

Abstract
Glucocorticoids are widely used for the suppression of inflammation, but evidence is growing that they can have rapid, non‐genomic actions that have been unappreciated. Diverse cell signaling effects have been reported for glucocorticoids, leading us to hypothesize that glucocorticoids alone can swiftly increase the 3′,5′‐cyclic adenosine monophosphate (cAMP) production. We found that prednisone, fluticasone, budesonide, and progesterone each increased cAMP levels within 3 minutes without phosphodiesterase inhibitors by measuring real‐time cAMP dynamics using the cAMP difference detector in situ assay in a variety of immortalized cell lines and primary human airway smooth muscle (HASM) cells. A membrane‐ impermeable glucocorticoid showed similarly rapid stimulation of cAMP, implying that responses are initiated at the cell surface. siRNA knockdown of Gαs virtually eliminated glucocorticoid‐stimulated cAMP responses, suggesting that these drugs activate the cAMP production via a G protein‐coupled receptor. Estradiol had small effects on cAMP levels but G protein estrogen receptor antagonists had little effect on responses to any of the glucocorticoids tested. The genomic and non‐genomic actions of budesonide were analyzed by RNA‐Seq analysis of 24 hours treated HASM, with and without knockdown of Gαs. A 140‐gene budesonide signature was identified, of which 48 genes represent a non‐genomic signature that requires Gαs signaling. Collectively, this non‐genomic cAMP signaling modality contributes to one‐third of the gene expression changes induced by glucocorticoid treatment and shifts the view of how this important class of drugs exerts its effects.
Keywords: airway smooth muscle, corticosteroids, G protein-coupled receptors, membrane glucocorticoid receptor, RNA sequencing
Abbreviations
- ßAR
ß‐adrenergic receptor
- cAMP
3′,5′‐cyclic adenosine monophosphate
- CS
connectivity score
- Fsk
forskolin
- GPCR
G protein‐coupled receptor
- GPER
G protein estrogen receptor
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- HASM
airway smooth muscle
- IBMX
3‐isobutyl‐1‐methylxanthine
- Iso
isoproterenol
- mGR
membrane‐bound glucocorticoid receptor
- PDE
phosphodiesterase
1. INTRODUCTION
Glucocorticoids are used in the treatment of a wide array of diseases, including rheumatoid arthritis, autoimmune disorders, allergy, cancer, and respiratory syndromes. The occurrence of side effects and glucocorticoid resistance, particularly with systemic use, greatly hamper their use.1 The combination of glucocorticoids ß‐agonists has long been considered the most effective means for controlling asthma and chronic obstructive pulmonary disease, even more than using either alone.2, 3, 4, 5 While the mechanisms underlying glucocorticoid genomic effects on enhancing the clinical efficacy of ß‐agonist have been previously studied,6 the possibility that non‐genomic signaling by glucocorticoids enhance the clinical efficacy of ß‐agonists has not been investigated due to the limited tools to measure real time kinetics of intracellular changes in 3′,5′‐cyclic adenosine monophosphate (cAMP) concentration.
Conventional thought suggests that glucocorticoids alter the cell function through changes in the gene expression that occur via activation of ubiquitously expressed intracellular glucocorticoid receptors (GR).7 In the absence of glucocorticoid, the GR resides in the cytoplasm then translocates to the cell nucleus upon binding of ligand. Nuclear GR then interacts with glucocorticoid response elements (GREs) to alter the gene expression. Various reports, some published over 25 years ago, suggest that glucocorticoids also induce rapid alterations in various signaling processes that appear to be non‐genomic in nature.5, 8, 9 The human skin blanching assay (often called the vasoconstrictor assay) has been used for nearly 50 years as a means of qualitatively assessing the topical availability and potency of glucocorticoids.10 This test characterizes the potency of glucocorticoids through non‐genomic effects on vasoconstriction. Some of these non‐genomic effects may require specific interactions with membrane‐bound versions of GR (mGR) or other undefined membrane components.5 Short duration treatment of various cell types with glucocorticoid affect many different signaling events, including agonist‐induced calcium release, reactive oxygen species, and arachidonic acid release.11, 12, 13, 14, 15, 16 BSA‐conjugated cortisol, a steroid unable to cross the cell membrane, has been used as a tool to differentiate plasma membrane GR effects from those of the cytosolic GR. For instance, the effects of short (5 to 90 minutes) exposure of BSA‐cortisol on leukemia cells was studied using proteomic tools and 128 unique proteins were found to be specifically upregulated.17 Interestingly, the putative mGR may interact with the NMDA receptor or may directly activate a G protein‐coupled receptor (GPCR) coupled to Gαs and/or Gq/11.18 While these and other studies support the idea that glucocorticoids possess the ability to modulate rapid, non‐genomic signaling, none reveal the specific signaling pathways or receptor(s) responsible.
Our studies demonstrate that glucocorticoids rapidly increase the cAMP levels in a variety of cell types. In human airway smooth muscle (HASM) glucocorticoids trigger this non‐genomic signaling via binding to an extracellular site and activating the stimulatory G protein, Gαs. While a GPCR might be involved, our data suggest that the G protein estrogen receptor (GPER) does not mediate this response. Removal of this rapid, non‐genomic signal via siRNA knockdown of Gαs modifies the transcriptomic response to the glucocorticoid, budesonide. Out of a 140 gene budesonide signature, the alteration of 48 of these genes was dependent upon the Gαs‐cAMP signal. Thus, of all the canonical changes in gene transcription by glucocorticoid, a full one‐third of them require this rapid, non‐genomic signal.
2. MATERIALS AND METHODS
2.1. Materials
Forskolin (Fsk) was purchased from LC Laboratories. Cell culture media and components were purchased from Thermo Fisher. Fetal bovine serum was purchased from Atlanta Biologicals. siRNA construct for silencing GNAS were obtained from Dharmacon. The sense sequence used was CGAUGUGACUGCCAUCAUCUU. Secondary antibodies were obtained from Santa Cruz Biotechnology. All other drugs and chemicals were purchased from Sigma‐Aldrich unless otherwise noted.
2.2. Cell culture
HASM cells were isolated from deceased, de‐identified lung donors by enzymatic dissociation in accordance with Institutional Review Board approval and as described previously.19 HASM cells were grown in Ham's F‐12 media (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, pen/strep, 25 mM HEPES, 1.7 mM CaCl2, and l‐glutamine. Cells were kept at 5% CO2 and 37°C. Experiments were performed on cells from passage 3‐7 using cells from 10 different donors in total, and at least three different donors for each study. Patient demographics are described in Table 1. Human fetal lung (HFL‐1) fibroblasts (American Type Culture Collection) were grown in Ham's F12 medium with 10% fetal bovine serum and 1% antibiotic‐antimycotic solution. HEK‐293 cells (American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum. All cells were kept in a humidified incubator with 5% CO2 at 37°C.
Table 1.
HASM cell patient demographics
| Designation | Age | Sex | Race |
|---|---|---|---|
| N100217 | 39 | M | Black |
| N041717 | 19 | M | Caucasian |
| N021014 | 54 | M | Caucasian |
| N101317K | 54 | F | Hispanic |
| N012317 | 29 | F | Caucasian |
| N012414 | 20 | F | Black |
| N030116 | 69 | M | Caucasian |
| N062017 | 47 | M | Hispanic |
| N080817 | 23 | M | Black |
| N012518K | 18 | M | Caucasian |
| N112017K | 53 | M | Asian |
| N011118K | 14 | M | Caucasian |
HASM cells were derived from the following patients who had no history of asthma or chronic illness. RNA‐seq analysis was performed using the cell lines from the first six rows.
2.3. Transfection
To transfect HASM cells with siRNA, 250 000 cells were incubated with 100 nM siRNA (target or scrambled) for 30 minutes at room temperature using HiPerFect transfection reagent (Qiagen) following the manufacturer's instructions. Cells were then transferred to 6‐well plates. After 5 hours incubation at 37°C with 5% CO2, HASM growth media containing 5% fetal bovine serum was added for 48 hours. Media was replaced with serum‐free media for 24 hours prior to drug treatment or assay.
2.4. cADDis cAMP assays
We performed kinetic measurements of cAMP production in live cells using the green cAMP difference detector in situ (cADDis) cAMP sensor (Montana Molecular, Bozeman, MT) as described previously.20 Briefly, sub‐confluent HASM, HFL‐1, or HEK‐293 cells were plated on a black‐walled, clear flat bottom 96‐well plates along with recombinant BacMam virus expressing the cADDis sensor and 1 µM trichostatin‐A. Cells were grown overnight at 5% CO2 and 37°C. Media was aspirated and replaced with 180 µL per well of 1X Dulbecco's Phosphate Buffered Saline Solution without calcium and magnesium. The 96‐well plate was covered with aluminum foil and incubated at room temperature for 30 minutes. Cell fluorescence was read from the plate bottom using excitation/emission wavelengths of 494 and 522 nm, respectively, using a SpectraMax M5 plate reader (Molecular Devices). A 5 minutes kinetic read on unstimulated cells was monitored until the variability in each well's fluorescence was ≤5%. Cells were then stimulated with the indicated drug and fluorescence changes in each well were read at 30 seconds intervals for 30 minutes. Data were transformed to the change in fluorescence over the initial fluorescence (ΔF/F 0) then plotted and fit to a single site decay model using GraphPad Prism 8.0 (GraphPad Software). The K value (slope) and the plateau from this one‐site decay fit are reported. To create a concentration‐response curve, the K was multiplied by the plateau for each drug concentration and plotted on a log scale.
2.5. Immunoblot analysis
Whole cell lysates were obtained by scraping cells in modified RIPA lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Igepal CA‐630, plus mammalian protease inhibitor cocktail). Whole cell lysates were separated on 10% SDS‐polyacrylamide gels by electrophoresis before being transferred to PVDF membranes (Millipore) by electroblotting. ß‐actin (Santa Cruz Biotechnology sc‐47778, 1:1000 dilution) and Gαs (Santa Cruz Biotechnology sc‐135914, 1:500 dilution) antibodies were simultaneously incubated overnight at 4°C following block in nonfat milk. The appropriate secondary antibodies with conjugated horseradish peroxidase were purchased from Santa Cruz Biotechnology. Images were captured using a BioRad Gel Doc system then the alignment, exposure, and contrast of each image was optimized using Adobe Photoshop CS4. Immunoreactive bands were analyzed by densitometric analysis using the volume plus density method and normalized to ß‐actin, as described previously.21
2.6. RNA‐Seq
Control or Gαs‐knockdown HASM cells (see transfection procedure above) from six different donors (see Table 1) at the same passage number were treated with either vehicle or 1 µM budesonide for 24 hours. Total RNA was extracted using the miRNAeasy mini kit (Qiagen). Approximately 1 µg of RNA from each sample was used to generate RNA‐Seq cDNA libraries for sequencing using the TruSeq RNA Sample Prep Kit v2 (Illumina, Inc). Sequencing of 100 bp single‐end reads was performed with an Illumina HiSeq 4000 instrument at the University of California, Irvine Genomics High‐Throughput Facility.
2.7. Data analysis and statistics
Data analysis and statistics: Standard curves were fit and unknown values were extrapolated using GraphPad Prism 8.0. Data were presented as the mean ± SEM. Statistical comparisons (t tests and one‐way analysis of variance) were performed and graphics were generated using GraphPad Prism 8.0.
RNA‐Seq alignment and quantification: RNA‐Seq data quality was checked using FastQC and all samples had high quality score (Phred score >28) for all nucleotides sequenced. FastQC analysis showed Illumina TrueSeq adapters were overrepresented in two samples. Cutadapt software was used to remove the identified adapters and reads were filtered for a minimum length of 20 bp. The Rsubread R package (version v1.30.6; Liao et al22) was used to align the reads and to produce the gene‐level summarized values using hg38 annotation from the Rsubread package. Integer‐based gene counts were generated using the featureCounts function in the Rsubread package.22, 23 limma 24 and edgeR (version v3.22.3) 25, 26 ) packages were used to calculate FPKM values27 and a custom script to convert FPKM to TPM values.28 Ensembl Genome Reference Consortium Human Build 38 patch 12 (GRCh38.p12) database was used to convert gene IDs to Hugo Gene Nomenclature Commitee (HGNC) HCNC gene symbols.29 RNA‐Seq data was available in GEO under accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130715. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94335, an independent dataset including 34 samples from fatal‐asthma and non‐asthma donors treated with control and budesonide,40 was also processed with the same alignment and quantification pipeline to minimize technical and analytical bias.
Differential gene expression and pathway analysis: Genes with less than 100 counts in 50% of our samples were filtered out prior to any analysis. Principal Component Analysis (PCA) was used to investigate interpatient variability compared to treatment‐specific variability. The prcromp function from the stats R package was used to compute PCAs. Plotting of first two PCAs showed intended treatment‐specific variability was more dominant than interpatient variability. Therefore, no adjustment was necessary.
DESeq2 R package was used to generate differential gene‐lists between various treatment conditions: (a) vehicle‐treated and Gαs‐knockdown vehicle‐treated HASM cells, (b) budesonide‐treated and Gαs‐knockdown budesonide‐treated HASM cells, (c) vehicle‐treated and budesonide‐treated HASM cells, and (d) Gαs‐knockdown vehicle‐treated and Gαs‐knockdown budesonide‐treated HASM cells. Gene‐lists from (a) and (b) were used for in silico validation of Gαs (GNAS gene) knockdown. Differential gene‐lists from (c) and (d) represent the budesonide induced transcriptional activity in control (genomic + non‐genomic) and Gαs‐knockdown (genomic only) HASM cells, respectively. (c) and (d) were compared to previously published budesonide‐associated differentially expressed genes by Himes et al30 for validation of our budesonide signature (Figure S1). Overlap analysis of signature gene‐lists was performed using a Venn diagram. Then, ASSIGN, a pathway profiling toolkit, was used to evaluate the gene‐lists (c) and (d) in predicting budesonide‐induced transcriptional activity in HASM.31 (c) and (d) gene‐lists were budesonide signatures due to Gαs‐independent and ‐dependent transcriptional changes due to 24‐hour post‐budesonide treatment, respectively. An independent HASM dataset, GSE94335,40 was used to validate both budesonide signatures. Predicted budesonide activity was correlated using Pearson’s correlation to evaluate budesonide and budesonide‐Gαs knockdown signatures.
Gene set enrichment analysis: Using fgsea function, a gene set enrichment analyses were performed against KEGG molecular pathways and gene ontology gene annotations for both budesonide signatures. Cutoff values of P < .05 and a false discovery rate (FDR) < 0.05 were used to assess significant enrichment.
Analysis of budesonide transcriptional activity in publicly available data: Connectivity scores (CSs) were assessed using the gene‐list that was unique to differentially expressed gene‐ list (3) using a ConnectivityMap (CMAP) query to identify most similar and dissimilar perturbagen signatures in a publicly available database.32
All RNA‐Seq data analyses except the CMAP query were performed in R version 3.6.0 and Bioconductor version 3.733 (R Core Team, 2014; http://www.R-project.org/). All codes are available at https://github.com/mumtahena/gluc_HASMs.
3. RESULTS
3.1. Rapid effect of glucocorticoids on cellular production of cAMP
Since glucocorticoids have been found to rapidly activate different signaling pathways in neurons,9, 18 we hypothesized that glucocorticoids stimulate the cAMP production in mammalian cells. Using a highly sensitive cAMP biosensor capable of displaying rapid cAMP kinetics in live HEK‐293 cells (cADDis, Montana Molecular), we examined responses to two commonly prescribed glucocorticoids. As shown in Figure 1A, the addition of 10 µM fluticasone increased cAMP levels (reflected as a decay in cADDis fluorescence) within 30‐60 seconds of drug exposure. This cAMP response reached a plateau at approximately 12 minutes and the maximal effect was nearly as efficacious as the response to a maximal concentration of the direct adenylyl cyclase activator, Fsk (10 µM, Figure 1A). A 10‐fold lower concentration of fluticasone (1 µM) also stimulated the cAMP levels, but at a somewhat slower rate of decay and smaller plateau. Fluticasone concentrations lower than 1 µM induced responses that were not statistically significant when compared to vehicle control (0.1 µM fluticasone is shown). The addition of budesonide (0.1, 1, or 10 µM) elicited responses similar to fluticasone (Figure 1B). We also expressed the cADDis sensor in HFL‐1 cells, a human fetal lung fibroblast cell line, and measured cAMP responses to glucocorticoids. Both budesonide and fluticasone induced rapid increases in cAMP levels in HFL‐1 cells that were similar to that seen in HEK‐293 cells (10 µM budesonide plateau was −0.251 in HFL‐1 cells compared to −0.395 in HEK‐293 cells; 10 µM fluticasone plateau was −0.341 in HFL‐1 cells compared −0.457 in HEK‐293 cells). Thus, two different glucocorticoids induce rapid increases in cAMP levels in two different cell lines (HEK‐293 and HFL‐1 cells) and these responses were large enough to observe without the presence of phosphodiesterase (PDE) inhibitors.
Figure 1.
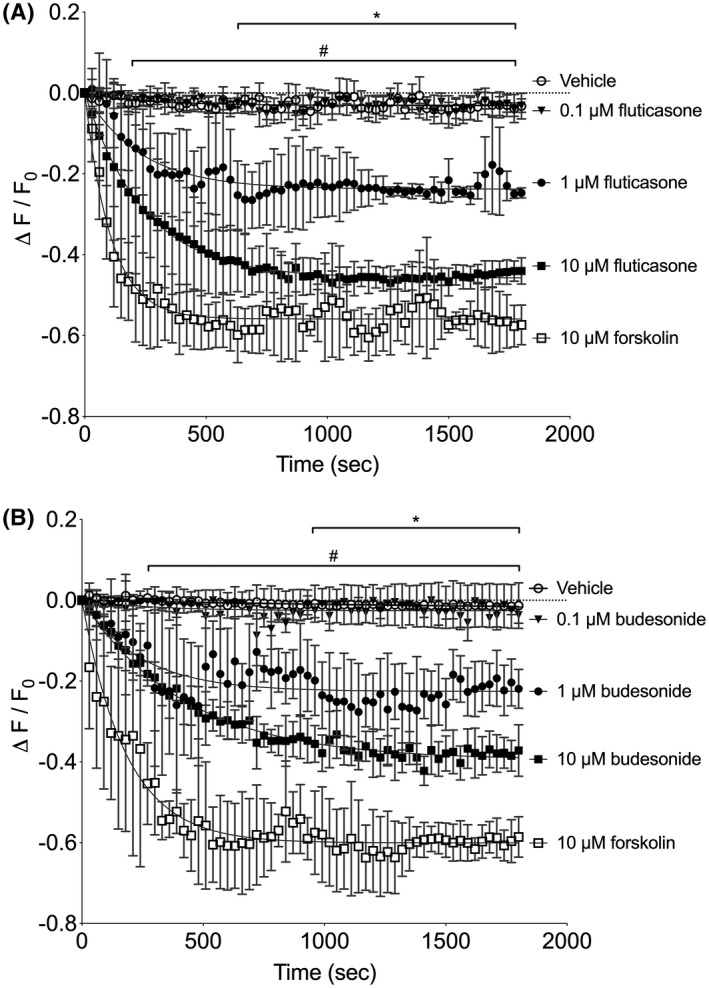
Glucocorticoids stimulate rapid cAMP responses in HEK‐293 cells. Cells were incubated with recombinant BacMam virus expressing the cADDis cAMP sensor. After establishing baseline, fluorescence decay was monitored for 30 minutes after addition of drug. cADDis sensor fluorescent decay curves elicited by 1 or 10 µM fluticasone (A) 1 or 10 µM budesonide (B) are shown. Fluorescence decay curves elicited by vehicle and 10 µM forskolin are shown as reference to the minimal and maximal responses. Each point represents the mean ± SEM of n = 5 experiments and lines represent the fit by one‐phase decay non‐linear regression analysis. * denotes P < .05 for the 1 µM glucocorticoid conditions at the indicated time points, # denotes P < .05 for the 10 µM glucocorticoid conditions at the indicated time points compared to vehicle using multiple t tests and the Holm‐Sidak method for correction of multiple comparisons. The 0.1 µM glucocorticoid conditions were not significantly different than vehicle
When primary cultured HASM cells obtained from several donors were treated with various glucocorticoids, a rapid production of cAMP was again observed. Prednisone (Figure 2A), fluticasone (Figure 2C), and budesonide (Figure 2D) elicited cAMP responses in HASM within minutes of drug addition. Prednisone induced smaller responses than fluticasone or budesonide, but significantly increased cAMP within 8 minutes of treatment. We also examined other steroids and found that progesterone (Figure 2B) stimulated the cAMP production in HASM cells. Estradiol did not stimulate the cAMP responses that were significantly different than vehicle (Figure 4A). To determine if glucocorticoids increase cAMP via inhibition of PDEs, we preincubated HASM with a broad‐spectrum PDE inhibitor, 3‐isobutyl‐1‐methylxanthine (IBMX). Fluticasone retained cAMP stimulating activity in the presence of 10 µM IBMX (Figure 2E). IBMX stimulated cADDis responses on its own, so once the baseline was set following IBMX addition, the maximal response to Fsk was diminished as compared to control (shown in Figure 2C). All these responses occurred within minutes of drug addition, indicating a non‐genomic mode of action.
Figure 2.
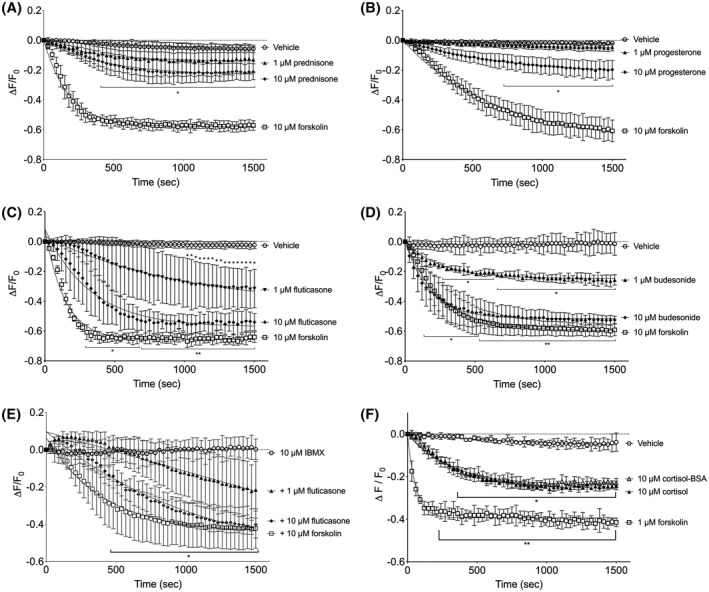
Glucocorticoids stimulate rapid cAMP responses in HASM. Primary HASM cells were incubated with recombinant BacMam virus expressing the cADDis cAMP sensor. After establishing baseline, fluorescence decay was monitored for 30 minutes after addition of drug. cADDis sensor fluorescent decay curves elicited by 1 or 10 µM prednisone (A), 1 or 10 µM progesterone (B), 1 or 10 µM fluticasone (C), 1 or 10 µM budesonide (D), and 1 or 10 µM fluticasone in cell preincubated with 10 µM IBMX (E). Fluorescence decay curves elicited by 10 µM forskolin are shown in each panel as reference to the maximal response. Fluorescence decay by cADDis was monitored for 30 minutes after addition of either vehicle, 1 µM forskolin, 10 µM cortisol, or 10 µM cortisol‐BSA (F). A different Y axis scale is used on panel E to better visualize these responses. Each point represents the mean ± SEM of n = 4‐6 donors and lines represent the fit by one‐phase decay non‐linear regression analysis. * denotes P < .05, ** denotes P < .01 of each time point compared to vehicle using multiple t tests and the Holm‐Sidak method for correction of multiple comparisons
Figure 4.
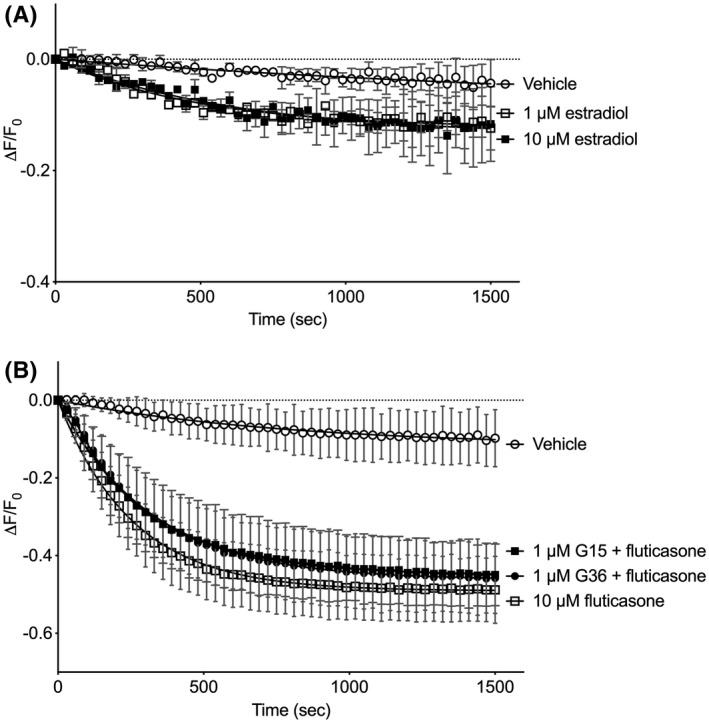
GPER antagonists do not block glucocorticoid‐stimulated cAMP responses. Primary HASM cells were incubated with recombinant BacMam virus expressing the cADDis cAMP sensor. After establishing baseline, fluorescence decay was monitored for 30 minutes after addition of drug. cADDis sensor fluorescent decay curves elicited by 1 or 10 µM estradiol (A) or 10 µM fluticasone (C). A different Y axis scale is used on panel A to better visualize these responses. Each point represents the mean ± SEM of n = 5 donors and lines represent the fit by one‐phase decay non‐linear regression analysis. No significant differences are seen comparing fluticasone alone to either condition where a GPER antagonist (G15 or G36) was included at any time point using multiple t tests and the Holm‐Sidak method for correction of multiple comparisons
We have recently observed that this non‐genomic action of glucocorticoids in HASM is blocked by RU486 but not altered by knockdown of GRα.34 Therefore, we posited that this rapid stimulation of cAMP levels involves glucocorticoid binding to a plasma membrane receptor. Addition of 10 µM cortisol or 10 µM cortisol‐BSA conjugate (the latter drug is unable to cross cell membranes) elicited identical cAMP responses in HASM (Figure 2F). Cortisol and cortisol‐BSA induced significant reductions in cADDis fluorescence within 4 minutes (as compared to vehicle) and reached a maximal response that was about half of that induced by a near‐maximal concentration of Fsk (1 µM). These data were consistent with the idea that glucocorticoids activate a membrane‐bound receptor via an outward‐facing binding site to stimulate cAMP production.
3.2. Role of Gαs in mediating glucocorticoid effects on production of cAMP
Since the stimulation of cAMP by glucocorticoids is rapid and plasma membrane delimited, we hypothesized that the response involves the direct activation of the stimulatory G protein, Gαs. To this end, we used siRNA strategy to knockdown the expression of Gαs in HASM cells. HASM cells were transfected with validated siRNA sequences specific for GNAS or scrambled siRNA.35 As shown in Figure 3A, immunoblot analysis indicated a reduction in expression of both the long and short forms of Gαs in siRNA transfected HASM as compared to scrambled control. We consistently observed a reduction in the cADDis sensor expression levels following the transfection procedure, which resulted in the maximal cADDis responses being reduced by about 50% (comparing the response to 10 µM Fsk in Figure 2 vs Figure 3). cAMP responses to vehicle or Fsk (10 µM) were unaffected by Gαs knockdown, indicating that adenylyl cyclase expression and total activity were unaffected (Figure 3B). In contrast, cAMP responses to 100 nM formoterol (a long‐acting ß2‐adrenoceptor agonist approved for the treatment of asthma) were significantly reduced in Gαs‐knockdown HASM as compared to control, consistent with ß‐adrenoceptors stimulating cAMP production via activation of Gαs (Figure 3C). cAMP responses to budesonide (10 µM, Figure 3D) or fluticasone (10 µM, Figure 3E) were also significantly diminished in cells with Gαs knockdown. Taken together, these results indicate that glucocorticoids activate a rapid, non‐genomic signaling pathway that stimulates Gαs and the production of cAMP.
Figure 3.
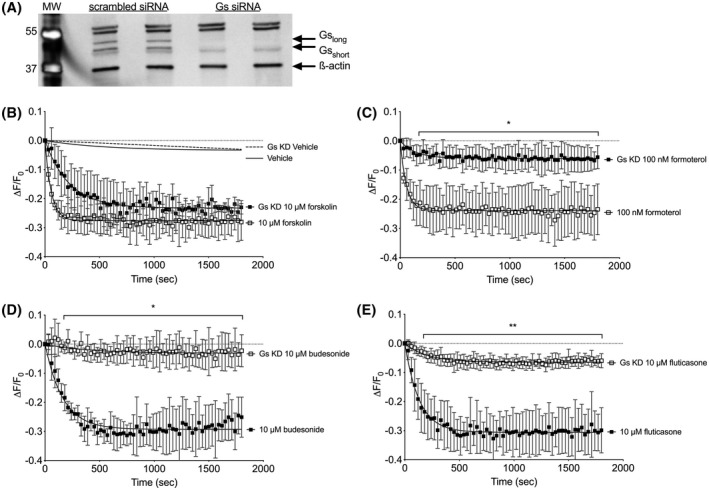
Glucocorticoid stimulation of cAMP depends upon Gαs expression. A, HASM were transfected with siRNA specific for GNAS or scrambled control for 48 hours and lysates analyzed by SDS‐PAGE and immunoblotting simultaneously with antibodies specific for Gαs and ß‐actin. Image is representative of n = 3 experiments on separate donor cells. RNA sequencing revealed GNAS transcript was reduced 6.07 ± 0.35 fold (n = 6) following transfection with siRNA. B‐E, cADDis sensor was expressed in control or Gαs‐knockdown HASM using a recombinant BacMam virus then responses to vehicle, forskolin (C), formoterol (C), budesonide (D), or fluticasone (E) were measured. Each point represents the mean ± SEM of n = 4‐5 donors. * denotes P < .05, ** denotes P < .01 of each time point
3.3. Role of GPER in mediating glucocorticoid effects on production of cAMP
Given that glucocorticoids act via an extracellular binding site and depend upon Gαs, we hypothesize that a GPCR is involved. The GPER is a known GPCR that is activated by specific steroids and has been reported to couple to Gαs in certain cells.36, 37, 38, 39 1 and 10 µM estradiol stimulated small cAMP responses that were not significantly different than vehicle (Figure 4A). GPER mRNA is detected in a wide array of cell types, including HEK‐293, HFL‐1, and HASM cells (GEO accession numbers http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128076, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73555, and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52778). We examined budesonide cAMP responses in cells preincubated with selective GPER antagonists to determine if glucocorticoids stimulated cAMP via activation of GPER. In HASM cells, the addition of 1 µM G36 had no effect on fluticasone stimulation of cAMP responses (Figure 4B). Another GPER antagonist, G15 (1 µM), also had no effect on fluticasone responses (Figure 4C). These results are consistent with the idea that the GPER is not involved in mediating the rapid cAMP signaling stimulated by glucocorticoids.
3.4. Transcriptional effects of glucocorticoid non‐genomic signaling through Gαs
Glucocorticoid effects on cells are attributed to their genomic actions, so the role that this rapid, non‐genomic signal plays in regulating cell function is relatively under studied.5 To investigate the transcriptional effects of the non‐genomic signaling through Gαs, we performed an RNA‐Seq analysis from HASM cells treated with 1 µM budesonide, comparing transcripts from control and Gαs‐knockdown samples 24 hours post‐treatment. We used HASM cell lines isolated from six different donors at the same passage. Each donor cell line was subjected to four conditions: scrambled siRNA treated with vehicle, scrambled siRNA treated with 1 µM budesonide, GNAS siRNA treated with vehicle, GNAS siRNA treated with 1 µM budesonide. We chose a 24‐hour drug treatment duration in order to capture the classical genomic effects of glucocorticoids. We chose to use 1 µM budesonide since this concentration elicited a sub‐maximal cAMP response that could be completely abolished by Gαs knockdown. In addition, this lower concentration is more consistent with other studies of the genomic effects of glucocorticoid treatment. With this approach, any observed differences in the budesonide transcriptomes between control and Gαs‐knockdown cells would reflect the role of the non‐genomic glucocorticoid signaling. We performed quantitative analysis of the transcripts that were altered by budesonide acting through both genomic and non‐genomic signaling (control) or acting through only genomic signaling (Gαs knockdown).
We began our analysis with the hypothesis that the non‐genomic transcriptional activities are due to signaling via Gαs. Two novel budesonide signatures were generated using the differential gene expression lists: (a) a 140‐gene genomic and non‐genomic signature of budesonide effect in control cells, and (b) a 121‐gene genomic only signature of budesonide in Gαs‐knockdown cells (Figure 5; Supplemental Tables S1 and S2). ASSIGN, a pathway analysis toolkit, was used to assess the predictive ability of these gene‐sets in determining budesonide induced transcriptional activity. As an internal validation, we used the signatures to predict budesonide activity in all 24 HASM samples using ASSIGN. Predictions show that both signatures were able to correctly identify budesonide induced transcriptional activity in all 24 HASM samples (Pearson's correlation 0.9977; P < .0001; Supplemental Figure S4). To validate these signatures further, we applied these two signatures to a previously published independent dataset of 34 HASM samples derived from asthma and non‐asthma donors with either control or budesonide treatment40 (GEO dataset accession http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94335). Since there are variation in budesonide concentration and duration for the treatment conditions between the datasets (100 nM for 1 hour vs 1 µM for 24 hours of budesonide treatment), we used ASSIGN’s background adaptive feature. Thus, the background gene expression differences between our signature (training) and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94335 (test) samples were adjusted to provide more accurate prediction. Despite the slight difference in predicted genomic + non‐genomic and genomic only budesonide activity, both signatures accurately estimated high budesonide activity in budesonide‐treated HASMs derived from both asthma and non‐asthma donors (Pearson's correlation 0.9995; P < .0001, Supplemental Table S1).40 This similarity in predicted budesonide activity validates the robustness of both types of budesonide signatures in capturing transcriptional activity regardless of Gαs status 24 hours post‐budesonide treatment. Although genes within each signature are different, the ubiquitous effects of the canonical genomic signaling pathway activated by budesonide, along with a large number of genes in the signatures, maintains the robustness of the predictive ability.
Figure 5.
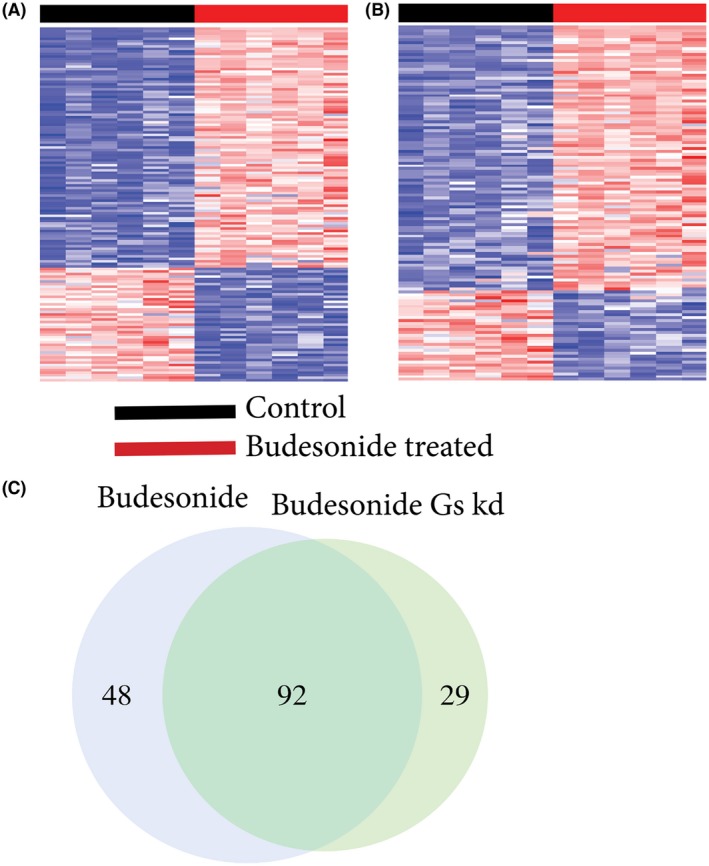
The transcriptional activity due to budesonide treatment in HASM. A, 140 gene budesonide signature representing the transcriptional activity (genomic + non‐genomic) variation between control and budesonide treated HASM. B, 121 gene budesonide Gαs knockdown signature showing the transcriptional activity (genomic only) variation between Gαs knockdown‐control and budesonide‐treated HASM. For both (A) and (B), each row represents a gene, and each column represents a sample. The red cell color represents level of overexpression and the blue cell color represents levels of low expression. Brighter the red, higher the gene expression and darker the blue, lower the expression. C, Comparison of genes from each budesonide signatures show 94 genes were shared between budesonide (out of 140 genes) and budesonide‐Gαs knockdown (out of 121 genes) signatures. 48 genes unique to budesonide signature represent Gαs dependent activity
To evaluate the roles of these signature genes in known cellular processes, a gene set enrichment analysis was performed with databases, particularly, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Gene Ontology (GO) annotations.41 These databases provide curated gene‐sets for understanding the functions and utilities of biological systems, cellular components, and molecular functions of a given test gene‐set. There was no significant enrichment of any KEGG or GO gene‐sets for the budesonide signature gene‐lists after adjusting the p‐value for multiple comparisons. This implies that these budesonide signatures present unique transcriptional activity not represented by the gene‐sets in these databases.
Next, we analyzed the overlapping genes from the two budesonide signatures to evaluate the genomic and non‐genomic interplay of these signature genes. There were 92 genes shared between budesonide and budesonide Gαs‐knockdown signatures (Figure 5C). Forty‐eight genes were unique to the budesonide signature, indicating these genes (26 upregulated and 22 downregulated) are dependent on Gαs‐dependent budesonide transcriptional activity (Table 2,Supplemental Figure S2). We then used an unsupervised hierarchical clustering method to investigate how the transcriptional activity attributed to this non‐genomic signature affected the HASM without knowing the treatment conditions. Essentially, we evaluated these 48 genes in their ability to differentiate the treatment conditions. The hierarchical clustering clearly differentiates the two budesonide treatment conditions (Table 2, Figure S2). The clustering also shows two distinct sub‐clusters that identify the non‐genomic signature.
Table 2.
List of 48 genes representing the non‐genomic budesonide transcriptional activity in HASM cells
| Gene symbols | Gene name |
|---|---|
| ADARB1 | adenosine deaminase, RNA‐specific, B1 |
| ADH1B | alcohol dehydrogenase 1B (class I), beta polypeptide |
| ANGPTL1 | angiopoietin‐like 1 |
| APBB2 | amyloid beta (A4) precursor protein‐binding, family B, member 2 |
| ARID5A | AT rich interactive domain 5A (MRF1‐like) |
| ARMC8 | armadillo repeat containing 8 |
| ARNTL | aryl hydrocarbon receptor nuclear translocator‐like |
| CCDC102B | coiled‐coil domain containing 102B |
| CCND3 | cyclin D3 |
| CHST7 | carbohydrate (N‐acetylglucosamine 6‐O) sulfotransferase 7 |
| CIART | Circadian Associated Repressor of Transcription |
| FADS1 | fatty acid desaturase 1 |
| FSTL3 | follistatin‐like 3 (secreted glycoprotein) |
| GAL | galanin prepropeptide |
| GPR1 | G protein‐coupled receptor 1 |
| HMGA1 | high mobility group AT‐hook 1 |
| IER5L | immediate early response 5‐like |
| IL16 | interleukin 16 |
| LY96 | lymphocyte antigen 96 |
| MAP3K7CL | NAMAP3K7 C‐Terminal Like |
| MEX3B | mex‐3 homolog B (C. elegans) |
| MMD | monocyte to macrophage differentiation‐associated |
| NNMT | nicotinamide N‐methyltransferase |
| NR1D1 | nuclear receptor subfamily 1, group D, member 1 |
| NR1D2 | nuclear receptor subfamily 1, group D, member 2 |
| NRG1 | neuregulin 1 |
| PDE5A | phosphodiesterase 5A, cGMP‐specific |
| PDLIM1 | PDZ and LIM domain 1 |
| PER1 | period homolog 1 (Drosophila) |
| PKDCC | protein kinase domain containing, cytoplasmic homolog (mouse) |
| PLA2G4A | phospholipase A2, group IVA (cytosolic, calcium‐dependent) |
| PLXNA2 | plexin A2 |
| PRRX1 | paired related homeobox 1 |
| PTPRG | protein tyrosine phosphatase, receptor type, G |
| PTX3 | pentraxin 3, long |
| RAB7B | RAB7B, member RAS oncogene family |
| RGMB | RGM domain family, member B |
| SEMA3A | sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3A |
| SH3PXD2B | SH3 and PX domains 2B |
| SHISAL1 | Shisa Like 1 |
| SLC19A2 | solute carrier family 19 (thiamine transporter), member 2 |
| SQOR | Sulfide Quinone Oxidoreductase |
| SSX2IP | synovial sarcoma, X breakpoint 2 interacting protein |
| STOM | stomatin |
| SYNJ2 | synaptojanin 2 |
| TEF | thyrotrophic embryonic factor |
| TMEM158 | transmembrane protein 158 (gene/pseudogene) |
| ZBTB16 | zinc finger and BTB domain containing 16 |
To find similar patterns of non‐genomic transcriptional activity in existing data sets, a gene expression query was performed using these 48 genes in the CMAP database. CMAP is a publicly available independent dataset housing over one million differential gene expression signatures. A CMAP query is used to find similarities and dissimilarities across the curated expression profiles of various perturbations, including compounds, overexpressions, and knockdowns.32 CS is a quantitative score between a query gene‐list and a perturbagen that ranges from −100 (opposing signature) to 100 (same signature). CS from a query with 48 Gαs‐dependent budesonide induced gene‐list across the CMAP database was the highest (98.97) for “GR agonist” among the 171 pharmacologic classes available in the CMAP database. CSs for GR agonists including budesonide investigated in different cell lines including prostate (PC3), melanoma (A375), lung (A549), and hepatocellular cancer (HA1E, HCC515) show high similarity (CSs>90) (Supplemental Figure S3, Table S2). This high similarity across various cell lines and glucocorticoid agonists indicates that the non‐genomic budesonide signature, as represented by these 48 genes, extends beyond HASM and is significantly associated with the GR agonists including, but not limited to, budesonide.
4. DISCUSSION
A number of glucocorticoids cause rapid increases in cAMP levels in multiple cell types (Figures 1 and 2). These responses appear to be mediated by a plasma membrane‐associated receptor with an outward facing binding site since albumin‐conjugated cortisol, which can't cross the plasma membrane, retains activity (Figure 2F). The increase in cAMP levels requires stimulation of Gαs, as knockdown of GNAS via siRNA drastically reduces cAMP signaling by glucocorticoids. Since our cAMP assays are performed in the absence of PDE inhibitors there is a possibility that glucocorticoids could work via inhibition of PDE activity. However, inclusion of a broad spectrum PDE inhibitor did not impinge upon fluticasone responses, implying that the glucocorticoid works independently of PDE inhibition. Glucocorticoids can also inhibit uptake of ß‐adrenergic receptor (ßAR) agonists by cells42 but our assays are performed in cultured cells with no other drugs or serum present. In these conditions, glucocorticoids would not increase cAMP within seconds via inhibition of uptake of other drugs, even if the cells were producing autocrine‐acting agonists. Glucocorticoids are also known to increase expression of ßAR43 but this occurs via genomic action so also can't explain our observations.
Based on these findings we speculate that these glucocorticoids activate a GPCR of unknown identity. This mechanism appears to be highly conserved through evolution since both insect and amphibian genomes appear to contain GPCRs that respond to glucocorticoids.9, 44 The canonical nuclear GR appears to be dispensable for the response as its knockdown via siRNA had little effect on these responses.34 RU‐486, a progesterone and GR antagonist, blocks the non‐genomic signaling but this chemical likely has the structural elements needed to antagonize glucocorticoid action at non‐GR binding sites.34 RU‐486 can also stimulate GR responses so this chemical can act as both an antagonist and an agonist of GR.45, 46 GR has been shown to exist in the plasma membrane of various cells.17 Plasma membrane GR could directly activate Gαs or may do so via transactivation of a GPCR. However, the concentrations required to activate cAMP signaling are much higher than those that activate genomic signaling, implying that these occur via different binding sites. It does not appear that glucocorticoids work through binding to GPER since two different antagonists had no effect on glucocorticoid cAMP responses (Figure 4). More work is needed to thoroughly understand the receptor mechanism involved in glucocorticoid stimulation of cAMP.
A number of investigators have reported rapid actions of glucocorticoids in various cells and tissues. These effects include modulation of basal and stimulated intracellular calcium levels, increases in reactive oxygen species and reactive nitrogen species, increased inflammatory and apoptotic pathways and reduced skeletal and smooth muscle tone (recently reviewed in 5). Gαs‐cAMP signaling has numerous and diverse effects in all cells so many of these prior observations of rapid glucocorticoid signaling may be mediated via increased cAMP. Efforts to understand the non‐genomic effects of glucocorticoids have focused on using the membrane‐impermeable cortisol‐BSA conjugate.17, 18 However, this agent is a weak activator of cAMP signaling (Figure 2F) so this is not an optimal approach. Gαs knockdown is a more specific and effective intervention that will be a more powerful means for understanding this non‐genomic signaling. This approach will be useful for understanding the divergent actions of glucocorticoids in other cells and tissues (at least until the membrane receptor involved is identified).
Other work demonstrates the physiological relevance of this non‐genomic signaling by glucocorticoids. We demonstrate that budesonide‐stimulated cAMP augments formoterol‐induced bronchodilation in human small airways when combined with formoterol, a long‐acting ßAR agonist.34 It is interesting to note that in the present study the glucocorticoids most efficacious at stimulating cAMP production were budesonide and fluticasone, two drugs developed for inhaled use in asthma. The clinical utility of these inhaled glucocorticoids is clearly enhanced by this unappreciated non‐genomic signaling that enhances bronchodilation. While success of these important drugs may have been aided by this additional activity, a complete understanding of this alternate signaling paradigm may inform future development of new drugs. Can drugs be designed with different mixtures of genomic and non‐genomic actions for specific clinical applications? Some side effects of glucocorticoids may be dependent on one signaling pathway or another, allowing improved drug design. Finally, a novel membrane receptor capable of augmenting bronchodilation in small airways could provide a much‐needed alternative drug target to existing ßAR agonists, which suffer from issues with tachyphylaxis and potential maladaptive responses after long‐term use.
Confirmation that glucocorticoids act via both rapid, non‐genomic, and canonical genomic signaling mechanisms led us to ask what the role of each might be in context of the known effects of these drugs on the gene transcription. We hypothesized that the cAMP signal serves as a primer to subsequent genomic effects, shaping the size, and/or direction of the transcription of particular genes. Using RNA‐seq and comparing the budesonide treatment transcriptomes from control cells and cells with Gαs knockdown allowed identification of the effects of both the genomic and non‐genomic signals. Comparing our data to existing data sets demonstrate that the gene expression changes we observe are highly reproducible across other cell types and other glucocorticoids. We identified a panel of 140 differentially expressed genes that characterize the effects of budesonide in HASM. Of these, 94 genes were shared between the control and Gαs knockdown conditions and 48 were unique to the control. These 48 genes (Table 2) depend upon the non‐genomic cAMP signal for their differential expression since they were not significantly altered by budesonide treatment in cells lacking Gαs. Thus, one‐third of the genes altered by budesonide treatment depended upon the non‐genomic signal in some way. These results show the importance of this alternate signaling mechanism and force a rethinking of how glucocorticoids act to alter cell function. We propose that this non‐genomic site of action is not an off‐target effect, but rather an integral part of canonical glucocorticoid action.
Our study does have some limitations that require further research. In particular, we chose a 24 hours time point to study the transcriptional effects of budesonide in order to capture the classical genomic effects. A rapid increase in cAMP upon adding glucocorticoid to HASMs suggests that the non‐genomic signaling could begin to alter the transcriptional activity soon after (within an hour or two) glucocorticoid application. Therefore, the transcriptional activity we observe at the 24 hours time point may be too late to capture the full effect of the non‐genomic signaling events. Additional studies are needed to capture RNA‐Seq datapoints from early glucocorticoid treatments to better understand non‐genomic activity attributed to glucocorticoids. Even with this limitation, our data makes clear that the non‐genomic signaling via Gαs and cAMP contributes substantially to the regulation of certain glucocorticoid‐responsive genes. We imagine that the non‐genomic signal integrates with the genomic signal via the convergence of transcription factors regulated by cAMP, including but not limited to CREB, with classical GRE at the promotor of certain genes (Figure 6). More research is needed to understand how these very different signaling modalities integrate at the level of the gene transcription.
Figure 6.
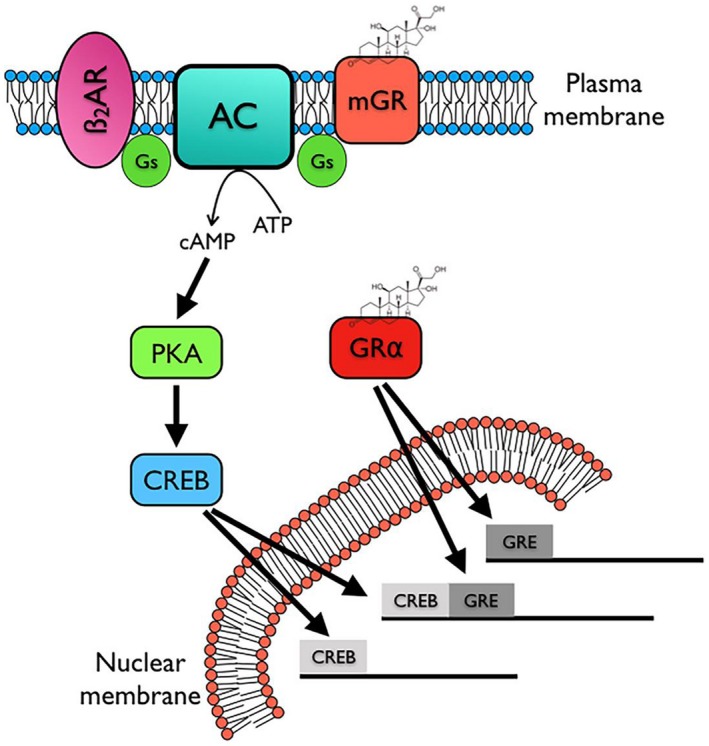
Schematic diagram of proposed signaling by a putative membrane glucocorticoid receptor (mGR). We propose two signaling pathways mediated by glucocorticoids, the canonical cytosolic GRα receptor mediating direct genomic effects via GRE and a second, non‐genomic activation of Gαs and cAMP signaling (illustrated here as signaling via CREB for simplicity, but other transcription factors are likely involved). The latter can have both rapid effects and effects that contribute to the genomic actions of glucocorticoids
Our study is also limited to a few cell types (just HASM in the case of RNA‐seq) so broad conclusions about the relevance non‐genomic cAMP signaling by glucocorticoids won't be appreciated until other cell types can be studied. However, our analysis of the 48 gene non‐genomic signature in the existing gene expression datasets indicate that many cell types contain this signature. It will be of particular importance to understand how immune cells, major targets of glucocorticoid therapy, respond in terms of both genomic, and non‐genomic signaling. The concentrations required for glucocorticoids to induce cAMP signaling are higher than those associated with activation of nuclear glucocorticoid receptors. While this highlights that a different receptor may be involved, one might question the clinical relevance of the glucocorticoid concentrations we use. While local tissue concentrations of budesonide following inhalation dosing are difficult to determine, plasma concentrations of glucocorticoid following oral dosing of 80 mg in humans reaches peaks of nearly 4 µM.47 Thus, the responses we observe are relevant to therapeutic uses of glucocorticoids.
Our study identifies a rapid, non‐genomic signaling mechanism by glucocorticoids that contributes to the known gene expression changes induced by these drugs. By acting on a membrane receptor and stimulating Gαs, glucocorticoids increase the cAMP levels in cells within seconds. We hypothesize that these signals get integrated into the networks that alter the expression of many genes along with the canonical genomic signaling by nuclear GR. This non‐genomic signal can also contribute to rapid effects such a smooth muscle relaxation and likely many others. These findings help explain the clinical utility of inhaled glucocorticoids on an acute basis but also provide a specific mechanism that can be leveraged for the development of future glucocorticoid analogs.
CONFLICT OF INTEREST
None of the authors have any conflicts of interest to report.
AUTHOR CONTRIBUTIONS
T.B. Johnstone, C. Koziol‐White, R.A. Panettieri, R.S. Ostrom conception and design; F.J. Nuñez, T.B. Johnstone, M.L. Corpuz, A.G. Kazarian, N.N. Mohajer conducted experiments; F.J. Nuñez, T.B. Johnstone, O. Tliba, R.A. Panettieri, C. Koziol‐White, M.R. Roosan, R.S. Ostrom analysis and interpretation; F.J. Nuñez, T.B. Johnstone, O. Tliba, R.A. Panettieri, C. Koziol‐White, M.R. Roosan, R.S. Ostrom drafting the manuscript for important intellectual content; all authors approved the final version.
Supporting information
ACKNOWLEDGMENTS
The authors thank Drs. Raymond Penn and Tonio Pera, Thomas Jefferson University, and Dr Gaoyuan Cao and Brian Deeney, Rutgers University, for isolating and providing HASM cells. This work was made possible, in part, through access to the Genomics High Throughput Facility Shared Resource of the Cancer Center Support Grant (P30CA‐062203) at the University of California, Irvine and NIH shared instrumentation grants 1S10RR025496‐01, 1S10OD010794‐01, and 1S10OD021718‐01.
Nuñez FJ, Johnstone TB, Corpuz ML, et al. Glucocorticoids rapidly activate cAMP production via Gαs to initiate non‐genomic signaling that contributes to one‐third of their canonical genomic effects. The FASEB Journal. 2020;34:2882–2895. 10.1096/fj.201902521R
Francisco J. Nuñez and Timothy B. Johnstone contributed equally to this work.
REFERENCES
- 1. Timmermans S, Souffriau J, Libert C. A general introduction to glucocorticoid biology. Front Immunol. 2019;10:1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scicchitano R, Aalbers R, Ukena D, et al. Efficacy and safety of budesonide/formoterol single inhaler therapy versus a higher dose of budesonide in moderate to severe asthma. Curr Med Res Opin. 2004;20:1403‐1418. [DOI] [PubMed] [Google Scholar]
- 3. Rabe KF, Pizzichini E, Stallberg B, et al. Budesonide/formoterol in a single inhaler for maintenance and relief in mild‐to‐moderate asthma: a randomized, double‐blind trial. Chest. 2006;129:246‐256. [DOI] [PubMed] [Google Scholar]
- 4. O'Byrne PM, Bisgaard H, Godard PP, et al. Budesonide/formoterol combination therapy as both maintenance and reliever medication in asthma. Am J Respir Crit Care Med. 2005;171:129‐136. [DOI] [PubMed] [Google Scholar]
- 5. Panettieri RA, Schaafsma D, Amrani Y, Koziol‐White C, Ostrom R, Tliba O. Non‐genomic effects of glucocorticoids: an updated view. Trends Pharmacol Sci. 2019;40:38‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Newton R, Giembycz MA. Understanding how long‐acting beta2 ‐adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids in asthma—an update. Br J Pharmacol. 2016;173:3405‐3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lemanske RF Jr., Busse WW. 6. Asthma. J Allergy Clin Immunol. 2003;111:S502‐S519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song IH, Buttgereit F. Non‐genomic glucocorticoid effects to provide the basis for new drug developments. Mol Cell Endocrinol. 2006;246:142‐146. [DOI] [PubMed] [Google Scholar]
- 9. Moore FL, Orchinik M. Membrane receptors for corticosterone: a mechanism for rapid behavioral responses in an amphibian. Horm Behav. 1994;28:512‐519. [DOI] [PubMed] [Google Scholar]
- 10. Smit P, Neumann HA, Thio HB. The skin‐blanching assay. J Eur Acad Dermatol Venereol. 2012;26:1197‐1202. [DOI] [PubMed] [Google Scholar]
- 11. Urbach V, Harvey BJ. Rapid and non‐genomic reduction of intracellular [Ca(2+)] induced by aldosterone in human bronchial epithelium. J Physiol. 2001;537:267‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Urbach V, Verriere V, Grumbach Y, Bousquet J, Harvey BJ. Rapid anti‐secretory effects of glucocorticoids in human airway epithelium. Steroids. 2006;71:323‐328. [DOI] [PubMed] [Google Scholar]
- 13. Flaherty RL, Owen M, Fagan‐Murphy A, et al. Glucocorticoids induce production of reactive oxygen species/reactive nitrogen species and DNA damage through an iNOS mediated pathway in breast cancer. Breast Cancer Res. 2017;19:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han JZ, Lin W, Chen YZ. Inhibition of ATP‐induced calcium influx in HT4 cells by glucocorticoids: involvement of protein kinase A. Acta Pharmacol Sin. 2005;26:199‐204. [DOI] [PubMed] [Google Scholar]
- 15. Buttgereit F, Krauss S, Brand MD. Methylprednisolone inhibits uptake of Ca2+ and Na+ ions into concanavalin A‐stimulated thymocytes. Biochem J. 1997;326(Pt 2):329‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Croxtall JD, Choudhury Q, Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor‐dependent, transcription‐independent mechanism. Br J Pharmacol. 2000;130:289‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vernocchi S, Battello N, Schmitz S, et al. Membrane glucocorticoid receptor activation induces proteomic changes aligning with classical glucocorticoid effects. Mol Cell Proteomics. 2013;12:1764‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Sheng H, Qi J, et al. Glucocorticoid acts on a putative G protein‐coupled receptor to rapidly regulate the activity of NMDA receptors in hippocampal neurons. Am J Physiol Endocrinol Metab. 2012;302:E747‐E758. [DOI] [PubMed] [Google Scholar]
- 19. Panettieri RA, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI. A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol. 1989;256:C329‐C335. [DOI] [PubMed] [Google Scholar]
- 20. Johnstone TB, Smith KH, Koziol‐White CJ, et al. PDE8 is expressed in human airway smooth muscle and selectively regulates cAMP signaling by beta2‐adrenergic receptors and adenylyl cyclase 6. Am J Respir Cell Mol Biol. 2018;58:530‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu X, Li F, Sun SQ, et al. Fibroblast‐specific expression of AC6 enhances beta‐adrenergic and prostacyclin signaling and blunts bleomycin‐induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;298:L819‐L829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923‐930. [DOI] [PubMed] [Google Scholar]
- 23. Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:1‐25. [DOI] [PubMed] [Google Scholar]
- 25. Nikolayeva O, Robinson MD. edgeR for differential RNA‐seq and ChIP‐seq analysis: an application to stem cell biology. Methods Mol Biol. 2014;1150:45‐79. [DOI] [PubMed] [Google Scholar]
- 26. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA‐seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281‐285. [DOI] [PubMed] [Google Scholar]
- 29. Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754‐D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Himes BE, Jiang X, Wagner P, et al. RNA‐Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One. 2014;9:e99625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen Y, Rahman M, Piccolo SR, et al. ASSIGN: context‐specific genomic profiling of multiple heterogeneous biological pathways. Bioinformatics. 2015;31:1745‐1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437‐1452.e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koziol‐White C, Johnstone TB, Corpuz ML, et al. Budesonide enhances agonist‐induced bronchodilation in human small airways by increasing cAMP production in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2019. Manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garcia‐Murillas I, Sharpe R, Pearson A, et al. An siRNA screen identifies the GNAS locus as a driver in 20q amplified breast cancer. Oncogene. 2013;33:2478‐2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu X, Stallone JN, Heaps CL, Han G. The activation of G protein‐coupled estrogen receptor induces relaxation via cAMP as well as potentiates contraction via EGFR transactivation in porcine coronary arteries. PLoS One. 2018;13:e0191418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu T, Yang G, Hou Y, et al. Cytoplasmic GPER translocation in cancer‐associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene. 2017;36:2131‐2145. [DOI] [PubMed] [Google Scholar]
- 38. Lindsey SH, Liu L, Chappell MC. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids. 2014;81:99‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu X, Li F, Klussmann E, Stallone JN, Han G. G protein‐coupled estrogen receptor 1 mediates relaxation of coronary arteries via cAMP/PKA‐dependent activation of MLCP. Am J Physiol Endocrinol Metab. 2014;307:E398‐E407. [DOI] [PubMed] [Google Scholar]
- 40. Kan M, Koziol‐White C, Shumyatcher M, et al. Airway smooth muscle‐specific transcriptomic signatures of glucocorticoid exposure. Am J Respir Cell Mol Biol. 2019;61:110‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Horvath G, Mendes ES, Schmid N, et al. Rapid nongenomic actions of inhaled corticosteroids on long‐acting beta(2)‐agonist transport in the airway. Pulm Pharmacol Ther. 2011;24:654‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Collins S, Caron MG, Lefkowitz RJ. Beta‐adrenergic receptors in hamster smooth muscle cells are transcriptionally regulated by glucocorticoids. J Biol Chem. 1988;263:9067‐9070. [PubMed] [Google Scholar]
- 44. Ishimoto H, Wang Z, Rao Y, Wu CF, Kitamoto T. A novel role for ecdysone in Drosophila conditioned behavior: linking GPCR‐mediated non‐canonical steroid action to cAMP signaling in the adult brain. PLoS Genet. 2013;9:e1003843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang S, Jonklaas J, Danielsen M. The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids. 2007;72:600‐608. [DOI] [PubMed] [Google Scholar]
- 46. Meyer ME, Pornon A, Ji JW, Bocquel MT, Chambon P, Gronemeyer H. Agonistic and antagonistic activities of RU486 on the functions of the human progesterone receptor. Embo J. 1990;9:3923‐3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gai MN, Pinilla E, Paulos C, Chavez J, Puelles V, Arancibia A. Determination of prednisolone and prednisone in plasma, whole blood, urine, and bound‐to‐plasma proteins by high‐performance liquid chromatography. J Chromatogr Sci. 2005;43:201‐206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials