophozoites of E. histolytica strains HM‐1:IMSS cl6 (Diamond, Mattern, & Bartgis, 1972) and G3 (Bracha, Nuchamowitz, & Mirelman, 2003) were axenically cultured in BI‐S‐33 medium (BIS) at 35.5°C as previously described (Diamond et al., 1978). CHO cells were maintained in F‐12 medium (Invitrogen‐Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum in 25 cm2 tissue culture flasks (IWAKI, Shizuoka, Japan) under 5% CO2 at 37°C. All chemicals of analytical grade were purchased from Sigma‐Aldrich (Missouri, USA) unless otherwise stated. The anti‐HA (11MO) and anti‐myc (9E10) monoclonal antibodies were purchased from Covance (Princeton, NJ, USA). The anti‐GFP antibody was purchased from Sigma‐Aldrich. The production of rabbit polyclonal antibodies against EhCS1, EhCP‐A5, EhVps26, and EhVps35 were previously described (Nakada‐Tsukui et al., 2005; Nakada‐Tsukui et al., 2009; Nozaki et al., 1999).
4.2. Plasmid construction
Standard techniques were used for DNA manipulation, subcloning, and plasmid construction. Plasmid vectors to produce E. histolytica lines that express EhSNX1 (EHI_060320) and EhSNX2 (EHI_004400) with HA‐ or GFP‐tag fused at the amino and carboxyl termini were constructed as follows. The full‐length protein coding region of these genes was amplified by polymerase chain reaction (PCR) with following primers: 5′‐GAACCCGGGATGGGAGATAATAAAGAAGATATTAT‐3′ and 5′‐GAACTCGAGTTATTCTGATGATGATGAACTTTCAT‐3′ (EhSNX1) or 5′‐GAACCCGGGATGAGTAATTATGCACAGTATGAATA‐3′ and 5′‐GAACTCGAGTTACCAACTAGTAACTTCCCAATTGT‐3′ (EhSNX2), where the restriction enzyme sites are underlined. PCR‐amplified fragments were digested by XmaI and XhoI and ligated into pEhEx‐HA (Nakada‐Tsukui et al., 2009) and pEhEx‐GFP vectors (Somlata et al., 2017 ) that were predigested with the two enzymes and purified to construct the plasmid to produce HA‐tagged and GFP‐fused SNXs at the amino terminus. The resultant plasmids were designated as pEhExHA‐SNX1, 2 and pEhExGFP‐SNX1, 2, respectively. Note that pEhExGFP contains a linker corresponding to five repeats of GA dipeptides) after GFP. To construct plasmids to express EhSNX1 and EhSNX2 with HA tag at the carboxyl terminus, the full‐length protein coding sequence of these genes was amplified by PCR with the following primers: 5′‐GAAAGATCTATGGGAGATAATAAAGAAGATATTAT‐3′ and 5′‐GAAAGATCTTTCTGATGATGATGAACTTTCATCAC‐3′ (EhSNX1); 5′‐GAAAGATCTATGAGTAATTATGCACAGTATGAATA‐3′ and 5′‐GAAAGATCTCCAACTAGTAACTTCCCAATTGTCTT‐3′ (EhSNX2), where restriction enzyme sites are underlined. Amplified fragments were digested by BglII and ligated into pEhEx‐HA. To construct plasmids for gene silencing of EhSNX1 and EhSNX2 gene, initial 420 bp of the EhSNX1 coding region and full length (426 bp) of EhSNX2 were PCR amplified with the following primers: 5′‐ GAAAGGCCTATGGGAGATAATAAAGAAGATATTAT‐3′ and 5′‐GAAGAGCTCTTGACTTCTTAATGACTGAAATTGAG‐3′ (EhSNX1); 5′‐GAAAGGCCTATGAGTAATTATGCACAGTATGAATA‐3′ and 5′‐GAAGAGCTCTTACCAACTAGTAACTTCCCAATTGT‐3′ (EhSNX2). These PCR fragments were digested by StuI and SacI and ligated into StuI‐ and SacI‐double digested psAP2‐Gunma (Mi‐ichi, Makiuchi, Furukawa, Sato, & Nozaki, 2011; Penuliar, Furukawa, Sato, & Nozaki, 2011). These plasmids were designated as psAP2‐SNX1 and psAP2‐SNX2. To construct vectors to express EhArpC3 with FLAG tag fused at the amino terminus, the full length protein coding sequence of these genes was amplified by PCR with the following primers: 5′‐GAACCCGGGATGACCACAATTGCCAAACAAGATAA‐3′ and 5′‐GAACTCGAGTTAAATGGTTTTGTTTAAGAACTTTC‐3′ (EhArpC3), where restriction enzyme sites are underlined. Amplified fragments were digested by XmaI and XhoI, and ligated into pEhEx‐FLAG, to produce pEhEx‐FLAG‐ArpC3. To expresses GFP‐HrsFYVE with c‐myc tag at the amino terminus under tetracycline induction in E. histolytica, a plasmid was generated by subcloning the ORF of myc‐GFP‐HrsFYVE PCR‐amplified from pKT‐MG‐Hrs‐FYVE (Nakada‐Tsukui, et al., 2009) to KpnI‐ and BamHI‐double‐digested pEhHygtetR‐O‐Cass vector, a kind gift from Dr. William A. Petri (Hamann, Buss, & Tannich, 1997; Ramakrishnan, Vines, Mann, & Petri, 1997) as previously described (Nakada‐Tsukui et al., 2009).
4.3. Establishment of E. histolytica transformants
The trophozoites of HM‐1:IMSS cl6 were transfected with pEhExHA‐SNX1, 2, pEhExGFP‐SNX1, 2, pEhEx‐FLAG‐ArpC3, pEhExHA, pEhExGFP, and pEhExFLAG, whereas the trophozoites of G3 strain were transfected with psAP2‐SNX1, psAP2‐SNX2, and psAP2 by lipofection as previously described (Nozaki et al., 1999). Geneticin was added at a concentration of 1 μg/ml at 24 hr after transfection then gradually until the geneticin concentration reached 10 μg/ml.
4.4. Immunoblot analysis
Approximately 1 × 105 trophozoites were harvested in the exponential growth phase, washed twice with phosphate buffer saline (PBS), pH 7.4, and resuspended in 50 μl of lysis buffer (50‐mM Tris‐HCl, pH 7.5, 150‐mM NaCl, 1% Triton X‐100, containing 50 μg/ml of E‐64, and complete mini). Approximately 20 μg of the total cell lysates were separated on 12% SDS‐polyacrylamide gels and subsequently electrotransferred onto nitrocellulose membranes. The membranes were incubated with 5% non‐fat dried milk in TBS‐T (50‐mM Tris‐HCl, pH 8.0, 150‐mM NaCl and 0.05% Tween‐20) for 30 min. The proteins were reacted with primary mouse antibodies specific for HA (with the dilution of 1:1000), myc (1:1000), GFP (1:100), FLAG (1:1000) rabbit antisera against EhVps26 (1:100), EhVps35 (1:500), EhCP‐A5 (1:1000), and CS1 (1:1000) at 4°C overnight. After the reaction with the primary antibodies, the membranes were washed with TBS‐T three times; they were further reacted with horseradish peroxidase (HRP)‐conjugated anti‐mouse or anti‐rabbit IgG antiserum (1:6000 or 1:8000, respectively) at room temperature for 1 hr. After washing with TBS‐T three times, the specific proteins were visualised by chemiluminescence detection using Immobilon Western Chemiluminescent HRP Substrate (Millipore corporation, Massachusetts, USA) according to the manufacturer's protocol.
4.5. Phagosome purification
Paramagnetic Dynabeads of 2.8‐μm diameter (catalog number 14203; Invitrogen, California, USA) were incubated with human serum (Sigma‐Aldrich) for 16 hr at 4°C, washed, and resuspended in transfection medium (OPTI‐MEM I medium [Thermo Fisher, Massachusetts, USA], adjusted to pH 6.8, containing 5‐mg/ml l‐cysteine and 1‐mg/ml ascorbic acid). Approximately 2.4 × 107 trophozoites transferred to each well on six‐well plates (Corning, New York, USA) and incubated at 35.5°C for 30 min to allow the trophozoites to attach to the surface of the well. After the medium was removed, a 2‐ml transfection medium containing approximately 1 × 107 human serum‐coated beads was added to each well (at the ratio 10 beads per cell). Beads were immediately sedimented on the surface of the amoebae seeded on the well by centrifugation at 100 × g at room temperature for 5 min. OPTI‐MEM was removed and 3 ml of worm BIS was added. The plates were further incubated at 35.5°C for 30 min. After incubation, the medium was removed and 3 ml of cold PBS was added to each well of the plates. The plates were put on ice for 10 min to detach the amoebae, and the amoebae were washed twice with cold PBS. After centrifugation at 800 × g at 4°C for 3 min, the supernatant was removed and the amoebae were resuspended in 500 μl of 8 mg/ml dithiobis (succinimidyl propionate; DSP) solution (Thermo Fisher, Massachusetts, USA), containing 0.5 mM of β‐glycerophosphate, 0.1 mM of ethylenediaminetetraacetic acid (EDTA), 10‐mM sodium fluoride, and 1‐mM sodium orthovanadate. The mixture was incubated on the rotator (10 rpm) at 4°C for 30 min. To quench the reaction, 50 μl of 1 M Tris‐HCl, pH 7.5 was added and the mixture was further incubated as above for 10 min. After the amoebae were treated with DSP, they were washed with homogenisation buffer (250‐mM sucrose, 3‐mM imidazol, in PBS, pH 7.4, and complete mini [Sigma Aldrich]). Cells were mechanically disrupted by applying 25–70 strokes of Dounce homogeniser. The homogenate was centrifuged at 800 × g at 4°C for 3 min to remove unbroken cells. Bead‐containing phagosomes were concentrated from the lysates by magnetic separation on PureProteome Magnetic Stand (Merck). The phagosomes were washed five times with 500 μl of homogenisation buffer containing protease inhibitors and lysed with 50 μl of lysis buffer (50‐mM Tris‐HCl, pH 7.5, 150‐mM NaCl, 1% Triton X‐100, containing 50 μg/ml of E‐64, and complete mini). For immunoblot analysis, approximately 20 μg of phagosomes were resuspended in SDS‐PAGE sample buffer (0.25‐M Tris‐HCl, pH 6.8, 8% SDS, 8% 2‐mercaptoethanol, 40% Glycerol, 0.004% Bromophenol Blue), boiled for 5 min, and subjected to SDS‐PAGE.
4.6. Lipid overlay assay
Approximately 6 × 106 cells were harvested and washed with PBS. Approximately 150 μl of lysis buffer (50‐mM Tris‐HCl, pH7.5, 15‐mM NaCl, containing 1% Triton X‐100, 0.05‐mg/ml E‐64, and complete mini) was added to an approximately 100‐μl bed volume of the pellet. The mixtures were incubated on ice for 30 min and centrifuged at 16,000 × g for 5 min. The supernatant was saved after centrifugation and used as the total lysate. Nitrocellulose membranes on which various phosphoinositides and other lipids had been spotted (PIP strips: P‐6001, Echelon Biosciences, Salt Lake City, USA) were treated with 1% fatty acid‐free bovine serum albumin (Sigma–Aldrich) in PBS‐T (PBS containing 0.05% Tween 20) for 1 hr at room temperature for blocking. The membranes were incubated with 2 ml of the lipid‐binding solution (PBS‐T containing 1% fatty acid free bovine serum albumin and complete mini) containing 20‐fold diluted amebic lysates for 2 hr at 4°C. After incubation with amoebic lysates, the membranes were washed with PBS‐T three times at 4°C. The membranes were then reacted with anti‐HA (with the dilution of 1:500), anti‐myc mouse monoclonal (1:500), anti‐GFP mouse monoclonal (1:100) antibodies, anti‐EhVps26 (1:100), and anti‐EhVps35 rabbit antisera (1:500), at 4°C for 3 hr. After the membranes were washed twice with PBS‐T at 4°C, they were further reacted with HRP‐conjugated anti‐mouse or anti‐rabbit IgG antiserum (1:6000 or 1:8000, respectively) at 4°C for 1 hr. After washing three times with PBS‐T at 4°C, the specific proteins were visualised by chemiluminescence detection using Immobilon Western Chemiluminescent HRP Substrate (Millipore corporation, Massachusetts, USA) according to the manufacturer's protocol.
4.7. Quantitative real‐time PCR
The levels of EhSNX1 and EhSNX2 gene transcripts were analysed by quantitative real‐time PCR. PolyA RNA was extracted, cDNA was prepared, and PCR was performed with cDNA as a template, as previously described (Mitra, Saito‐Nakano, Nakada‐Tsukui, Sato, & Nozaki, 2007). RNA polymerase II served as an internal control (GenBank accession number, XP_649091; Gilchrist et al., 2006). PCR was performed using the following primer sets: sense primer, 5′‐CCTGGTTATAGATTTAAACCAGAATTTACTGAACA‐3′ and antisense primer, 5′‐GAACTCGAGTTATTCTGATGATGATGAACTTTCAT‐3′ for EhSNX1; sense primer 5′‐TCGGTACAATCCCACCTCTTCCCTGGAGATACAATC‐3′ and antisense primer 5′‐GAACTCGAGTTACCAACTAGTAACTTCCCAATTGT‐3′ for EhSNX2 and the following cycling parameters: an initial step of denaturation at 95°C for 20 s, followed by 40 cycles of denaturation at 95°C for 3 s, annealing and extension at 60°C for 30 s.
4.8. Staining of CHO cells
After CHO cells were cultured as described above on 10‐cm diameter plastic dishes (IWAKI) for 2 days (before reaching confluence), they were incubated with CellTracker Blue CMAC Dye (Thermo Fisher) at the final concentration of 10 μM at 37°C for 30 min. CHO cells were detached from the surface of the flasks by treating with PBS containing 0.5‐mg/ml trypsin and 0.2‐mM EDTA at 37°C for 5 min, washed twice with PBS, and resuspended in 600 μl of transfection medium per 3 × 106 CHO cells harvested from one dish.
4.9. Immunoflorescence assay
Approximately 5 × 103 E. histolytica trophozoites were incubated with 5 × 104 Dynabeads M‐280 Tosylactivated beads (Invitrogen, California, USA) or 5 × 104 CHO cells, which had been prestained with CellTracker Blue as described below, in 50‐μl BI‐S‐33 medium in 8‐mm round wells on a slide glass at 35.5°C for 15 min. Cells were fixed with 3.7% paraformaldehyde and subsequently permeabilised with 0.2% saponin in PBS containing 1% bovine serum albumin for 10 min each at room temperature. The cells were reacted with anti‐HA mouse antibody (with the dilution of 1:1000) or anti‐Vps26 rabbit antibody (1:1000) as previously described (Nakada‐Tsukui et al., 2005). After washing with PBS three times, the cells were reacted with Alexa Fluor‐488 anti‐mouse secondary antibody (1:1000) or Alexa Fluor‐568 anti‐rabbit secondary antibody (1:1000; Thermo Fisher), respectively. The samples were observed using Carl‐Zeiss LSM 780 Meta lase‐scanning confocal microscope. The resultant images were further analysed using Zen software (Carl Zeiss, Oberkochen, Germany).
4.10. Live imaging and quantification of GFP‐fused EhSNXs
Approximately 7 × 104 trophozoites of the amoeba transformants transfected with pEhExGFP‐SNX1, 2, or pEhExGFP were cultured on 3.5‐cm diameter glass bottom dish in BIS medium under reduced oxygen using Anaerocult (Merck). After the medium was removed, approximately 7 × 105 CHO cells were added to the dish at the ratio of 10 CHO cells per amoeba, and the center part of the glass bottom dish was covered by a cover slip. Live imaging was performed on Carl Zeiss LSM780 (Oberkochen, Germany) with default settings on the time series mode. Images captured were analysed by ZEN software (Carl Zeiss). The recruitment of GFP‐EhSNX1/2 to phagosomes and trogosomes was estimated by quantitating the fluorescent intensity of a region of interest on the phagosome membrane and the neighboring cytoplasm. The average intensity per pixel of the region of interest was calculated. The ratio of the fluorescence intensity of GFP‐EhSNX1/2 on the phagosome membrane against that in the cytosol was calculated and referred as the relative fluorescent signal on the membrane.
4.11. Cross‐linking and immunoprecipitation of HA‐tagged EhSNXs and native EhVps26
Approximately 1 × 106 trophozoites of the amoeba transformants transfected with pEhExHA‐SNX1, pEhExHA‐SNX2, pEhExSNX1‐HA, pEhExSNX2‐HA, or pEhExHA were cultured on a 10‐cm diameter dish in BIS medium with reduced oxygen using Anaerocult (Merck). CHO cells were added to the dish at the ratio of 10 CHO cells per amoeba, and the dish was incubated at 35.5°C for 30 min. After the medium containing uningested CHO cells was removed, the amoebae were detached from the surface of the dishes by adding cold PBS into the dishes and incubating them on ice for 10 min. After the trophozoites were washed with PBS three times, the cell pellet was cross‐linked with DSP as described above. After washing twice with PBS, the cells were lysed with 800 μl of lysis buffer (50‐mM Tris‐HCl, pH 7.5, 15‐mM NaCl, containing 1% Triton X‐100, 0.05 mg/ml E‐64, and complete mini). After the debris was removed by centrifugation at 16,000 × g at 4°C for 5 min, the lysates were mixed and incubated with 50 μl of protein G Sepharose beads (GE Healthcare, Illinoi, USA; 80 % slurry) at 4°C for 1 hr. After centrifugation at 800 × g at room temperature for 3 min, the supernatant was transferred to a new 1.5‐ml microtube containing either approximately 50 μl (80% slurry) of anti‐HA monoclonal antibody produced in mouse, clone HA‐7, purified immunoglobulin conjugated to agarose beads (Sigma‐Aldrich) or 50 μl of protein G Sepharose (GE Healthcare), which was treated with 250 μl of PBS, 2 μl of anti‐Vps26 rabbit antiserum for 1 hr at 4°C, was incubated with inversion at 4°C for 3.5 hr. The mixture was centrifuged at 800 × g to separate unbound proteins. The beads were washed three times with 1 ml of lysis buffer. After washing, the beads were resuspended in 180 μl of lysis buffer containing HA‐peptide at the final concentration of 0.2 mg/ml and the mixture was incubated at 4°C overnight to elute the bound proteins for HA‐EhSNXs or directly boiled with 50 μl of 2x SDS‐PAGE sample buffer for EhVps26.
4.12. Quantification of trogocytosis and phagocytosis using image cytometer
Approximately 3 × 105 trophozoites were incubated in 6 ml of BIS medium containing 10‐μM CellTracker Green for 30 min at 35.5°C. CHO cells were stained with 10‐μM CellTracker Blue as described above. Approximately 1 × 105 trophozoites were seeded onto a 3.5‐cm diameter glass bottom dish in 2 ml of BIS medium and incubated for 20 min. For trogocytosis measurement, after removing the supernatant, approximately 1 × 105 CHO cells in 90‐μl BIS medium were added onto the 1.0‐cm diameter glass central pit of the dish. For phagocytosis measurement, CHO cells were killed by incubating at 55°C for 15 min and added to the pit of the dish. After the pit was covered with a cover glass, the mixture was cultivated at 35.5°C under anaerobic conditions. The images were taken on a Confocal Quantitative Image Cytometer, CQ1 (Yokogawa Electric Corporation, Tokyo, Japan) every 10 min for 80 min. The volume of the ingested CHO cells (blue) was calculated using three‐dimensionally reconstituted data. The data shown are the average of the volumes of all trogosomes per amoeba in three wells each. Amoebae were mixed with CHO cells prepared as described above, and images were captured on CQ1. To analyse these data using three‐dimensional structured images, CQ1 was set as below. At first, the top and bottom positions of a Z‐stack contain almost all phagosomes. The number of slices depends on the distance between layers, which should be less than 4 μm. The images were further analysed to quantify the volume and the fluorescence intensity of the ingested CHO cells using the CellPathfinder high content analysis software from the manufacturer of CQ1.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
FUNDING INFORMATION
Grants‐in‐Aid for Scientific Research (B) and Challenging Research (Exploratory) (KAKENHI JP26293093, JP17K19416, and JP18H02650 to T.N.) and Grants‐in‐Aid for Scientific Research (B) and (C) (JP16K08766 and JP19H03463 to K.N‐T.) from the Japan Society for the Promotion of Science.
Grant for research on emerging and re‐emerging infectious diseases from Japan Agency for Medical Research and Development (AMED, JP18fk0108046 to T.N.; 17fk0108122j1101, 18fk0108049j1102, and 19fk0108049j1103 to K.N‐T)
Grant for US‐Japan Cooperative Medical Science Program from AMED (JP17jk0210018 to K.N‐T.)
Grant from the National Center for Global Health and Medicine Intramural Research Fund (29–2013 to T.N.)
Grant for Science and Technology Research Partnership for Sustainable Development (SATREPS) from AMED and Japan International Cooperation Agency (JICA) (T.N.).
Supporting information
Figure S1.
Expression of GFP‐HrsFYVE under tetracycline induction. Approximately 1 × 105 of trophozoites were cultivated with 10 μg/ml tetracycline for 0‐48 hr, harvested, and subjected to SDS‐PAGE and immunoblot analyses. Approximately 20 μg protein from lysates at each time point was electrophoresed. An arrowhead indicates GFP‐HrsFYVE.
Figure S2.
Effects of phosphatase inhibitors and cross linker on the stability of GFP‐HrsFYVE. Approximately 1 × 107 of trophozoites were cultivated with 10 μg/ml tetracycline for 24 hr and harvested. Cells were treated or untreated with DSP, β‐glycerophosphate, EDTA, sodium fluoride, and sodium orthovanadate. They were homogenised by a Dounce homogeniser and separated into cytosol and membrane fractions by centrifugation at 100,000 × g for 1 hr. The fractions were subjected to SDS‐PAGE and immunoblot analyses. Approximately 20 μg protein from fractions was electrophoresed. An arrowhead indicates GFP‐HrsFYVE.
Figure S3.
Amino acid alignment of the PX domain of HsSNX1‐3 and EhSNX1‐2. The residues conserved in EhSNX1‐2 and HsSNX3, but not in HsSNX1‐2, are shown with yellow background. Residue numbers by amino acid sequences correspond to those in each protein.
Figure S4.
The effects of gene silencing of EhSNX2 gene on phagocytosis of prekilled CHO cells. Trophozoites of EhSNX2gs and control strains stained with CellTracker Green were incubated with prekilled CHO cells stained with CellTracker Blue. The images were taken on CQ1 every 10 min for 80 min. The volume of the ingested dead CHO cells (blue) was calculated using three dimensionally reconstituted data. Bars indicate standard errors. Asterisks indicate statistical significance by Tukey‐test (p<0.05). Experiments were conducted three times independently, and a representative data set is shown.
Movie S1.
Movie S2.
Table S1.
A list of proteins detected from bead‐containing phagosomes isolated from GFP‐HrsFYVE‐expressing transformants in the presence or absence of tetracycline induction. Protein name, accession number, predicted molecular weight, quantitative value (QV), the ratio of QV in the absence of tetracycline to QV in the presence of tetracycline, and category of each protein detected are shown. Only the proteins that were detected >2‐fold in the absence of tetracycline compared to those detected in the presence of tetracycline are listed.
Table S2.
A list of proteins detected from the unique band specifically recognised from HA‐EhSNX1‐expressing transformants by immunoprecipitation using anti‐HA antibody. The values indicate the relative frequency of the detected peptides corresponding to each protein. “HA‐EhSNX1/HA” indicates the division of the value of HA‐EHhSNX1 by the value of HA control. This is a whole list of the detected proteins from the specific bands.
Watanabe N, Nakada‐Tsukui K, Nozaki T. Two isotypes of phosphatidylinositol 3‐phosphate‐binding sorting nexins play distinct roles in trogocytosis in Entamoeba histolytica . Cellular Microbiology. 2020;22:e13144 10.1111/cmi.13144
Natsuki Watanabe and Kumiko Nakada‐Tsukui contributed equally to this paper.
REFERENCES
- Boettner, D. R. , Huston, C. D. , Linford, A. S. , Buss, S. N. , Houpt, E. , Sherman, N. E. , & Petri, W. a. (2008). Entamoeba histolytica phagocytosis of human erythrocytes involves PATMK, a member of the transmembrane kinase family. PLoS Pathogens, 4, 122–133. 10.1371/journal.ppat.0040008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino, J. S. , & Hurley, J. H. (2008). Retromer. Current Opinion in Cell Biology, 20, 427–436. 10.1016/j.ceb.2008.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracha, R. , Nuchamowitz, Y. , & Mirelman, D. (2003). Transcriptional silencing of an amoebapore gene in Entamoeba histolytica: Molecular analysis and effect on pathogenicity. Eukaryotic Cell, 2, 295–305. 10.1128/EC.2.2.295-305.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman, C. , & Ktistakis, N. T. (2010). Regulation of autophagy by phosphatidylinositol 3‐phosphate. FEBS Letters, 584, 1302–1312. 10.1016/j.febslet.2010.01.011 [DOI] [PubMed] [Google Scholar]
- Byekova, Y. A. , Powell, R. R. , Welter, B. H. , & Temesvari, L. A. (2010). Localization of phosphatidylinositol (3,4,5)‐trisphosphate to phagosomes in Entamoeba histolytica achieved using glutathione S‐transferase‐ and green fluorescent protein‐tagged lipid biosensors. Infection and Immunity, 78, 125–137. 10.1128/IAI.00719-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, R. Y. R. , & Wong, S. H. (2013). SNX3 recruits to phagosomes and negatively regulates phagocytosis in dendritic cells. Immunology, 139(1), 30–47. 10.1111/imm.12051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen, P. (2008). Endosomal sorting and signalling: An emerging role for sorting nexins. Nature Reviews Molecular Cell Biology, 9, 574–582. http://www.nature.com/nrm/journal/v9/n7/abs/nrm2427.html;jsessionid=2B6040A2FF75C8EA809BE493E30C2D52, 10.1038/nrm2427 [DOI] [PubMed] [Google Scholar]
- Diamond, L. S. , Mattern, C. F. T. , & Bartgis, I. L. (1972). Viruses of Entamoeba histolytica. I. Identification of transmissible virus‐like agents. Journal of Virology, 9, 326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, L. S. , Harlow, D. R. , & Cunnik, C. C. , (1978). A new medium for the axenic cultivation of Entamoeba histolytica and other Entamoeba. Trans R Soc Trop Med Hyg, 72, 431–432. [DOI] [PubMed] [Google Scholar]
- Furukawa, A. , Nakada‐tsukui, K. , & Nozaki, T. (2013). Cysteine protease‐binding protein family 6 mediates the trafficking of amylases to phagosomes in the enteric protozoan Entamoeba histolytica . Infection and Immunity, 81, 1820–1829. 10.1128/IAI.00915-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist, C. A. , Houpt, E. , Trapaidze, N. , Fei, Z. , Crasta, O. , Asgharpour, A. , … Petri, W. A. (2006). Impact of intestinal colonization and invasion on the Entamoeba histolytica transcriptome. Molecular and Biochemical Parasitology, 147, 163–176. 10.1016/j.molbiopara.2006.02.007 [DOI] [PubMed] [Google Scholar]
- Goley, E. D. , & Welch, M. D. (2006). The ARP2/3 complex: An actin nucleator comes of age. Nature Reviews Molecular Cell Biology, 7, 713–726. 10.1038/nrm2026 [DOI] [PubMed] [Google Scholar]
- Hamann, L. , Buss, H. , & Tannich, E. , (1997). Tetracycline controlled gene expression in Entamoeba histolytica. MolBiochem Parasitol, 84, 83–91. [DOI] [PubMed] [Google Scholar]
- Lenoir, M. , Ustunel, C. , Rajesh, S. , Kaur, J. , Moreau, D. , Gruenberg, J. , & Overduin, M. (2018). Phosphorylation of conserved phosphoinositide binding pocket regulates sorting nexin membrane targeting. Nature Communications, 9, 1–12. 10.1038/s41467-018-03370-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano, R. , Naghavi, M. , Foreman, K. , Lim, S. , Shibuya, K. , Aboyans, V. , … Murray, C. J. L. (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. The Lancet, 380, 2095–2128. 10.1016/S0140-6736(12)61728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion, S. , Laurent, C. , & Guillén, N. (2005). Signalization and cytoskeleton activity through myosin IB during the early steps of phagocytosis in Entamoeba histolytica: a proteomic approach . Cellular Microbiology, 7, 1504–1518. 10.1111/j.1462-5822.2005.00573.x [DOI] [PubMed] [Google Scholar]
- Meng, X. , & Ji, Y. (2013). Modern computational techniques for the HMMER sequence analysis. ISRN Bioinformatics, 2013, 1–13. 10.1155/2013/252183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi‐ichi, F. , Makiuchi, T. , Furukawa, A. , Sato, D. , & Nozaki, T. (2011). Sulfate activation in mitosomes plays an important role in the proliferation of Entamoeba histolytica . PLoS Neglected Tropical Diseases, 5, 1–7. 10.1371/journal.pntd.0001263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra, B. N. , Saito‐Nakano, Y. , Nakada‐Tsukui, K. , Sato, D. , & Nozaki, T. (2007). Rab11B small GTPase regulates secretion of cysteine proteases in the enteric protozoan parasite Entamoeba histolytica . Cellular Microbiology, 9, 2112–2125. 10.1111/j.1462-5822.2007.00941.x [DOI] [PubMed] [Google Scholar]
- Nakada‐Tsukui, K. , Okada, H. , Mitra, B. N. , & Nozaki, T. (2009). Phosphatidylinositol‐phosphates mediate cytoskeletal reorganization during phagocytosis via a unique modular protein consisting of RhoGEF/DH and FYVE domains in the parasitic protozoon Entamoeba histolytica . Cellular Microbiology, 11, 1471–1491. 10.1111/j.1462-5822.2009.01341.x [DOI] [PubMed] [Google Scholar]
- Nakada‐Tsukui, K. , Saito‐nakano, Y. , Ali, V. , & Nozaki, T. (2005). A retromerlike complex is a novel Rab7 effector that is involved in the transport of the virulence factor cysteine protease in the enteric protozoan parasite Entamoeba histolytica . Molecular Biology of the Cell, 16, 5294–5303. 10.1091/mbc.E05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki, T. , Asai, T. , Sanchez, L. B. , Kobayashi, S. , Nakazawa, M. , & Takeuchi, T. (1999). Characterization of the gene encoding serine acetyltransferase, a regulated enzyme of cysteine biosynthesis from the protist parasites Entamoeba histolytica and Entamoeba dispar: Regulation and possible function of the cysteine biosynthetic pathway in Entamoeba . Journal of Biological Chemistry, 274, 32445–32452. 10.1074/jbc.274.45.32445 [DOI] [PubMed] [Google Scholar]
- Okada, M. , Huston, C. D. , Oue, M. , Mann, B. J. , Petri, W. A. , Kita, K. , & Nozaki, T. (2006). Kinetics and strain variation of phagosome proteins of Entamoeba histolytica by proteomic analysis. Molecular and Biochemical Parasitology, 145, 171–183. 10.1016/j.molbiopara.2005.10.001 [DOI] [PubMed] [Google Scholar]
- Penuliar, G. M. , Furukawa, A. , Sato, D. , & Nozaki, T. (2011). Mechanism of trifluoromethionine resistance in Entamoeba histolytica . Journal of Antimicrobial Chemotherapy, 66, 2045–2052. 10.1093/jac/dkr238 [DOI] [PubMed] [Google Scholar]
- Picazarri, K. , Nakada‐Tsukui, K. , Tsuboi, K. , Miyamoto, E. , Watanabe, N. , Kawakami, E. , & Nozaki, T. (2015). Atg8 is involved in endosomal and phagosomal acidification in the parasitic protist Entamoeba histolytica . Cellular Microbiology, 17, 1510–1522. 10.1111/cmi.12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston, K. S. (2015). Taking a bite: Amoebic trogocytosis in Entamoeba histolytica and beyond. Current Opinion in Microbiology, 28, 26–35. 10.1016/j.mib.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston, K. S. , Solga, M. D. , MacKey‐Lawrence, N. M. , Bhattacharya, A. , & Petri, W. A. (2014). Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Nature, 508, 526–530. 10.1038/nature13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan, G. , Vines, R. R. , Mann, B. J. , & Petri, W. A., Jr. (1997). A tetracycline‐inducible gene expression systemin Entamoeba histolytica. Mol Biochem Parasitol, 84, 93–100. [DOI] [PubMed] [Google Scholar]
- Seaman, M. N. J. (2012). The retromer complex—Endosomal protein recycling and beyond. Journal of Cell Science, 125, 4693–4702. 10.1242/jcs.103440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlata , Nakada‐Tsukui, K. , & Nozaki, T. (2017). AGC family kinase 1 participates in trogocytosis but not in phagocytosis in Entamoeba histolytica . Nature Communications, 8, 1–12. 10.1038/s41467-017-00199-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, V. K. , Yadav, R. , Watanabe, N. , Tomar, P. , Mukherjee, M. , Gourinath, S. , … Datta, S. (2017). Structural and thermodynamic characterization of metal binding in Vps29 from Entamoeba histolytica: Implication in retromer function. Molecular Microbiology, 106, 562–581. 10.1111/mmi.13836 [DOI] [PubMed] [Google Scholar]
- Worby, C. A. , & Dixon, J. E. (2002). Sorting out the cellular functions of sorting nexins. Nature Reviews . Molecular and Cellular Biology, 3, 919–931. [DOI] [PubMed] [Google Scholar]
- Yeung, T. , & Sergio, G. (2007). Lipid signaling and the modulation of surface charge during phagocytosis. Immunological Reviews, 219, 17–36. 10.1111/j.1600-065X.2007.00546.x [DOI] [PubMed] [Google Scholar]
- Yousuf, M. A. , Mi‐Ichi, F. , Nakada‐Tsukui, K. , & Nozaki, T. (2010). Localization and targeting of an unusual pyridine nucleotide transhydrogenase in Entamoeba histolytica . Eukaryotic Cell, 9, 926–933. 10.1128/EC.00011-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Expression of GFP‐HrsFYVE under tetracycline induction. Approximately 1 × 105 of trophozoites were cultivated with 10 μg/ml tetracycline for 0‐48 hr, harvested, and subjected to SDS‐PAGE and immunoblot analyses. Approximately 20 μg protein from lysates at each time point was electrophoresed. An arrowhead indicates GFP‐HrsFYVE.
Figure S2.
Effects of phosphatase inhibitors and cross linker on the stability of GFP‐HrsFYVE. Approximately 1 × 107 of trophozoites were cultivated with 10 μg/ml tetracycline for 24 hr and harvested. Cells were treated or untreated with DSP, β‐glycerophosphate, EDTA, sodium fluoride, and sodium orthovanadate. They were homogenised by a Dounce homogeniser and separated into cytosol and membrane fractions by centrifugation at 100,000 × g for 1 hr. The fractions were subjected to SDS‐PAGE and immunoblot analyses. Approximately 20 μg protein from fractions was electrophoresed. An arrowhead indicates GFP‐HrsFYVE.
Figure S3.
Amino acid alignment of the PX domain of HsSNX1‐3 and EhSNX1‐2. The residues conserved in EhSNX1‐2 and HsSNX3, but not in HsSNX1‐2, are shown with yellow background. Residue numbers by amino acid sequences correspond to those in each protein.
Figure S4.
The effects of gene silencing of EhSNX2 gene on phagocytosis of prekilled CHO cells. Trophozoites of EhSNX2gs and control strains stained with CellTracker Green were incubated with prekilled CHO cells stained with CellTracker Blue. The images were taken on CQ1 every 10 min for 80 min. The volume of the ingested dead CHO cells (blue) was calculated using three dimensionally reconstituted data. Bars indicate standard errors. Asterisks indicate statistical significance by Tukey‐test (p<0.05). Experiments were conducted three times independently, and a representative data set is shown.
Movie S1.
Movie S2.
Table S1.
A list of proteins detected from bead‐containing phagosomes isolated from GFP‐HrsFYVE‐expressing transformants in the presence or absence of tetracycline induction. Protein name, accession number, predicted molecular weight, quantitative value (QV), the ratio of QV in the absence of tetracycline to QV in the presence of tetracycline, and category of each protein detected are shown. Only the proteins that were detected >2‐fold in the absence of tetracycline compared to those detected in the presence of tetracycline are listed.
Table S2.
A list of proteins detected from the unique band specifically recognised from HA‐EhSNX1‐expressing transformants by immunoprecipitation using anti‐HA antibody. The values indicate the relative frequency of the detected peptides corresponding to each protein. “HA‐EhSNX1/HA” indicates the division of the value of HA‐EHhSNX1 by the value of HA control. This is a whole list of the detected proteins from the specific bands.