

Abstract
In vitro data support involvement of cytochrome P450 (CYP)2C8 and CYP3A4 in the metabolism of the anaplastic lymphoma kinase inhibitor brigatinib. A 3‐arm, open‐label, randomized, single‐dose, fixed‐sequence crossover study was conducted to characterize the effects of the strong inhibitors gemfibrozil (of CYP2C8) and itraconazole (of CYP3A) and the strong inducer rifampin (of CYP3A) on the single‐dose pharmacokinetics of brigatinib. Healthy subjects (n = 20 per arm) were administered a single dose of brigatinib (90 mg, arms 1 and 2; 180 mg, arm 3) alone in treatment period 1 and coadministered with multiple doses of gemfibrozil 600 mg twice daily (BID; arm 1), itraconazole 200 mg BID (arm 2), or rifampin 600 mg daily (QD; arm 3) in period 2. Compared with brigatinib alone, coadministration of gemfibrozil with brigatinib did not meaningfully affect brigatinib area under the plasma concentration‐time curve (AUC0–inf; geometric least‐squares mean [LSM] ratio [90%CI], 0.88 [0.83‐0.94]). Coadministration of itraconazole with brigatinib increased AUC0–inf (geometric LSM ratio [90%CI], 2.01 [1.84‐2.20]). Coadministration of rifampin with brigatinib substantially reduced AUC0–inf (geometric LSM ratio [90%CI], 0.20 [0.18‐0.21]) compared with brigatinib alone. The treatments were generally tolerated. Based on these results, strong CYP3A inhibitors and inducers should be avoided during brigatinib treatment. If concomitant use of a strong CYP3A inhibitor is unavoidable, the results of this study support a dose reduction of brigatinib by approximately 50%. Furthermore, CYP2C8 is not a meaningful determinant of brigatinib clearance, and no dose modifications are needed during coadministration of brigatinib with CYP2C8 inhibitors.
Keywords: brigatinib, CYP2C8, CYP3A, drug‐drug interactions, induction, inhibition, non–small cell lung cancer
Brigatinib (ARIAD Pharmaceuticals, Inc, Cambridge, Massachusetts, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited), a next‐generation anaplastic lymphoma kinase (ALK) tyrosine kinase inhibitor, received accelerated approval in the United States and approval in Canada and the European Union for the treatment of patients with ALK‐positive non–small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib. Approval was based on a randomized phase 2 trial (ALTA, NCT02094573), in which brigatinib demonstrated substantial efficacy with an acceptable safety profile post‐crizotinib.1 More recently, a phase 3 trial in crizotinib‐naive patients with ALK‐positive NSCLC showed that progression‐free survival was significantly longer among patients who received brigatinib than among those who received crizotinib (12‐month progression‐free survival, 67% versus 43%; hazard ratio, 0.49 [95%CI, 0.33‐0.74]; P < .001).2
The recommended dose of brigatinib is 90 mg orally once daily for the first 7 days of treatment, which, if tolerated, is followed by escalation to 180 mg once daily. Brigatinib single‐ and repeat‐dose systemic exposures increased dose‐proportionally following administration in patients with cancer across the dose range of 60‐240 mg once daily.3 After administration of 180 mg brigatinib once daily in patients with cancer, the mean plasma elimination half‐life was 25 hours, with a corresponding steady‐state apparent oral clearance (CL/F) of 12.7 L/h.3, 4 A study in healthy volunteers demonstrated that consumption of a high‐fat meal decreased brigatinib peak concentration (Cmax) by 13% and delayed median time to Cmax (tmax) from 2 hours to 5 hours compared with fasted‐state administration, but it had no impact on total systemic exposure.5 Therefore, brigatinib can be administered with or without food.5
Following administration of a single 180‐mg oral dose of [14C]‐brigatinib to healthy volunteers, 65% and 25% of the administered dose were recovered in feces and urine, respectively.4 Metabolic clearance of brigatinib was primarily via N‐demethylation (to N‐desmethyl brigatinib) and cysteine conjugation.4 The major circulating radioactive components were unchanged brigatinib (92%) and its primary metabolite N‐desmethyl brigatinib (3.5%), which inhibited ALK with approximately 3‐fold lower potency than brigatinib in vitro.4 Steady‐state exposure (area under the plasma concentration‐time curve [AUC]) of the primary metabolite in patients was less than 10% of brigatinib exposure.3, 4 Taken together with the 3‐fold lower potency of this minor circulating active metabolite, it can be inferred that the parent drug is the principal contributor to the overall ALK inhibitory pharmacologic effect of orally administered brigatinib.
In human liver microsomes only cytochrome P450 (CYP)‐selective inhibitors of CYP2C8 and CYP3A were shown to inhibit the formation of the primary metabolite, N‐desmethyl brigatinib, by at least 10% (data on file). Additionally, in vitro reaction phenotyping experiments using individual recombinant CYP enzymes indicated that the metabolism of brigatinib was primarily catalyzed by CYP2C8 and CYP3A4, and to a much lesser extent by CYP3A5 (data on file). At clinically relevant concentrations, brigatinib did not inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or CYP3A4/5 activity in human liver microsomes (data on file). Brigatinib, at clinically relevant concentrations, induced CYP3A expression in human hepatocytes via activation of the pregnane X receptor,4 although the clinical pharmacokinetics (PK) of brigatinib are time‐independent following repeat‐dose administration at doses of 180 mg/day, suggesting the lack of autoinduction at therapeutic doses.3 A clinical drug‐drug interaction (DDI) study between brigatinib and the CYP3A substrate midazolam is ongoing (NCT03420742).
Because brigatinib is primarily metabolized by CYP2C8 and CYP3A in vitro, this multi‐arm DDI study was conducted to evaluate the effects of a strong index inhibitor of CYP2C8 (gemfibrozil) or CYP3A (itraconazole) and a strong inducer of CYP3A (rifampin) on the single‐dose PK of brigatinib. The results of this study were intended to provide guidance with regard to concomitant medication use during brigatinib administration.
Methods
Subjects
The protocol and consent form were approved by the institutional review board of the study center (Ontario Institutional Review Board, Aurora, Ontario, Canada) before the study initiation. All subjects provided written informed consent. The study was performed at the phase 1 unit of INC Research Toronto, Inc (Toronto, Ontario, Canada) in accordance with the requirements of the Declaration of Helsinki, the International Council for Harmonisation guidelines for Good Clinical Practice, and other applicable regulatory requirements.
Eligible subjects were nonsmoking healthy men or women 18 to 65 years of age with a body mass index of 18 to 33 kg/m2 and a minimum weight of 50 kg at screening. Subjects were excluded from study participation if they had a clinically significant abnormality as assessed by physical examination, medical history, 12‐lead ECG, vital signs, or laboratory values; a history of any clinically significant illness; evidence of clinically significant hepatic or renal impairment; any condition that could potentially alter the absorption, metabolism, or excretion of the study drug; or received treatment with an investigational drug within 5 times the elimination half‐life, if known, or within 30 days, if unknown, before first administration of the study drug.
Study Design
This was a phase 1, single‐center, 3‐arm, open‐label, randomized, single‐dose, fixed‐sequence crossover study conducted in healthy subjects in accordance with US Food and Drug Administration guidelines for the assessment of drug interactions.6 The primary objective was to evaluate the effects of multiple doses of gemfibrozil, itraconazole, and rifampin on the single‐dose PK of brigatinib in healthy subjects. The secondary objective was to evaluate the safety and tolerability of a single 90‐mg or 180‐mg dose of brigatinib administered alone or after coadministration with gemfibrozil, itraconazole, or rifampin.
The study included the screening visit, 2 treatment periods separated by a washout period of at least 16 days, and a follow‐up visit 14 (±3) days after the last dose of brigatinib. Subjects were randomized to 1 of 3 study arms to evaluate the effects of coadministration of gemfibrozil (arm 1), itraconazole (arm 2), or rifampin (arm 3) on brigatinib PK.
Subjects randomized to the gemfibrozil DDI arm (Figure 1A) received a single 90‐mg oral dose of brigatinib on day 1 of treatment period 1. During treatment period 2, subjects received gemfibrozil 600 mg twice daily (BID) for 4 days (days 17‐20). On the morning of day 21, subjects received gemfibrozil 600 mg together with a single 90‐mg dose of brigatinib. Twice‐daily dosing of gemfibrozil continued through day 25. Subjects received the single doses of brigatinib following an overnight fast of at least 10 hours at approximately the same time on each dosing day followed by a standardized meal at least 2 hours postdose. On days when gemfibrozil was administered alone, subjects had fasted for at least 4 hours before dosing and were provided with a light snack/meal approximately 30 minutes postdose.
Figure 1.
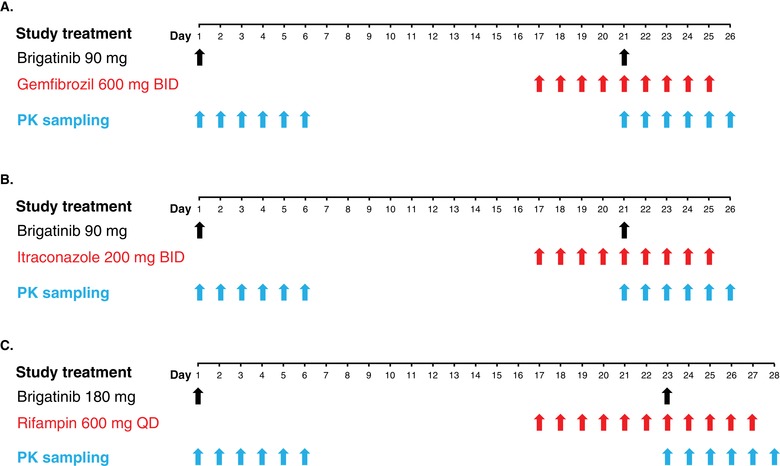
DDI study designs: study treatment and PK sampling for (A) the gemfibrozil DDI study arm (n = 20), (B) the itraconazole study arm (n = 20), and (C) the rifampin study arm (n = 20). BID indicates twice daily; DDI, drug‐drug interaction; PK, pharmacokinetics; QD, every day.
In the itraconazole DDI arm (Figure 1B), subjects received a single 90‐mg oral dose of brigatinib on day 1 in treatment period 1. During treatment period 2, subjects received itraconazole 200 mg BID for 4 days (days 17‐20). On the morning of day 21, subjects received itraconazole 200 mg together with a single 90‐mg dose of brigatinib. Twice‐daily dosing of itraconazole continued through day 25. Subjects received the single doses of brigatinib following an overnight fast of at least 10 hours at approximately the same time on each dosing day followed by a standardized meal at least 2 hours postdose. On days when itraconazole was administered alone, subjects had fasted for at least 4 hours before dosing and were provided with a light snack/meal approximately 30 minutes postdose.
In the rifampin DDI arm (Figure 1C), subjects received a single 180‐mg oral dose of brigatinib on day 1 of treatment period 1. During treatment period 2, subjects received rifampin 600 mg once daily (QD) for 6 days (days 17‐22), and on the morning of day 23, they received rifampin 600 mg together with a single 180‐mg dose of brigatinib; QD dosing of rifampin continued through day 27. Subjects received the single doses of brigatinib following an overnight fast of at least 10 hours at approximately the same time on each dosing day followed by a standardized meal at least 2 hours postdose. On days when rifampin was administered alone, subjects had fasted for at least 4 hours before dosing and were provided with a light snack/meal approximately 1 hour postdose.
For all 3 study arms, in both treatment periods, water was provided ad libitum except for 1 hour before and 1 hour after brigatinib administration.
Assessments
For both treatment periods in each study arm, venous blood samples were collected predose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 36, 48, 60, 72, 96, and 120 hours postdose to measure plasma concentrations of brigatinib. Brigatinib was extracted from K2 EDTA (dipotassium ethylenediaminetetraacetic acid)‐containing plasma samples by protein precipitation. Chromatographic separation was accomplished using ACE C18 50 × 2.1 mm, 3‐µm columns (Advanced Chromatography Technologies, Aberdeen, Scotland). Mobile phase A consisted of 2 mmol/L ammonium acetate in water, and mobile phase B consisted of 2 mmol/L ammonium acetate in methanol. A gradient method was employed, starting at 5% mobile phase B and increasing to 98% over 2.5 minutes at a flow rate of 0.6 mL/min. Compounds were then ionized using positive ion atmospheric pressure chemical ionization with an API‐5500 mass spectrometer (Sciex, Redwood City, California) in multiple reaction–monitoring scan mode and m/z mass range parameters (parent/product) of 584.3/418.0 for brigatinib and 588.3/484.2 for the internal standard AP29109 (D4‐brigatinib).
Two validated methods were used to analyze brigatinib. The first, a single‐range method developed to allow analysis of plasma samples with concentrations exceeding 500 ng/mL, had a lower limit of quantitation of 25 ng/mL (calibration range, 25‐2500 ng/mL using a 50‐µL plasma sample). Any sample with a brigatinib concentration lower than 25 ng/mL was reanalyzed using the second method, which was a dual‐range assay with a lower limit of quantitation of 0.100 ng/mL and an upper limit of 500 ng/mL using a 100‐µL plasma sample. In the single‐range method, intra‐run precision for brigatinib in human plasma samples ranged from 1.0% to 12.9% coefficient of variation (CV), with a bias of –11.6% to 5.6%, and inter‐run precision ranged from 1.7% to 10.1% CV, with a bias of –2.5% to 1.1%. For the dual‐range method, intra‐run precision for brigatinib in human plasma samples for the high‐range and low‐range assays ranged from 3.3% to 10.0% and 1.1% to 12.7% CV, respectively, with a bias of –15.8% to 0.3% and –8.7% to –2.3% CV. Inter‐run precision ranged from 4.8% to 8.8% and 3.1% to 8.4% CV, with a bias of –6.3% to –1.3% and –7.3% to –4.2% CV.
PK parameters of brigatinib for the primary end‐point analysis included Cmax, AUC from time 0 to 120 hours (AUC0‐120), AUC from time 0 to infinity (AUC0‐inf), CL/F, tmax, and elimination half‐life (t½).
Safety evaluations included adverse events, vital signs, clinical laboratory assessments (hematology, chemistry, urinalysis), physical examinations, and 12‐lead ECG. Adverse events were evaluated throughout the study and up to 14 days after the last dose of brigatinib.
PK and Statistical Analyses
All patients who received at least 1 dose of brigatinib were included in the safety population. The PK‐evaluable population was defined as all subjects who complied with the protocol and had an evaluable PK profile (ie, exposure to treatment, no emesis within 4 hours of dosing, availability of sufficient plasma drug concentration‐time data to permit the calculation of PK parameters, and absence of major protocol violations). The plasma PK parameters for brigatinib were calculated using noncompartmental methods with Phoenix WinNonlin version 6.4 (Certara, Princeton, New Jersey). All PK and safety parameters were summarized using descriptive statistics. Brigatinib concentrations that were below the lower limit of quantification were imputed as 0.
Statistical analyses were performed using SAS (version 9.3 or higher; SAS Institute, Cary, North Carolina). For the estimation of the effects of gemfibrozil, itraconazole, or rifampin on the PK of brigatinib, PK parameters (natural log–transformed [ln]) of brigatinib, except tmax, were compared between the 2 treatment conditions (brigatinib coadministered with a strong CYP inhibitor/inducer versus brigatinib administered alone) using an analysis of variance model that included treatment as a fixed effect. Each analysis of variance calculated the least‐squares mean (LSM), the difference between treatment LSM values, and the standard error associated with the difference. Ratios of LSM were calculated using the exponential of the difference between treatment LSM values from the analyses on the ln‐transformed Cmax, AUC0‐inf, AUC0‐120, and CL/F values. The tmax values were not ln‐transformed and were analyzed using nonparametric analysis (Walsh averages and the appropriate quartile of the Wilcoxon signed rank test statistic).
Results
Subjects
A total of 60 subjects were randomized (20 per study arm) and received at least 1 dose of brigatinib (safety and PK populations). Demographic and baseline characteristics of the randomized populations are presented in Table 1. One subject each in the gemfibrozil and rifampin DDI study arms discontinued in treatment period 2. A total of 58 subjects completed the study (gemfibrozil DDI arm, n = 19; itraconazole DDI arm, n = 20; rifampin DDI arm, n = 19).
Table 1.
Demographics and Baseline Characteristics in Each DDI Study Arm
| Characteristic | Gemfibrozil DDI Arm (n = 20) | Itraconazole DDI Arm (n = 20) | Rifampin DDI Arm (n = 20) |
|---|---|---|---|
| Age,a y | |||
| Mean (SD) | 44 (11) | 45 (13) | 43 (12) |
| Range | 23‐65 | 23‐60 | 24‐62 |
| Sex, n (%) | |||
| Male | 12 (60) | 14 (70) | 11 (55) |
| Female | 8 (40) | 6 (30) | 9 (45) |
| Race, n (%) | |||
| White | 14 (70) | 15 (75) | 8 (40) |
| Black/African American | 4 (20) | 5 (25) | 9 (45) |
| Asian | 1 (5) | 0 | 2 (10) |
| Other | 1 (5) | 0 | 1 (5) |
| Ethnicity, n (%) | |||
| Non‐Hispanic/non‐Latino | 16 (80) | 19 (95) | 18 (90) |
| Hispanic or Latino | 4 (20) | 1 (5) | 2 (10) |
| Weight, kg | |||
| Mean (SD) | 75 (13) | 78 (13) | 78 (14) |
| Range | 50‐102 | 52‐105 | 51‐104 |
| Height, cm | |||
| Mean (SD) | 170.5 (9.0) | 173.6 (9.8) | 170.5 (11.7) |
| Range | 150.3‐185.0 | 157.7‐193.6 | 149.2‐188.1 |
| BMI, kg/m2 | |||
| Mean (SD) | 25.7 (2.9) | 25.9 (2.4) | 26.7 (3.3) |
| Range | 19.2‐29.9 | 21.0‐29.6 | 19.1‐30.4 |
BMI indicates body mass index; DDI, drug‐drug interaction.
Age at the time of informed consent.
Pharmacokinetics
Effect of the Strong CYP2C8 Inhibitor Gemfibrozil on the PK of Brigatinib
Mean brigatinib plasma concentration‐time profiles with and without coadministration of gemfibrozil are presented in Figure 2A. When brigatinib was coadministered with gemfibrozil in period 2, the geometric mean AUC0‐inf of brigatinib was reduced by 12% compared with administration of brigatinib alone (geometric LSM ratio [90%CI], 0.88 [0.83‐0.94]) (Table 2). Mean brigatinib plasma concentrations in the periabsorptive phase were lower when it was coadministered with gemfibrozil than when brigatinib was administered alone, resulting in an approximately 40% reduction in peak concentrations (geometric LSM ratio [90%CI] for Cmax of 0.59 [0.54‐0.65]). The median tmax of brigatinib was not affected by coadministration of gemfibrozil (2.0 hours for both treatment periods). The geometric mean t½ of brigatinib was 26.5 hours when it was administered alone and 33.9 hours when coadministered with gemfibrozil. Although brigatinib peak concentrations were reduced during gemfibrozil coadministration, the overall effect of gemfibrozil was inferred not to be clinically relevant, as total systemic exposure of brigatinib was not meaningfully altered.
Figure 2.
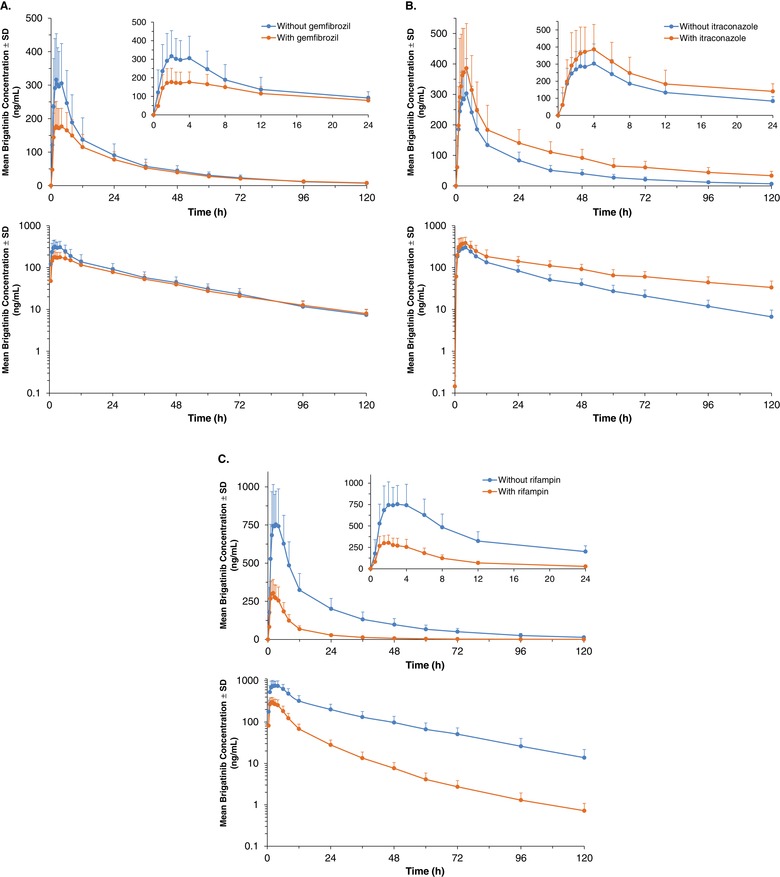
Mean (±SD) plasma brigatinib concentration‐time profiles (linear and log‐linear plots) with and without coadministration of (A) gemfibrozil, (B) itraconazole, and (C) rifampin (PK‐evaluable population). Insets show the PK profiles for the first 24 hours after dosing. PK indicates pharmacokinetics.
Table 2.
Plasma PK Parameters of Brigatinib With (Test Condition) and Without (Reference Condition) Coadministration of Gemfibrozil, Itraconazole, and Rifampin
| Study Arm/Parameter | Test Condition | Reference Condition | Geometric LS Mean Ratio (90%CI) (Test vs Reference) | |
|---|---|---|---|---|
| Gemfibrozil DDI arm | ||||
| n = 19 | n = 20 | 0.59 (0.54‐0.65) | ||
| Cmax, ng/mL | Geometric mean (% CV) | 198.9 (35.8) | 347.8 (40.5) | |
| Mean (SD) | 209.7 (66.1) | 373.2 (140.9) | ||
| n = 19 | n = 20 | 0.85 (0.80‐0.91) | ||
| AUC0‐120, h·ng/mL | Geometric mean (% CV) | 5340 (27.5) | 6488 (34.2) | |
| Mean (SD) | 5520 (1432) | 6839 (2318) | ||
| n = 19 | n = 19 | 0.88 (0.83‐0.94) | ||
| AUC0‐inf, h·ng/mL | Geometric mean (% CV) | 5742 (25.7) | 6875 (33.9) | |
| Mean (SD) | 5913 (1451) | 7233 (2371) | ||
| n = 19 | n = 19 | 1.13 (1.06‐1.20) | ||
| CL/F, L/h | Geometric mean (% CV) | 15.7 (25.7) | 13.1 (33.9) | |
| Mean (SD) | 16.2 (4.3) | 13.8 (4.6) | ||
| n = 19 | n = 20 | Test ‐ Reference | ||
| tmax, h | Median (range) | 2.0 (1.0‐6.0) | 2.0 (1.0‐4.0) | 0.3 (–3.0 to 4.0) |
| n = 19 | n = 19 | |||
| t½, h | Mean (SD) | 34.3 (6.1) | 26.7 (4.0) | |
| Itraconazole DDI arm | ||||
| n = 20 | n = 20 | 1.21 (1.13‐1.30) | ||
| Cmax, ng/mL | Geometric mean (% CV) | 401.4 (38.4) | 331.3 (31.9) | |
| Mean (SD) | 428.7 (163.1) | 347.2 (111.9) | ||
| n = 20 | n = 20 | 1.82 (1.72‐1.93) | ||
| AUC0‐120, h·ng/mL | Geometric mean (% CV) | 11 178 (31.2) | 6139 (28.5) | |
| Mean (SD) | 11 690 (3697) | 6382 (1918) | ||
| n = 11 | n = 20 | 2.01 (1.84‐2.20) | ||
| AUC0‐inf, h·ng/mL | Geometric mean (% CV) | 13 501 (35.6) | 6452 (28.7) | |
| Mean (SD) | 14 235 (4822) | 6709 (2006) | ||
| n = 11 | n = 20 | 0.50 (0.45‐0.54) | ||
| CL/F, L/h | Geometric mean (% CV) | 6.67 (35.6) | 14.0 (28.7) | |
| Mean (SD) | 7.05 (2.54) | 14.46 (3.84) | ||
| n = 20 | n = 20 | Test ‐ Reference | ||
| tmax, h | Median (range) | 2.6 (1.5‐6.0) | 2.8 (1.5‐4.0) | 0.0 (–2.5 to 2.0) |
| n = 11 | n = 20 | |||
| t½, h | Mean (SD) | 44.9 (8.4) | 30.5 (6.8) | |
| Rifampin DDI arm | ||||
| n = 19 | n = 20 | 0.40 (0.37‐0.44) | ||
| Cmax, ng/mL | Geometric mean (% CV) | 333.9 (29.2) | 825.9 (31.2) | |
| Mean (SD) | 346.6 (94.9) | 863.3 (265.2) | ||
| n = 19 | n = 20 | 0.20 (0.19‐0.22) | ||
| AUC0‐120, h·ng/mL | Geometric mean (% CV) | 3019 (25.7) | 15 143 (33.8) | |
| Mean (SD) | 3111 (783) | 15 907 (4948) | ||
| n = 19 | n = 20 | 0.20 (0.18‐0.21) | ||
| AUC0‐inf, h·ng/mL | Geometric mean (% CV) | 3042 (25.8) | 15 616 (34.4) | |
| Mean (SD) | 3136 (793) | 16 429 (5195) | ||
| n = 19 | n = 20 | 5.11 (4.73‐5.51) | ||
| CL/F, L/h | Geometric mean (% CV) | 59.2 (25.8) | 11.5 (34.4) | |
| Mean (SD) | 61.0 (15.4) | 12.2 (4.3) | ||
| n = 19 | n = 20 | Test – Reference | ||
| tmax, h | Median (range) | 2.0 (1.0‐4.0) | 2.5 (1.5‐6.0) | –0.5 (–2.1 to 1.0) |
| n = 19 | n = 20 | |||
| t½, h | Mean (SD) | 23.7 (3.2) | 25.1 (4.1) | |
AUC0‐120 indicates area under the plasma concentration‐time curve from time 0 to 120 hours; AUC0‐inf, area under the plasma concentration‐time curve from time 0 to infinity; CL/F, apparent oral clearance; Cmax, peak plasma concentration; CV, coefficient of variation; DDI, drug‐drug interaction; LS, least squares; PK, pharmacokinetics; t½, elimination half‐life; tmax, time to peak plasma concentration.
Test = brigatinib + CYP inhibitor/inducer; Reference = brigatinib alone.
Effect of the Strong CYP3A Inhibitor Itraconazole on the PK of Brigatinib
Mean brigatinib plasma concentration‐time profiles with and without coadministration of itraconazole are presented in Figure 2B. Although the geometric mean Cmax of brigatinib increased minimally (by 21%) with coadministration of itraconazole (Table 2), there was a nearly 50% prolongation in the geometric mean t½ of brigatinib (30 hours when administered alone and 44 hours when coadministered with itraconazole). This resulted in the percentage AUC extrapolated being greater than 20% in some subjects; hence, both AUC0‐120 and AUC0‐inf are reported. Mean brigatinib plasma concentrations increased when brigatinib was coadministered with itraconazole. In the presence of itraconazole, geometric mean AUC0‐120 and AUC0‐inf for brigatinib increased by 82% (geometric LSM ratio [90%CI], 1.82 [1.72‐1.93]) and 101% (geometric LSM ratio [90%CI], 2.01 [1.84‐2.20]), respectively (Table 2).
Effect of the Strong CYP3A Inducer Rifampin on the PK of Brigatinib
Mean brigatinib plasma concentration‐time profiles with and without coadministration of rifampin are presented in Figure 2C. During coadministration with the strong metabolic inducer rifampin, brigatinib plasma concentrations were substantially reduced. In the presence of rifampin the geometric mean AUC0‐inf of brigatinib was reduced by 80% (geometric LSM ratio [90%CI], 0.20 [0.18‐0.21]), reflecting an approximately 5‐fold increase in CL/F, and the geometric mean Cmax of brigatinib decreased by 60% (geometric LSM ratio [90%CI], 0.40 [0.37‐0.44]; Table 2). The geometric mean t½ for brigatinib was similar in the presence (23.4 hours) and absence (24.8 hours) of rifampin.
Safety
Table 3 shows the overall incidence of treatment‐emergent adverse events (TEAEs) by DDI study arm. Most TEAEs were mild in intensity, with no severe TEAEs reported. The most common TEAEs (≥10%) were cough (20%) and throat irritation (10%) when brigatinib was coadministered with gemfibrozil and cough (10%) when brigatinib was coadministered with rifampin. No TEAEs occurred in ≥10% of subjects during brigatinib coadministration with itraconazole. The only treatment‐related TEAE reported in more than 1 patient was cough (brigatinib + gemfibrozil, n = 4; brigatinib + rifampin, n = 2; brigatinib + itraconazole, n = 0). No deaths or serious TEAEs were reported. Two subjects discontinued treatment in treatment period 2 due to adverse events (gemfibrozil arm, n = 1, emesis; rifampin arm, n = 1, elevated aspartate aminotransferase and alanine aminotransferase levels).
Table 3.
Summary of Treatment‐Emergent Adverse Events Within Each of the DDI Study Arms
| Gemfibrozil | Itraconazole | Rifampin | |
|---|---|---|---|
| DDI Arm | DDI Arm | DDI Arm | |
| TEAE | (n = 20) | (n = 20) | (n = 20) |
| Any TEAE | 9 (45) | 2 (10) | 7 (37) |
| Any severe TEAE | 0 | 0 | 0 |
| Any serious TEAE | 0 | 0 | 0 |
| AE leading to discontinuation | 1 (5) | 0 | 1 (5) |
| On‐study deaths | 0 | 0 | 0 |
AE indicates adverse event; DDI, drug‐drug interaction; TEAE, treatment‐emergent adverse event.
Data are n (%).
There were no apparent treatment‐related changes in mean laboratory parameters. One subject had clinically significant urinalysis results associated with a mild TEAE of urinary tract infection 1 day after receiving brigatinib and gemfibrozil, and 1 subject discontinued the study due to a mild increase in hepatic enzymes following pretreatment with rifampin. There were no clinically significant abnormalities in vital signs, ECGs, or physical examination findings during the study.
Discussion
In vitro reaction phenotyping studies have shown that brigatinib is primarily metabolized by CYP2C8 and CYP3A4. Accordingly, this 3‐arm study examined the effects of gemfibrozil (a strong CYP2C8 inhibitor), itraconazole (a strong CYP3A inhibitor), and rifampin (a strong CYP3A inducer) on the PK of brigatinib in healthy subjects.
Coadministration of gemfibrozil 600 mg BID with a single 90‐mg dose of brigatinib did not produce a clinically relevant effect on the total systemic exposure (AUC0‐inf) of brigatinib. However, an approximately 40% reduction in peak concentrations was observed. The effect of gemfibrozil on the PK of brigatinib was unexpected, and the underlying mechanism for decreased Cmax in the presence of gemfibrozil is unknown. A similar finding was reported for imatinib, where coadministration with gemfibrozil reduced the geometric mean Cmax and AUC0–12 of imatinib by 35% and 23% (P <.001), respectively; the authors concluded this was likely due to inhibition of a transporter such as OATP1A2 or OATP2B1 that may be important for imatinib uptake.7 The lack of a meaningful effect of the strong CYP2C8 inhibitor gemfibrozil on the total systemic exposure of brigatinib indicates that CYP2C8‐mediated metabolism does not meaningfully contribute to the clearance of brigatinib in vivo. Therefore, brigatinib can be coadministered with strong CYP2C8 inhibitors without the need for dose modifications.
When a single 90‐mg dose of brigatinib was coadministered with multiple 200‐mg BID doses of itraconazole, brigatinib exposure was increased, as indicated by an approximate doubling of total systemic exposure characterized by a 101% increase in geometric mean AUC0‐inf. Strong CYP3A inhibition by itraconazole resulted in a 21% increase in geometric mean Cmax, but in a nearly 50% prolongation of half‐life. These observations suggest that the mechanism of the itraconazole‐brigatinib interaction mainly involves reduction of systemic (hepatic) clearance with only a minimal impact on presystemic extraction and oral bioavailability.8
Brigatinib is administered in clinical practice at a daily dose of 90 mg for 1 week followed by escalation, if tolerated, to a dose of 180 mg per day. This dosing regimen was approved based on pivotal evidence of a favorable benefit/risk profile in a phase 2 trial in crizotinib‐refractory ALK+ NSCLC.1 A maximum tolerated dose was not formally established in the first‐in‐human phase 1 dose‐escalation study of brigatinib in patients with cancer, which evaluated doses ranging from 30 to 300 mg per day.3 However, 240 mg per day is considered to be at the upper end of the acceptable tolerated dose range.3 Therefore, it can be inferred that a doubling of systemic exposure of brigatinib in the setting of daily repeat‐dose administration with a strong CYP3A inhibitor is likely to result in steady‐state brigatinib exposures that would exceed the established acceptable tolerated range. Thus, concomitant use of strong CYP3A inhibitors (eg, boceprevir, cobicistat, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, clarithromycin, itraconazole, ketoconazole, posaconazole, voriconazole, and conivaptan) with brigatinib is not recommended. If concomitant use of a strong CYP3A inhibitor is unavoidable, the results of this study support a dose reduction of brigatinib by approximately 50% considering the available tablet strengths (ie, from 180 mg to 90 mg, or from 90 mg to 60 mg). After discontinuation of the strong CYP3A inhibitor, brigatinib treatment may be resumed at the dose that was tolerated prior to initiation of treatment with the strong CYP3A inhibitor.
As expected, coadministration of multiple doses of rifampin 600 mg QD with a single 180‐mg brigatinib dose substantially reduced brigatinib exposure, with an 80% reduction observed in brigatinib geometric mean AUC0‐inf. In clinical practice, successive dose reductions to 120 mg, 90 mg, and 60 mg brigatinib are recommended for patients unable to tolerate daily doses of 180 mg. As a result, 60 mg per day represents the lower end of the therapeutic dose range. The 80% reduction of brigatinib systemic exposure by rifampin, a representative strong CYP3A inducer, may translate to decreased efficacy, as it would result in systemic exposures well below that observed on average in patients treated with 60‐mg daily doses of brigatinib. Based on these considerations, the concomitant use of strong CYP3A inducers (eg, rifampin, carbamazepine, and phenytoin) should be avoided in patients treated with brigatinib.
Single oral doses of brigatinib 90 mg (in the gemfibrozil and itraconazole arms) and 180 mg (in the rifampin arm), administered in this study alone or concomitantly with gemfibrozil, itraconazole, or rifampin, were generally well tolerated in healthy subjects. Most TEAEs were of mild intensity, and only 2 subjects discontinued treatment due to TEAEs (1 subject during coadministration of brigatinib and gemfibrozil due to emesis and 1 subject during coadministration of brigatinib and rifampin due to elevated liver enzymes).
Conclusions
The results of these DDI studies indicate that coadministration of a strong CYP2C8 inhibitor has no clinically meaningful effect on the PK of brigatinib. Strong CYP2C8 inhibitors can therefore be administered without any brigatinib dose adjustments. Strong CYP3A inhibitors should be avoided, as systemic exposure of brigatinib was increased 2‐fold by itraconazole. If concomitant use of brigatinib and a strong CYP3A inhibitor is unavoidable, the dose of brigatinib should be reduced by approximately 50%. Once the strong CYP3A inhibitor is discontinued, brigatinib treatment should be resumed at the dose tolerated before the initiation of treatment with the strong CYP3A inhibitor. Concomitant administration of brigatinib with strong CYP3A inducers should be avoided, as brigatinib systemic exposure was reduced by 80% during coadministration with rifampin. These DDI study findings are reflected in the prescribing information for brigatinib.
Acknowledgments
This study was funded by ARIAD Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. The authors wish to thank the Principal Investigator of the study, Luis Robles, MD, of INC Research Toronto, Inc, Toronto, Ontario, Canada. Professional medical writing assistance was provided by Lauren Gallagher, RPh, PhD, and Lela Creutz, PhD, of Peloton Advantage, LLC, an OPEN Health company, Parsippany, New Jersey, and funded by ARIAD Pharmaceuticals, Inc.
Author Disclosures
Meera Tugnait: employment, stock (ARIAD). Neeraj Gupta: employment (Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited). Michael J. Hanley: employment (Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited). Daryl Sonnichsen: consultant (ARIAD). David Kerstein: employment, stock, and other ownership interests (ARIAD). David J. Dorer: employment, stock, and other ownership interests (ARIAD). Karthik Venkatakrishnan: employment (Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited). Narayana Narasimhan: employment, stock, and other ownership interests (ARIAD).
Data Sharing Statement
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met, as set forth in Takeda's Data Sharing Policy (see http://www.TakedaClinicalTrials.com/Approach for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.
Current affiliations: Tugnait, Blueprint Medicines, Cambridge, MA, USA; Kerstein, Anchiano Therapeutics, Cambridge, MA, USA; Dorer, Dorer Statistical Consulting Company, Brookline, MA, USA; Narasimhan, Aileron Therapeutics, Cambridge, MA, USA
References
- 1. Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib‐refractory anaplastic lymphoma kinase‐positive non‐small‐cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490‐2498. [DOI] [PubMed] [Google Scholar]
- 2. Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus crizotinib in ALK‐positive non–small‐cell lung cancer. N Engl J Med. 2018;379(21):2027‐2039. [DOI] [PubMed] [Google Scholar]
- 3. Gettinger SN, Bazhenova LA, Langer CJ, et al. Activity and safety of brigatinib in ALK‐rearranged non‐small‐cell lung cancer and other malignancies: a single‐arm, open‐label, phase 1/2 trial. Lancet Oncol. 2016;17(12):1683‐1696. [DOI] [PubMed] [Google Scholar]
- 4. Hirota T, Muraki S, Ieiri I. Clinical pharmacokinetics of anaplastic lymphoma kinase inhibitors in non‐small‐cell lung cancer. Clin Pharmacokinet. 2019;58(4):403‐420. [DOI] [PubMed] [Google Scholar]
- 5. Tugnait M, Gupta N, Hanley MJ, et al. The effect of a high‐fat meal on the pharmacokinetics of brigatinib, an oral anaplastic lymphoma kinase inhibitor, in healthy volunteers. Clin Pharmacol Drug Dev. 2019;8(6):734‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guidance for industry. Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. Rockville, MD: US Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research; 2012. [Google Scholar]
- 7. Filppula AM, Tornio A, Niemi M, Neuvonen PJ, Backman JT. Gemfibrozil impairs imatinib absorption and inhibits the CYP2C8‐mediated formation of its main metabolite. Clin Pharmacol Ther. 2013;94(3):383‐393. [DOI] [PubMed] [Google Scholar]
- 8. Wilkinson GR, Shand DG. Commentary: a physiological approach to hepatic drug clearance. Clin Pharmacol Ther. 1975;18(4):377‐390. [DOI] [PubMed] [Google Scholar]