

Abstract
CD8+ Tregs have been long described and significant progresses have been made about their phenotype, their functional mechanisms, and their suppressive ability compared to conventional CD4+ Tregs. They are now at the dawn of their clinical use. In this review, we will summarize their phenotypic characteristics, their mechanisms of action, the similarities, differences and synergies between CD8+ and CD4+ Tregs, and we will discuss the biology, development and induction of CD8+ Tregs, their manufacturing for clinical use, considering open questions/uncertainties and future technically accessible improvements notably through genetic modifications.
Keywords: CD8+ Treg, therapy, tolerance
1. INTRODUCTION
CD8+ Tregs were the first suppressive cells reported but studies were then focused on CD4+ Tregs.1 Over the last two decades, we, along with others, deeply investigated the CD8+ Tregs.2 To date, significant progress have been made about their phenotype, their functional mechanisms, and their suppressive ability compared to conventional CD4+ Tregs.3 Through this in‐depth fundamental research, it has been learned more about their origin and development and such advances will lead to improvement in their generation including from stem cells (unpublished work discussed in this review).
Cell therapy has developed in recent years in the field of transplantation and autoimmune diseases with promising results. Current clinical trials use T cells and non‐T cells, and among Tregs, FOXP3+ and FOXP3− CD4+ Tregs (phase I ONE, ALT‐TEN, phase I/IIa CATS1), but not CD8+ Tregs. We are at the dawn of the first in human clinical trial using CD8+ Treg‐cell therapy in transplanted patients (https://www.reshape-h2020.eu/). Experience from previous and current clinical trials using T cell therapy, combined with our extensive knowledge of CD8+ Tregs, incremented by the technical revolution of T Cell Receptor (TCR) and chimeric antigen receptor (CAR) engineering, in synergy with Treg promoting drugs, led us to the Eight‐Treg first in human clinical trial.
In this review, we will discuss the biology and development of CD8+ Tregs, their contribution to the tolerance status apart from CD4+ Tregs, their manufacturing for clinical use, considering open questions/uncertainties and future technically accessible betterments notably through genetic modifications.
2. MAIN CHARACTERISTICS OF CD8+ REGULATORY T CELLS
Despite a streamline work from several groups, the definition of CD8+ Tregs remains the major problem of this cell subset population and the reason for them not being assessed in clinical protocols. This lack in assessment is making marker discovery even more problematic and slow, leading to a vicious circle. The fact that, at steady state, expression of FOXP3, a sensitive marker of Treg, is lower in the CD8+ T cell population compared to the CD4+ T cell population in mouse, rat and human,3 has contributed to this disregard of CD8+ Treg's potential and interest, despite in vitro and in vivo data demonstrating their suppressive capacity. In addition, the methylation status of FOXP3 in human CD8+ Tregs showed an intermediate level of demethylation compared to one of the CD4+ Tregs for some locus of the FOXP3 gene.3 We showed that following ex vivo stimulation, human CD8+ Tregs sorted based on the expression of CD45RC, a splicing variant of the CD45 molecule key in the signaling of T cells that we use as a marker to isolate CD8+ Tregs4, 5, 6; FOXP3 is expressed by all expanded CD8+ Tregs that display highly upregulated levels of expression vs. blood unstimulated CD8+FOXP3+, suggesting that FOXP3 can correlate with the in vivo and in vitro suppressive potential of human CD8+ Tregs.3 In contrast, in a rat model of transplantation tolerance following costimulation blockade inducing in vivo CD8+ Tregs, FOXP3 was not upregulated,4 nor was it in a model of tolerance in rat using donor antigen therapy.7 FOXP3 is also not found particularly expressed in mouse CD8+ Tregs, and rather the transcription factor HELIOS or the surface marker CD122+ or CD28‐ are used to identify CD8+ Tregs in mice.8, 9 Other surface markers were also suggested and among the most frequently used CD25, CD127, GITR, CD39, Lag3, and CTLA‐4 that are used in different contexts and different species to identify CD8+ Tregs and analyze their suppressive activity in vitro and in vivo in autoimmune diseases, immune reactions to non‐self, cancer and materno‐fetal tolerance.2, 10 Indeed, using a combination of these markers to identify CD8+ Tregs, their suppressive action has been demonstrated in autoimmune diseases such as in the experimental autoimmune encephalomyelitis model,11, 12, 13 multiple sclerosis,14, 15, 16 systemic lupus erythematosus (SLE)17, 18 and primary biliary cirrhosis,19 in infection in humans with mycobacteria, human immunodeficiency virus or epstein‐barr virus,20, 21, 22, 23 in cancer in humans and mice24, 25, 26 or in transplantation in humans, mice and rats.3, 4, 27, 28, 29, 30, 31, 32 CXCR5 was used to identify antibody‐suppressor FOXP3−CD8+ cells in mice and essential in their function of suppression of humoral immunity, reducing germinal center B cells and CD4+ T follicular helper cells in the context of hepatocyte transplantation.33
A very recent study of single cell RNA sequencing on CD4+ Tregs has highlighted the importance of some transcription factors and extra‐membrane molecules.34 This work showed that CD4+ Tregs and CD4+ Tconvs can be distinguished by the ubiquitous expression of FOXP3, IKZF2 (IKAROS), TNFR2, IL2RA and IL2RB in mice and humans. Analysis using DGE‐RNA sequencing of fresh and activated CD8+CD45RClow/‐ Tregs allowed us to show the increase in FOXP3 expression and suppressive molecules after activation. However, nowadays no single cell RNA sequencing study is being published on CD8+ Tregs. Our analysis of single cell RNA sequencing data on fresh CD8+CD45RClow/‐ cells from healthy volunteers confirms the expression of molecules known in CD4+ Tregs such as TNFR2 but never clearly showed before in CD8+ Tregs (unpublished data).
Soluble factors can also be used to identify and even sort CD8+ Tregs since the development of kit using bispecific antibodies allows the sorting of live cytokine‐secreting cells. We have shown that sorted human IFNγ+IL‐10+CD8+CD45RClow/‐ Tregs were more potent suppressor cells than the rest of the CD8+CD45RClow/‐ Tregs3 and this was also indicative of the role of these cytokines in CD8+ Treg suppressive activity, in particular IFNγ (Figure 1). Our interest in IFNγ came from the observations that blocking its activity abrogated the CD8+CD45RClow/‐ Treg suppressive activity in a model of allogeneic cardiac transplantation.4 We further demonstrated that indeed, IFNγ induced indoleamine 2,3‐dioxygenase (IDO) production by endothelial cells (ECs) of the graft and plasmacytoid dendritic cells, an enzyme catabolizing tryptophan essential for effector T cells proliferation.4, 35 IFNγ was also involved in fibroleukin‐2 (FGL2) induction, a cytokine acting further through regulatory B cell induction.36 We have also shown that IL‐34, a recently discovered cytokine involved in monocyte/macrophage differentiation, was secreted by approximately half of FOXP3+CD8+ and CD4+ Tregs.37 We showed that IL‐34 was involved in FOXP3+ Treg suppressive activity and acted as a suppressive cytokine since administration in a model of allogeneic cardiac transplantation in combination with a suboptimal dose of rapamycin‐induced transplant tolerance. This was the first description of a role for this cytokine in T cell biology and transplantation.38 Thus, a complex regulatory network of cells around CD8+ Tregs is acting to sustain tolerance.39 Other more classical cytokines such as TGFβ have also been described as playing a role in CD8+ Treg suppressive activity.2
Figure 1.
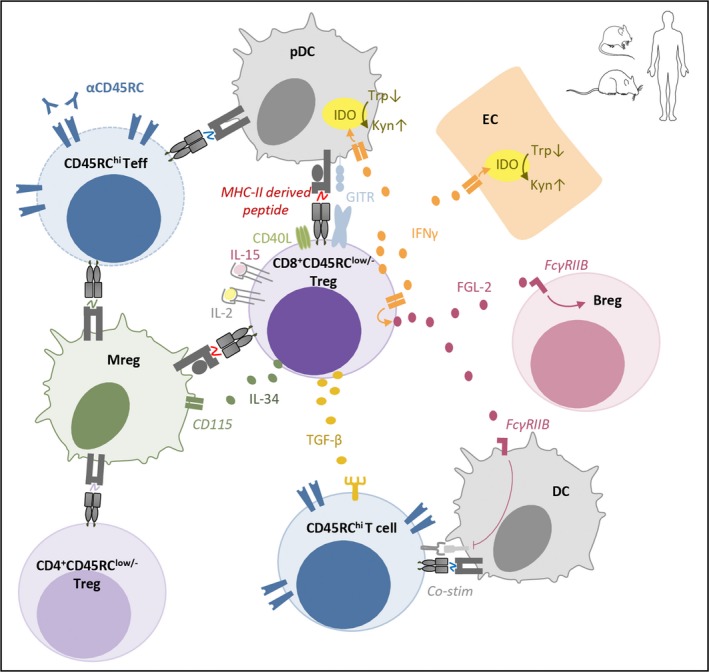
Schematic depicting identified mechanisms of action and markers of rat, mouse and human CD8+CD45RClow/- Tregs. Breg, regulatory B cell; Co‐stim, costimulatory molecules; DC, dendritic cell; EC, endothelial cell; IDO, indoleamine 2,3‐dioxygenase; Kyn, kynurenin; Mreg, regulatory macrophages; pDC, plasmacytoid dendritic cell; Trp, tryptophan. Bended arrows indicate conversion or induction. Up and down arrows indicate increase and decrease, respectively
Some of these molecules or mechanisms of action have been evidenced by study of the fetal‐maternal interface.40, 41 Indeed, in maternal tolerance where the allogeneic fetal trophoblast invades maternal tissues and interacts with maternal leukocytes, the immune system needs to be regulated to tolerate the presence of the fetus. CD8+ T cells are the main component of decidual T cells and CD8+ Tregs have been evidenced as important in the maintenance of a normal pregnancy.42, 43, 44 Decreased number and altered function of Tim‐3+CTLA‐4+CD8+ T cells correlated with miscarriage and blockade of Tim‐3 and CTLA‐4 inhibited production of anti‐inflammatory cytokines and were detrimental in the maintenance of the pregnancy.42 Allogeneic fetal trophoblasts express the non‐classical major histocompatibility complex (MHC) class I molecule human leukocyte antigen G which is needed for immune tolerance establishment and was shown to increase the number of Tregs.45, 46
MHC restriction preference of CD8+ Tregs has been shown as important for their suppressive activity and for some subpopulations of CD8+ Tregs different from what has been described for CD8+ Teff cells. Particularly in mice and humans, the best described population of mouse CD8αα+ TCRαβ+ Tregs or human CD8+ Tregs (no specific phenotype associated except pMHC restriction) has been described to preferentially recognize non‐classical MHC class I molecules Qa‐1 or HLA‐E that are orthologous genes.47, 48, 49, 50 These non‐classical MHC class I restricted populations have the property to recognize TCR, MHC or heat shock protein derived peptides (ie Qdm, HSP60sp) presented by Qa‐1 or HLA‐E.50, 51 We have also shown that this capacity to recognize MHC‐derived peptide is not restricted to non‐classical MHC class I molecule but also true for classical MHC class I molecule. We showed that peptides derived from a polymorphic region of the β1domain of MHC class II molecules from the donor and presented by recipient classical MHC I molecules can expand CD8+CD45RClow/‐ Tregs and induce tolerance in transplantation in an antigen‐specific manner in rats7, 52 and humans (unpublished data).
In this context, the identification of new membrane markers for CD8+ Treg populations to better define a consensus phenotype is a major challenge and objective for the future. In addition, a better understanding of the mechanisms involved in their development and function would allow to better understand and optimize their clinical potential.
3. SIMILARITIES, DIFFERENCES AND SYNERGIES BETWEEN CD8+ AND CD4+ TREGS
A major aspect that remains to be properly addressed given the lack of assessment of CD8+ Tregs in diseases is the similarities, differences and synergies that could exist between CD8+ and CD4+ Tregs that could emphasize the interest in CD8+ Tregs (Table 1).
Table 1.
Differences between CD8+ and CD4+ Tregs
| CD8+ Treg | CD4+ Treg | |
|---|---|---|
| MHC | Class I, classical7 and non‐classical47 | Class II53 |
| Composition | Mostly memory3 | Naive and memory213 |
| Overall suppressive capacity | CD8> or =CD43, 32, 54 | |
| Suppression of memory and/or naive T cell responses | Memory and naive58 | Naive57 |
| Interferon gamma mediated suppression | Repeatedly described3, 4, 65, 73, 74, 75, 76, 77 | Rarely described70, 78 |
| IL‐2 induced proliferation | CD8> or =CD432, 103, 104, 105, 106, 107 | |
| IL‐15 induced proliferation | CD8> or =CD43, 54, 61, 111, 112, 113, 114 | |
| Preferred polyclonal stimulation | mAbs3 | Beads146, 147, 148, 149 |
3.1. Function and mechanisms of action
A very obvious and important difference is that CD8+ and CD4+ Tregs recognize their cognate antigens presented by MHC‐I or MHC‐II molecules, respectively. Therefore, CD8+ Tregs will activate their suppressive activity on virtually all cells whereas CD4+ Tregs activation will occur only on cells expressing MHC‐II molecules. In the case of solid organ transplantation, CD8+ Tregs will activate on not only all donor graft cells but also on dendritic cells through indirect donor alloantigen presentation on recipient MHC‐I molecules.7 Thus, for both CD8+ and CD4+ Tregs, bystander inhibition of T and B cell activation will occur through antigen presenting cells (APCs) which express MHC‐I+ and MHC‐II+ molecules. In inflamed tissues non‐immune cells upon activation by IFNγ and/or TNFα may express MHC‐II molecules, but MHC‐I expression is constitutive by virtually all cells and its expression is also upregulated by IFNγ and/or TNFα. Thus, CD8+ Tregs will be advantageous, particularly in organ transplantation or graft versus host disease (GVHD) since suppression will additionally occur in all allogeneic cells, including ECs of vascularized grafts or host organs in GVHD. Another potential area in which MHC‐I antigen recognition might be useful is in the case of using in the future allogeneic off‐the shelf Tregs. Recipient cells attacking allogeneic CD8+ Tregs may be suppressed through MHC‐I recognition whereas CD4+ Tregs could only do it if the recipient cells express MHC‐II molecules, which can be the case for human T cells but this demands high levels of IFNγ‐mediated activation. Although a group recently published the structure of an induced CD4+ Tregs' TCR binding a pMHC,53 little is known about CD4+ Tregs pMHC restriction in contrast to CD8+ Tregs. This is probably due to the easiness to generate MHC class I/peptide tetramers that led to the opportunity to better identify and understand the TCR constraints and antigen recognition of CD8+ Tregs.
A direct comparison of CD8+ and CD4+ Tregs' overall suppressive capacity has not been done in many settings but in solid organ transplant54 and GVHD32 models in mice, suppression by CD8+ Tregs was superior to the one of CD4+ Tregs. We have observed that fresh, in vitro expanded human polyclonal and donor‐specific CAR engineered CD8+ Tregs were all superior to their CD4+ Treg counterparts (unpublished data and Ref. 3). Importantly, in certain tolerance models one or the other or both Treg subsets can be induced and play a role. As examples, in the same rat heart allotransplantation tolerance model, CD8+ and/or CD4+ Tregs were differently involved depending on the treatment used: blocking CD40‐CD40L induced tolerance that was entirely dependent and transferable by CD8+ Tregs,4 whereas donor‐specific blood transfusion as well as anti‐CD45RC MAb or IL‐34 administration tolerance was dependent on both CD8+ and CD4+ Tregs.37, 55 In these two last models adoptive transfer of each of the CD8+ or CD4+ Treg subsets resulted in tolerance in a fraction of the recipients that was lower than the one obtained when all Tregs were transferred.37, 55 Similarly, in a mouse model, GVHD was most efficiently inhibited when both CD4+ and CD8+ Tregs were used associated and the presence of CD8+ Tregs preserved a Teff‐mediated graft versus leukemia effect that was lost when CD4+ Treg cells were used alone.56 Interestingly in the perspective of simultaneously using CD8+ and CD4+ Tregs, both Treg subsets could be produced in vitro at the same time, such as when they are induced in the presence of APCs treated with IL‐34.37 Finally, the suppressive capacity of CD8+ and CD4+ Tregs to suppress naive and memory CD4+ T effector response has been compared in the transplantation field by different groups. This is a very clinically relevant situation to analyze the role of naive vs. memory CD4+ T effector responses, since both are activated at different time points during transplantation and memory T cell response is described as particularly difficult to control with usual immunotherapeutic or even immunosuppression protocols. Both naive and memory CD4+ Treg cells only suppressed naive and not memory CD4+ and CD8+ T responses.57 In contrast, CD8+ Treg cells suppressed both naive and memory effector CD4+ T cell responses.58
As far as mechanisms of action are concerned, several cytokines have been described as playing a role for both CD8+ and CD4+ Tregs, although not necessarily in all models or in vivo or in vitro situations. As an example, IL‐10‐mediated suppression has been described for CD8+CD122+ Tregs in mice 54 but not for human CD8+CD45RClow/− Tregs.3
Similarly, TGFβ has been implicated in the suppressive function of some CD4+ Tregs and at least some types of CD8+ Tregs, such as mouse peptide‐specific CD8+ iTreg cells,59 CD8+FOXP3+CD25+ Tregs induced by corneal ECs60 and CD8+CD45RClow/‐ human Treg cells3 but not of CD8+CD45RA+CCR7+FOXP3+ Treg cells.61
IL‐34 is a cytokine with immunoregulatory properties by at least inducing M2 macrophages38 and that is expressed by both CD8+ and CD4+ Tregs and is able to prolong heart allograft survival.37
FGL2 immunoregulatory properties were first described in CD4+ Treg cells,62 FGL2 expression was then reported in mouse CD8αα Treg cells63 and in rat CD40Ig‐induced CD8+CD45RClow/‐ Treg cells functions. FGL2 function is at least mediated through the FcγRIIB receptor inhibiting dendritic cell maturation, inducing B cell apoptosis64 and generating regulatory B cells through unknown mechanisms.36 IFNγ is a mechanism of suppression not only of CD8+ Tregs3, 65, 66, 67 but also of CD4+ Tregs.68, 69, 70 The mechanism of suppression by IFNγ includes actions on endothelial and antigen presenting cells, such as induction of IDO and FGL2.
IL‐35 has been previously described as expressed by CD4+ Tregs71 but it has also been described as expressed and playing a suppressive role for tumor‐associated CD8+ Tregs.66, 72
Although IFNγ has proinflammatory consequences it has also been described as having pro‐tolerogenic actions, through the production of molecules such as IDO, iNOs and FGL2.68 IFNγ has been described as produced by several CD8+ Treg types 3, 4, 65, 73, 74, 75, 76, 77 although the function of IFNγ in CD8+FOXP3+ cells has not been specifically analyzed. In contrast, and although suppression through IFNγ has been described by CD4+ Tregs in certain models70, 78 CD4+FOXP3+ Tregs in general produce low levels of IFNγ.
PD‐1 has been described in mice to be expressed at lower levels in CD8+ Tregs vs CD4+ Tregs32 but PD‐1 expression is important for CD8+ Treg function,79, 80 as it is for CD4+ Tregs.81, 82 Additionally, PD‐L1 expressed by CD4+ Tregs83 and by human CD8+ Tregs84 suppress CD4+ Teff responses directly through PD‐1 or also through PD‐L1 expressed by DC and macrophages. Therefore, the use of PD‐L1‐Fc or agonistic anti‐PD‐1 antibodies would inhibit immune responses by both promoting Treg activity and by inhibiting Teff responses directly, as well as by inducing tolerogenic APCs.
Although cytotoxicity could be expected to be a widespread mechanism of suppression by CD8+ Treg cells and not of CD4+ Treg cells, only in some cases CD8+ Tregs have been described as cytotoxic 85, 86, 87, 88 and CD4+ Tregs have also been shown in some instances as cytotoxic,89, 90, 91, 92, 93, 94 thus this mechanism is shared by both types of Tregs.
3.2. Metabolic activity
Mechanistic target of rapamycin (mTOR) through mTOR complex 1 (mTORC1) and mTORC2 regulates activation and proliferation through the phosphorylation of several transcription factors.95 Mechanistic target of rapamycin signaling is high and increases the inflammatory response in Teff cells maintaining glycolysis whereas mTOR activity is low in CD4+ Tregs.96 Inhibitors of mTOR such as rapamycin directly bind and inhibit mTORC1. Activity of mTORC1 is necessary for CD4+ Treg cell function to maintain their metabolic status through lipogenesis.97 Therefore, rapamycin at moderate concentrations inhibits CD4+ Teff growth but promotes CD4+ Treg growth and function.98, 99 Although metabolic activity and mTOR function have not been directly analyzed in CD8+ Tregs, the metabolic status of memory CD8+ cells is similar to that of CD4+ Tregs100, 101 and different types of human CD8+ Tregs having a memory phenotype have been shown to expand in the presence of rapamycin,3, 102 thus, inclusion of rapamycin in the in vitro expansion of clinically applicable human CD8+ Tregs as for CD4+ Tregs is a logical approach to increase safety.
3.3. Proliferation in vitro and in vivo
Freshly analyzed human and mouse CD8+FOXP3+ Tregs have been reported as IL2Rα/CD25high and CD127low as CD4+FOXP3+ Tregs103 whereas human CD8+CD45RClow/‐ have a more complex phenotype.3 Transgenic rats with green fluorescent protein under control of the FOXP3 promoter are also CD25highCD127− (unpublished data). The other chains of the IL2R are expressed in a large proportion of fresh human CD8+FOXP3+ Tregs comparable to the ones of CD4+FOXP3+ Tregs (unpublished data). With this expression of the IL2R chains similar to those of CD4+FOXP3+ Tregs it is logical that low dose IL‐2 administration in vivo to mice and human healthy volunteers expanded CD8+FOXP3+ Tregs in both species and even in a higher proportion in humans (6.7 ± 4.3 fold) as compared to CD4+FOXP3+ Tregs (1.8 ± 0.7 fold). The suppression capacity after IL‐2 treatment was also higher in CD8+FOXP3+ Tregs vs. CD4+FOXP3+ Tregs.103 Administration of low dose IL‐2 in cynomolgus monkeys expanded both CD8+FOXP3+ and CD4+FOXP3+ Tregs 10‐fold and 15‐fold, respectively when compared to the baseline levels and the suppressive effect after IL‐2 treatment was comparable for CD8+ and CD4+ FOXP3+ Tregs.104 Similar increase in CD8+ and CD4+ Tregs (11 to 14‐fold) was observed when using an IL‐2 mutein molecule.105 Increase in CD8+FOXP3+ Tregs along with CD4+FOXP3+ Tregs was also observed in patients with hepatitis C virus‐induced vasculitis 106 and type 1 diabetic patients 107 treated with low doses of IL‐2. In a model of GVHD in mice, administration of IL‐2 antibody complexes and rapamycin was synergic to CD8+FOXP3+ Tregs increase up to 30‐fold whereas CD4+FOXP3+ Tregs increased 4‐fold.32 In other studies, in patients treated with low doses of IL‐2, CD8+ Tregs were not reported,108, 109 although only a few of them described extensive immunophenotyping.109
Although IL‐15 favors thymic development of CD4+ Tregs along with IL‐2 through the IL‐2Rβ/γc cytokine receptor complex,110 IL‐15Ra is not expressed by CD4+ Tregs and these cells respond to IL‐15 only when soluble IL‐15Ra binds IL‐15 and presents it in trans.111 This explains why adult CD4+ Tregs do not respond to IL‐15 in vitro while they do in vivo.111, 112 In contrast, IL‐15 is necessary for optimal CD8+ Tregs expansion in vivo113, 114 and in vitro.3, 54, 61
In vitro expansion of CD8+ Tregs in the same conditions used to expand CD4+ Tregs has shown lower suppression activity in some cases but this may be related to the use of culture conditions that are optimal for CD4+ and not CD8+ Tregs.115 In this regard, we have observed that the use of beads coated with anti‐CD3 and anti‐CD28 MAbs ideal for the culture of CD4+ Tregs is toxic for human CD8+ Tregs while being well‐tolerated by mouse CD8+ Tregs (manuscript in preparation). It is then important when comparing in vitro cultured CD8+ and CD4+ Tregs to use the best culture conditions for each cell type.
4. BIOLOGY AND DEVELOPMENT OF CD8+ TREGS
4.1. Origin
The thymic origin of CD8+ nTregs is however still poorly described. In 2016, the presence of CD8+CD28low Treg cells in peripheral blood mononuclear cells (PBMCs) and in children's thymuses was described and also their thymic origin demonstrated.9, 116 CD8+ Tregs are associated with different phenotypes depending on the studies (CD122, CD28, CD45RC, CD103, PD‐1) but mostly their phenotype is rather associated to a differentiation status of central memory cells or effectors memory as characterized by the absence of CD28, CD62L or CD122 expression.3, 117 Other teams describe them rather through the expression of CD44, CCR7 and CD62L which are markers of central memory cells 3, 73 or CD122+ and CD28+.32, 54 When activated, CD8+ Tregs lose the expression of the CD45RA, distinguishing the naive state, to express CD45RO, a marker of memory state, and are dependent on IL‐2 and IL‐15. Naive CD8+ Tregs leave the thymus and such natural CD8+ Tregs have been shown by us with a limited suppressive capacity in both rat and human.3, 4 Indeed, adoptively transferred naive rat CD8+CD45RClow/‐ Tregs cannot inhibit acute transplant rejection in a fully allogeneic model of transplantation4 and human CD8+CD45RClow/‐ Tregs from blood of healthy individuals are more potent suppressor cells in vitro following anti‐CD3 and anti‐CD28 stimulation.3 Effector and memory Tregs have a much more potent suppressive capacity than naive Tregs in mice and humans.118 Effector Tregs are found in the blood and in secondary lymphoid organs, while memory Tregs are found mainly in peripheral tissues and in secondary lymphoid organs.
Transcription factors can identify the thymic origin of CD8+ Tregs. Some were suggested for a long time as reliable markers of Tregs from thymic origin. Indeed, HELIOS, a member of Ikaros family of zinc finger transcription factor, is expressed by 100% of CD4+FOXP3+ Tregs in the thymus and 70% of CD4+FOXP3+ Tregs in peripheral lymphoid tissues.119 HELIOS seems to be involved in the stabilization of the phenotype of CD4+ and CD8+ Tregs in an inflammatory context.8 In a mouse model deficient for HELIOS, CD8+ and CD4+ Tregs were not able to control effector T cells responses. This demonstrates HELIOS' involvement in suppressive functions, differentiation and survival of Tregs.8, 120 Human CD8+CD45RClow/‐ cells co‐express HELIOS and FOXP3.3 FOXP3 seems to be a key transcription factor in the development of Tregs including CD8+ Tregs since silencing FOXP3 with siRNA abrogated the ability of CD8+ Tregs to suppress anti‐DNA antibodies in a lupus model in mice.121 Although adoptive transfer of CD4+FOXP3+ Tregs can cure immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome in FOXP3‐deficient mice122 this does not exclude the possibility that transfer of CD8+FOXP3+ Tregs could also have a similar effect. This is an important experiment that has not been reported and would confirm a physiological role in immune homeostasis for CD8+FOXP3+ Treg. Induced CD8+ Tregs express less FOXP3 than CD4+ Tregs.123 However, we have shown a specific expression of FOXP3 in natural CD8+CD45RClow/‐ Tregs while we do not find expression in CD8+CD45RChigh effector T cells. This expression of FOXP3 is all the more important after cell activation.3 This expression is correlated with the expression of suppressive molecules such as CTLA‐4, GITR, IFNγ and TGFβ. In mice, other transcription factors have been described as involved in the development and function of CD4+ Tregs but they have not yet been described for CD8+ Tregs. IFR4 was shown essential for the expression of Blimp‐1 in the differentiation of CD4+ effector Tregs124; the canonical pathway nuclear factor‐kappa B is also involved in the development and function of CD4+ Tregs by performing inducible deletions of some subunits of the complex. The c‐Rel subunit was critical in thymic development, while the p65 subunit was essential for maturing Tregs and maintaining immune tolerance.125
4.2. Targeted therapies modulate or synergize with CD8+ Tregs
Tregs have been well described to be induced or synergize with antibody or cytokine therapies, to be the main effector regulatory mechanisms boosting the efficacy of such therapies and being key to long‐term sustained effect. Thus, new therapies should strategically aim to promote Tregs to induce a satisfactory control of immune responses. Strategies of costimulation blockade have been shown to efficiently induce and mediate their tolerogenic effect through CD8+ Tregs. Blockade of the CD40/CD40L costimulatory pathway is a well‐known strategy in transplantation for its potential in animal models in tolerance induction, although its translation to the clinic has been slowed down due to thromboembolic events.126 We have shown that blockade of the CD40/CD40L pathway using an adenovirus encoding CD40Ig, a chimeric molecule, in a model of fully incompatible cardiac allotransplantation in rats resulted in tolerance induction dependent on CD8+CD45RClow/− Tregs.2, 4, 35, 127 Blockade of the 4‐1BB/4‐1BBL costimulatory pathway together with immunization in mice was also shown to induce antigen‐specific CD8+ Tregs acting in a IFNγ and TGFβ dependent manner.59 In this model IFNγ directly stimulates CD8+ Tregs to induce a TGFβ‐based suppression.59 In contradiction, 4‐1BB has been shown as important for the suppressive activity of CD8+ Tregs in a model of allergic inflammation.128 We and others showed that IFNγ and TGFβ were key mediators of the suppressive activity of CD8+ Tregs.4, 68, 129, 130 Blockade of ICOS/B7h costimulation also showed potent induction of alloantigen‐specific CD8+PD1+ Tregs in vivo in a model of heart grafts in mice.131 More surprisingly, targeting CD3, a molecule expressed by definition by Treg, has shown effectiveness in inducing both CD4+ and CD8+ Tregs in a collagen induced arthritis model of rheumatoid arthritis in mice.132, 133 In this model, CD8+ Tregs, unlike CD4+ Tregs, potently inhibited Th17 responses, thereby inhibiting a wider range of inflammatory pathways. The authors also showed that monocyte membrane bound TNFα increased FOXP3 expression in CD8+ T cells. Administration of hOKT3g1 (Ala‐Ala) in patients with type 1 diabetes mallitus halted disease progression for >1 year and was associated with increased CD8+ FOXP3+ Tregs.134 Induction of CD8+ Tregs was dependent on TNF and associated with TNFR2 expression by the CD8+ Tregs.135 There is a ying and yang action observed by several groups for cytokines such as TNFα and IFNγ. Anti‐TNF antibodies are used in Crohn's disease and have shown effectiveness in management of the disease but also common failure.136 The importance of action of these cytokines on and by CD8+ Tregs, but also more recently by CD4+ Tregs,137 should certainly be taken into consideration in the potential limited effect of cytokine‐targeting drugs in autoimmune diseases.
Co‐administration of CD8+ Treg therapy with costimulation blockade is also showing synergistic efficacy. In a model of skin transplantation in mice, CD8+CD122+PD‐1+ Tregs expanded ex vivo combined with costimulation blockade of CD40/CD154, but not of B7/CD28, synergizing to prolong the allograft survival in an IL‐10‐dependent manner.113 In fact, blockade of the B7/CD28 costimulation pathway has been also shown by us as being detrimental to CD8+ Treg‐mediated tolerance. Simultaneous blockade of the CD40/CD40L and CD28/B7 interactions using CD40Ig and anti‐CD28 mAbs abrogated in 50% of the recipients tolerance, inhibited CD8+ Treg induction and modified the regulatory mechanisms taking place in the remaining recipients that did not reject their allograft,138 suggesting that the B7/CD28 costimulation is required for CD8+ Treg expansion and function.
CD8+ Treg induction following administration of depleting, modulating or blocking drugs has been very poorly addressed. There are several strategies under investigation for their potential in modulating the Teff/Treg balance, that are not directly targeting costimulation blockade but rather eliminating Teff of activating Tregs. We have shown that modulation of the Teff/Treg balance using anti‐CD45RC mAbs depleting CD45RChigh cells, ie naive and TEMRA cells, but preserving CD45RClow/− cells, ie Tregs, can efficiently induce transplant tolerance in rats and humanized immune mice.55 The mechanisms of tolerance involved the transient depletion of naive and TEMRA cells at the time of transplantation and during 10‐20 days associated with a transient increase of CD4+ and CD8+ Tregs. In this model we demonstrated that upon arrest of the treatment and return to normal level, CD4+ and CD8+ Tregs gained antigen specificity and were functionally superior as shown by in vitro suppressive assays, in vivo adoptive cell transfer in transplanted recipients and transcriptomic analysis. We have similar unpublished results in a model of GVHD in rats and nod scid gamma (NSG) mice and in a model of lupus (unpublished data). In a lupus model in mice, administration of T cell targeting nanoparticles loaded with IL‐2 and TGFβ significantly expanded CD4+ and CD8+ Tregs that could reduce the disease.139 Cytokine therapies such as the one using IL‐34 has been shown to also modulate the Treg/Teff balance by differentiating monocytes into regulatory macrophages that could in turn induce CD8+ and CD4+ Tregs responsible for the long‐term tolerogenic effect.37
5. CD8+ TREG CLINICAL TRIAL IN KIDNEY TRANSPLANTED PATIENTS
While CD8+ T cells have been used in clinic as effector cells to treat cancer and infectious diseases (NCT00791037, NCT01325636, NCT01475058, NCT00110578), CD8+ Tregs have never been used as suppressive cells to treat autoimmune diseases or to control transplant rejection. Indeed, the lack of interest for CD8+ Tregs due to the difficulties of identification and characterization of these cells, in contrast with the extensive knowledge acquired for years on CD4+ Tregs, and the urgency for HSC‐grafted patients with fatal outcome supported rapid translation of CD4+ Treg to the clinic for cell‐based therapies.1 The new interest for CD8+ Tregs over the past two decades has strengthened their therapeutic potential, leading to a future first in human phase I CD8+ Treg‐cell therapy in kidney transplanted patients, named Eight‐Treg, supported by the ReSHAPE consortium, and that will take place in 20212 (https://www.reshape-h2020.eu/) (Figure 2). The primary objective of the Eight‐Treg Phase I trial will be to determine the safety and dose of CD8+ Treg cells. CD8+ Tregs showed no cytotoxicity in vitro or in vivo in NSG mouse models and we observed comparable suppressive activity for CD8+ Tregs and CD4+ Tregs in vitro.3 Currently, the phase I clinical trials using CD4+ Tregs evaluate the cell toxicity by a dose escalation from 5 × 105 to 2 × 107 cells/kg in solid organ transplanted (SOT) patients and from 1 × 105 to 3 × 107 cells/kg in hematopoietic stem cell transplantation (HSCT) patients (NCT02371434 ONEnTreg13, NCT03444064, NCT01050764, NCT00725062, NCT03198234). In particular, the previous ONETreg1 clinical trial showed the safety of a 1 × 107/kg CD4+ Tregs infusion in patients, and the downstream Two Study is underway to evaluate the efficacy of this dose in the treatment of kidney transplanted patients (NCT02129881, 2017‐001421‐41). Even the highly cytotoxic tumor infiltrating lymphocytes (TIL) CD8+ cells infused up to 3x109 cells/kg in patients with melanoma did not induce serious adverse events (NCT01118091, NCT01236573).140 Altogether, these results suggest a low risk in receiving a high dose of CD8+ Tregs for patients. Therefore, the Eight‐Treg protocol is designed for the administration of escalating cell dose every three patients, from 3 × 105 to 2 × 107 CD8+ Tregs/kg in 12 kidney transplanted patients (https://www.reshape-h2020.eu).
Figure 2.
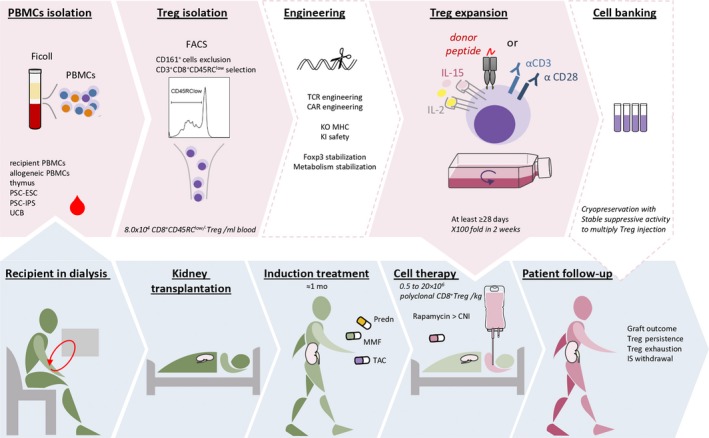
CD8+ Treg‐based therapy process. In vitro steps are indicated in pink background, optional steps in blank background and clinical steps in blue background. CNI, calcineurin inhibitor; KI, knock‐in; KO, knock‐out; MMF, mycophenolate mofetil; Prdn, prednisolone; TAC, tacrolimus; UCB, umbilical cord blood
The type and dose of immunosuppressive drug treatments are critical to ensure patient safety whether the cell therapy is not effective, but should not be deleterious to the adoptively transferred CD8+ Treg cells. We have recently shown that mycophenolic acid stopped their proliferation in contrast to rapamycin.3 In addition, we observed that rapamycin promoted CD8+ Treg proliferation and suppression capacities.3 Similarly, calcineurin inhibitor drugs (CNIs) were deleterious to the persistence of CD4+ Tregs in patients up to 1 year after kidney transplantation, while rapamycin monotherapy promoted CD4+ Tregs in SOT and HSCT patients141, 142, 143, 144 (NCT00803010). Nowadays, some clinical trials focus on the impact of different IS drugs, including rapamycin, on the frequency and function of CD4+ Tregs in SOT patients, and the inclusion of the analysis of CD8+ Tregs counterpart would be of great interest (NCT01014234, NCT01640743). In ongoing clinical trials in SOT, induction treatment is generally composed of tacrolimus + MMF+prednisolone for 1 month. Meanwhile, CD4+ Tregs are transferred adoptively rather after the critical rejection period to avoid any erroneous conclusion about any cytotoxic effect induced by the injected cells, between 1 week and 6 months after transplantation. Thus, ongoing clinical trials switch from tacrolimus to rapamycin one month after SOT to spare CD4+ Tregs at least and maybe to promote them (TASK, DelTa).145 This rational strategy will be applied to the Eight‐Treg clinical trial.
It is important to note that the Treg cells product, with or without genetic modifications, is classified as an Advanced Therapy Medicinal Product and regulated by the directive 1394/2007, aiming at standardizing the manufacturing process. The purity of cell product, the expression of key markers, and the cytotoxic and suppressive functions will be controlled. Fortunately, the manufacturing processes of CD4+ Tregs and CD8+ TILs used in ongoing clinical trials are helpful to set up the good manufacturing practices manufacturing of CD8+ Tregs product.140, 146, 147, 148, 149, 150 In addition, these processes can be upgraded by technical advances, such as clinical grade flow cytometry cell sorting. With this equipment, we can isolate 8.0 × 104 CD8+ Tregs/mL blood, based on negative expression of CD161, low/neg expression of CD45RC and positive expression of CD8 marker from peripheral blood lymphocytes. Then, the cells can be highly expanded, about 200 fold in 2 weeks, while maintaining their suppressive capacity and without acquiring cytotoxic function.3 Although beads coated with anti‐CD3/anti‐CD28 mAbs are generally used to expand CD4+ Tregs, CD8+ Tregs are rather stimulated using the OKT3 clone, as CD8+ TIL for cell therapy in cancer, associated with CD28 stimulation using the CD28.2 mAb clone.140 Besides, many ongoing clinical trials are evaluating the therapeutic potential of antigen‐specific Tregs, such as donor alloantigen‐specific Tregs in SOT (DarTregs in the delta, NJLT001, DART, ARTEMIS, LITTMUS‐MGH) or tumor cells specific Teffs (TILs) in cancer (NCT01118091, NCT01236573, NCT03610490, NCT03068624), and TCR and CAR engineering is a promising alternative strategy for antigen‐specific cell therapy in transplantation and autoimmune diseases.151 Actually, we observed higher efficacy of donor MHC‐I‐specific CAR CD8+ Tregs than polyclonal ones in MHC‐I restricted transplantation models in NSG mice in which this MHC‐I antigen was incompatible (unpublished data). Our current manufacturing process would allow us to infuse up to 1 × 108 polyclonal CD8+ Tregs/kg or 1 × 107 CAR‐CD8+ Tregs/kg in patients.
An important problem for the success of Treg cell‐based therapies is the persistence of the infused cells in the patient. However, the lack of specific markers for CD8+ Tregs is a limitation for their tracking in blood and protocol biopsies of patients. Indeed, FOXP3 has been shown to be transiently overexpressed in Teffs when stimulated while there are technical problems in analyzing treg‐specific demethylated region methylations in biopsies.152, 153 There are also technical problems in detecting key cytokines to track IL‐34+IFNγ+TGFβ+CD8+ Tregs. Thus, new methods of systematic proteomic and transcriptomic analyses are emerging. Notably, deuterium is currently used in the TASK clinical trial (NCT02088931) to track adoptively transferred CD4+ Tregs in blood and in the grafted kidney, and this method might be considered to track CD8+ Tregs in patients.
Early exhaustion of adoptively transferred Tregs would affect their persistence and thus limit their therapeutic effect. Exhaustion of Tconv is reported as the expression of a combination of inhibitory receptors, activation and memory markers, such as PD‐1, TIM‐3, LAG‐3, CTLA‐4, CD160, TIGIT, BTLA‐4, and correlates with improved outcome of transplanted patients.154, 155 However, PD‐1 is not only a discriminant marker but also plays a functional role in the suppressive activity of both CD4+ and CD8+ Treg subsets,84 while the CTLA‐4 and LAG3 markers of human CD8+ Tregs may have a role.27, 28, 156, 157 It is important to note that the CD8+ Tregs used for the Eight‐Treg trial show no phenotypic sign of exhaustion after 2 weeks of culture, and survive and still proliferate for at least 1 month in vitro.3
Co‐treatment with IL‐2 is considered to promote the persistence of CD8+ Tregs in patients, as required for ex vivo culture.3, 103, 104, 105, 106, 107 Encouraging results were obtained in HSCT patients treated with low dose IL‐2, expanding a functional CD4+Foxp3+ Treg subset associated with a lower incidence of GVHD while maintaining a low viral infection incidence158, 159 (NCT00539695). However, soluble circulating IL2‐R can mediate sequestration of IL‐2 and limit its effect 160 (NCT01927120). Further investigations regarding the dose and duration of IL‐2 administration are required. Here again, rapamycin can promote the persistence and stability of CD8+ Tregs face to an inflammatory environment.3, 161
To cope with the lack of persistence of the infused Tregs, we could multiply the injections of CD8+ Tregs as in CD4+ Treg‐based clinical trials (RSMU‐001: NCT01446484, Treg: NCT01624077, NCT02749084). This process requires the production of a large amount of Tregs and reliable storage with a stable suppressive function.162 Long‐term culture and stable cryopreservation of CD8+ Tregs are possible.3
6. NEW SOURCES OF TREGS: DIFFERENTIATION FROM PLURIPOTENT STEM CELLS
Although cell therapy with Tregs is very promising for treating GVHD, rejection after transplantation and autoimmune diseases, one of the limitations is the source of cells to be injected. The cell source must be easily accessible and allow a large number of cells to be donated. PBMCs and cord blood cells are the most traditional sources. They also allow autologous treatments. However, the prevalence of Tregs in these sources is quite low and requires more rounds cell expansion.163
Recently a study has shown the possibility of using Tregs from the thymus of children recovered after cardiac surgery. Indeed, cardiac intervention in children routinely requires the removal of the thymus and these thymuses are then thrown away. However, there are more Tregs cells in a child's thymus than in an adult's entire blood volume and 100 times more than in a unit of cord blood, making it a very interesting source.164 Nevertheless, this source requires a very complex organization between surgical teams and teams of biologists to collect the thymus and extract the cells and the Treg product would be from allogeneic sources to the recipient.
Since 1998, human embryonic stem cell has been the ultimate source of cells for regenerative medicine. Thanks to their pluripotent characteristic, they can differentiate into any cell in the body and are able to renew themselves indefinitely. More recently, induced pluripotent stem cells (IPSC) were obtained by reprogramming of adult somatic cells using transduction of four factors essential for pluripotency in mice and then in humans.165, 166 This IPSC could lead to tailor‐made regenerative medicine. This avoids alloreactivity and therefore potentially reduces the doses of immunosuppressive treatment, which has many side effects. In recent years, studies on the differentiation of T cells from embryonic stem (ES) or induced pluripotent stem (iPS) cells have been carried out mainly in the field of oncology. In 2009, Timmermans et al were the first to demonstrate T cell differentiation from ES using an OP9 cell co‐culture protocol.167 They reported CD3 and TCR expression and IFNγ secretion after stimulation of their cells via TCR. Nowadays, several groups focus on obtaining T cells with antigenic specificity and have shown the possibility of reprogramming specific antigen T cells into IPSC: T‐IPS. During the differentiation of these T‐IPS the antigenic specificity is preserved.168, 169, 170, 171 All these studies showed the great potential of IPS‐derived T cells as an alternative source for T cell‐based immunotherapy in cancer treatments. This is why the use of ES‐ or IPSC‐derived Tregs is also considered with great interest in the treatment of GVHD or in autoimmune diseases. In 2012, R. Haque et al showed that the transduction of FOXP3 during the differentiation of IPSC into T cells led to the production of functional CD4+FOXP3+ Tregs in mice.172 Indeed, these CD4+FOXP3+ Tregs derived from IPSC were able to secrete suppressive molecules such as IL‐10 and TGFβ and they controlled autoimmunity in an arthritic mouse model. The same group showed in 2016 the use of Tregs derived from antigen‐specific IPSC in the same arthritic mouse model.173 Specific tissues or organ antigen targeting by IPSC‐derived CD4+ Tregs allowed a faster cell response on the inflammation site. They also showed that the adoptive transfer of autoantigen‐specific IPSC‐CD4+ Tregs significantly reduced the CD8+/CD4+ ratio in the pancreas of diabetic mice.174 Our own protocol of fully functional CD4+ and CD8+ Treg differentiation from human ES or IPS using FOXP3 transduction during the differentiation process allowed efficient generation of FOXP3+CD4+ and FOXP3+CD8+ Tregs (unpublished data). In addition, recently the knockdown of the polycomb group protein EZH1 was shown to activate lymphoid differentiation potential from pluripotent stem cells (PSCs) and this combined to FOXP3 transduction could improve Treg differentiation from PSCs.175
All these studies demonstrate the proof of concept that Tregs derived from ES cells or IPSC are an alternative source of T and Treg cells in immunotherapy.
7. ENGINEERING OF CD8+ TREGS
7.1. TCR/CAR and others
In recent years, many clinical trials have used polyclonal Tregs or expanded Tregs.176 However, polyclonal Tregs are non‐specific and can potentially cause global immunosuppression. Several studies have demonstrated that the use of antigen‐specific Tregs was much more effective than polyclonal Tregs in animal models.7, 52, 177, 178 Specific antigen Tregs can be generated by cultivating them with APCs containing specific antigens or using TCR engineering techniques. However, expansion with APCs can remain quite ineffective because of the few specific antigen Tregs present in the original polyclonal cells and difficulty to translate to the clinic. Therefore, TCR engineering appears as a promising technique to obtain antigen‐specific Tregs.179, 180, 181 Nevertheless, the use of TCRs remains restricted to a given MHC and TCR cannot be identified in all diseases, this is why another strategy providing antigen specificity emerged a few years ago with spectacular results in cancer: the CAR. The extracellular part of this synthetic receptor binds to surface molecules on the cells independently of the MHC, unlike TCRs, and independently of the patient's HLA haplotype since the specificity is provided by antibody sequences.182 The manufacture of CARs has evolved over the years, today there are four generations of CARs. The 1st generation contains a single chain variable fragment of an extracellular monoclonal antibody, a transmembrane domain and finally an intracellular domain CD3z to conduct the signal.183 For the second generation, a costimulation domain linked to CD3z has been added: CD28 (cytotoxic potential) or 4‐1BB (persistence and decrease exhaustion).184 The third generation contains the CD3z, CD28 and 4‐1BB domains which improve persistence and anti‐tumor activity.185 Finally, the fourth generation called T cell redirected for universal cytokine‐mediated killing (TRUCKs) for TRUCKs allows the production of inducible cytokines after the recognition of the molecule of interest by the CAR. This system could make it possible to recruit non‐CAR cells on the tumor site, for example through the secretion of chemokines to promote their elimination.186 Cells are now mainly used for cancer immunotherapy. CAR‐T cells targeting CD19 are effective in the treatment of malignant hematological diseases in preclinical and clinical trials.182, 187, 188 They were approved by the US food and drug administration for clinical treatments in 2017. This technology has shown its full potential in cancer therapy, it is now extended to therapies using Tregs. CAR Tregs' ability to control an inflamatory bowel disease in mice189 and in another model of colitis in mice190 has been demonstrated. Clinical studies show the use of Tregs to prevent GVHD after allogeneic hematopoietic stem cell transplantation.191, 192, 193 However, one study shows that the transfer of Tregs after hematopoietic stem cell transplantation can lead to overall immunosuppression and thus an increase in viral infections in the patient.194 This is why a more specific therapy is being sought and the use of antigen‐specific CAR Tregs is on the rise. HLA‐A2 has been shown to be a common antigen of incompatibility in transplantation with a prevalence of approximately 50% in European phenotype patients. Over the last 3 years, three groups have used anti‐HLA‐A2 antibody sequences to generate CARs and shown that anti‐HLA‐A2 CAR‐transduced CD4+ Tregs in xeno‐GVHD and skin grafts in NSG mice humanized with PBMCs had a higher suppressive potential than control CD4+ Tregs.178, 195, 196, 197 This year, our group has shown for the first time the proof of concept that CD8+CD45RClow/‐ Tregs expressing a CAR directed against HLA‐A2 are significantly more suppressive on CD4+ and CD8+ effector T cell in vitro than polyclonal Tregs and are able to inhibit graft rejection and GVHD more effectively than polyclonal Tregs in vivo in a model of HLA‐A2+ skin graft rejection and HLA‐A2+ PBMC GVHD in NSG mice (manuscript submitted). The transfer of antigen‐specific Tregs offers a promising strategy against pathologies characterized by aberrant immune activation such as transplant rejection and autoimmune diseases. In addition, the antigenic specificity provided by the CAR allows a much more targeted therapy and thus avoids the side effects of an overall immunosuppression of the patient.
In addition, insertion of a CNI resistance gene into Tregs is an interesting option considered for future clinical trials (the TacRes trial). Indeed, the stability of CD8+ Treg function may be optimized by genetic modifications. The CRISPR/Cas9 system opens new possibilities for stabilizing Foxp3 expression in Tregs.198 Besides, demethylation of Foxp1 could help Foxp3 DNA binding and increase CD8+ Treg suppressive function as reported for CD4+ Tregs.199 Otherwise, targeting genes such as HIF‐1alpha KO would switch Treg metabolism from glycolytic‐driven migration to an oxidative phosphorylation‐driven immunosuppression.200 On the other hand, overexpression of Hdr1 could help Tregs to face to ER stress response to an inflammatory environment.201 Further studies are required to assess the relevance of targeting these genes to improve the stability of CD8+ Tregs.
7.2. Safety system
To avoid any adverse event, genetic modifications to introduce a safety switch into the cells prior to the adoptive transfer have been developed. There are several techniques involving suicide genes. The two most commonly used techniques are Herpes Simplex virus thymidine kinase (HSV‐TK) and human inducible caspase 9 (iCasp9). HSV‐TK is the first suicide gene studied. The interaction of HSV‐TK with Ganciclovir leads to cell suicide by polymerase and DNA synthesis disruption.202 This technique has been tested in clinical phases I and II in France.203 While effective, this approach is limited by the immunogenicity of HSV‐TK expression with resultant rejection of modified cells.204 iCasp9 is very effective, 30 minutes after the injection of the molecule allowing his dimerization (CID) there is elimination of 90% of the T cells modified for iCasp9. The dimerization of iCasp9 with CID results in a cascade of caspases that is responsible for cell death by apoptosis.204, 205, 206, 207 However, this approach also has disadvantages such as the possible toxicity of the remaining cells and the possible autonomous dimerization of iCasp9 leading to the elimination of the cells before their action. An iCasp9 gene has been introduced into the CAR construct for effector T cell‐based therapy clinical trials in cancer (NCT03696784, NCT03016377, NCT03721068, NCT01822652).208
Other approaches such as controlling the intensity or toxicity of T or Treg cells by adding an “on switch” system via the CAR are being studied. The T cell response can be controlled by the recognition of two antigens simultaneously.209, 210, 211 Kloss et al showed in a mouse prostate cancer model that T cells co‐transduced with a CAR against two antigenic prostate tumors (prostate‐specific membrane antigen and prostate stem cell antigen) were able to destroy tumors expressing both antigens. However, these cells have no effect on cells with only one of the two antigens.210 Conversely, it is possible to transduce T cells with an inhibitory CTLA‐4‐ or PD‐1‐based iCAR to limit cytokine secretion, cytotoxicity, and proliferation.212 Overall, all this work provides an opportunity to specify selectivity and mimic the activity of modified T cells in order to reconcile processing power and safety.
8. CONCLUSION
Altogether, we believe that CD8+ Tregs are powerful players in immune tolerance that should be developed further through careful and systematic analyses in rodent models and human diseases. Although there is always some way to go, all these recent advances and upheavals will bring us to a better understanding of the CD8+ Tregs and the next generation CD8+ Tregs usable for cell therapy in transplantation and autoimmune diseases.
ACKNOWLEDGMENT
We thank the Labex IGO program supported by the National Research Agency via the investment of the future program ANR‐11‐LABX‐0016‐01 and the IHU‐Cesti project program ANR‐10‐IBHU‐005. The IHU‐Cesti project is also supported by Nantes Metropole and the Pays de la Loire Region. We also thank the foundation Progreffe, the Fondation pour la Recherche Médicale, the Etoiles Montantes of Pays de la Loire and the Fondation du rein for personal support. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 825392. CG, IA and SB have patents that have been licensed to TxCell SA, a Sangamo company.
Flippe L, Bézie S, Anegon I, Guillonneau C. Future prospects for CD8+ regulatory T cells in immune tolerance. Immunol Rev. 2019;292:209–224. 10.1111/imr.12812
This article is part of a series of reviews covering Tolerance and Exhaustion in Peripheral T and B cells appearing in Volume 292 of Immunological Reviews.
Contributor Information
Ignacio Anegon, Email: ignacio.anegon@univ-nantes.fr.
Carole Guillonneau, Email: carole.guillonneau@univ-nantes.fr.
REFERENCES
- 1. Möller G. Do suppressor T cells exist? Scand J Immunol. 1988;27(3):247‐250. [DOI] [PubMed] [Google Scholar]
- 2. Bézie S, Anegon I, Guillonneau C. Advances on CD8+ Treg cells and their potential in transplantation. Transplantation. 2018;102(9):1467‐1478. [DOI] [PubMed] [Google Scholar]
- 3. Bézie S, Meistermann D, Boucault L, et al. Ex vivo expanded human non‐cytotoxic CD8+CD45RClow/− Tregs efficiently delay skin graft rejection and GVHD in humanized mice. Front Immunol. 2017;8:2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guillonneau C, Hill M, Hubert F‐X, et al. CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN‐gamma, and indoleamine 2,3‐dioxygenase. J Clin Invest. 2007;117(4):1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xystrakis E, Cavailles P, Dejean AS, et al. Functional and genetic analysis of two CD8 T cell subsets defined by the level of CD45RC expression in the rat. J Immunol. 2004;173(5):3140‐3147. [DOI] [PubMed] [Google Scholar]
- 6. Xystrakis E, Dejean AS, Bernard I, et al. Identification of a novel natural regulatory CD8 T‐cell subset and analysis of its mechanism of regulation. Blood. 2004;104(10):3294‐3301. [DOI] [PubMed] [Google Scholar]
- 7. Picarda E, Bézie S, Venturi V, et al. MHC‐derived allopeptide activates TCR‐biased CD8+ Tregs and suppresses organ rejection. J Clin Invest. 2014;1(124):2497‐2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim H‐J, Barnitz RA, Kreslavsky T, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science. 2015;350(6258):334‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vuddamalay Y, Attia M, Vicente R, et al. Mouse and human CD8(+) CD28(low) regulatory T lymphocytes differentiate in the thymus. Immunology. 2016;148(2):187‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vieyra‐Lobato MR, Vela‐Ojeda J, Montiel‐Cervantes L, López‐Santiago R, Moreno‐Lafont MC. Description of CD8+ regulatory T lymphocytes and their specific intervention in graft‐versus‐host and infectious diseases, autoimmunity, and cancer. J Immunol Res. 2018;2018:3758713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saligrama N, Zhao F, Sikora MJ, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature. 2019;572(7770):481–487. 10.1038/s41586-019-1467-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Najafian N, Chitnis T, Salama AD, et al. Regulatory functions of CD8+CD28− T cells in an autoimmune disease model. J Clin Invest. 2003;112(7):1037‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu P, Bamford RN, Waldmann TA. IL‐15‐dependent CD8+ CD122+ T cells ameliorate experimental autoimmune encephalomyelitis by modulating IL‐17 production by CD4+ T cells: immunomodulation. Eur J Immunol. 2014;44(11):3330‐3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu G‐Z, Fang L‐B, Hjelmström P, Gao X‐G. Increased CD8+ central memory T cells in patients with multiple sclerosis. Mult Scler J. 2007;13(2):149‐155. [DOI] [PubMed] [Google Scholar]
- 15. Aristimuño C, de Andrés C, Bartolomé M, et al. IFNβ‐1a therapy for multiple sclerosis expands regulatory CD8+ T cells and decreases memory CD8+ subset: a longitudinal 1‐year study. Clin Immunol. 2010;134(2):148‐157. [DOI] [PubMed] [Google Scholar]
- 16. Negrini S, Fenoglio D, Parodi A, et al. Phenotypic alterations involved in CD8+ Treg impairment in systemic sclerosis. Front Immunol. 2017;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L, Bertucci AM, Ramsey‐Goldman R, Harsha‐Strong ER, Burt RK, Datta SK. Major pathogenic steps in human lupus can be effectively suppressed by nucleosomal histone peptide epitope‐induced regulatory immunity. Clin Immunol. 2013;149(3):365‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L, Bertucci AM, Ramsey‐Goldman R, Burt RK, Datta SK. Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF‐producing CD8+ Treg cells are associated with immunological remission of lupus. J Immunol. 2009;183(10):6346‐6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bernuzzi F, Fenoglio D, Battaglia F, et al. Phenotypical and functional alterations of CD8 regulatory T cells in primary biliary cirrhosis. J Autoimmun. 2010;35(3):176‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boer MC, van Meijgaarden KE, Joosten SA, Ottenhoff T. CD8+ regulatory T cells, and not CD4+ T cells, dominate suppressive phenotype and function after in vitro live Mycobacterium bovis‐BCG activation of human cells. PLoS ONE. 2014;9(4):e94192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boer MC, Prins C, van Meijgaarden KE, van Dissel JT, Ottenhoff T, Joosten SA. Mycobacterium bovis BCG vaccination induces divergent proinflammatory or regulatory T cell responses in adults. Clin Vaccine Immunol. 2015;22(7):778‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fenoglio D, Dentone C, Signori A, et al. CD8+ CD28− CD127lo CD39+ regulatory T‐cell expansion: a new possible pathogenic mechanism for HIV infection? J Allergy Clin Immunol. 2018;141(6):2220‐2233.e4. [DOI] [PubMed] [Google Scholar]
- 23. Popescu I, Macedo C, Abu‐Elmagd K, et al. EBV‐specific CD8+ T cell reactivation in transplant patients results in expansion of CD8+ type‐1 regulatory T cells. Am J Transplant. 2007;7(5):1215‐1223. [DOI] [PubMed] [Google Scholar]
- 24. Chaput N, Louafi S, Bardier A, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009;58(4):520‐529. [DOI] [PubMed] [Google Scholar]
- 25. Kiniwa Y, Miyahara Y, Wang HY, et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13(23):6947‐6958. [DOI] [PubMed] [Google Scholar]
- 26. Canale FP, Ramello MC, Núñez N, et al. CD39 expression defines cell exhaustion in tumor‐infiltrating CD8+ T cells. Can Res. 2018;78(1):115‐128. [DOI] [PubMed] [Google Scholar]
- 27. Boor P, Metselaar HJ, de Jonge S, Mancham S, van der Laan L, Kwekkeboom J. Human plasmacytoid dendritic cells induce CD8+ LAG‐3+ Foxp3+ CTLA‐4+ regulatory T cells that suppress allo‐reactive memory T cells. Eur J Immunol. 2011;41(6):1663‐1674. [DOI] [PubMed] [Google Scholar]
- 28. Zheng J, Liu Y, Liu Y, et al. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Sci Transl Med. 2013;5(168):168ra9. [DOI] [PubMed] [Google Scholar]
- 29. Barbon CM, Davies JK, Voskertchian A, et al. Alloanergization of human T cells results in expansion of alloantigen‐specific CD8(+) CD28(−) suppressor cells. Am J Transplant. 2014;14(2):305‐318. [DOI] [PubMed] [Google Scholar]
- 30. Avivi I, Stroopinsky D, Rowe JM, Katz T. A subset of CD8+ T cells acquiring selective suppressive properties may play a role in GvHD management. Transpl Immunol. 2013;28(1):57‐61. [DOI] [PubMed] [Google Scholar]
- 31. Beres AJ, Haribhai D, Chadwick AC, Gonyo PJ, Williams CB, Drobyski WR. CD8+ Foxp3+ regulatory T cells are induced during graft‐versus‐host disease and mitigate disease severity. J Immunol. 2012;189(1):464‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robb RJ, Lineburg KE, Kuns RD, et al. Identification and expansion of highly suppressive CD8+FoxP3+ regulatory T cells after experimental allogeneic bone marrow transplantation. Blood. 2012;119(24):5898‐5908. [DOI] [PubMed] [Google Scholar]
- 33. Zimmerer JM, Ringwald BA, Elzein SM, et al. Antibody‐suppressor CD8+ T cells require CXCR5. Transplantation. 2019;103(9):1809‐1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single‐cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. 2018;19(3):291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li XL, Ménoret S, Bezie S, et al. Mechanism and localization of CD8 regulatory T cells in a heart transplant model of tolerance. J Immunol. 2010;185(2):823‐833. [DOI] [PubMed] [Google Scholar]
- 36. Bézie S, Picarda E, Tesson L, et al. Fibrinogen‐like protein 2/fibroleukin induces long‐term allograft survival in a rat model through regulatory B Cells. PLoS ONE. 2015;10(3):e0119686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bézie S, Picarda E, Ossart J, et al. IL‐34 is a Treg‐specific cytokine and mediates transplant tolerance. J Clin Invest. 2015;125(10):3952‐3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guillonneau C, Bézie S, Anegon I. Immunoregulatory properties of the cytokine IL‐34. Cell Mol Life Sci. 2017;74(14):2569‐2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bézie S, Picarda E, Ossart J, Martinet B, Anegon I, Guillonneau C. Compensatory regulatory networks between CD8 T, B, and myeloid cells in organ transplantation tolerance. J Immunol. 2015;195(12):5805‐5815. [DOI] [PubMed] [Google Scholar]
- 40. Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191‐1193. [DOI] [PubMed] [Google Scholar]
- 41. Lindau R, Mehta RB, Lash GE, et al. Interleukin‐34 is present at the fetal–maternal interface and induces immunoregulatory macrophages of a decidual phenotype in vitro. Hum Reprod. 2018;33(4):588‐599. [DOI] [PubMed] [Google Scholar]
- 42. Wang S, Sun F, Li M, et al. The appropriate frequency and function of decidual Tim‐3+CTLA‐4+CD8+ T cells are important in maintaining normal pregnancy. Cell Death Dis. 2019;10(6):407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tilburgs T, Strominger JL. CD8+ effector T cells at the fetal‐maternal interface, balancing fetal tolerance and antiviral immunity. Am J Reprod Immunol. 2013;69(4):395‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tilburgs T, Roelen DL, van der Mast BJ, et al. Differential distribution of CD4(+)CD25(bright) and CD8(+)CD28(−) T‐cells in decidua and maternal blood during human pregnancy. Placenta. 2006;27(Suppl A):S47‐S53. [DOI] [PubMed] [Google Scholar]
- 45. Crespo ÂC, van der Zwan A, Ramalho‐Santos J, Strominger JL, Tilburgs T. Cytotoxic potential of decidual NK cells and CD8+ T cells awakened by infections. J Reprod Immunol. 2017;119:85‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tilburgs T, Crespo ÂC, van der Zwan A, et al. Human HLA‐G+ extravillous trophoblasts: immune‐activating cells that interact with decidual leukocytes. Proc Natl Acad Sci USA. 2015;112(23):7219‐7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu D, Ikizawa K, Lu L, Sanchirico ME, Shinohara ML, Cantor H. Analysis of regulatory CD8 T cells in Qa‐1‐deficient mice. Nat Immunol. 2004;5(5):516‐523. [DOI] [PubMed] [Google Scholar]
- 48. Varthaman A, Khallou‐Laschet J, Clement M, et al. Control of T cell reactivation by regulatory Qa‐1‐restricted CD8+ T cells. J Immunol. 2010;184(12):6585‐6591. [DOI] [PubMed] [Google Scholar]
- 49. Tang X, Maricic I, Purohit N, et al. Regulation of immunity by a novel population of Qa‐1‐restricted CD8αα+TCRαβbeta+ T cells. J Immunol. 2006;177(11):7645‐7655. [DOI] [PubMed] [Google Scholar]
- 50. Jiang H, Canfield SM, Gallagher MP, et al. HLA‐E‐restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest. 2010;120(10):3641‐3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li J, Goldstein I, Glickman‐Nir E, Jiang H, Chess L. Induction of TCR Vbeta‐specific CD8+ CTLs by TCR Vbeta‐derived peptides bound to HLA‐E. J Immunol. 2001;167(7):3800‐3808. [DOI] [PubMed] [Google Scholar]
- 52. Picarda E, Anegon I, Guillonneau C. T‐cell receptor specificity of CD8(+) Tregs in allotransplantation. Immunotherapy. 2011;3(4 Suppl):35‐37. [DOI] [PubMed] [Google Scholar]
- 53. Beringer DX, Kleijwegt FS, Wiede F, et al. T cell receptor reversed polarity recognition of a self‐antigen major histocompatibility complex. Nat Immunol. 2015;16(11):1153‐1161. [DOI] [PubMed] [Google Scholar]
- 54. Dai Z, Zhang S, Xie Q, et al. Natural CD8+CD122+ T cells are more potent in suppression of allograft rejection than CD4+CD25+ regulatory T cells: CD8+CD122+ cells are supreme T suppressors. Am J Transplant. 2014;14(1):39‐48. [DOI] [PubMed] [Google Scholar]
- 55. Picarda E, Bézie S, Boucault L, et al. Transient antibody targeting of CD45RC induces transplant tolerance and potent antigen‐specific regulatory T cells. JCI Insight. 2017;2(3):e90088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heinrichs J, Li J, Nguyen H, et al. CD8(+) Tregs promote GVHD prevention and overcome the impaired GVL effect mediated by CD4(+) Tregs in mice. Oncoimmunology. 2016;5(6):e1146842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang J, Brook MO, Carvalho‐Gaspar M, et al. Allograft rejection mediated by memory T cells is resistant to regulation. Proc Natl Acad Sci USA. 2007;104(50):19954‐19959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Long X, Cheng Q, Liang H, et al. Memory CD4+ T cells are suppressed by CD8+ regulatory T cells in vitro and in vivo. Am J Transl Res. 2017;9(1):63‐78. [PMC free article] [PubMed] [Google Scholar]
- 59. Myers L, Croft M, Kwon BS, Mittler RS, Vella AT. Peptide‐specific CD8 T regulatory cells use IFN‐gamma to elaborate TGF‐beta‐based suppression. J Immunol. 2005;174(12):7625‐7632. [DOI] [PubMed] [Google Scholar]
- 60. Yamada Y, Sugita S, Horie S, Yamagami S, Mochizuki M. Mechanisms of immune suppression for CD8+ T cells by human corneal endothelial cells via membrane‐bound TGFbeta. Invest Ophthalmol Vis Sci. 2010;51(5):2548‐2557. [DOI] [PubMed] [Google Scholar]
- 61. Suzuki M, Jagger AL, Konya C, et al. CD8+CD45RA+CCR7+FOXP3+ T cells with immunosuppressive properties: a novel subset of inducible human regulatory T cells. J Immunol. 2012;189(5):2118‐2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shalev I, Wong KM, Foerster K, et al. The novel CD4+CD25+ regulatory T cell effector molecule fibrinogen‐like protein 2 contributes to the outcome of murine fulminant viral hepatitis. Hepatology. 2009;49(2):387‐397. [DOI] [PubMed] [Google Scholar]
- 63. Denning TL, Granger SW, Granger S, et al. Mouse TCRαβ+CD8αα intraepithelial lymphocytes express genes that down‐regulate their antigen reactivity and suppress immune responses. J Immunol. 2007;178(7):4230‐4239. [DOI] [PubMed] [Google Scholar]
- 64. Liu Y, Xu S, Xiao F, et al. The FGL2/fibroleukin prothrombinase is involved in alveolar macrophage activation in COPD through the MAPK pathway. Biochem Biophys Res Commun. 2010;396(2):555‐561. [DOI] [PubMed] [Google Scholar]
- 65. Paunicka K, Chen PW, Niederkorn JY. Role of IFN‐γ in the establishment of anterior chamber‐associated immune deviation (ACAID)‐induced CD8+ T regulatory cells. J Leukoc Biol. 2012;91(3):475‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Olson BM, Jankowska‐Gan E, Becker JT, Vignali D, Burlingham WJ, McNeel DG. Human prostate tumor antigen‐specific CD8+ regulatory T cells are inhibited by CTLA‐4 or IL‐35 blockade. J Immunol. 2012;189(12):5590‐5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Balashov KE, Khoury SJ, Hafler DA, Weiner HL. Inhibition of T cell responses by activated human CD8+ T cells is mediated by interferon‐gamma and is defective in chronic progressive multiple sclerosis. J Clin Invest. 1995;95(6):2711‐2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang J. Yin and yang interplay of IFN‐gamma in inflammation and autoimmune disease. J Clin Invest. 2007;117(4):871‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Konieczny BT, Dai Z, Elwood ET, et al. IFN‐gamma is critical for long‐term allograft survival induced by blocking the CD28 and CD40 ligand T cell costimulation pathways. J Immunol. 1998;160(5):2059‐2064. [PubMed] [Google Scholar]
- 70. Sawitzki B, Kingsley CI, Oliveira V, Karim M, Herber M, Wood KJ. IFN‐γ production by alloantigen‐reactive regulatory T cells is important for their regulatory function in vivo. J Exp Med. 2005;201(12):1925‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Collison LW, Workman CJ, Kuo TT, et al. The inhibitory cytokine IL‐35 contributes to regulatory T‐cell function. Nature. 2007;450(7169):566‐569. [DOI] [PubMed] [Google Scholar]
- 72. Zhang J, Zhang Y, Wang Q, et al. Interleukin‐35 in immune‐related diseases: protection or destruction. Immunology. 2019;157(1):13‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Krupnick AS, Lin X, Li W, et al. Central memory CD8+ T lymphocytes mediate lung allograft acceptance. J Clin Invest. 2014;124(3):1130‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Takahashi T, Hsiao HM, Tanaka S, et al. PD‐1 expression on CD8+ T cells regulates their differentiation within lung allografts and is critical for tolerance induction. Am J Transplant. 2018;18(1):216‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zimmerer JM, Pham TA, Wright CL, et al. Alloprimed CD8+ T cells regulate alloantibody and eliminate alloprimed B cells through perforin‐ and FasL‐dependent mechanisms: CD8+ T cells kill IgG1‐producing B cells. Am J Transplant. 2014;14(2):295‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bjarnadottir U, Lemarquis AL, Halldorsdottir S, Freysdottir J, Ludviksson BR. The suppressive function of human CD8(+) iTregs is inhibited by IL‐1β and TNFα. Scand J Immunol. 2014;80(5):313‐322. [DOI] [PubMed] [Google Scholar]
- 77. Choi BK, Asai T, Vinay DS, Kim YH, Kwon BS. 4–1BB‐mediated amelioration of experimental autoimmune uveoretinitis is caused by indoleamine 2,3‐dioxygenase‐dependent mechanisms. Cytokine. 2006;34(5–6):233‐242. [DOI] [PubMed] [Google Scholar]
- 78. Wang Z. Role of IFN‐g in induction of Foxp3 and conversion of CD4+CD25− T cells to CD4+ Tregs. J Clin Invest. 2006;116(9):2434‐2441. 10.1172/JCI25826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dai H, Wan N, Zhang S, Moore Y, Wan F, Dai Z. Cutting edge: programmed death‐1 defines CD8+CD122+ T cells as regulatory versus memory T cells. J Immunol. 2010;185(2):803‐807. [DOI] [PubMed] [Google Scholar]
- 80. Chikuma S, Terawaki S, Hayashi T, et al. PD‐1‐mediated suppression of IL‐2 production induces CD8+ T cell anergy in vivo. J Immunol. 2009;182(11):6682‐6689. [DOI] [PubMed] [Google Scholar]
- 81. van der Merwe M, Abdelsamed HA, Seth A, Ong T, Vogel P, Pillai AB. Recipient myeloid‐derived immunomodulatory cells induce PD‐1 ligand‐dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor‐recipient immune tolerance after murine nonmyeloablative bone marrow transplantation. J Immunol. 2013;191(11):5764‐5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Francisco LM, Salinas VH, Brown KE, et al. PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015‐3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Amarnath S, Costanzo CM, Mariotti J, et al. Cells and human myeloid dendritic cells promote tolerance via programmed death ligand‐1. PLoS Biol. 2010;8(2):e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Machicote A, Belén S, Baz P, Billordo LA, Fainboim L. Human CD8+HLA‐DR+ regulatory T cells, similarly to classical CD4+Foxp3+ cells, suppress immune responses via PD‐1/PD‐L1 axis. Front Immunol. 2018;9:2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lu L, Kim H‐J, Werneck M, Cantor H. Regulation of CD8+ regulatory T cells: interruption of the NKG2A‐Qa‐1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc Natl Acad Sci USA. 2008;105(49):19420‐19425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mu C, Zhang X, Wang L, et al. Enhanced suppression of polyclonal CD8(+)25(+) regulatory T cells via exosomal arming of antigen‐specific peptide/MHC complexes. J Leukoc Biol. 2017;101(5):1221‐1231. [DOI] [PubMed] [Google Scholar]
- 87. Akane K, Kojima S, Mak TW, Shiku H, Suzuki H. CD8+CD122+CD49dlow regulatory T cells maintain T‐cell homeostasis by killing activated T cells via Fas/FasL‐mediated cytotoxicity. Proc Natl Acad Sci USA. 2016;113(9):2460‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen Z, Han Y, Gu Y, et al. CD11c(high)CD8+ regulatory T cell feedback inhibits CD4 T cell immune response via Fas ligand‐Fas pathway. J Immunol. 2013;190(12):6145‐6154. [DOI] [PubMed] [Google Scholar]
- 89. Daniel V, Sadeghi M, Wang H, Opelz G. CD4+CD25+Foxp3+IFNγ+CD178+ human induced Treg (iTreg) contribute to suppression of alloresponses by apoptosis of responder cells. Hum Immunol. 2013;74(2):151‐162. [DOI] [PubMed] [Google Scholar]
- 90. Boissonnas A, Scholer‐Dahirel A, Simon‐Blancal V, et al. Foxp3+ T cells induce perforin‐dependent dendritic cell death in tumor‐draining lymph nodes. Immunity. 2010;32(2):266‐278. [DOI] [PubMed] [Google Scholar]
- 91. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21(4):589‐601. [DOI] [PubMed] [Google Scholar]
- 92. Guipouy D, Gertner‐Dardenne J, Pfajfer L, German Y, Belmonte N, Dupré L. Granulysin‐ and granzyme‐dependent elimination of myeloid cells by therapeutic ova‐specific type 1 regulatory T cells. Int Immunol. 2019;31(4):239‐250. [DOI] [PubMed] [Google Scholar]
- 93. Gondek DC, Lu L‐F, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact‐mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B‐dependent, perforin‐independent mechanism. J Immunol. 2005;174(4):1783‐1786. [DOI] [PubMed] [Google Scholar]
- 94. Cao X, Cai SF, Fehniger TA, et al. Granzyme B and perforin are important for regulatory T cell‐mediated suppression of tumor clearance. Immunity. 2007;27(4):635‐646. [DOI] [PubMed] [Google Scholar]
- 95. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. 2016;12(3):169‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish Treg‐cell function. Nature. 2013;499(7459):485‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Battaglia M, Stabilini A, Roncarolo M‐G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105(12):4743‐4748. [DOI] [PubMed] [Google Scholar]
- 99. Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178(1):320‐329. [DOI] [PubMed] [Google Scholar]
- 100. Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299‐3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Coe DJ, Kishore M, Marelli‐Berg F. Metabolic regulation of regulatory T cell development and function. Front Immunol. 2014;5:590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sun J, Yang Y, Huo X, et al. Efficient therapeutic function and mechanisms of human polyclonal CD8+ CD103+ Foxp3+ regulatory T cells on collagen‐induced arthritis in mice. J Immunol Res. 2019;19(2019):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Churlaud G, Pitoiset F, Jebbawi F, et al. Human and mouse CD8(+)CD25(+)FOXP3(+) regulatory T cells at steady state and during interleukin‐2 therapy. Front Immunol. 2015;6:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Aoyama A, Klarin D, Yamada Y, et al. Low‐dose IL‐2 for in vivo expansion of CD4+ and CD8+ regulatory T cells in nonhuman primates. Am J Transplant. 2012;12(9):2532‐2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Peterson LB, Bell C, Howlett SK, et al. A long‐lived IL‐2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun. 2018;95:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Saadoun D, Rosenzwajg M, Joly F, et al. Regulatory T‐cell responses to low‐dose interleukin‐2 in HCV‐induced vasculitis. N Engl J Med. 2011;365(22):2067‐2077. [DOI] [PubMed] [Google Scholar]
- 107. Rosenzwajg M, Churlaud G, Mallone R, et al. Low‐dose interleukin‐2 fosters a dose‐dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun. 2015;58:48‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. He J, Zhang X, Wei Y, et al. Low‐dose interleukin‐2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med. 2016;22(9):991‐993. [DOI] [PubMed] [Google Scholar]
- 109. Hirakawa M, Matos T, Liu H, et al. Low‐dose IL‐2 selectively activates subsets of CD4+ Tregs and NK cells. JCI Insight. 2016;1(18):e89278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Apert C, Romagnoli P, van Meerwijk JPM. IL‐2 and IL‐15 dependent thymic development of Foxp3‐expressing regulatory T lymphocytes. Protein Cell. 2018;9(4):322‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Waickman AT, Ligons DL, Hwang SuJin, et al. CD4 effector T cell differentiation is controlled by IL‐15 that is expressed and presented in trans. Cytokine. 2017;99:266‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL‐2 is essential for TGF‐beta to convert naive CD4+CD25− cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178(4):2018‐2027. [DOI] [PubMed] [Google Scholar]
- 113. Liu H, Qiu F, Wang Y, et al. CD8+CD122+PD‐1+ Tregs synergize with costimulatory blockade of CD40/CD154, but Not B7/CD28, to prolong murine allograft survival. Front Immunol. 2019;10:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang L‐X, Li Y, Yang G, et al. CD122+CD8+ Treg suppress vaccine‐induced antitumor immune responses in lymphodepleted mice. Eur J Immunol. 2010;40(5):1375‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mayer CT, Floess S, Baru AM, Lahl K, Huehn J, Sparwasser T. CD8+ Foxp3+ T cells share developmental and phenotypic features with classical CD4+ Foxp3+ regulatory T cells but lack potent suppressive activity. Eur J Immunol. 2011;41(3):716‐725. [DOI] [PubMed] [Google Scholar]
- 116. Vuddamalay Y, van Meerwijk J. CD28− and CD28lowCD8+ regulatory T cells: of mice and men. Front Immunol. 2017;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liu Q, Zheng H, Chen X, et al. Human mesenchymal stromal cells enhance the immunomodulatory function of CD8(+)CD28(−) regulatory T cells. Cell Mol Immunol. 2015;12(6):708‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lei H, Kuchenbecker L, Streitz M, et al. Human CD45RA(−) FoxP3(hi) memory‐type regulatory T cells show distinct TCR repertoires with conventional T cells and play an important role in controlling early immune activation. Am J Transplant. 2015;15(10):2625‐2635. [DOI] [PubMed] [Google Scholar]
- 119. Thornton AM, Korty PE, Tran DQ, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic‐derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184(7):3433‐3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sebastian M, Lopez‐Ocasio M, Metidji A, Rieder SA, Shevach EM, Thornton AM. Helios controls a limited subset of regulatory T cell functions. J Immunol. 2016;196(1):144‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Singh RP, La Cava A, Wong M, Ebling F, Hahn BH. CD8+ T cell‐Mediated suppression of autoimmunity in a murine lupus model of peptide‐induced immune tolerance depends on Foxp3 expression. J Immunol. 2007;178(12):7649‐7657. [DOI] [PubMed] [Google Scholar]
- 122. Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF‐β‐induced Foxp3+ regulatory T cells rescue scurfy mice. Eur J Immunol. 2008;38(7):1814‐1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lin W, Haribhai D, Relland LM, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8(4):359‐368. [DOI] [PubMed] [Google Scholar]
- 124. Cretney E, Xin A, Shi W, et al. The transcription factors Blimp‐1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. 2011;12(4):304‐311. [DOI] [PubMed] [Google Scholar]
- 125. Oh H, Grinberg‐Bleyer Y, Liao W, et al. An NF‐κB transcription‐factor‐dependent lineage‐specific transcriptional program promotes regulatory T cell identity and function. Immunity. 2017;47(3):450‐465.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhang T, Pierson RN, Azimzadeh AM. Update on CD40 and CD154 blockade in transplant models. Immunotherapy. 2015;7(8):899‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Guillot C, Guillonneau C, Mathieu P, et al. Prolonged blockade of CD40‐CD40 ligand interactions by gene transfer of CD40Ig results in long‐term heart allograft survival and donor‐specific hyporesponsiveness, but does not prevent chronic rejection. J Immunol. 2002;168(4):1600‐1609. [DOI] [PubMed] [Google Scholar]
- 128. Tsai Y, Yang KD, Wen Y, Hung C, Chien J, Lin C. Allergen‐specific immunotherapy enhances CD8+ CD25+ CD137+ regulatory T cells and decreases nasal nitric oxide. Pediatr Allergy Immunol. 2019;30:531‐539. [DOI] [PubMed] [Google Scholar]
- 129. Zhang S, Ke X, Zeng S, et al. Analysis of CD8+ Treg cells in patients with ovarian cancer: a possible mechanism for immune impairment. Cell Mol Immunol. 2015;12(5):580‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chen M‐L, Yan B‐S, Kozoriz D, Weiner HL. Novel CD8+ Treg suppress EAE by TGF‐beta‐ and IFN‐gamma‐dependent mechanisms. Eur J Immunol. 2009;39(12):3423‐3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Izawa A, Yamaura K, Albin MJ, et al. A novel alloantigen‐specific CD8+PD1+ regulatory T cell induced by ICOS‐B7h blockade in vivo. J Immunol. 2007;179(2):786‐796. [DOI] [PubMed] [Google Scholar]
- 132. Notley CA, McCann FE, Inglis JJ, Williams RO. ANTI‐CD3 therapy expands the numbers of CD4+ and CD8+ treg cells and induces sustained amelioration of collagen‐induced arthritis. Arthritis Rheum. 2010;62(1):171‐178. [DOI] [PubMed] [Google Scholar]
- 133. Ellis S, McGovern JL, van Maurik A, Howe D, Ehrenstein MR, Notley CA. Induced CD8+FoxP3+ Treg cells in rheumatoid arthritis are modulated by p38 phosphorylation and monocytes expressing membrane tumor necrosis factor α and CD86. Arthritis Rheumatol. 2014;66(10):2694‐2705. [DOI] [PubMed] [Google Scholar]
- 134. Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti‐CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J Clin Invest. 2005;115(10):2904‐2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ablamunits V, Bisikirska B, Herold KC. Acquisition of regulatory function by human CD8(+) T cells treated with anti‐CD3 antibody requires TNF. Eur J Immunol. 2010;40(10):2891‐2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kennedy NA, Heap GA, Green HD, et al. Predictors of anti‐TNF treatment failure in anti‐TNF‐naive patients with active luminal Crohn's disease: a prospective, multicentre, cohort study. Lancet Gastroenterol Hepatol. 2019;4(5):341‐353. [DOI] [PubMed] [Google Scholar]
- 137. Pierini A, Strober W, Moffett C, et al. TNF‐α priming enhances CD4+FoxP3+ regulatory T‐cell suppressive function in murine GVHD prevention and treatment. Blood. 2016;128(6):866‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Guillonneau C, Séveno C, Dugast A‐S, et al. Anti‐CD28 antibodies modify regulatory mechanisms and reinforce tolerance in CD40Ig‐treated heart allograft recipients. J Immunol. 2007;179(12):8164‐8171. [DOI] [PubMed] [Google Scholar]
- 139. Horwitz DA, Bickerton S, Koss M, Fahmy TM, La Cava A. Suppression of murine lupus by cd4+ and cd8+ Treg cells induced by T cell‐targeted nanoparticles loaded with interleukin‐2 and transforming growth factor β. Arthritis Rheumatol. 2019;71(4):632‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhang L, Morgan RA, Beane JD, et al. Tumor‐infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin‐12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21(10):2278‐2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Segundo DS, Ruiz JC, Izquierdo M, et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4+CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation. 2006;82(4):550‐557. [DOI] [PubMed] [Google Scholar]
- 142. Zeiser R, Leveson‐Gower DB, Zambricki EA, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111(1):453‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Pidala J, Kim J, Jim H, et al. A randomized phase II study to evaluate tacrolimus in combination with sirolimus or methotrexate after allogeneic hematopoietic cell transplantation. Haematologica. 2012;97(12):1882‐1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ramírez E, Morales JM, Lora D, et al. Peripheral blood regulatory T cells in long‐term kidney transplant recipients. Transpl Proc. 2009;41(6):2360‐2362. [DOI] [PubMed] [Google Scholar]
- 145. Mathew JM, H‐Voss J, LeFever A, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. 2018;8(1):7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Fraser H, Safinia N, Grageda N, et al. A rapamycin‐based GMP‐compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol Ther Methods Clin Dev. 2018;8:198‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Marín Morales JM, Münch N, Peter K, et al. Automated clinical grade expansion of regulatory T cells in a fully closed system. Front Immunol. 2019;10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Safinia N, Vaikunthanathan T, Fraser H, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget. 2016;7(7):7563‐7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Putnam AL, Safinia N, Medvec A, et al. Clinical grade manufacturing of human alloantigen‐reactive regulatory T cells for use in transplantation: clinical grade alloantigen‐reactive Tregs. Am J Transplant. 2013;13(11):3010‐3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Fuchs A, Gliwiński M, Grageda N, et al. Minimum information about T regulatory cells: a step toward reproducibility and standardization. Front Immunol. 2017;8:1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhang Q, Lu W, Liang C‐L, et al. Chimeric antigen receptor (CAR) Treg: a promising approach to inducing immunological tolerance. Front Immunol. 2018;9:2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wieczorek G, Asemissen A, Model F, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Can Res. 2009;69(2):599‐608. [DOI] [PubMed] [Google Scholar]
- 153. Allan SE, Crome SQ, Crellin NK, et al. Activation‐induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19(4):345‐354. [DOI] [PubMed] [Google Scholar]
- 154. Fribourg M, Anderson L, Fischman C, et al. T‐cell exhaustion correlates with improved outcomes in kidney transplant recipients. Kidney Int. 2019;96:436‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Fuertes Marraco SA, Neubert NJ, Verdeil G, Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front Immunol. 2015;6:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Yu Y, Zitzner JR, Houlihan J, et al. Common gamma chain cytokines promote rapid in vitro expansion of allo‐specific human CD8+ suppressor T cells. PLoS ONE. 2011;6(12):e28948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Sega EI, Leveson‐Gower DB, Florek M, Schneidawind D, Luong RH, Negrin RS. Role of lymphocyte activation gene‐3 (Lag‐3) in conventional and regulatory T cell function in allogeneic transplantation. PLoS ONE. 2014;9(1):e86551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Kennedy‐Nasser AA, Ku S, Castillo‐Caro P, et al. Ultra low‐dose IL‐2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res. 2014;20(8):2215‐2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Brehm MA, Kenney LL, Wiles MV, et al. Lack of acute xenogeneic graft‐ versus ‐host disease, but retention of T‐cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J. 2019;33(3):3137‐3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Betts BC, Pidala J, Kim J, et al. IL‐2 promotes early Treg reconstitution after allogeneic hematopoietic cell transplantation. Haematologica. 2017;102(5):948‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Scotta C, Esposito M, Fazekasova H, et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4(+)CD25(+)FOXP3(+) T regulatory cell subpopulations. Haematologica. 2013;98(8):1291‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Gołąb K, Grose R, Placencia V, et al. Cell banking for regulatory T cell‐based therapy: strategies to overcome the impact of cryopreservation on the Treg viability and phenotype. Oncotarget. 2018;9(11):9728‐9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Brunstein CG, Miller JS, McKenna DH, et al. Umbilical cord blood‐derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127(8):1044‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Dijke IE, Hoeppli RE, Ellis T, et al. Discarded human thymus is a novel source of stable and long‐lived therapeutic regulatory T cells. Am J Transplant. 2016;16(1):58‐71. [DOI] [PubMed] [Google Scholar]
- 165. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663‐676. [DOI] [PubMed] [Google Scholar]
- 166. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861‐872. [DOI] [PubMed] [Google Scholar]
- 167. Timmermans F, Velghe I, Vanwalleghem L, et al. Generation of T cells from human embryonic stem cell‐derived hematopoietic zones. J Immunol. 2009;182(11):6879‐6888. [DOI] [PubMed] [Google Scholar]
- 168. Vizcardo R, Masuda K, Yamada D, et al. Regeneration of human tumor antigen‐specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell. 2013;12(1):31‐36. [DOI] [PubMed] [Google Scholar]
- 169. Wakao H, Yoshikiyo K, Koshimizu U, et al. Expansion of functional human mucosal‐associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12(5):546‐558. [DOI] [PubMed] [Google Scholar]
- 170. Maeda T, Nagano S, Ichise H, et al. Regeneration of CD8αβ T cells from T‐cell–derived iPSC imparts potent tumor antigen‐specific cytotoxicity. Can Res. 2016;76(23):6839‐6850. [DOI] [PubMed] [Google Scholar]
- 171. Kawamoto H, Masuda K, Nagano S, Maeda T. Cloning and expansion of antigen‐specific T cells using iPS cell technology: development of “off‐the‐shelf” T cells for the use in allogeneic transfusion settings. Int J Hematol. 2018;107(3):271‐277. [DOI] [PubMed] [Google Scholar]
- 172. Haque R, Lei F, Xiong X, et al. Programming of regulatory T cells from pluripotent stem cells and prevention of autoimmunity. J Immunol. 2012;189(3):1228‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Haque M, Song J, Fino K, et al. Stem cell‐derived tissue‐associated regulatory T cells ameliorate the development of autoimmunity. Sci Rep. 2016;6(1):20588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Haque M, Lei F, Xiong X, et al. Stem cell–derived tissue‐associated regulatory T cells suppress the activity of pathogenic cells in autoimmune diabetes. JCI Insight. 2019;4(7):126471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Vo LT, Kinney MA, Liu X, et al. Regulation of embryonic haematopoietic multipotency by EZH1. Nature. 2018;553(7689):506‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Saudemont A, Jespers L, Clay T. Current status of gene engineering cell therapeutics. Front Immunol. 2018;9:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Kim YC, Zhang A‐H, Su Y, et al. Engineered antigen‐specific human regulatory T cells: immunosuppression of FVIII‐specific T‐ and B‐cell responses. Blood. 2015;125(7):1107‐1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. MacDonald KG, Hoeppli RE, Huang Q, et al. Alloantigen‐specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126(4):1413‐1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Brusko TM, Koya RC, Zhu S, et al. Human antigen‐specific regulatory T cells generated by T cell receptor gene transfer. PLoS ONE. 2010;5(7):e11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Hull CM, Nickolay LE, Estorninho M, et al. Generation of human islet‐specific regulatory T cells by TCR gene transfer. J Autoimmun. 2017;79:63‐73. [DOI] [PubMed] [Google Scholar]
- 181. Wright GP, Notley CA, Xue S‐A, et al. Adoptive therapy with redirected primary regulatory T cells results in antigen‐specific suppression of arthritis. Proc Natl Acad Sci USA. 2009;106(45):19078‐19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Rivière I, Sadelain M. Chimeric antigen receptors: a cell and gene therapy perspective. Mol Ther. 2017;25(5):1117‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody‐binding domains and the gamma or zeta subunits of the immunoglobulin and T‐cell receptors. Proc Natl Acad Sci USA. 1993;90(2):720‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zhao Z, Condomines M, van der Stegen S, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Van Schandevyl S, Kerre T. Chimeric antigen receptor T‐cell therapy: design improvements and therapeutic strategies in cancer treatment. Acta Clin Belg. 2018;13:1‐7. [DOI] [PubMed] [Google Scholar]
- 187. Gauthier J, Yakoub‐Agha I. Chimeric antigen‐receptor T‐cell therapy for hematological malignancies and solid tumors: clinical data to date, current limitations and perspectives. Curr Res Transl Med. 2017;65(3):93‐102. [DOI] [PubMed] [Google Scholar]
- 188. Jain MD, Davila ML. Concise review: emerging principles from the clinical application of chimeric antigen receptor T cell therapies for B cell malignancies: principles of CAR T therapy. Stem Cells. 2018;36(1):36‐44. [DOI] [PubMed] [Google Scholar]
- 189. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology. 2008;134(7):2014‐2024. [DOI] [PubMed] [Google Scholar]
- 190. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen‐specific regulatory T cells. Mol Ther. 2014;22(5):1018‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Trzonkowski P, Bieniaszewska M, Juścińska J, et al. First‐in‐man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin Immunol. 2009;133(1):22‐26. [DOI] [PubMed] [Google Scholar]
- 192. Di Ianni M, Falzetti F, Carotti A, et al. Tregs prevent GVHD and promote immune reconstitution in HLA‐haploidentical transplantation. Blood. 2011;117(14):3921‐3928. [DOI] [PubMed] [Google Scholar]
- 193. Brunstein CG, Miller JS, Cao Q, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Brunstein CG, Blazar BR, Miller JS, et al. Adoptive transfer of umbilical cord blood‐derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. 2013;19(8):1271‐1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Boardman DA, Philippeos C, Fruhwirth GO, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. 2017;17(4):931‐943. [DOI] [PubMed] [Google Scholar]
- 196. Noyan F, Zimmermann K, Hardtke‐Wolenski M, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC‐specific chimeric antigen receptor. Am J Transplant. 2017;17(4):917‐930. [DOI] [PubMed] [Google Scholar]
- 197. Pierini A, Iliopoulou BP, Peiris H, et al. T cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight. 2017;2(20):92865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Okada M, Kanamori M, Someya K, Nakatsukasa H, Yoshimura A. Stabilization of Foxp3 expression by CRISPR‐dCas9‐based epigenome editing in mouse primary T cells. Epigenetics Chromatin. 2017;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, Rudensky AY. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol. 2019;20(2):232‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Miska J, Lee‐Chang C, Rashidi A, et al. HIF‐1α is a metabolic switch between glycolytic‐driven migration and oxidative phosphorylation‐driven immunosuppression of Tregs in glioblastoma. Cell Rep. 2019;27(1):226‐237.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Xu Y, Melo‐Cardenas J, Zhang Y, et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine–induced ER stress response. JCI Insight. 2019;4(5):121887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Bordignon C, Bonini C, Verzeletti S, et al. Transfer of the HSV‐tk Gene into donor peripheral blood lymphocytes for in vivo modulation of donor anti‐tumor immunity after allogeneic bone marrow transplantation. The San Raffaele Hospital, Milan, Italy. Hum Gene Ther. 1995;6(6):813‐819. [DOI] [PubMed] [Google Scholar]
- 203. Tiberghien P, Cahn J‐Y, Brion A, et al. Use of donor T‐lymphocytes expressing herpes‐simplex thymidine kinase in allogeneic bone marrow transplantation: a phase I‐II study. Laboratorie d'Histocompatibilité et Thérapeutique Immuno‐Moléculaire, Besançon, France. Hum Gene Ther. 1997;8(5):615‐624. [DOI] [PubMed] [Google Scholar]
- 204. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T‐cell therapy. Mol Ther Oncolytics. 2016;3:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Tey S‐K, Dotti G, Rooney CM, Heslop HE, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2007;13(8):913‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Zhou X, Di Stasi A, Tey S‐K, et al. Long‐term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123(25):3895‐3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Straathof KC. An inducible caspase 9 safety switch for T‐cell therapy. Blood. 2005;105(11):4247‐4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Minagawa K, Al‐Obaidi M, Di Stasi A. Generation of suicide gene‐modified chimeric antigen receptor‐redirected T‐cells for cancer immunotherapy. Methods Mol Biol. 2019;1895:57‐73. [DOI] [PubMed] [Google Scholar]
- 209. Wilkie S, van Schalkwyk M, Hobbs S, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059‐1070. [DOI] [PubMed] [Google Scholar]
- 210. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Roybal K, Rupp L, Morsut L, et al. Precision tumor recognition by T cells with combinatorial antigen‐sensing circuits. Cell. 2016;164(4):770‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Fedorov VD, Themeli M, Sadelain M. PD‐1‐ and CTLA‐4‐based inhibitory chimeric antigen receptors (iCARs) divert off‐target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Rosenblum MD, Way SS, Abbas AK. Regulatory T cell memory. Nat Rev Immunol. 2016;16(2):90‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]