

Abstract
Macrophages (Mϕ) have been reported to downmodulate the cytotoxicity of natural killer (NK) cell against solid tumor cells. However, the collaborative role between NK cells and Mϕ remains underappreciated, especially in hematological cancers, such as chronic myeloid leukemia (CML). We observed a higher ratio of innate immune cells (Mϕ and NK) to adaptive immune cells (T and B cells) in CML bone marrow aspirates, prompting us to investigate the roles of NK and Mϕ in CML. Using coculture models simulating the tumor inflammatory environment, we observed that Mϕ protects CML from NK attack only when CML was itself mycoplasma‐infected and under chronic infection–inflammation condition. We found that the Mϕ‐protective effect on CML was associated with the maintenance of CD16 level on the NK cell membrane. Although the NK membrane CD16 (mCD16) was actively shed in Mϕ + NK + CML trioculture, the NK mCD16 level was maintained, and this was independent of the modulation of sheddase by tissue inhibitor of metalloproteinase 1 or inhibitory cytokine transforming growth factor beta. Instead, we found that this process of NK mCD16 maintenance was conferred by Mϕ in a contact‐dependent manner. We propose a new perspective on anti‐CML strategy through abrogating Mϕ‐mediated retention of NK surface CD16.
Keywords: Chronic infection, inflammation, chronic myeloid leukemia, macrophages, maintenance of NK mCD16, natural killer cells, tumor environment
The collaborative role between natural killer (NK) cells and macrophages (Mϕ) remains unappreciated in hematological tumors. Here, we found that mycoplasma‐infected chronic myeloid leukemia (CML) cells were protected by Mϕ from NK cytotoxicity and this protective effect was associated with Mϕ‐mediated maintenance of NK membrane CD16 level in a contact‐dependent manner. Further studies would shed light into potential therapeutics for CML via modulation of interactions between innate immune cells, NK and Mϕ.
Introduction
Natural killer (NK) cell spontaneously recognizes and eliminates cancer or pathogen‐infected cells without prior sensitization.1 This is attributed to the expression of germline‐encoded activating and inhibitory receptors through which the NK cell receives signals to make a cellular decision on launching cytotoxicity.2 CD16 is a well‐recognized NK activating receptor that mediates antibody‐dependent cellular cytotoxicity against target cells coated (or opsonized) with specific antibodies.3 In addition, CD16 is involved in nonantibody‐dependent cellular cytotoxicity‐dependent NK functions through (a) direct lysis of target cells in the absence of antibody coating,4 (b) boosting serial engagement of target cells through CD16 shedding,5 and (c) coupling of NK CD3ζ adaptor molecule to CD2 activation mediator upon CD16 cleavage.6 As the nonantibody‐dependent cellular cytotoxicity function of NK CD16 is less explored and could be potentially important, we were prompted to further investigate such a function of NK CD16.
Upon activation, NK cell secretes cytotoxic molecules, such as perforin and granzymes, to induce target cell death.7 This innate capability of NK cell makes it a key player in cancer immunosurveillance. Indeed, individuals with low NK cell counts have increased risk of developing leukemia and other cancers.8 It is therefore unclear how cancer cells thrive despite the presence of such effective killer cells.9, 10
The most abundant immune cell population present in the tumor environment is the macrophage (Mϕ).11 Depending on environmental cues, Mϕ can be polarized into classically activated proinflammatory M1 or the alternatively activated anti‐inflammatory M2 phenotypes.12 In the tumor environment, Mϕ more closely resembles the “healing” M2 type, which is protumorigenic and immunosuppressive.13 In solid tumor environments, it was proposed that Mϕ renders NK cell dysfunctional14 through soluble factors such as transforming growth factor beta (TGFβ) and prostaglandin E2, and through ligand–receptor interactions via HLAG‐ILT2 and CD48‐2B4.15, 16, 17, 18 However, the collaborative role of NK cell and Mϕ in hematological cancers remains unclear. Here, we query how Mϕ would modulate NK activity in chronic myeloid leukemia (CML), a hematological cancer arising from the bone marrow, characterized by abnormal proliferation of granulocytes (neutrophils, basophils, eosinophils) and myeloid progenitors as a result of constitutive activation of BCR‐Abl tyrosine kinase.19
Inflammation is identified as one of the key hallmarks of cancer,20 characterized by the presence of inflammatory cytokines such as interleukin‐6 (IL‐6), IL‐8 and tumor necrosis factor‐alpha (TNFα) in the cancer environment.21 This could be a result of chronic infection condition. Notably, mycoplasma is detected in 50% of myeloid leukemia patients,22 and other patients with solid cancers.23, 24, 25 In cell cultures, the presence of mycoplasma creates a low‐grade chronic inflammatory condition without compromising cell viability, ideal for promoting cancer transformation.26 Moreover, there are several reports supporting mycoplasma‐induced production of inflammatory cytokines, such as IL‐6, IL‐8 and TNFα.27, 28 How these infection–inflammation conditions may promote clinical cancer progression is hitherto unexplored.
In this study, we sought to understand whether Mϕ could alter NK activation in CML, particularly through the modulation of NK CD16 level, in the presence or absence of mycoplasma‐induced inflammatory conditions. To investigate the specific interactions between CML, NK and Mϕ, we performed ex vivo coculture of primary NK cell and Mϕ (derived from healthy blood donors) with CML cell lines. By systematically delineating findings under mycoplasma negative (myco−) and mycoplasma positive (myco+) conditions, we further defined specific contributions from mycoplasma‐induced inflammation.
Results
CML cells showed inflammation induced by acute and chronic mycoplasma infection
The tumor environment of CML patients is characterized by inflammation, and mycoplasma is also detected in bone marrow samples of myeloid leukemia patients.22, 29 Hence, to model inflammation condition in CML, we infected CML cell lines with mycoplasma, using two strategies: (1) short‐term (acute) mycoplasma‐infected CML cells (referred to as myco tx) that were experimentally infected with mycoplasma through addition of mycoplasma‐containing culture medium for up to 7 days, and (2) long‐term (chronic) mycoplasma‐infected CML cells (referred to as myco+ and annotated L for long‐term), which were cells carrying latent infection with mycoplasma for many passages. Noninfected cultures were annotated as myco−. We determined that CML cells acutely and chronically infected with mycoplasma were mycoplasma positive (Figure 1a). In the figure, the nonspecific band detected in infected CML cell lines, but absent in noninfected controls, could be attributed to nonspecific amplification of a conserved portion of the mycoplasma genome, either from the primer sets that were used or from priming by the mycoplasma PCR products.
Figure 1.
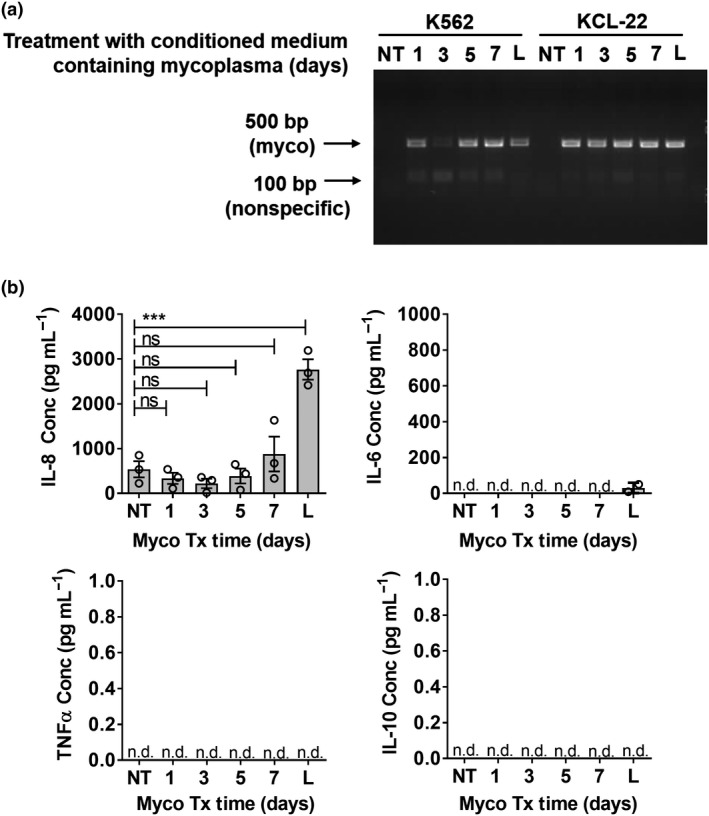
Increased production of interleukin‐8 (IL‐8) by chronic myeloid leukemia (CML) with chronic and acute infection of mycoplasma. Noninfected K562 cells were treated with mycoplasma‐containing culture supernatant for 1, 3, 5 and 7 days (myco tx). These acutely infected cultures were compared with noninfected (NT) and chronically infected CML cultures (L). (a) Cell culture supernatants were tested for presence of mycoplasma via PCR. DNA bands were visualized via UV transillumination (Bio‐Rad imager and Syngene Genesnap software) of SYBR safe‐stained agarose gel. (b) Mycoplasma‐infected K562 cells were seeded at 1 million cells mL–1 and incubated overnight. Culture supernatants were tested for presence of IL‐8, IL‐6, tumor necrosis factor‐α (TNF‐α) and IL‐10 using ELISA. Results shown are mean ± s.e.m. of three independent experiments (donors). See Supplementary figure 1 for individual replicate experiments. Statistical significance was determined using repeated measures one‐way ANOVA followed by Tukey's test. ***P < 0.001. L, CML cells that were long‐term mycoplasma infected because of tissue culture procedures; n.d., nondetectable; ns, nonsignificant; NT, nontreated CML cells that were mycoplasma free.
To determine the inflammation status, we tested for inflammatory cytokines (IL‐8/IL‐6/TNFα/IL‐10) produced into the culture supernatant of myco− (NT), myco tx (days 1, 3, 5, 7) and myco+ CML (annotated L for long term) cells. Of the four cytokines tested, only IL‐8 was produced at detectable levels, with significantly increased production by CML cells which were chronically infected with mycoplasma (Figure 1b and Supplementary figure 1). IL‐6/TNFα/IL‐10 were nondetectable (n.d.), except for trace level of IL‐6 in chronically infected culture. The species of mycoplasma infecting and resulting in the increased IL‐8 production were determined to be Mycoplasma fermentans and Mycoplasma hyorhinis (Supplementary figure 2).
Taken together, mycoplasma infection of K562 CML cells induced high production of IL‐8. This was consistent with the reported upregulation of IL‐8 in the serum of CML patients.29, 30 Hence, to simulate the inflammatory condition in the CML environment, we employed the strategy of using chronically infected (myco+) CML cells compared with noninfected counterparts (myco−) in subsequent coculture experiments with primary Mϕ and NK cells.
Mϕ protected mycoplasma‐infected CML from NK cytotoxicity
To determine the influence of Mϕ and NK in CML survival, we first queried the change in proportion of innate Mϕ and NK cells compared with adaptive T and B cells in bone marrow aspirates of CML patients and nonleukemia orthopedic patient controls (Supplementary figure 3). We observed a low ratio of [Mϕ‐NK]:[T‐B] cells in nonleukemia controls of about 0.1, that is, the proportion of adaptive T and B cells were 10‐fold more than innate Mϕ and NK cells. However, CML patients in the more severe “accelerated and blast crisis phase” showed significant (P = 0.033) increase in the ratio of [Mϕ‐NK]:[T‐B] cells. This suggested a shift in the immunological profile in patients with severe CML toward antigen‐independent innate immunity, prompting us to further determine the functional roles of Mϕ and NK in CML survival.
We performed ex vivo mono, duo and triocultures using primary Mϕ and NK cells from the peripheral blood of healthy donors, and myco−/myco+ K562 CML cells (Figure 2a). All treatments were normalized to cancer alone (C) because of the background viability of C sample. In the duoculture of NK + CML (NC), we observed a decrease in CML survival under both myco− and myco+ conditions, demonstrating NK‐mediated killing of CML (Figure 2b, c and Supplementary figure 4). In the absence of mycoplasma infection, a comparison between Mϕ + NK + CML (MNC) and Mϕ + NK (NC) showed no further modulation of myco− CML survival mediated by Mϕ. Interestingly, with mycoplasma‐infected CML, the NK‐mediated killing of myco+ CML was attenuated in the presence of Mϕ (Figure 2b, c and Supplementary figure 4), suggesting that Mϕ protected myco+ CML from NK killing. We also observed that duoculture of Mϕ + CML (MC) did not reduce CML survival, indicating that the killing activity against CML was mediated by NK, and not Mϕ. We further cocultured NK and Mϕ with another CML cell line, KCL‐22 (Supplementary figure 5). When KCL‐22 was myco−, there was further decrease in KCL‐22 survival in MNC trio cultures. However, when KCL‐22 was myco+, the Mϕ appeared to protect KCL‐22 from NK killing, consistent with that of K562. Taken together, our findings suggest that there is no observable Mϕ protection of myco− CML cells from NK cytotoxicity. Mycoplasma infection of the CML cells prompted Mϕ‐mediated protection from NK cytotoxicity.
Figure 2.
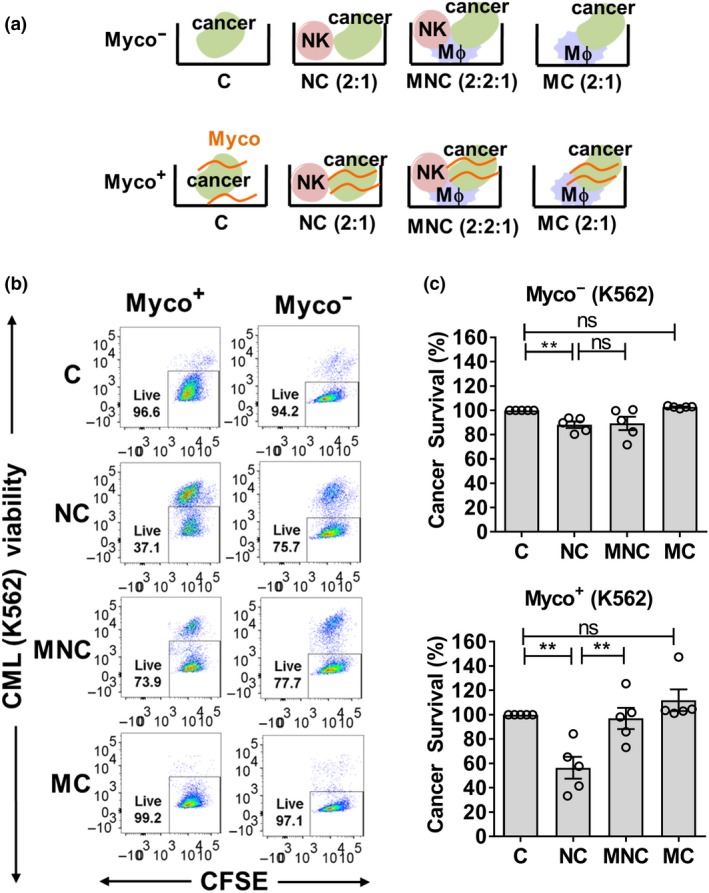
Macrophage (Mϕ) attenuated natural killer (NK) cell cytotoxicity against mycoplasma‐infected chronic myeloid leukemia (CML). (a) Experimental setup of mono, duo and triocultures with Mϕ, NK and CML cells (cell density ratios in parentheses). Cells were cocultured for 24 h prior to measurement of CML survival based on flow cytometry. Chronically infected cultures were annotated myco+, whereas noninfected cultures were annotated myco−. (b) Representative flow cytometry plots showing carboxyfluorescein succinimidyl ester‐stained cancer cells and % live cells based on negative staining for fixable viability dye. (c) CML survival measured under myco+ and myco− conditions, upon coculture with Mϕ, NK cells and normalized to cancer‐alone control. Results shown are mean ± s.e.m. of five independent experiments (donors). See Supplementary figure 4 for individual replicate experiments. Statistical significance was determined using repeated measures one‐way ANOVA followed by Fisher's LSD test. **P < 0.01. C, CML alone; MC, Mϕ + CML; MNC, Mϕ + NK + CML; NC, NK + CML; ns, nonsignificant.
Only myco+ CML cells were protected by Mϕ from NK cytotoxicity
Henceforth, we performed further studies using one of the two CML cell lines, K562, simply referred to as CML. To confirm that the Mϕ‐protective effect was specific to mycoplasma infection, we tested cocultures of (1) 7‐day myco treatment (tx) CML cells with Mϕ and NK cells and (2) CML cells cleared of mycoplasma infection by ciprofloxacin treatment, with Mϕ and NK cells. Supplementary figure 6 shows the efficacy of ciprofloxacin treatment which eliminated mycoplasma infection in CML cells. Consistent with the observation for chronically infected CML (Figure 2c), we also observed Mϕ protection of 7‐day myco tx CML from NK killing (Figure 3a, b and Supplementary figure 7a, b). On the contrary, when chronically infected CML was cleared of mycoplasma by ciprofloxacin treatment (6‐day cipro tx), the Mϕ protective effect was abrogated (Figure 3c, d and Supplementary figure 7c, d), supporting the importance of mycoplasma infection condition in mediating Mϕ protection of CML from NK killing.
Figure 3.
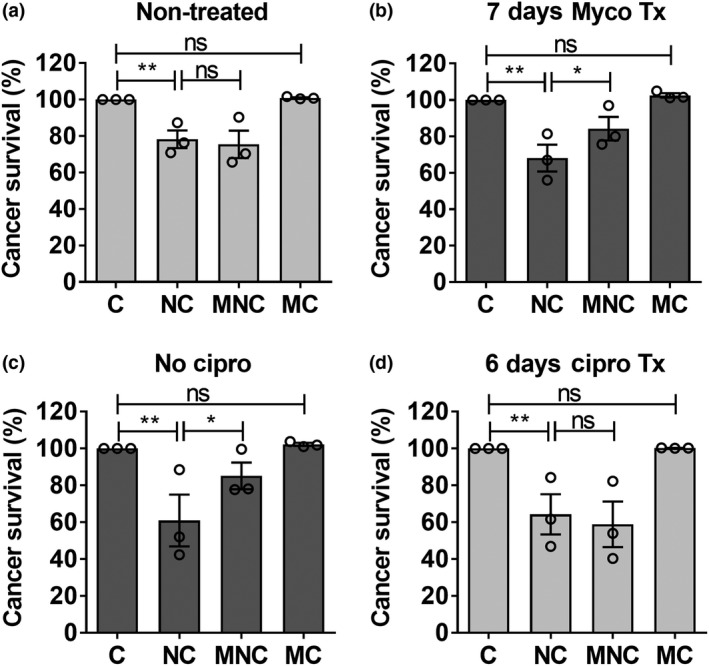
Macrophage (Mϕ) protection of chronic myeloid leukemia (CML) was specific to infection–inflammation condition induced by mycoplasma. Natural killer (NK) cells and Mϕ were cocultured with (a, b) noninfected CML treated with mycoplasma‐containing medium, or (c, d) chronically infected CML treated with 10 μg mL−1 ciprofloxacin. CML survival was subsequently measured based on negative staining for fixable viability dye and normalized to cancer alone control. Results shown are mean ± s.e.m. of three independent experiments (donors). See Supplementary figure 7 for individual replicate experiments. Statistical significance was determined using repeated measures one‐way ANOVA followed by Fisher's least significant difference test. *P < 0.05; **P < 0.01. C, CML alone; MNC, Mϕ + NK + CML; MC, Mϕ + CML; NC, NK + CML; ns, nonsignificant.
We further queried whether inflammation induced by lipopolysaccharide (LPS) or poly(I:C) would create an environment conducive to Mϕ protection of CML from NK cytotoxicity (refer to Supplementary methods). Stimulation of inflammation in the CML by LPS or poly(I:C) was confirmed by induced IL‐8 production by CML cells (Supplementary figures 8a, b and 9a, b). We found that Mϕ protection was only conferred on mycoplasma‐infected CML but not on LPS‐ or poly(I:C)‐treated CML (Supplementary figures 8c and 9c).
Altogether, we have demonstrated that Mϕ protection of CML from NK cytotoxicity was specific to mycoplasma‐induced inflammation condition because (1) infection of “clean” CML cells with mycoplasma‐containing medium resulted in Mϕ protection of the myco tx CML from NK cytotoxicity, (2) there was abrogation of Mϕ‐protective effect of CML when the mycoplasma was eradicated by ciprofloxacin treatment of CML, and (3) inflammation induced by treatment of CML with LPS or poly(I:C) alone did not confer Mϕ protection of CML from NK cytotoxicity.
IL‐8 was not involved in Mϕ protection of myco+ CML from NK cytotoxicity
The high production of IL‐8 by myco+ CML and the high level of IL‐8 in the serum of CML patients29, 30 led us to query whether IL‐8 was involved in the observed Mϕ‐mediated protection of myco+ CML from NK cytotoxicity (Figure 2c). We thus attempted to neutralize IL‐8 in trioculture of Mϕ, NK and myco+ CML (MNC). However, we did not observe modulation of Mϕ‐protective effect toward myco+ CML with increasing doses of IL‐8 neutralizing antibody (Supplementary figure 10), indicating that IL‐8 does not contribute directly to the Mϕ‐mediated protection of CML.
NK degranulation was suppressed by Mϕ in MNC trioculture
The changes in CML survival observed in mono, duo and triocultures of Mϕ, NK and CML were interpreted as modulation of NK cytotoxicity against CML (Figure 2c). To further substantiate that NK activation (and hence cytotoxicity) was modulated, we measured NK degranulation marker, CD107a, under mono, duo and triocultures over two time points (4 and 24 h). The 4‐h incubation is the commonly reported time point for degranulation assays.31 Because we performed cocultures of Mϕ, NK and CML cells for 24 h, we also performed the degranulation assay during the last 4 h of the 24‐h coculture, as outlined in Figure 4a and Supplementary figure 11a. At the 24‐h time point, comparing NC duoculture with N alone culture, there was an increase in the proportion of CD107a+ NK, suggesting an increase in NK activation and degranulation in NC coculture. By contrast, there was no significant increase in CD107+‐activated NK cells in cocultures with myco− CML (Figure 4a), consistent with the higher NK cytotoxicity observed for myco+ than myco− CML cells (Figure 2c). At the 4‐h time point, the increase in the proportion of CD107a+ NK in NC duo culture was also observed (Figure 4b and Supplementary figure 11b).
Figure 4.
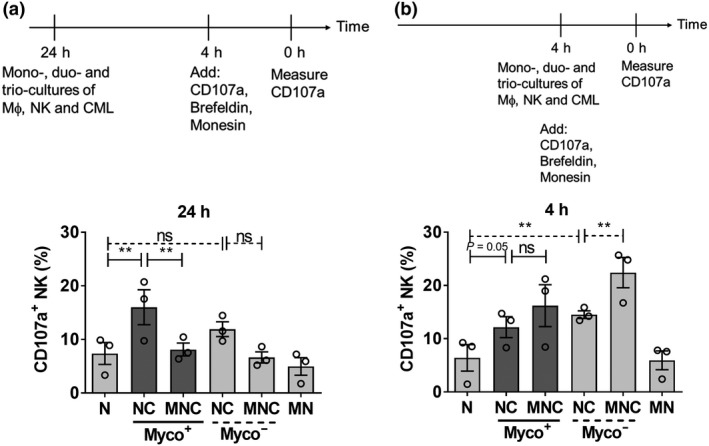
Macrophage (Mϕ) suppressed natural killer (NK) degranulation over time in MNC trioculture. NK, Mϕ, and myco− and myco+ chronic myeloid leukemia (CML) were incubated in mono, duo and triocultures according to the experimental strategies outlined in (a) (24 h) and (b) (4 h). CFSE−CD14−CD56+ NK cells were then gated and CD107a+ NK cells were determined. Results shown are mean ± s.e.m. of three independent experiments (donors). See Supplementary figure 11 for individual replicate experiments. Statistical significance was determined using repeated measures one‐way ANOVA followed by Tukey's test. **P < 0.01. CFSE, carboxyfluorescein succinimidyl ester; MN, Mϕ + NK; MNC, Mϕ + NK + CML; N, NK alone; NC, NK + CML; ns, nonsignificant.
The increase in CD107a+ NK in the NC duo culture was attenuated under MNC triocultures at the 24‐h time point (Figure 4a), suggesting a suppression of NK activation in the presence of Mϕ. This corroborates the observation that Mϕ protected myco+ CML from NK cytotoxicity (Figure 2c). Interestingly, for the 4‐h time point in MNC triocultures, we observed a trend toward further increase in CD107a+ NK instead of attenuation, suggesting that there was a gradual Mϕ‐mediated suppression of NK activity over time.
Activated NK are also known to secrete cytokines and chemokines such as interferon‐γ (IFNγ), TNFα and macrophage inflammatory protein‐1α (MIP‐1α). Hence, we also tested for these secreted NK factors in the mono, duo and triocultures. However, we found no significant modulation of NK IFNγ, TNFα and MIP‐1α production (Supplementary figure 12a–c). Taken together, the data suggested that (1) the decrease in CML survival in NC duo cultures (Figure 2c) was due to increase in NK degranulation and cytotoxicity (Figure 4a); (2) the increase in CML survival in MNC triocultures compared with NC duo cultures (Figure 2c) was due to suppression of NK degranulation and cytotoxicity mediated by Mϕ (Figure 4) and (3) NK IFNγ, TNFα and MIP‐1α cytokines production did not influence the killing of CML in the coculture system. The observed Mϕ protection of CML from NK cytotoxicity was mediated by the suppression of NK degranulation capability, prompting us to further query the mechanism of NK suppression.
Maintenance of NK membrane CD16 in MNC trioculture
To clarify the mechanism of Mϕ‐mediated suppression of NK killing ability, we first characterized the changes in the expression of NK‐activating membrane receptors, membrane NKp46 and membrane CD16 (mCD16), in triocultures of NK, Mϕ and CML cells (MNC). There were marginal changes in NKp46+ NK in mono, duo and triocultures regardless of myco+ or myco− conditions (Supplementary figure 13). Comparing NC with N alone, the proportion of mCD16+ NK was significantly reduced in NC, and the reduction was higher for myco+ than for myco− CML (Figure 5a and Supplementary figure 14a). As shedding of surface CD16 on NK cells has previously been associated with elevated NK cell effector responses,32, 33 we interpreted the reduction in mCD16+ NK to be an increase NK activation. We found that the reduction in NK mCD16 level in NC was rescued in MNC, and again to a higher extent for myco+ CML than for myco− CML cultures, suggesting a suppression of NK activation in MNC compared with NC. We also observed a reciprocal pattern for CD16− NK (Figure 5b and Supplementary figure 14b). In summary, Mϕ mediated a rescue of NK mCD16 level in MNC trioculture.
Figure 5.
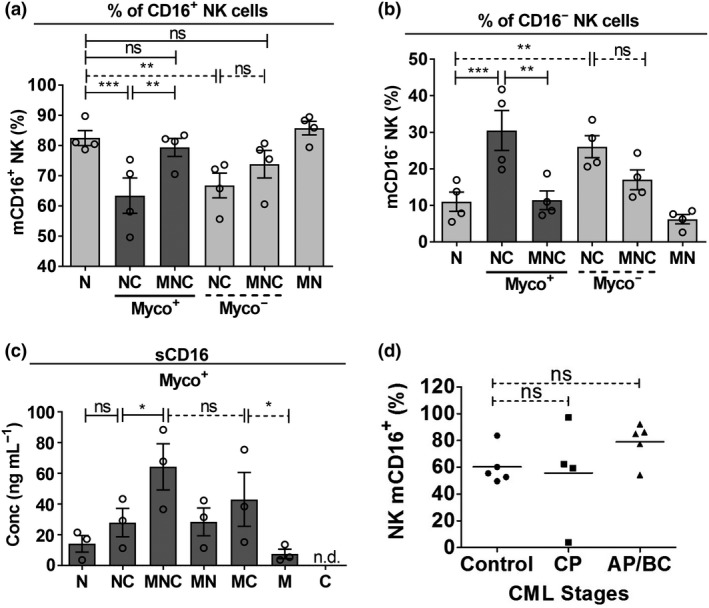
Natural killer (NK) cell membrane CD16 (mCD16) level was maintained in vitro in MNC trioculture under infection–inflammation condition of chronic myeloid leukemia (CML) cells and in vivo in CML patients. (a, b) NK, macrophage (Mϕ) and myco−/myco+ CML were incubated in mono, duo and triocultures. CFSE−CD14−CD56+ NK cells were gated. Percentage of (a) CD16+ and (b) CD16− NK were determined on NK upon coculture with Mϕ and CML. NK mCD16 level was determined specifically on CD56dim NK, which is the cytotoxic counterpart of the NK population. (c) NK, Mϕ and myco− and myco+ CML cells were incubated in mono, duo and triocultures. Culture supernatants were collected and measured for concentration (conc) of soluble CD16 (sCD16) using ELISA. For (a–c), results shown are mean ± s.e.m. of three or four independent experiments (donors). See Supplementary figure 14 for individual replicate experiments. (d) % mCD16+ of NK cells in bone marrow exudates of CML patients at various stages (CP, AP and BC) compared with that in control, which represents non‐CML patients' bone marrow. Each data point represents information extracted from one patient. Closed circles represent non‐CML orthopedic controls; closed squares represent CP patients; closed triangles represent AP/BC patients. Control patients n = 5; CP chronic phase patients n = 4; AP/BC accelerated phase/blast crisis patients n = 5. All statistical significance was determined using repeated measures one‐way ANOVA followed by Tukey's test. *P < 0.05; **P < 0.01; ***P < 0.001. AP, accelerated phase; BC, blast crisis phase; CFSE, carboxyfluorescein succinimidyl ester; CP, chronic phase; MN, Mϕ + NK; MNC, Mϕ + NK + CML; N, NK alone; NC, NK + CML; ns, nonsignificant.
The increase in shedding of CD16 from NK indicates NK activation, whereas a decrease in shedding indicates suppression of NK activity. NK mCD16 is regulated post‐translationally by metalloproteinases (MMPs, also functioning as sheddase), which mediates cleavage upon activation of NK.33, 34, 35, 36 Hence, we queried whether the Mϕ‐mediated perturbations in NK mCD16 level were due to modulation of NK CD16 shedding.32 As a proxy for MMP–sheddase activity, we measured changes in the expression level of membrane CD62L (mCD62L) on NK cells, a sheddase substrate that is often comodulated with CD16.32 We found a significant decrease in NK mCD62L in NC duo culture (Supplementary figure 15), corroborating the drop in NK mCD16 (Figure 5a), and a recovery of NK mCD62L when in MNC trioculture compared with NC duo culture, and this was consistent with the rescue of NK mCD16 level in MNC. The data for NK mCD62L appeared to suggest the potential involvement of MMP–sheddase in modulating the NK mCD16 level. Therefore, we measured soluble CD16 (sCD16) in the culture supernatants of the cocultures of NK, Mϕ and myco+ CML. Consistent with reduced mCD16 level in NC compared with N alone (Figure 5a), a trend toward increased CD16 shedding was observed (Figure 5c and Supplementary figure 14c). Owing to the rescue of NK mCD16 in MNC compared with NC (Figure 5a), we further envisaged a decrease in the level of NK sCD16 shedded into MNC culture supernatant compared with that in NC. Instead, we observed a further increase in sCD16 in MNC compared with NC (Figure 5c). It is plausible that despite continuous shedding of CD16 from NK in MNC triocultures, the level of mCD16 on NK was actively maintained.
Both the Mϕ and NK could have contributed to the pool of sCD16 in the culture supernatant, as evidenced by the increased shedding of sCD16 in (1) NC compared with N and (2) MC compared with M (Figure 5c). Thus, we queried whether the surge in sCD16 detected in MNC (compared with MC or NC) was solely resulting from Mϕ. We observed that the shedding of Mϕ sCD16 in the MC culture supernatant could also be attributed to the increase in mCD16 expression on Mϕ in MC compared with that in M (Supplementary table 3). Interestingly, there was no further increase in the mCD16 level on Mϕ in MNC compared MC (Supplementary table 3). Hence, we reasoned that the increased level of sCD16 in the MNC trioculture supernatant is attributed to the combined shedding of CD16 from both Mϕ and NK.
In MNC, the NK mCD16 level was not downregulated and was maintained compared with baseline (N alone sample; Figure 5a). Such maintenance in the NK mCD16 level was also observed on the NK cells from CML patients' bone marrow compared with nonleukemia individual's bone marrow, regardless of the stage of CML (Figure 5d). Taken together, we propose that mycoplasma‐infected CML cells likely escape NK cytotoxicity through Mϕ‐mediated suppression of NK activity, which is associated with the maintenance of the NK mCD16 level.
Mϕ protection of CML from NK killing was independent of tissue inhibitor of metalloproteinase 1
To understand how Mϕ mediates the maintenance of NK mCD16 in MNC trioculture, we investigated the potential of tissue inhibitor of metalloproteinase (TIMP) in modulating the NK mCD16 level. TIMPs are endogenous inhibitors of MMP–sheddase, which are involved in post‐translational regulation of the NK mCD16 level.33, 34 TIMPs are also detected in mononuclear cells isolated from myeloid leukemia patients37 and in Mϕ.38 We therefore sought to verify whether modulation of sheddase activity by TIMPs may have an influence on NK activation. We first determined the level of Mϕ TIMP in cocultures with NK and myco+/myco− CML (Supplementary figure 16a). Interestingly, Mϕ TIMP‐1 expression was increased in MNC only under the myco+ condition. Thus, we asked whether manipulation of the level of TIMP‐1 secreted by Mϕ would influence the observed Mϕ protection of myco+ CML from NK cytotoxicity. We neutralized the secreted TIMP‐1 with TIMP‐1 neutralizing antibody (refer to Supplemental methods) in MNC and determined changes in Mϕ protection of myco+ CML (Supplementary figure 16b, white bars). Comparing between MNC trioculture treated with TIMP‐1 neutralizing antibody and trioculture treated with immunoglobulin G (IgG) isotype control, no difference in CML survival was observed. Because it has been suggested that Mϕ suppresses NK activity via TGFβ,15, 17 we also treated MNC trioculture with TGFβ neutralizing antibody, which showed an observable but insignificant decrease in CML survival compared with IgG isotype control‐treated trioculture (Supplementary figure 16b, gray bars). Taken together, the data suggest that the maintenance of NK mCD16 level is independent of TGFβ and Mϕ TIMP‐1 modulation of MMP–sheddase activity.
Mϕ protection of CML and maintenance of NK CD16 level was contact dependent
We further clarified whether the mechanism of Mϕ protection of CML from NK cytotoxicity was contact dependent. We disrupted contact between Mϕ and NK + CML by using transwell assays. Comparing Figure 6a, b (also Supplementary figure 17a, b), we found that the Mϕ protection of CML from NK cytotoxicity was abrogated in the presence of transwell, as there was no increase in CML survival in MNC compared with NC. However, with myco− CML, we observed no difference in Mϕ protection, regardless of contact (no transwell; Figure 6c and Supplementary figure 17c) or without contact (with transwell; Figure 6d and Supplementary figure 17d).
Figure 6.
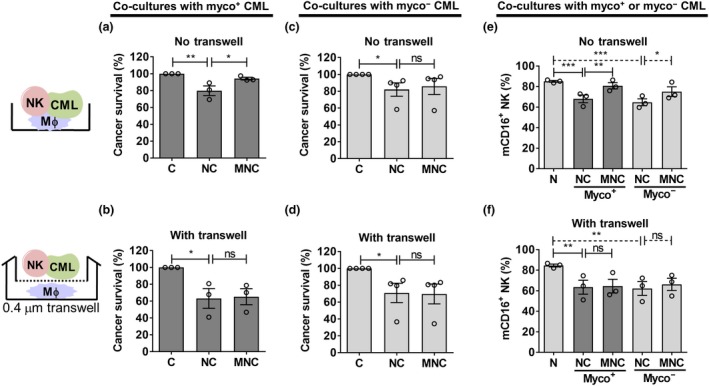
Macrophage (Mϕ) protection of chronic myeloid leukemia (CML) from natural killer (NK) cell cytotoxicity was abrogated when cell–cell contact interactions were disrupted. Transwell assay was performed as illustrated in this figure. (a, b) Mϕ, NK cells and myco+ CML cells were cocultured in the presence and absence of transwell for 24 h. CML survival was then assessed based on negative staining for viability dye and normalizing to cancer‐alone control. (c, d) Mϕ, NK cells and myco− CML were assessed as described in a and b. For a–d, see Supplementary figure 17 for individual replicate experiments. (e, f) Mϕ, NK cells and myco+ or myco− CML were cocultured in the presence and absence of transwell for 24 h and then assessed for percentage mCD16+ NK cells by flow cytometry. Results shown are mean ± s.e.m. of three or four independent experiments (donors). Statistical significance was determined using repeated measures one‐way ANOVA followed by Fisher's least significant difference test (a–d) or Tukey's test (e, f). *P < 0.05; **P < 0.01; ***P < 0.001. C, CML alone; mCD16, membrane CD16; MC, Mϕ + CML; MNC, Mϕ + NK + CML; N, NK alone; NC, NK + CML; ns, nonsignificant.
Because we have shown the association between the Mϕ‐protective effect of CML cells and the maintenance of NK CD16 level (Figure 5), we further queried whether the Mϕ‐mediated maintenance of NK CD16 level was contact dependent. We performed the same transwell assay and measured the percentage of CD16+ NK cells in cocultures of NK, Mϕ and myco+/myco− CML. We found that the maintenance of NK CD16 level in MNC trioculture (Figure 6e and Supplementary figure 17e) was abolished when the contact interaction between Mϕ and NK‐CML was disrupted (Figure 6f and Supplementary figure 17f), suggesting that Mϕ‐mediated maintenance of NK CD16 level was contact dependent. We therefore propose that under the inflammatory condition induced by mycoplasma infection, the CML cells escape NK cytotoxicity through Mϕ‐mediated maintenance of NK mCD16 level in a contact‐dependent manner.
Discussion
We aimed to understand the influence of Mϕ and NK cells on CML survival, particularly on how Mϕ modulates NK cytotoxicity. We focused on CML because of the lack of studies on Mϕ–NK interactions in hematological tumors, and that solid tumors are well known to be resistant to immunotherapeutic efforts, whereas hematological malignancies are responsive owing to the accessibility of immune cells to the leukemic cells.39, 40 Moreover, we found an increase in the ratio of innate to adaptive immune cells in CML bone marrow compared with nonleukemia controls, further prompting us to focus our investigation of Mϕ and NK cells on CML.
The key findings that led us to propose the mechanism for Mϕ protection of CML from NK cytotoxicity are shown in Figure 7. Mϕ protection was demonstrated when the reduction in cancer survival between NC and C (i.e. NK killing) was attenuated in the presence of Mϕ (in MNC trioculture). The attenuation was only observed in MNC for myco+ K562 CML cells. When myco+ K562 CML cells were treated with ciprofloxacin to eliminate mycoplasma infection, the attenuation of NK killing was not observed in MNC. These two observations led us to reason that mycoplasma infection prompted Mϕ‐mediated protection of CML from NK killing. We found that Mϕ protected mycoplasma‐infected CML from NK cytotoxicity through attenuating NK degranulation. We further observed that the protection of CML from NK killing in MNC trioculture was associated with the maintenance of NK mCD16 level in a contact‐dependent manner. This suggested that Mϕ in the MNC culture might have signaled the restoration of NK mCD16 level, leading us to inquire how modulation of mCD16 might influence the NK activation level.
Figure 7.
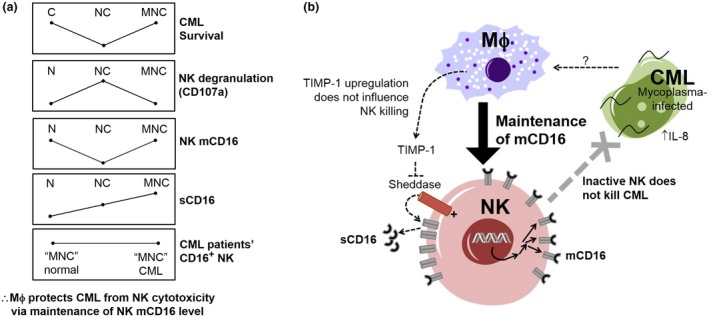
Proposed mechanism of macrophage (Mϕ) inhibition of natural killer (NK) cell cytotoxicity through modulation of NK membrane CD16 (mCD16) level. (a) Comparison of trends of chronic myeloid leukemia (CML) survival relative to NK activation (based on degranulation marker, CD107a), NK mCD16 level, soluble CD16 (sCD16) and NK CD16+ cells in CML patients' bone marrow. The protection of CML survival in MNC trioculture is associated with reduced NK degranulation and maintenance of NK mCD16 level. (b) A hypothetical model of NK mCD16 maintenance as a mechanism of Mϕ‐mediated suppression of NK activation, and hence, protection of myco+ CML. We have also found that maintenance of NK mCD16 level is independent of Mϕ tissue inhibitor of metalloproteinase 1 (TIMP‐1) modulation of NK sheddase activity. It was proposed that modulation of NK mCD16 influences NK activation level via either regulation of NK attachment and detachment from target cells or activating mediators in NK activation pathway. IL, interleukin.
To determine whether Mϕ protection of mycoplasma‐infected CML from NK cytotoxicity is specific to inflammation induced by mycoplasma, we have further used LPS and poly(I:C) to induce a “general”/sterile inflammation. We found that Mϕ did not protect LPS‐ or poly(I:C)‐treated CML from NK cytotoxicity, further suggesting that Mϕ‐mediated protection might be specific to inflammation induced by mycoplasma infection. Future studies may involve the use of other chronic infectious pathogens such as lymphocytic choriomeningitis virus,41 and bacteria associated with persistent infections42 to determine whether the phenomenon extends beyond mycobacterium‐induced inflammation.
The loss of CD16 (cleavage) in activated NK cells was reported.33 Hence, by extension, the maintenance of mCD16 could be viewed as a “by‐product” of a diminished activation status,33 rather than a mechanism of active repression. However, if the modulation of CD16 is a by‐product of NK activation, we would expect that when NK is activated to kill CML cells (in NC duo culture), the level of sCD16 would increase, and when NK was suppressed by Mϕ from killing CML (in MNC trio‐culture), the sCD16 level would decrease compared with NC. However, the latter was not observed in our study. With reference to Figures 5a, c, the anticipated decrease in sCD16 in MNC compared with NC did not occur. Instead, a further increase in sCD16 level (threefold in MNC compared with NC) was detected in the culture supernatant. As such, the NK activation level did not concur with the level of NK CD16 shedding. This led us to propose that the maintenance of the level of NK CD16 (despite active shedding) could be a mechanism through which NK activation is suppressed.
There are several proposals on correlations of NK mCD16 with NK activation level. First, it was recently reported that the presence of CD16 on NK membrane at the immunological synapse results in a tight binding between the effector (NK) and the target (cancer).5 The cleavage of CD16 is mandatory for NK to be freed to attack its next target. Therefore, the maintenance of NK mCD16 level would lower NK cytotoxicity. Second, it was proposed that CD16 couples CD3ζ, an adaptor molecule associated with the CD16 cytoplasmic domain, to CD2, an adhesion molecule capable of stimulating NK activation.6 The cleavage of NK mCD16 was proposed to facilitate the coupling of CD3ζ to CD2 molecule, which consequently enhances NK activation via CD2.43 Thus, the maintenance of NK mCD16 level would presumably attenuate NK activation. However, we did not observe changes in the phosphorylation level of CD3ζ in NK cells under mono, duo and triocultures. Future investigations into the potential of these CD16 post‐translational regulations in modulating NK activation would be beneficial.
The mechanism of Mϕ‐mediated maintenance of NK mCD16 level is hitherto underappreciated. A plausible mechanism is through the upregulation of NK mCD16 expression in NK cells, although further exploration is warranted because the transcriptional regulation of NK CD16 expression through epigenetic modification or cytokine treatment (e.g. TGFβ‐mediated downregulation)44, 45 was only recently investigated. We observed that Mϕ protected CML via a TGFβ‐/TIMP‐1‐independent pathway. However, we found that the Mϕ‐protective effect was contact dependent, suggesting the plausible involvement of membrane‐bound factors on Mϕ in the modulation of NK cytotoxicity and Mϕ‐protective effect on CML. It was reported that Mϕ suppressed NK activity through ligand–receptor interactions via HLAG‐ILT2 and CD48‐2B4,17, 18 but whether these interactions influence the level of NK mCD16 remain to be determined.
Mϕ only conferred protection on myco+ CML but not on myco− CML, suggesting that the signal from mycoplasma or the mycoplasma‐modulated CML “educates” Mϕ to confer protection on CML from NK cytotoxicity. One such potential signal was purported to be IL‐8, which was the key cytokine induced in the myco+ CML cells (Figure 1b). However, neutralizing IL‐8 in MNC trioculture did not modulate the Mϕ‐protective effect on mycoplasma‐infected CML cells. Thus, other signals contributing to Mϕ protection of CML may need to be tested in future, for example, IL‐4 and IL‐13, which were reported to polarize Mϕ into immunosuppressive protumor M2 subtype and separately, reported to be induced upon mycoplasma infection.12, 46 Nevertheless, we observed that disruption of contact between Mϕ from CML and NK cells in transwell assays abrogated mCD16 maintenance and Mϕ‐mediated protection. Therefore, it appears that the effect of Mϕ‐mediated protection is contact dependent, perhaps through membrane ligand–receptor interactions, rather than being primarily reliant on soluble factors.
Based on our findings, we propose a mechanism of Mϕ suppression of NK cytotoxicity on CML under infection–inflammation condition, through maintenance of the NK mCD16 level. While the current study has focused on mycoplasma infections in CML, it aims to suggest the broader concept that infection‐induced inflammation provokes cancer progression through Mϕ‐mediated NK repression. The findings from this study hope to prompt further similar studies involving other bacterial infections and cancers.
Methods
Isolation of primary cells and Mϕ differentiation
Apheresis cone from healthy donors were acquired upon donor's consent from Health Sciences Authority (HSA, Singapore) under approved HSA and NUS‐Institutional Review Board protocols (201706‐06 and H‐17‐028, respectively). Peripheral blood mononuclear cells were isolated using Ficoll‐Paque (GE Healthcare, Chicago, IL, USA) gradient centrifugation. Peripheral blood mononuclear cells were apportioned for monocytes and NK enrichment. NK and monocytes were enriched from peripheral blood mononuclear cells using magnetic negative selection following manufacturer's protocol (Stem Cell Technologies, Vancouver, Canada). Enriched NK was kept frozen until autologous monocytes were differentiated into Mϕ. Enriched monocytes were differentiated into Mϕ by 7‐day treatment with 75 ng mL−1 recombinant human macrophage colony‐stimulating factor (Thermo Fisher Scientific, Waltham, MA, USA) in Roswell Park Memorial Institute medium‐1640 (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (GE Healthcare), 1% v/v penicillin–streptomycin (Thermo Fisher Scientific), 1 mm sodium pyruvate and 0.1 mm non‐essential amino acids (Sigma‐Aldrich, St Louis, MO, USA).
Patients' samples
Acquisition of patients' samples was approved by Domain Specific Review Board (ref no: 2016/00698). All patients/representatives gave written informed consent according to the Domain Specific Review Board guidelines. Deidentified CML patients' samples were acquired from the Cancer Science Institute of Singapore Tissue Bank. Deidentified nonleukemia bone marrow samples from total knee arthroplasty procedures were acquired from the Department of Orthopaedics, National University Hospital, Singapore. Peripheral blood mononuclear cells were extracted using Ficoll‐Paque gradient centrifugation and were frozen prior to being used for polychromatic flow cytometry staining.
Cell culture
CML cell lines (K562 and KCL‐22) were cultured in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum and penicillin–streptomycin at 37°C in 5% CO2 under humidified conditions.
PCR test for mycoplasma
The presence of mycoplasma was generally detected by PCR,47 using forward and reverse primers listed in Supplementary table 1. A sample was determined to be mycoplasma positive (myco+) on detection of a 500‐bp band. The specific species of mycoplasma were determined using species‐specific primers, also listed in Supplementary table 1. Culture supernatants were heated prior to PCR. PCR products were electrophoresed on 1.5% agarose gel and visualized using gel imager (Bio‐Rad Laboratories, Hercules, CA, USA).
Mycoplasma infection and ciprofloxacin treatment of CML cells
To infect clean CML cultures with mycoplasma, 30% noninfected CML culture medium was replaced with mycoplasma‐containing medium and treated for 1, 3, 5 and 7 days. To clear infected cultures of mycoplasma, CML cultures were replaced with culture medium containing 10 μg mL−1 ciprofloxacin and treated for 2, 4 and 6 days. The cell cultures were replaced with clean medium post‐treatment.
ELISA
Culture supernatants from the CML cells were tested for IL‐6, IL‐8, TNFα and IL‐10 according to manufacturer's protocol (Becton Dickinson, Franklin Lakes, NJ, USA). The detection limits of IL‐6, IL‐8, TNFα and IL‐10 were 4.7, 3.1, 7.8 and 7.8 pg mL−1, respectively.
Soluble CD16 measurement
sCD16 was measured in cell coculture supernatants following a reported protocol.33 In brief, 96‐well microplate (NUNC MaxiSorp, NUNC, Rochester, NY, USA) was coated overnight at 4°C with 100 μL of 10 μg mL−1 anti‐CD16 (Becton Dickinson, clone 3G8) in phosphate‐buffered saline. The plates were washed with phosphate‐buffered saline containing 0.05% Tween‐20 and blocked with 200 μL phosphate‐buffered saline containing 2% bovine serum albumin at 37°C for 1 h. Subsequently, 100 μL samples/standards (Thermo Fisher Scientific) were incubated for 2 h at room temperature. The plates were then incubated with 100 μL of 0.5 μg mL−1 of biotinylated anti‐CD16 (Bio‐Rad Laboratories, clone DJ130c) for 1 h at room temperature. About 100 μL of streptavidin–horseradish peroxidase (BioLegend, San Diego, CA, USA) was added for 1 h at room temperature in the dark. Finally, tetramethyl benzidine/H2O2 substrate (Sigma‐Aldrich) was added and incubated for 30 min at 37°C. The reaction was terminated with 50 μL of 1 m H2SO4 and absorbance at 450 nm was read with correction at 570 nm using a microplate spectrophotometer (BioTek, Winooski, VT, USA).
NK cytotoxicity assay
CML cells were stained with 10 μm carboxyfluorescein succinimidyl ester (Sigma‐Aldrich) for 30 min at 37°C. Following coculture (of CML, Mϕ and NK), cells were stained with viability dye [7‐aminoactinomycin D or fixable viability dye; Thermo Fisher Scientific] before analysis using flow cytometry. All carboxyfluorescein succinimidyl ester+ events represented CML. To determine the specific % cancer cell survival, the following formula was used: % cancer cell survival = [(number of carboxyfluorescein succinimidyl ester+fixable viability dye− events)/(total number of carboxyfluorescein succinimidyl ester+ events)] × 100%.
Polychromatic flow cytometry
Following coculture (of CML, Mϕ and NK), cells were stained with fixable viability dye. Then, the cells were blocked with Fc receptor inhibitor (Thermo Fisher Scientific), stained with primary conjugated antibodies (Supplementary table 2) and fixed with 4% paraformaldehyde. For staining intracellular proteins, IFNγ, TNFα, MIP‐1α and TIMP‐1, the coculture samples were incubated with GolgiPlug (Becton Dickinson) 4 h prior to immunostaining. Cells were fixed and permeabilized using a Fixation/Permeabilisation Solution Kit (Becton Dickinson) and stained with primary conjugated antibodies (Supplementary table 2).
NK degranulation
Coculture (of CML, Mϕ and NK) was performed for 24 h. At the last 4 h of the coculture, anti‐CD107a (Supplementary table 2) and protein transport inhibitor, monensin (Thermo Fisher Scientific), were added. The standard 4‐h degranulation assay was also performed, where anti‐CD107a and monensin were added to the cocultures and incubated for 4 h.31, 48 At the end of the assay, cells were harvested and processed for flow cytometry.
Statistical analysis
At least 5000 events of the flow cytometry‐gated populations were collected. Flow cytometry data were acquired on BD LSRFortessa and analyzed using FlowJo V10 software. Statistical tests were performed using GraphPad Prism 8.0.2 and described in respective figure legends. P‐value < 0.05 was determined to be significant. Overall results were assessed based on reproducible statistical trends across at least three independent experiments.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Acknowledgments
This work was supported by a grant from the Ministry of Education (MOE, R‐154‐000‐A76‐114). QWWC is a PhD research scholar supported by the National University of Singapore Graduate School for Integrative Sciences and Engineering (NGS). We thank Woei‐Shyang Loh, Thomas Kwok Seng Loh, Department of Otolaryngology (Head and Neck Surgery), Bryan Koh, Department of Orthopaedics, NUHS, Chor Hiang Siow and Han Lee Goh for guidance and administrative management of Domain Specific Review Board (DSRB: 2016/00698) for National Health Group Research.
References
- 1. Baginska J, Viry E, Paggetti J, et al The critical role of the tumor microenvironment in shaping natural killer cell‐mediated anti‐tumor immunity. Front Immunol 2013; 4: 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sabry M, Lowdell MW. Tumor‐primed NK cells: waiting for the green light. Front Immunol 2013; 4: 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 2013; 31: 10.1146/annurev‐immunol‐020711‐075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mandelboim O, Malik P, Davis DM, Jo CH, Boyson JE, Strominger JL. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc Natl Acad Sci USA 1999; 96: 5640–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Srpan K, Ambrose A, Karampatzakis A, et al Shedding of CD16 disassembles the NK cell immune synapse and boosts serial engagement of target cells. J Cell Biol 2018; 217: 3267–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grier JT, Forbes LR, Monaco‐Shawver L, et al Human immunodeficiency‐causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest 2012; 122: 3769–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 2015; 15: 388–400. [DOI] [PubMed] [Google Scholar]
- 8. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol 2013; 132: 515–525; quiz 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor‐infiltrating/tumor‐associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst 2014; 106: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pierson BA, Miller JS. CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood 1996; 88: 2279–2287. [PubMed] [Google Scholar]
- 11. Biswas SK, Allavena P, Mantovani A. Tumor‐associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol 2013; 35: 585–600. [DOI] [PubMed] [Google Scholar]
- 12. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010; 11: 889–896. [DOI] [PubMed] [Google Scholar]
- 13. Schultze JL, Schmieder A, Goerdt S. Macrophage activation in human diseases. Semin Immunol 2015; 27: 249–256. [DOI] [PubMed] [Google Scholar]
- 14. Krneta T, Gillgrass A, Ashkar AA. The influence of macrophages and the tumor microenvironment on natural killer cells. Curr Mol Med 2013; 13: 68–79. [PubMed] [Google Scholar]
- 15. Krneta T, Gillgrass A, Poznanski S, et al M2‐polarized and tumor‐associated macrophages alter NK cell phenotype and function in a contact‐dependent manner. J Leukoc Biol 2017; 101: 285–295. [DOI] [PubMed] [Google Scholar]
- 16. Young MR, Endicott RA, Duffie GP, Wepsic HT. Suppressor alveolar macrophages in mice bearing metastatic Lewis lung carcinoma tumors. J Leukoc Biol 1987; 42: 682–688. [DOI] [PubMed] [Google Scholar]
- 17. Nunez SY, Ziblat A, Secchiari F. Human M2 macrophages limit NK cell effector functions through secretion of TGF‐β and engagement of CD85j. J Immunol 2018; 200: 1008–1015. [DOI] [PubMed] [Google Scholar]
- 18. Wu Y, Kuang D‐M, Pan W‐D, et al Monocyte/macrophage‐elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013; 57: 1107–1116. [DOI] [PubMed] [Google Scholar]
- 19. Hanlon K, Copland M. Chronic myeloid leukaemia. Medicine 2017; 45: 287–291. [Google Scholar]
- 20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 21. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014; 2014: 149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barile MF, Bodey GP, Snyder J, Riggs DB, Grabowski MW. Isolation of Mycoplasma orale from leukemic bone marrow and blood by direct culture. J Natl Cancer Inst 1966; 36: 155–159. [Google Scholar]
- 23. Kidder M, Chan PJ, Seraj IM, Patton WC, King A. Assessment of archived paraffin‐embedded cervical condyloma tissues for mycoplasma‐conserved DNA using sensitive PCR‐ELISA. Gynecol Oncol 1998; 71: 254–257. [DOI] [PubMed] [Google Scholar]
- 24. Quirk JT, Kupinski JM, DiCioccio RA. Detection of mycoplasma ribosomal DNA sequences in ovarian tumors by nested PCR. Gynecol Oncol 2001; 83: 560–562. [DOI] [PubMed] [Google Scholar]
- 25. Sasaki H, Igaki H, Ishizuka T, Kogoma Y, Sugimura T, Terada M. Presence of Streptococcus DNA sequence in surgical specimens of gastric cancer. Jpn J Cancer Res 1995; 86: 791–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsai S, Wear DJ, Shih JW, Lo SC. Mycoplasmas and oncogenesis: persistent infection and multistage malignant transformation. Proc Natl Acad Sci USA 1995; 92: 10197–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Namiki K, Goodison S, Porvasnik S, et al Persistent exposure to mycoplasma induces malignant transformation of human prostate cells. PLoS One 2009; 4: e6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shimizu T, Kimura Y, Kida Y, et al Cytadherence of Mycoplasma pneumoniae induces inflammatory responses through autophagy and toll‐like receptor 4. Infect Immun 2014; 82: 3076–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hantschel O, Gstoettenbauer A, Colinge J, et al The chemokine interleukin‐8 and the surface activation protein CD69 are markers for Bcr‐Abl activity in chronic myeloid leukemia. Mol Oncol 2008; 2: 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanmamed MF, Carranza‐Rua O, Alfaro C, et al Serum interleukin‐8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin Cancer Res 2014; 20: 5697–5707. [DOI] [PubMed] [Google Scholar]
- 31. Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods 2004; 294: 15–22. [DOI] [PubMed] [Google Scholar]
- 32. Lajoie L, Congy‐Jolivet N, Bolzec A, et al ADAM17‐mediated shedding of FcγRIIIA on human NK cells: identification of the cleavage site and relationship with activation. J Immunol 2014; 192: 741–751. [DOI] [PubMed] [Google Scholar]
- 33. Romee R, Foley B, Lenvik T, et al NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease‐17 (ADAM17). Blood 2013; 121: 3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peruzzi G, Femnou L, Gil‐Krzewska A, et al Membrane‐type 6 matrix metalloproteinase regulates the activation‐induced downmodulation of CD16 in human primary NK cells. J Immunol 2013; 191: 1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grzywacz B, Kataria N, Verneris MR. CD56dimCD16+ NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia 2007; 21: 356–359. [DOI] [PubMed] [Google Scholar]
- 36. Penack O, Gentilini C, Fischer L, et al CD56dimCD16– cells are responsible for natural cytotoxicity against tumor targets. Leukemia 2005; 19: 835–840. [DOI] [PubMed] [Google Scholar]
- 37. Ries C, Loher F, Zang C, Ismair MG, Petrides PE. Matrix metalloproteinase production by bone marrow mononuclear cells from normal individuals and patients with acute and chronic myeloid leukemia or myelodysplastic syndromes. Clin Cancer Res 1999; 5: 1115–1124. [PubMed] [Google Scholar]
- 38. Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol 2008; 28: 2108–2114. [DOI] [PubMed] [Google Scholar]
- 39. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol 2017; 10: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Katlinski KV, Gui J, Katlinskaya YV, et al Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 2017; 31: 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Snell LM, MacLeod BL, Law JC, et al CD8+ T cell priming in established chronic viral infection preferentially directs differentiation of memory‐like cells for sustained immunity. Immunity 2018; 49: 678–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grant SS, Hung DT. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 2013; 4: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabry M, Tsirogianni M, Bakhsh IA, et al Leukemic priming of resting NK cells is killer Ig‐like receptor independent but requires CD15‐mediated CD2 ligation and natural cytotoxicity receptors. J Immunol 2011; 187: 6227–6234. [DOI] [PubMed] [Google Scholar]
- 44. Otegbeye F, Ojo E, Moreton S, et al Inhibiting TGF‐beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS One 2018; 13: e0191358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Victor AR, Weigel C, Scoville SD, Chan WK. Epigenetic and posttranscriptional regulation of CD16 expression during human NK cell development. J Immunol 2018; 200: 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hao Y, Kuang Z, Jing J, et al Mycoplasma pneumoniae modulates STAT3‐STAT6/EGFR‐FOXA2 signaling to induce overexpression of airway mucins. Infect Immun 2014; 82: 5246–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uphoff CC, Drexler HG. Comparative PCR analysis for detection of mycoplasma infections in continuous cell lines. Vitro Cell Dev Biol Anim 2002; 38: 79–85. [DOI] [PubMed] [Google Scholar]
- 48. Rubio V, Stuge TB, Singh N, et al Ex vivo identification, isolation and analysis of tumor‐cytolytic T cells. Nat Med 2003; 9: 1377–1382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials