

ABSTRACT
BACKGROUND
The nucleic acid targeted pathogen reduction (PR) system utilizing amustaline (S‐303) and glutathione (GSH) is designed to inactivate blood‐borne pathogens and leukocytes in red blood cell concentrates (PR‐RBCC). Inactivation is attained after amustaline intercalates and forms covalent nucleic acid adducts preventing replication, transcription, and translation. After pathogen inactivation, amustaline spontaneously hydrolyzes to S‐300, the primary negatively charged reaction product; amustaline is below quantifiable levels in PR‐RBCC. GSH quenches free unreacted amustaline.
STUDY DESIGN AND METHODS
The genotoxic and carcinogenic potential of PR‐RBCC, the reaction by‐products, and S‐300 were assessed in accordance with the International Conference on Harmonization (ICH) guidelines and performed in compliance with the Food and Drug Administration (FDA) good laboratory practice standards, 21 CFR Part 58. in vitro bacterial reverse mutagenicity and chromosomal aberration assays were performed with and without exogenous S9 metabolic activation, and in in vivo clastogenicity and carcinogenic assays using validated murine models.
RESULTS
PR‐RBCCs were not genotoxic in vitro and in vivo and were non‐carcinogenic in p53+/− transgenic mice transfused over 26 weeks. Estimated safety margins for human exposure ranged from >90 to >36 fold for 2 to 5 PR‐RBCCs per day, respectively. PR‐RBCCs and S‐300 did not induce chromosome aberration in the in vivo murine bone marrow micronucleus assay at systemically toxic doses.
CONCLUSIONS
PR‐RBCCs did not demonstrate genotoxicity in vitro or in vivo and were not carcinogenic in vivo. These studies support the safety of PR‐RBCCs and suggest that there is no measurable genotoxic hazard associated with transfusion of PR‐RBCCs.
Red blood cell transfusion is a critical therapy for the support of acute anemia due to blood loss, short‐term bone marrow suppression and for longer term support of chronic anemia in patients with transfusion‐dependent thalassemia and sickle cell disease. Despite these therapeutic benefits, blood transfusion is associated with the risk of blood‐borne transfusion‐transmitted infection (TTI).1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 Pathogen reduction (PR) treatment of blood products provides a proactive approach to prevent TTI from a broad spectrum of pathogens and prevention of graft versus host disease from contaminating leukocytes.4, 15
Depending on the cause of bleeding, exposure may range from 1 to 4 RBC units per transfusion episode repeatedly for days to weeks.16 With massive hemorrhage, RBC transfusion may require ≥10 RBC units over 24 hours.17 Alternatively, chronic transfusion therapy for patients with hereditary disorders of erythropoiesis (Thalassemia, Sickle Cell Anemia) may require life‐long transfusion support with ~2‐3 RBC units every 2‐5 weeks placing the patient at greater risk of exposure to pathogens and transfusion reactions.18, 19
The INTERCEPT Blood System for PR of red blood cells concentrates (RBCCs) uses amustaline (S‐303) and glutathione (GSH). Amustaline is a modular compound consisting of three components: an acridine anchor, an effector and an ester linker (Fig. 1). Bifunctional alkylating agents like amustaline, form covalent bonds between two nucleic acid bases producing a cross‐link.20, 21, 22, 23, 24 Amustaline was designed to target and inactivate nucleic acids and then degrade to non‐reactive by‐products to achieve safety for transfusion. The anchor selectively targets nucleic acids by intercalation and reversibly binds to the helical regions of DNA and RNA. The effector reacts irreversibly with guanine bases creating adducts and crosslinks preventing nucleic acid replication, transcription, or translation. This mode of action allows inactivation in hours of both extracellular and cell‐associated (membrane‐bound and intracellular) pathogens as well as contaminating leukocytes. At physiological pH (7.35–7.45), amustaline degrades by hydrolysis of the linker with an initial half‐life of approximately 20 minutes in the presence of GSH and RBCs followed by a second half‐life of approximately 7 hours (Fig. 2), giving rise to the negatively charged degradant S‐300, other amustaline and GSH reaction products and a small chain fragment (Fig. 3, Table 1). The level of amustaline drops by 1000‐fold over the first 3 hours, by the end of the process “hold time” of 18–24 hours, and additive solution exchange, residual unreacted amustaline concentration is <1 nM. The GSH reaction products (9‐GA, 9‐amino GSH and 9‐amino GSSG) are found in the extracellular fraction and are mostly removed by the exchange step (Table 1). The non‐GSH reaction products (S‐300 and acridine) are found both intracellularly and extracellularly. The small chain fragment (Fig. 1) is bound to proteins in the intracellular RBC compartment whereas in the extracellular space the small chain fragment binds primarily to GSH.
Figure 1.
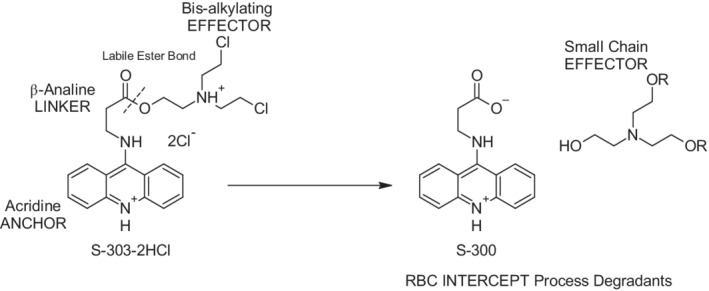
Structure of amustaline dihydrochloride and hydrolysis products; S‐300 and triethanolamine “small chain effector” fragment. Degradation of amustaline is a pH‐driven reaction in which the nucleic acid–targeting “anchor” is hydrolyzed from the crosslinking effector moiety at the labile ester bond in the “linker.” Amustaline degrades stoichiometrically to S‐300 and a small chain b‐substituted triethanolamine “effector” fragment.
Figure 2.
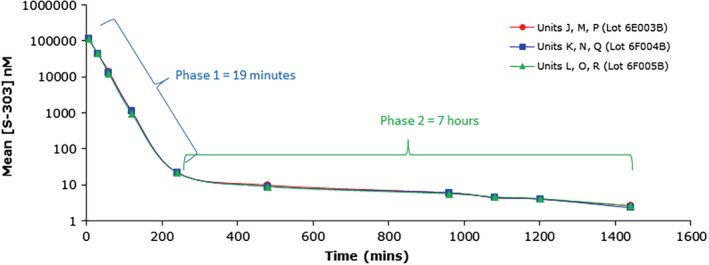
Degradation kinetics of amustaline in INTERCEPT‐treated SAG‐M RBC (N = 3). In the presence of GSH and RBCs, the degradation of amustaline is biphasic with an initial half‐life of approximately 20 minutes followed by a second half‐life of approximately 7 hours due to hydrolysis of the linker moiety giving rise to the nonreactive degradant S‐300 (the major decomposition product) and a small chain fragment. The time course of degradation of amustaline in human PR‐RBC treated with 0.2 mM amustaline/20 mM GSH shows a decrease of residual amustaline from ~115 μM at 6 minutes post addition of amustaline to 11.6 μM after 1 hour, 1.1 μM after 2 hours, and 0.009 μM after 8 hours. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 3.
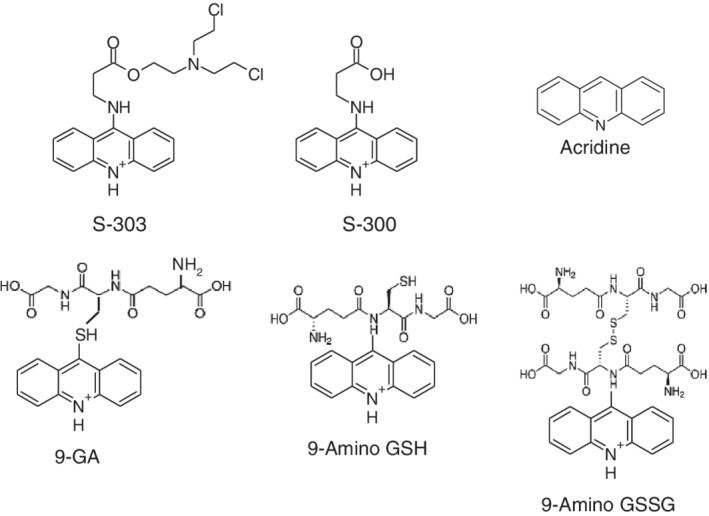
Structures of amustaline and major degradation compounds resulting from the INTERCEPT blood system for RBCs treatment process. Degradation products resulting from the INTERCEPT process include S‐300, the major amustaline degradation product with smaller amounts of acridine and glutathione ‐associated products. The small chain fragment is not measured or characterized.
Table 1.
Major degradation compounds in INTERCEPT RBCs
| Post‐INTERCEPT | ||
|---|---|---|
| n | Mean ± SD (min‐max) | |
| GSH (mM) | 213 | 5.48 ± 1.37 (3.32‐12.73) |
| GSSG (mM)* | 213 | 0.75 ± 0.04 (< 0.75‐1.34) |
| S‐300 (μM)† | 213 | 18.10 ± 3.21 (10.84‐26.06) |
| 9‐GA (μM)† | 213 | 4.11 ± 1.37 (< 1.00‐14.09) |
| 9A‐GSH (μM)† | 213 | 2.45 ± 0.75 (< 1.00‐5.45) |
| 9A‐GSSG (μM)† | 213 | 1.00 ± 0.00 (< 1.00‐1.00) |
| Acridine (μM)† | 127 | 1.00 ± 0.00 (< 1.00‐1.00) |
| Residual Amustaline (nM)‡ | 68 | 0.75 ± 0.01 (< 0.75‐0.81) |
For values reported as <0.75 mM, the limit of quantitation, 0.75 mM, was used to calculate the mean and SD.
For values reported as <1 μM, the limit of quantitation, 1 μM, was used to calculate the mean and SD.
For values reported as <0.75 nM, the limit of quantitation, 0.75 nM, was used to calculate the mean and SD.
Amustaline can react with non‐nucleic acid macromolecules including plasma proteins in the extracellular compartment and proteins on the surface of the RBC membrane, the quenching agent GSH in the PR process reduces non‐specific side reactions including amustaline binding to the RBC surface which has the potential to form an anti‐acridine immune response.25 GSH quenches extracellular reactions of amustaline without a significant impact on inactivation of intracellular pathogens. GSH is a naturally occurring tripeptide (γ‐glutamylcysteinylglycine), and the most abundant non‐protein thiol in cells. GSH is present at 1‐10 mM in most tissues where its primary physiological role is to protect cells from oxidative and chemical stresses by quenching reactive free radicals.18, 26 Transfused GSH does not penetrate cell‐membranes, and thus distributes only in the extracellular plasma space.16
The PR process for RBCs is designed to minimize recipient exposure to residual amustaline. Firstly, unreacted extracellular amustaline is quenched by 20 mM GSH (100‐fold higher level than amustaline). Secondly, the PR process includes an 18‐24 hour room temperature incubation period facilitating amustaline decomposition to non‐genotoxic degradants. Finally, the processing solution which may contain residual unreacted reagents, GSH, and treatment degradation products is replaced with fresh additive solution prior to storage of pathogen reduced RBCs. Following these steps, the residual amustaline is below the defined Threshold of Toxicological Concern (TTC; ICH M7 2015, Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk) of 1 nM based on a daily exposure to one unit of PR‐RBCC for a lifetime.
The systemic toxicology profile of the amustaline/GSH PR technology has been previously reported.17 We report here the detailed toxicological aspects with regards to the patient exposures.
To characterize the genetic toxicology profile of PR‐RBCC, in vitro and in vivo nonclinical safety assessments were performed to evaluate the genotoxic potential of PR‐RBCC. To provide a comprehensive assessment of genotoxic potential, studies were conducted using PR‐RBCC, which includes all acridine‐containing process degradants (Fig. 3) and other reaction by‐products (Fig. 1) in the extracellular and RBC compartments. Studies were also performed with S‐300, the major degradation product of the PR process and present in PR‐RBCCs. The studies included the Ames test for mutagenicity, both in vitro and in vivo chromosomal aberration clastogenicity assays, and an in vivo carcinogenicity assessment. All studies were conducted at GLP‐compliant laboratories in accordance with ICH and Organization for Economic Co‐operation and Development (OECD) guidelines and performed in compliance with the FDA good laboratory practice (GLP) standards, 21 CFR Part 58.18,19
MATERIALS AND METHODS
Study design overview
In vitro assessments included bacterial reverse mutation and chromosomal aberration assays performed with and without an exogenous S9 metabolic activation system obtained from rat liver previously treated with Arcolor 1254.19 In specific studies, lysates of PR‐RBCCs were used as RBC lysis would release any potentially genotoxic compound or degradant that may be concentrated in the red cells. in vivo studies evaluated clastogenic effects in a mouse RBC micronucleus assay. Carcinogenic potential of PR‐RBCCs was assessed in a heterozygous p53+/− murine model with added murine plasma prepared with 1 mM amustaline and 10 mM GSH to increase exposure to active agents (Table 2). Levels of amustaline higher than 1 mM could not be used due to adverse effects on the red cells. This level of GSH was selected to reduce quenching of amustaline further increasing exposure to unreacted amustaline at a level equivalent to the residual GSH in a unit of INTERCEPT RBC post‐treatment.
Table 2.
Summary of in vitro and in vivo genotoxicology studies with PR‐RBC and S‐300
| Test | Test System | Test article | Max. dose tested | Findings | |
|---|---|---|---|---|---|
| Mutagenicity | Ames assay +/− S9 | S. typhimurium | PR‐RBC | 0.2 mM amustaline (2‐20 mM GSH) | Negative at all doses tested |
| ‐TA1537 | |||||
| ‐TA100 | |||||
| ‐TA102 | |||||
| ‐TA1535 | |||||
| E. coli | S‐300 (DMSO) | 6.1 mg/mL | Positive | ||
| ‐WP2uvrA | |||||
| Chromosome aberration | In vitro clastogenicity assay +/− S9 | Human Peripheral Blood Lymphocytes (HPBL)* | PR‐RBCC | 0.1 mL/mL | Negative |
| S‐300 (DMSO) | 280 μg/mL | Negative | |||
| In vivo micronucleus assay to evaluate frequency of bone marrow MN‐PCE¥ | Intravenous injection | PR‐RBC | 0.2 mM amustaline/2 mM GSH ≤25 mL/kg | Negative | |
| No mortality | |||||
| No clinical signs | |||||
| S‐300 (5% dextrose) | 100 mg/kg | Negative | |||
| No mortality | |||||
| No clinical signs | |||||
| Carcinogenicity | p53+/− murine model | Intravenous injection |
Murine PR‐RBC 1 mM amustaline/10 mM GSH 1/week 26 weeks + Murine PR‐plasma 1 mM amustaline/10 mM GSH 2/week 26 weeks |
20 mL/kg* | Negative |
The dose volume of 20 mL/kg was chosen as twice the human clinical dose volume and is the maximum dose volume that could be repeatedly administered to a mouse by intravenous administration.
Test articles
PR‐RBCC
The most representative test article for clinical transfusion was prepared by treating RBCCs derived from whole blood collected in CPD re‐suspended in additive solutions (Erythro‐Sol or SAG‐M). The PR treatment was performed with either 0.3 mM amustaline/3 mM GSH, 0.2 mM S amustaline/2 mM GSH, 1 mM amustaline/10 mM GSH, or 0.2 mM amustaline/20 mM GSH followed by incubation from 2 to 24 hours (0.3 mM amustaline/3 mM GSH treatment only) at 20‐25°C, or 18‐24 hours at 20‐25°C. For preparation of these test articles, there was no exchange step, increasing exposure levels to process degradants.
S‐300
S‐300 was synthesized and suspended in DMSO as a saturated stock solution of S‐300 at 40 mg/mL used for the Ames assay with and without S9 activation. To increase sensitivity, test mixtures were incubated for both 20 and 60 minutes at 37°C prior to plating.27
Amustaline was included in these studies and was positive in the Ames test and chromosome aberration assays (amustaline is cytotoxic, limiting the levels tested). Amustaline tested in vivo showed toxicity at doses >5 mg/mL with a lowest observed effect level (LOEL) of 250 μg/kg.
Assessment of Genotoxicity
In vitro bacterial reverse mutagenicity using the Ames assay
Bacterial mutagenic potential was evaluated in a bacterial reverse mutation assay with 37°C pre‐incubation to enhance the sensitivity of the test strains.27 Due to the chemical nature and mode of action of amustaline, specific Salmonella typhimurium and Escherichia coli strains were selected for sensitivity to intercalating and alkylation agents. PR‐RBCCs were tested with and without S9 metabolic activation with a 60‐minute pre‐incubation period at 37°C. Testing included intact RBCs and RBC lysates. Due to the presence of excess histidine/tryptophan that would interfere with selective media or facilitate bacterial growth, a maximum plating volume of 200 μL was used. To mimic the impact of storage, PR‐RBCCs were evaluated after treatment at 18 hours, 1 day, 2 weeks, and 4‐6 weeks of 4°C storage. For studies using S‐300, the S‐300 was dissolved in DMSO at a stock concentration of 40 mg/mL, and the maximum dose tested was 6.1 mg/mL (4325 μg/plate).
Criteria for a positive mutagenic response required a dose‐related increase in mean bacterial revertants per plate of at least one tester strain with a minimum of two increasing concentrations and a three‐fold increase above the concurrent vehicle control in mean mutant colonies per plate for strains TA1535 and TA1537, and a two‐fold increase for all the other strains.
In vitro Clastogenicity using human peripheral blood lymphocytes
Clastogenic activity was evaluated using an in vitro cytogenetic assay in human peripheral blood lymphocytes (HPBL) with continuous exposure for 4 and 20 hours with and without S9 activation. After the 4‐hour incubation, PR‐RBCs were incubated for an additional 16‐18 hours at 21°C. Metaphase cells were harvested 20 hours after an initial 2 hour exposure to Colcemid (N‐methyl‐N‐deacetyl‐colchicine, Sigma Aldrich) which arrests cell cycle at the metaphase‐anaphase border. Metaphase spreads were stained with Giemsa and evaluated microscopically for chromatid and chromosome aberrations, a minimum of 200 cells/dose group. Fisher's exact test was used to compare pairwise the proportion (%) of aberrant cells in each treatment group with the vehicle control and the Cochran‐Armitage test was used to measure dose‐responsiveness. A test article was considered positive if a statistically significant and dose‐dependent increase in frequency of aberrant metaphases (p ≤ 0.05) was induced. If only one criterion was met, the result was considered equivocal. If neither criterion was met, the results were considered negative. Cytotoxicity was evaluated by the mitotic index (MI); the percent of cells in mitosis per 500 cells counted.
Two independent assessments of clastogenicity with PR‐RBCCs (intact RBCs and RBC lysates) were performed; the first using 0.2 mM S amustaline/2 mM GSH and the second using the system designed for clinical applications, which uses 0.2 mM amustaline/20 mM GSH followed by an overnight incubation at 21°C (pre‐exchange), at which point the processing solution (which contains unreacted reagents, GSH and treatment degradation products) is removed by centrifugation and replaced with new RBC additive solution (post‐solution exchange). As with the Ames assay, the volume of RBCs tested was limited due to interference with the stringent conditions of the assay: the maximum dose volume that would not cause excessive dilution to the cell culture medium with reduction in the mitotic index. Consequently, dose levels ranged from 0.025–0.1 mL test of solution per mL of HPBL culture with Mitomycin C as a positive control at 0.13 and 0.25 μg/mL (in the presence of S9) and 25 to 50 μg/mL (in the absence of S9).
For studies with S‐300, HPBLs were incubated with S‐300 for 4 and 20 hours at 37°C with S9 and for 4 hours at 37°C without S9.
In vivo Clastogenicity using the mouse erythrocyte micronucleus assay
In vivo clastogenic activity of S‐300 and human PR‐RBCCs was evaluated in a mouse bone marrow micronucleus assay. For each test article, pilot toxicity studies were used to set dose level tolerance. Mice were administered each test article by single slow bolus intravenous tail vein injection and bone marrow samples were collected 24 and 48 hours after treatment. Bone marrow was evaluated microscopically for the frequency of micronucleus polychromatic erythrocytes (MN‐PCEs) among 2000 PCE. The test article was considered positive if a statistically significant increase in the frequency of MN‐PCE relative to the vehicle control was induced (p ≤ 0.05 using Kastenbaum‐Bowman tables). Cyclophosphamide was the positive control article.
For PR‐RBCC, a pilot assay using untreated control human RBCs was conducted to set the tolerance of dose volumes to human RBCs in the mouse model. Male and female mice (ICR, Harlan Sprague Dawley) were dosed with control RBC at dose volumes of 10 and 25 mL/kg body weight. Although lethargy and piloerection were observed after dose administration, there was no mortality so the dose volume for the micronucleus study was set at 25 mL/kg. For the in vivo clastogenicity MN‐PCE assay, human RBC were treated with 0.2 mM amustaline/2 mM GSH, incubated overnight at 18°C and delivered to mice by single bolus tail vein intravenous injection at 6, 12, and 25 mL/kg and the vehicle control of untreated RBC was infused at 25 mL/kg.
For S‐300, a pilot toxicity assay using S‐300 dissolved in 5% dextrose and dosed to ICR mice at 10, 25, 50, 75, and 100 mg/kg body weight showed no mortality or clinical signs at any dose. Therefore, the maximum soluble dose for the micronucleus assay was set at 100 mg/kg.
Carcinogenicity using the heterozygous p53+/− mouse model
The heterozygous p53+/− mouse model with 5‐7 week old mice at start of dosing is responsive to genotoxic carcinogens.4, 28 Studies were conducted to test the carcinogenic potential of mouse allogeneic RBCs treated with 1 mM amustaline/10 mM GSH by slow bolus tail vein infusions of 20 mL/kg RBC. Male and female mice were assigned to 4 groups (20 mice per sex per group) and were treated for six months with 20 mL/kg of vehicle control (RBC in Erythrosol) once per week (Group 1), untreated control RBC once per week plus untreated mouse plasma twice per week (Group 2), 1 mM amustaline/10 mM GSH treated RBC once per week (Group 3), and 1 mM amustaline /10 mM GSH treated RBC once per week followed by 1 mM amustaline/10 mM GSH treated mouse plasma twice per week (Group 4). The positive control group received p‐cresidine by daily oral gavage at 400 mg/kg.
After 26 weeks of treatment, mice were bled for hematologic evaluations, euthanized, evaluated for macroscopic alterations, and tissues collected, stained with hematoxylin and eosin and examined microscopically for histopathologic neoplastic lesions.
RESULTS
In vitro assessment of mutagenicity using the Ames bacterial reverse mutation assay
PR‐RBCC treated with 0.3 mM amustaline and 3 mM GSH were used to assess the time course of mutagenicity in bacterial strain TA102 during the amustaline/GSH treatment process. As expected, bacterial inactivation was observed from 0 to 1 hour after addition of amustaline/GSH to RBCC. Mutagenicity in viable bacteria was observed from 2 to 5 hours following amustaline/GSH treatment of RBCC; and no mutagenicity was observed in viable bacterial after 6 hours of treatment. The level of amustaline was measured at each time point and it was determined that the TA102 strain was sensitive to amustaline in PR‐RBCC at a dose as low as 0.15 μg/plate.
In the three studies that evaluated PR‐RBCC, after the full PR process (18‐24 h incubation at 20‐25°C) with or without the processing solution removal step used in the clinical PR treatment process, PR‐RBCC were non‐mutagenic in all bacterial strains at all time points and at all amustaline/GSH concentrations in the presence and absence of S9 using both intact RBCs and red cell lysates (Table 1, available as supporting information in the online version of this paper).
The major process degradant, S‐300 was Ames positive (Table 2, available as supporting information in the online version of this paper). This was an expected result as S‐300 retains the acridine portion of the amustaline molecule that is responsible for nucleic acid intercalation and the TA 1537 strain is sensitive to intercalating agents.29
In vitro chromosome aberration
Human RBCs were treated with 0.2 mM amustaline/2 mM glutathione providing lower quenching capacity with potentially higher levels of residual amustaline and without the exchange step. Under these conditions treated PR‐RBCCs exhibited no statistically significant increases in structural or numerical chromosome aberrations in any test group relative to solvent control, regardless of dose level and time of exposure. There was a statistically significant increase (20.5%) in the percentage of structurally damaged cells in the positive control exposed to Mitomycin C. In a second study using 0.2 mM amustaline/20 mM glutathione, there were no statistically significant increases in structural or numerical chromosome aberrations for the 4 or 20 hour incubation groups, both in the presence and absence of S9. There was a statistically significant increase (23%) in the proportion of structurally damaged cells in the positive control exposed to Mitomycin C. PR‐RBCCs pre‐ and post‐storage solution exchange with up to 0.2 mM amustaline and 20 mM GSH were concluded to be negative for the induction of structural or numerical chromosome aberrations under the conditions of this assay (Table 3, available as supporting information in the online version of this paper).
Table 3.
Relationship of clinical exposure to S‐300 exposures in genotoxicity studies used to estimate safety margins
| Mean highest | Multiples of clinical exposure* | ||
|---|---|---|---|
| Non‐genotoxic | |||
| Study type | Dose level of S‐300 (μg/mL) | Two RBC components | Five RBC components |
| Ames | |||
| Activated | 117 | >4,300x | >1,700x |
| Non‐activated | 390 | >14,000x | >5,800x |
| HPBL | |||
| Non‐activated | 280† | >10,000x | >4,000x |
| Activated | 280† | >10,000x | >4,000x |
Ratio of highest non‐genotoxic S‐300 dose level to the predicted clinical peak plasma level of S‐300 from transfusion of 2 units of PR‐RBCC (26.83 ng/mL) and 5 PR‐RBCC units (67.08 ng/mL).
Negative at the highest dose tested.
For S‐300, the chromosome aberration assay was performed with S‐300 dose levels of 70, 140, and 280 μg/mL with HPBLs treated for 4 and 20 hours without S9 and for 4 hours with S9. There were no statistically significant increases in structural or numerical chromosome aberrations relative to solvent control in the 4 hour exposure groups regardless of dose level and presence or absence of S9 (Table S4, available as supporting information in the online version of this paper). There were no statistically significant increases in numerical chromosome aberrations observed in the non‐activated 20 hour continuous exposure group relative to solvent control. S‐300 was concluded to be negative for induction of chromosome aberrations.
In vivo mouse micronucleus
For PR‐RBCC, bone marrow cells collected 24 and 48 hours after transfusion showed slight reductions (up to 16%) in the ratio of polychromatic to total erythrocytes (indicating some slight cytoxicity in some of the test article groups), but no significant increases in MN‐PCE relative to vehicle control (p > 0.05, Kastenbaum‐Bowman) (Table S5A, available as supporting information in the online version of this paper). The positive control with Cyclophosphamide (40 mg/kg) induced a significant increase in the MN‐PCE control (p ≤ 0.05, Kastenbaum‐Bowman). All criteria for a valid test were met and PR‐RBCCs were concluded to be negative in the murine micronucleus test.
For S‐300, in the micronucleus assay, ICR mice dosed with 25‐100 mg/kg body weight at 25 mL/kg showed lethargy only at the 100 mg/kg dose; and this was only immediately after dosing. All other animals were normal. Bone marrow cells collected 24 and 48 hours after exposure showed some moderate reductions (up to 30%) in the ratio of polychromatic to total erythrocytes does not indicate significant cytotoxicity (which would be >50%), it does suggest some bioavailability of S‐300 to the bone marrow target. There were no significant increases in MN‐PCE relative to vehicle control in any group, and S‐300 was concluded to be negative for clastogenicity in this assay (Table S5B, available as supporting information in the online version of this paper).
Carcinogenicity assessment of PR‐RBCs
The carcinogenic potential of PR‐RBCC was assessed in a p53+/− deficient transgenic mouse model (C57Bl/6Tac‐Trp53 tml heterozygous for the wild‐type p53 gene). The positive control, p‐cresidine in corn oil was dosed orally at 400 mg/kg/day.
There was no evidence of an increase in incidence of tumors, no treatment‐related effects on mortality, clinical signs, body weights, or food consumption. In contrast, the positive control, p‐cresidine caused an expected increased incidence in urinary bladder transitional cell hyperplasia and transitional/squamous cell carcinoma, findings which are consistent with previous data for this compound in the same model Route of administration, test articles and dosing regimen provided chronic exposure to the full spectrum of amustaline, S‐300, and GSH reaction and degradation products. The highest dose group of mice (Group 4) received S‐300 at a mean dose of 14.4 mg/kg/week (based on the S‐300 molecular weight of 266.3) and a dose volume of 20 mL/kg for both the PR‐RBCC and the plasma test articles. This resulted in a human equivalent dose (HED) of 0.16 mg/kg (9.6 mg/person/day based on a 60 kg person).
S‐300 in PR‐RBCC and plasma was non‐carcinogenic with no effects on mortality, clinical signs, body weight, food consumption, or hematological parameters and no test article‐related macroscopic or histo‐morphological findings.
DISCUSSION
The objective of pathogen reduction (PR) of RBC components is to reduce the risk of transfusion‐transmitted infectious disease (TTID) and TA‐GVHD. Depending on the duration of transfusion exposure and the prevalence of blood borne pathogens in blood donor populations, the risk of TTID may be minimal or substantial during epidemics of newly emerging pathogens.4 PR should not contribute excess treatment related mortality (TRM) to blood transfusion and long term TRM due to genetic toxicity cannot be assessed from short term clinical trials. Therefore, it is important to design and conduct preclinical studies in relevant animal models to estimate clinical safety margins for transfusion of PR‐RBCCs based on estimated clinical exposures (Table 3).
Traditional non‐clinical safety programs use the active pharmaceutical ingredient (API) at dose levels calculated to achieve the highest no effect level, to define safety margins in relation to anticipated clinical exposures. However, this concept cannot be directly applied to the PR‐RBCC using amustaline and GSH because the process is performed ex vivo and is designed so that the acceptable level of residual amustaline of less than 1 nM at the completion of the process based on a lifetime, daily exposure to a genotoxic impurity using the threshold of toxicological concern (TTC) criteria as defined in ICH M7 2015 (Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk).
The preclinical toxicology program consisted of studies that evaluated PR‐RBCC; the most relevant test article because they are administered clinically and encompass all breakdown products present in PR‐RBCC and S‐300, the major process degradant. As expected, amustaline is a direct‐acting mutagen and is designed to degrade during the PR process. Consequently, the genotoxic potential of PR‐RBCCs treated with amustaline and GSH was the most appropriate article to be evaluated in in vitro and in vivo clastogenicity and mutagenicity assays, and carcinogenicity studies.
In the absence of human toxicokinetic data, a meaningful basis for genotoxic risk assessment can be predicted by simple allometry (FDA Guidance July 2005; Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers). Preclinical pharmacokinetic data from rats and dogs transfused with S‐300 or with amustaline/GSH RBC were used to determine systemic exposure (Table S6, available as supporting information in the online version of this paper).
Animals had daily dosing of 100 mg/kg for 7 days or 25 mg/kg for 28 days daily (Fischer rats) and 50 mg/kg (Beagle dogs) over 28 days predicting in an average Cmax of 11.07 ± 7.75 μg/mL. Exposure to the level of S‐300 in 2 PR‐RBCC units (0.107 mg/kg) predicted an average human Cmax of 5.23 ± 1.99 ng/mL (Table S6, available as supporting information in the online version of this paper). For S‐300 levels resulting from PR‐RBCC transfusion, allogeneic PR‐RBCC were transfused 3 times weekly for 4 weeks in rats and dogs or weekly for 39 weeks in dogs and 26 weeks in rats.17 These studies resulted in an average Cmax of 1.26 ± 0.86 μg/mL which again, using simple allometry predicted a human S‐300 Cmax of 26.83 ± 22.21 ng/mL (Table S6, available as supporting information in the online version of this paper).
Using the latter value (26.83 ng/mL) as the more conservative estimate, the ratios of highest non‐genotoxic dose to the predicted human clinical peak plasma level of S‐300 from an exposure to 5 RBC components ranged from >1,700 fold to >4,000 fold (Table 3). For the in vivo study in mice using S‐300, the maximum transfused was 100 mg/kg (HED of 8.13 mg/kg), the margin of safety was estimated to be >80 fold and >30 fold for transfusion of 2 and 5 PR‐RBCC. Due to the large ratios in vitro and the absence of any genotoxic effect in vivo, these findings are not considered to indicate toxicological risk for the proposed use of PR‐RBCC.
No carcinogenic risk of PR‐RBCC was observed using the p53+/− mouse model using amustaline/GSH treated RBCs and plasma. Based on the mouse dosed at 2.1 mg/kg/day (14.4 mg S‐300/kg/week), the HED of S‐300 was 0.16 mg/kg or 9.6 mg/person/day for a 60 kg person. A human RBC transfusion of two PR‐RBCCs in 1 day (0.107 mg/kg exposure) results in a margin of safety >90‐fold or >36‐fold for transfusion of 5 PR‐RBCC components.
Amustaline can bind to RBC membrane surface proteins creating the potential for an immune response, so the process was optimized with increased GSH minimizing these interactions. In a recent clinical trial in patients with transfusion‐dependent thalassemia receiving multiple PR‐RBCC components approximately every 2 weeks, there were no anti‐acridine antibodies detected.30
In summary, the genotoxicity program was designed to support clinical use of PR‐RBCCs by conducting comprehensive assessments of PR‐RBCC containing all of the PR process degradants and the primary process degradant S‐300. No genotoxicity was observed using either in vitro studies or in vivo studies. The negative carcinogenicity study also demonstrated the safety of the assessed product. These studies support the safety of PR‐RBCC and suggest that there is no measurable genotoxic hazard associated with transfusion of PR‐RBCC.
CONFLICT OF INTEREST
The authors are employees of Cerus Corporation.
Supporting information
Table S1. Ames bacterial reverse mutation test. PR‐RBCC; Evaluation and Dose Range Tested.
Table S2. Ames bacterial reverse mutation test. S‐300; Evaluation with Two Different Pre‐incubation Periods.
Table S3. Assessment of Clastogenicity of PR‐RBCC (0.2 mM amustaline/20 mM GSH) by in vitro Mammalian Chromosome Aberration Test in HPBL cells (in the absence of S9).
Table S4. Assessment of Clastogenicity of S‐300 by in vitro Mammalian Chromosome Aberration Test in HPBL cells.
Table S5. in vivo Assessment of Clastogenicity in a Bone Marrow Micronucleus Assay for (A) PR‐RBCC and (B) S‐300.
Table S6. Prediction of Human Cmax S‐300 following transfusion of 2 units of PR‐RBCC using Allometric Scaling
REFERENCES
- 1. Allain JP, Bianco C, Blajchman MA, et al. Protecting the blood supply from emerging pathogens: the role of pathogen inactivation. Transfus Med Rev 2005;19:110‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leiby DA. Babesiosis and blood transfusion: flying under the radar. Vox Sang 2006;90:157‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stramer SL, Hollinger FB, Katz LM, et al. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion 2009;49(Suppl 2):1S‐29S. [DOI] [PubMed] [Google Scholar]
- 4. Kleinman S, Stassinopoulos A. Risks associated with red blood cell transfusions: potential benefits from application of pathogen inactivation. Transfusion 2015;55:2983‐3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damgaard C, Magnussen K, Enevold C, et al. Viable bacteria associated with red blood cells and plasma in freshly drawn blood donations. PLoS One 2015;10:e0120826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Busch MP, Kleinman SH, Nemo GJ. Current and emerging infectious risks of blood transfusions. JAMA 2003;289:959‐62. [DOI] [PubMed] [Google Scholar]
- 7. Dodd RY. Bacterial contamination and transfusion safety: experience in the United States. Transfus Clin Biol 2003;10:6‐9. [DOI] [PubMed] [Google Scholar]
- 8. Bihl F, Castelli D, Marincola F, et al. Transfusion‐transmitted infections. J Transl Med 2007;5:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Candolfi E. [Transfusion‐transmitted malaria, preventive measures]. Transfus Clin Biol 2005;12:107‐13. [DOI] [PubMed] [Google Scholar]
- 10. Gubernot DM, Lucey CT, Lee KC, et al. Babesia infection through blood transfusions: reports received by the US Food and Drug Administration, 1997‐2007. Clin Infect Dis 2009;48:25‐30. [DOI] [PubMed] [Google Scholar]
- 11. Guinet F, Carniel E, Leclercq A. Transfusion‐transmitted Yersinia enterocolitica sepsis. Clin Infect Dis 2011;53:583‐91. [DOI] [PubMed] [Google Scholar]
- 12. Herwaldt BL, Linden JV, Bosserman E, et al. Transfusion‐associated babesiosis in the United States: a description of cases. Ann Intern Med 2011;155:509‐19. [DOI] [PubMed] [Google Scholar]
- 13. Holness LG. 24‐year review of fatalities due to contaminated blood and blood components. Transfusion 2005;45:51A. [Google Scholar]
- 14. Asad S, Sweeney J, Mermel LA. Transfusion‐transmitted babesiosis in Rhode Island. Transfusion 2009;49:2564‐73. [DOI] [PubMed] [Google Scholar]
- 15. Busch MP, Bloch EM, Kleinman S. Prevention of transfusion‐transmitted infections. Blood 2019;133:1854‐64. [DOI] [PubMed] [Google Scholar]
- 16. Kennett EC, Bubb WA, Bansal P, et al. NMR studies of exchange between intra‐ and extracellular glutathione in human erythrocytes. Redox Rep 2005;10:83‐90. [DOI] [PubMed] [Google Scholar]
- 17. North A, Ciaravino V, Mufti N, et al. Preclinical pharmacokinetic and toxicology assessment of red blood cells prepared with S‐303 pathogen inactivation treatment. Transfusion 2011;51:2208‐18. [DOI] [PubMed] [Google Scholar]
- 18. Mansoor MA, Svardal AM, Ueland PM. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Anal Biochem 1992;200:218‐29. [DOI] [PubMed] [Google Scholar]
- 19. Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use . ICH harmonized tripartite guideline 2011;S2(R1).
- 20. Brookes P, Lawley PD. Effects of Alkylating Agents on T2 and T4 Bacteriophages. Biochem J 1963;89:138‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohn KW, Spears CL, Doty P. Inter‐strand crosslinking of DNA by nitrogen mustard. J Mol Biol 1966;19:266‐88. [DOI] [PubMed] [Google Scholar]
- 22. Lawley PD, Brookes P. Interstrand cross‐linking of DNA by difunctional alkylating agents. J Mol Biol 1967;25:143‐60. [DOI] [PubMed] [Google Scholar]
- 23. Mattes WB. Use of [8‐3H]guanine‐labeled deoxyribonucleic acid to study alkylating agent reaction kinetics and stability. Anal Biochem 1992;206:161‐7. [DOI] [PubMed] [Google Scholar]
- 24. Mattes WB, Hartley JA, Kohn KW. DNA sequence selectivity of guanine‐N7 alkylation by nitrogen mustards. Nucleic Acids Res 1986;14:2971‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benjamin RJ, McCullough J, Mintz PD, et al. Therapeutic efficacy and safety of red blood cells treated with a chemical process (S‐303) for pathogen inactivation: a Phase III clinical trial in cardiac surgery patients. Transfusion 2005;45:1739‐49. [DOI] [PubMed] [Google Scholar]
- 26. Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol 2002;348:93‐112. [DOI] [PubMed] [Google Scholar]
- 27. Yahagi T, Nagao M, Seino Y, et al. Mutagenicities of N‐nitrosamines on Salmonella. Mutat Res 1977;48:121‐9. [DOI] [PubMed] [Google Scholar]
- 28. Nambiar PR, Turnquist SE, Morton D. Spontaneous tumor incidence in rasH2 mice: review of internal data and published literature. Toxicol Pathol 2012;40:614‐23. [DOI] [PubMed] [Google Scholar]
- 29. Ferguson LR, Denny WA. The genetic toxicology of acridines. Mutat Res 1991;258:123‐60. [DOI] [PubMed] [Google Scholar]
- 30. Aydinok Y, Piga A, Origa R, et al. Amustaline‐glutathione pathogen‐reduced red blood cell concentrates for transfusion‐dependent thalassaemia. Br J Haematol 2019;186:625‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Ames bacterial reverse mutation test. PR‐RBCC; Evaluation and Dose Range Tested.
Table S2. Ames bacterial reverse mutation test. S‐300; Evaluation with Two Different Pre‐incubation Periods.
Table S3. Assessment of Clastogenicity of PR‐RBCC (0.2 mM amustaline/20 mM GSH) by in vitro Mammalian Chromosome Aberration Test in HPBL cells (in the absence of S9).
Table S4. Assessment of Clastogenicity of S‐300 by in vitro Mammalian Chromosome Aberration Test in HPBL cells.
Table S5. in vivo Assessment of Clastogenicity in a Bone Marrow Micronucleus Assay for (A) PR‐RBCC and (B) S‐300.
Table S6. Prediction of Human Cmax S‐300 following transfusion of 2 units of PR‐RBCC using Allometric Scaling