

Abstract
Photon upconversion has enjoyed increased interest in the last years due to its high potential for solar‐energy harvesting and bioimaging. A challenge for triplet–triplet annihilation upconversion (TTA‐UC) processes is to realize these features in solid materials without undesired phase segregation and detrimental dye aggregation. To achieve this, we combine a palladium porphyrin sensitizer and a 9,10‐diphenylanthracene annihilator within a crystalline mesoporous metal–organic framework using an inverted design. In this modular TTA system, the framework walls constitute the fixed sensitizer, while caprylic acid coats the channels providing a solventlike environment for the mobile annihilator in the channels. The resulting solid material shows green‐to‐blue delayed upconverted emission with a luminescence lifetime of 373±5 μs, a threshold value of 329 mW cm−2 and a triplet–triplet energy transfer efficiency of 82 %. The versatile design allows straightforward changing of the acceptor amount and type.
Keywords: hybrid materials, metal–organic frameworks, porphyrins, triplet–triplet annihilation, upconversion
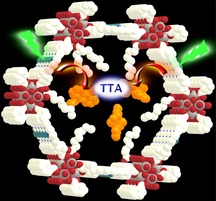
TTA upconversion: Hydrophobic coated channels of a mesoporous MOF with palladium porphyrin walls hosting anthracene acceptors furnish a stable crystalline material showing efficient triplet–triplet annihilation upconversion in the solid state (see figure).
Photon upconversion (UC), that is sometimes also referred to as anti‐Stokes‐shifted luminescence, spectrally converts two low energy photons to a single high energy one.1 UC applications range from bioimaging and sensing to solar energy conversion.2, 3, 4, 5 Classical UC materials comprise rare‐earth metal materials2, 3 and two‐photon absorbing dyes.6 The former materials suffer from low absorption cross‐sections while particularly the latter require very high excitation power densities for the simultaneous absorption of two photons.1 An increasingly used UC process is triplet–triplet annihilation upconversion (TTA‐UC) involving strongly absorbing and emitting molecules.7, 8, 9, 10 The sensitizer absorbs a low‐energy photon and undergoes intersystem crossing (ISC) from the S1 to the T1 state with a high quantum yield Φ ISC. This T1 state transfers its energy to an acceptor (annihilator) by triplet–triplet energy transfer (TTET) with a quantum yield Φ TTET (Figure 1 a). Finally, two acceptors in their T1 states undergo triplet–triplet annihilation (TTA) yielding one acceptor dye in its S0 state and one acceptor dye in its S1 state with a quantum yield Φ TTA. The S1 state emits a delayed fluorescence at higher energy with a quantum yield Φ f (Figure 1 a).8, 9, 10 The overall internal UC quantum yield is given by Equation (1).).
| (1).) |
Figure 1.
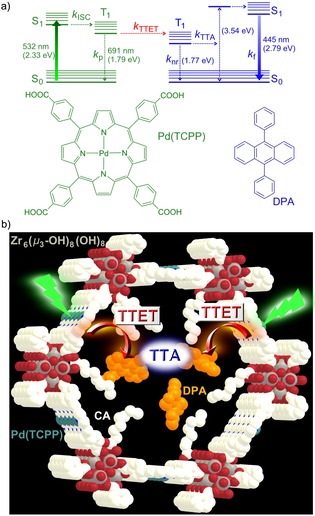
a) Jablonski diagram of TTA‐UC with Pd(TCPP) as a sensitizer and DPA as an annihilator and b) design concept for the CA/DPA@PCN‐222(Pd) TTA‐UC MOF; Pd(TCPP) as the linker and triplet donor; Zr6(μ 3‐OH)8(OH)8 as the secondary building units (SBUs); DPA as triplet acceptor; CA coordinated to the SBUs as a “solvent” in the hexagonal channels; grey (Zr), red (O), blue (N), white (C), light blue (Pd), H atoms omitted.
The factor accounts for the absorption of two photons that are converted to a single upconverted photon.
According to spin statistics for the encounter of two triplets, Φ TTA,max=1/9, although higher values have been reported.11 Provided that ISC, TTET, and fluorescence occur with unity quantum yields, the maximum internal efficiency for TTA‐UC can reach Φ UC=5.5 %. ISC and fluorescence efficiencies are intrinsic properties of the sensitizer and the annihilator, respectively. TTET requires an interaction between the sensitizer in its T1 state and the annihilator, whereas for TTA two annihilators in their T1 states must collide. Hence, Φ TTET and Φ TTA depend on the concentration of excited triplets T1 and thus on the excitation power density, as well as on T1–T1 distances and the annihilator mobility.8, 9, 10, 11 A general challenge of TTA‐UC is its oxygen sensitivity due to the efficient energy transfer from the T1 states to 3O2, generating reactive 1O2 and reducing Φ UC.12, 13, 14
Rationally designing an efficient TTA‐UC system requires sensitizers with Φ ISC close to unity like platinum and palladium porphyrins, such as PtOEP and PdOEP (H2OEP=2,3,7,8,12,13,17,18‐octaethylporphyrin) in combination with annihilators, which feature appropriate energy levels and very high fluorescence quantum yields like 9,10‐diphenylanthracene (DPA, Φ f(DPA)=95 %, Figure 1 a).8, 9, 10, 11 Such dye combinations result in high Φ UC for optimized concentrations in solution.
The design of energy conversion devices as well as sensors and reporter systems relies on UC in the solid state. For TTA‐UC, the chromophores are typically dispersed in soft materials, such as polymers, nanocapsules, or micelles.15, 16, 17, 18, 19 For example, drop‐cast films of PtOEP and DPA show TTA‐UC with threshold excitation power densities I thresh of several kW cm−2.20 Rapid drying of these materials yields a favorable distribution of the sensitizer in the annihilator matrix and lowers I thresh to 6.8 Wcm−2.21 Optimization of the annihilator further reduces phase segregation and leads to an even lower I thresh of 1.7 mW cm−2.22 However, the interface between the chromophores and the relative orientation of the chromophores is difficult to control in less ordered systems. In highly ordered systems, like crystalline materials, the diffusion length of the triplet excitons can, in principle, be controlled and optimized.23 Very promising materials and UC properties have been observed for sensitizer/annihilator systems incorporated spatially controlled into metal–organic frameworks (MOFs). Examples present MOFs with anthracene‐based linkers and surface‐bound palladium sensitizers (I thresh=104 mW cm−2; Φ UC=0.46 %),24 two MOFs connected by a heterojunction (I thresh=25–117 mW cm−2; Φ UC=0.1 %),25 and MOFs with mixed sensitizer/annihilator linkers (I thresh=2.5 mW cm−2; Φ UC=1.28 %).26 The confinement of the porphyrin sensitizers in the MOF reduces detrimental porphyrin dimerization/aggregation and hence aggregation‐induced luminescence quenching. Other examples combine MOF annihilators with external mobile sensitizers.27
In this study, we combine the sensitizer and the DPA annihilator within a crystalline mesoporous MOF using an inverted design, namely immobilizing palladium porphyrin linkers Pd(TCPP) as sensitizers in the channel walls and placing the mobile DPA in the channels of the MOF (Figure 1 b). To provide an optimum microenvironment for DPA enabling efficient TTET and TTA processes, the channels of the chosen MOF with Zr6(μ3‐OH)8(OH)8 SBUs and PCN‐222 structure28 were modified with caprylic acid (CA; octanoic acid) acting as a solvent for DPA (Figure 1 b).
The palladium derivative PCN‐222(Pd) of the well‐known thermally and chemically robust MOF PCN‐222(M) series (M=Mn, FeCl, Co, Ni, Cu and Zn)28b is prepared from Pd(TCPP) and ZrCl4 in a solvothermal reaction (Supporting Information). As determined by single‐crystal XRD, the hexagonal unit cell (a=b=42.455(7) Å; c=17.1037(2) Å) of the resulting needle‐shaped crystals (Figure 2 a) matches that of PCN‐222(FeCl) (a=b=41.968(7) Å; c=17.143(2) Å28b). In the PCN‐222(Pd) Kagomé structure, the Pd⋅⋅⋅Pd distances across the small trigonal pore, the large hexagonal pore and along the channels amount to ≈9.9, 16.9 and 19.8 Å, respectively, preventing porphyrin aggregation, yet enabling efficient energy transfer. The PXRD pattern of the bulk material matches the simulated pattern using the above unit‐cell metrics and Pd instead of FeCl (Figure 2 a). The results from the characterization of the PCN‐222(Pd) host obtained by IR (Figure 2 b), UV/Vis absorption, and emission spectroscopy are provided in the Supporting Information (Figures S1 and S2).
Figure 2.
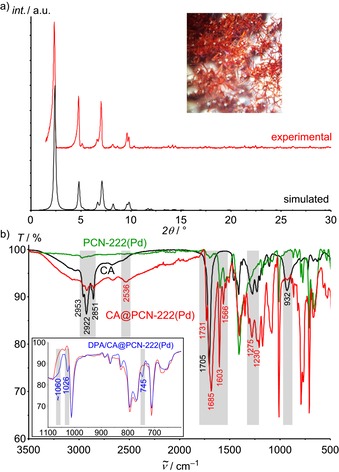
a)Experimental and simulated PXRD pattern (fwhm 2θ=0.2°) of PCN‐222(Pd); optical photograph of the crystals immersed in acetone (inset). b) IR spectra of CA (black), PCN‐222(Pd) (green), CA@PCN‐222(Pd) (red) and CA/DPA@PCN‐222(Pd) (blue) with the relevant vibrational bands highlighted.
Small and large molecules can penetrate into the large hexagonal channels of PCN‐222(M) (Ø 3.7 nm), allowing pH sensing,29 catalytic reactions,28b, 30, 31, 32, 33, 34 adsorptive removal of molecular species like dyes from solution,35 as well as loading and release of drugs36, 37 To probe the insertion of the annihilator into the channels of PCN‐222(Pd), we used NMR spectroscopy and a 19F‐labelled 1,2,3,4,5,6,7,8‐octafluoro‐9,10‐bis(4‐trifluoromethyl‐phenyl) anthracene (DPA‐F14). The 19F NMR resonances of DPA‐F14 shift from δ=−63 (CF3), −134 (CF) and −155 (CF) ppm in solution38 to δ=−64, −130/‐134 and −153 ppm in the crystalline state (Supporting Information, Figures S3 and S4). The splitting of the central resonance is attributed to different chemical environments in the crystals. DPA‐F14 hosted in DPA‐F14@PCN‐222(Pd) gives two discernable resonances in the 19F MAS NMR spectrum at δ=−67 and −122 ppm (Supporting Information, Figure S5). The latter can be ascribed to CF3 groups interacting with the MOF. The resonances of the CF moieties are not observed, which is ascribed to the dispersion of the resonances by different chemical environments. These results confirm the successful insertion of DPA‐F14 in the MOF channels35, 36 and suggest that DPA‐F14 is disordered and interacts with the MOF.
As DPA (Supporting Information, Figure S6) and DPA derivatives show an excimer emission at about 570 nm in neat films,39 we decided to provide the DPA molecules in the channel with a hydrophobic environment, such as a fatty acid to prevent dye aggregation and to increase the mobility of DPA.12 Hence, oleic acid [(9Z)‐octadec‐9‐enoic acid] was mixed with DPA and the fluorescence recorded. However, under laser irradiation, DPA and oleic acid seemed to react, possibly by a cycloaddition reaction.40 Hence, we employed the photostable saturated surfactant caprylic acid (CA, C8H16O2). In the as‐synthesized PCN‐222(Pd), the Zr6 cluster contains two terminal OH ligands, one of which points into the hexagonal channel, providing six OH groups per channel segment for the functionalization with carboxylic acids (Figure 1 b).33 To assess the attachment of CA to the SBUs, desolvated PCN‐222(Pd) was first treated at 70 °C with CA alone giving CA@PCN‐222(Pd) (Supporting Information). The IR spectrum of CA@PCN‐222(Pd) confirms coating the MOF channels with CA molecules, involving different binding modes to the SBU, that is, hydrogen and coordinative bonds.41 Only a few CA molecules exist as hydrogen‐bonded dimers42 in the channels (see the Supporting Information for detailed assignments). This provides a rather nonpolar solution‐like environment for the DPA acceptor in the channel enabling mobility of the acceptors in the channels (Figure 1 b).
CA and DPA were then co‐inserted into the channels of desolvated PCN‐222(Pd) at 70 °C yielding CA/DPA@PCN‐222(Pd). Thanks to the high thermal stability of the PCN‐222(M) MOFs,28b the morphology of the CA/DPA@PCN‐222(Pd) crystals and the powder XRD pattern remain intact under these conditions (Supporting Information, Figure S7). The IR spectrum reveals a superposition of the bands of the OH and the C=O stretching vibrations of CA/DPA@PCN‐222(Pd) and those of CA@PCN‐222(Pd), suggesting an identical coating of the channel walls and a solution‐like interior. Discernible IR bands of DPA appear at ≈1060, 1026 and 745 cm−1 in CA/DPA@PCN‐222(Pd) (Figure 2 b inset; Supporting Information, Figure S8) confirming the successful co‐uptake. UV/Vis absorption spectra of PCN‐222(Pd) and CA/DPA@PCN‐222(Pd) further corroborate the presence of Pd(TCPP) (526/555 nm) and DPA (397 nm), respectively (Supporting Information, Figure S9). DPA uptake by CA/DPA@PCN‐222(Pd) was determined photometrically to 0.29–0.66 mg DPA/ mg PCN‐222(Pd). The exact amount slightly depends on the insertion conditions, that is, concentration, temperature, and solvent.35, 36 Without coating the channel walls, loading of methylene blue and doxorubicin in to PCN‐222 gave 0.906 and 1.09 mg/ mg MOF, respectively.35, 36 Subsequent leaking of DPA from CA/DPA@PCN‐222(Pd) was studied in toluene and in water at 70 °C. In toluene, 0.37 mg DPA/mg CA/DPA@PCN‐222(Pd) were released after 5 h, while no release was observed in water after 2 h (Supporting Information), confirming the desired retention of the DPA guests in the hydrophobic host in water.
Oxygen can quench the phosphorescence of palladium porphyrin like Pd(TCPP) yielding 1O2.30, 34, 36 Under illumination with a 532 nm laser (5.25 mW) in the presence of oxygen in the MOF channels, this 1O2 formation leads to photobleaching of PCN‐222(Pd) and CA/DPA@PCN‐222(Pd). Sealing the MOF with an O2‐barrier polymer (polyvinyl alcohol) and applying Na2SO3 as O2 scavenger cannot completely eliminate Pd(TCPP) quenching by remaining traces of O2 in the MOF channels. However, long‐term illumination studies reveal, that after a certain period of time when all residual O2 in the channels was consumed, PCN‐222(Pd) becomes photostable (Supporting Information, Figure S10). To exclude even traces of O2 in the MOF channels, all synthetic post‐modification procedures (desolvation, CA and DPA loading) and optical measurements were performed under rigorous exclusion of O2. Under these conditions, PCN‐222(Pd) and CA/DPA@PCN‐222(Pd) are photostable (Supporting Information, Figure S11).
UC luminescence (UCL) spectra of CA/DPA@PCN‐222(Pd) show a blue DPA emission around 440 nm upon excitation at 532 nm (Figure 3 a and the video in the Supporting Information). Excitation‐power density dependent UCL measurements confirm the quadratic excitation‐power density dependency expected for TTA‐UC at lower power densities with a slope of 1.84±0.05 that eventually saturates (slope 1.01±0.05). At low excitation intensity (with low T1(DPA) concentration) the UC emission depends quadratically on the laser power densities (weak annihilation limit), while at high laser power density the UC emission scales linearly with the excitation power density (strong annihilation limit).9 These observations suggest that the DPA acceptor molecules diffuse essentially freely in the MOF channels leading to second and pseudo‐first order reactions under low and high laser intensity, respectively. The intersection gives a threshold value of I thresh=329 mW cm−2 (Figure 3 b). This I thresh is larger than that of the best systems reported (I thresh few mW cm−2), but much smaller than that of typical two‐component porphyrin‐DPA systems (I thresh several kW cm−2).20, 21, 22, 23, 24, 25, 26
Figure 3.
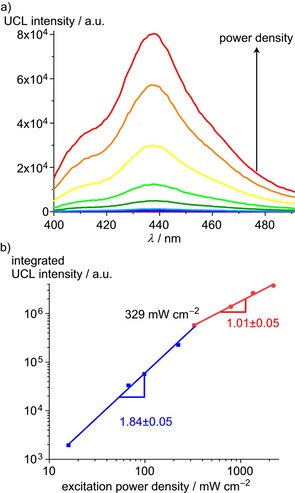
a) TTA‐UC luminescence (UCL) intensity of solid CA/DPA@PCN‐222(Pd) as function of increasing excitation power density (emission slit width 15 nm) and b) integrated UCL intensity of CA/DPA@PCN‐222(Pd) as a function of the excitation power density (λ exc=532 nm). To avoid direct excitation of DPA, a 495 nm long‐pass filter was placed between the 532 nm laser and the CA/DPA@PCN‐222(Pd) sample.
To verify that the blue DPA emission indeed arises from UC, O2 was introduced on purpose, which led to the immediate disappearance of the DPA emission band (Supporting Information, Figure S17). As a prompt DPA fluorescence is barely affected by oxygen, this O2‐sensitivity clearly evidences the upconverted emission from DPA.
To further confirm the delayed nature of the DPA fluorescence in CA/DPA@PCN‐222(Pd) originating from the population through longer‐lived triplet states, we measured the fluorescence lifetime of pristine DPA in the solid state (τ f=4.0±0.2 ns) and trapped in the MOF (Supporting Information, Figures S12–S14). Due to the TTA process in the MOF, the DPA lifetime strongly increases from 4.0 ns to τ UC=373±5 μs. Correspondingly, the lifetime of the porphyrin donor in PCN‐222(Pd) (τ p,0=702±1 μs) is reduced in CA/DPA@PCN‐222(Pd) (τ p=129±1 μs) due to the TTET process (Supporting Information, Figures S15–S16). The TTET efficiency amounts to Φ TTET=1–τ p/τ p,0=82 %. The steady‐state emission spectra measured at several points of the solid CA/DPA@PCN‐222(Pd) sample gave TTET efficiencies between 77–85 %, supporting the time‐resolved data (Supporting Information, Figure S18). According to Equation (1) and assuming Φ ISC=100 %, Φ TTA=11.1 % (conservative maximum theoretical limit under saturation conditions),11 and Φ f(DPA)=95 %, we estimate Φ UC,max to 4.3 % for CA/DPA@PCN‐222(Pd). The UC efficiency might be further increased in the future by reducing the reabsorption of the UC emission caused by Pd(TCPP) (Supporting Information, Figure S9).
In summary, we report a highly modular solid TTA‐UC system comprising of a crystalline, thermally stable PCN‐222(Pd) MOF with CA‐coated MOF channels and with a DPA annihilator embedded in a solution‐like environment in the MOF channels. This solid material displays blue upconverted delayed emission with a luminescence lifetime of 373±5 μs, a threshold value of 329 mW cm−2 and a triplet–triplet energy transfer efficiency of 82 % (Supporting Information, Figure S19; video). This optical application adds another facet to the versatile chemistry of PCN‐222 MOFs. The design concept is also applicable to other TTA‐UC pairs and enables tuning of the UCL color, for example, by replacing DPA with other dyes as exemplarily shown for 2,5,8,11‐tetra‐tert‐butyl‐perylene, that yields UCL at 450 nm (Supporting Information, Figure S20). Current work aims to reduce the oxygen sensitivity and to increase the retention of the trapped annihilators in organic environments, for example, by tuning the chain length of the carboxylic acid and by coating the MOF surface.43 In addition, the TTA‐UC efficiency will be further enhanced by reducing the reabsorption of the UC emission caused by Pd(TCPP) and by optimizing the sensitizer/annihilator interface.12, 22, 39, 44, 45
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Acknowledgements
S..G. acknowledges the Alexander von Humboldt Foundation for a Georg Forster Research Fellowship. This work was supported by the Deutsche Forschungsgemeinschaft (DFG; grants RE 1203/23‐1, RE 1203/12‐3). We thank Dr. Mihail Mondeshki (Mainz, Germany) for the MAS NMR measurements, Dr. Lothar Fink (Frankfurt, Germany) for powder XRD measurements, Dr. Dieter Schollmeyer (Mainz, Germany) for determinations of cell constants and Dr. Yi You and Boyang Xue (Berlin, Germany) for help with the video.
S. Gharaati, C. Wang, C. Förster, F. Weigert, U. Resch-Genger, K. Heinze, Chem. Eur. J. 2020, 26, 1003.
Contributor Information
Dr. Ute Resch‐Genger, Email: ute.resch@bam.de.
Prof. Dr. Katja Heinze, Email: katja.heinze@uni-mainz.de.
References
- 1. Auzel F., Chem. Rev. 2004, 104, 139–173. [DOI] [PubMed] [Google Scholar]
- 2. Haase M., Schäfer H., Angew. Chem. Int. Ed. 2011, 50, 5808–5829; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5928–5950. [Google Scholar]
- 3.
- 3a. Zhou J., Liu Q., Feng W., Sun Y., Li F., Chem. Rev. 2015, 115, 395–465; [DOI] [PubMed] [Google Scholar]
- 3b. Wang Y., Zheng K., Song S., Fan D., Zhang H., Liu X., Chem. Soc. Rev. 2018, 47, 6473–6485; [DOI] [PubMed] [Google Scholar]
- 3c. Zhu X., Su Q., Feng W., Li F., Chem. Soc. Rev. 2017, 46, 1025–1039. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Schulze T. F., Schmidt T. W., Energy Environ. Sci. 2015, 8, 103–125; [Google Scholar]
- 4b. Goldschmidt J. C., Fischer S., Adv. Opt. Mater. 2015, 3, 510–535. [Google Scholar]
- 5. Borisov S. M., Larndorfer C., Klimant I., Adv. Funct. Mater. 2012, 22, 4360–4368. [Google Scholar]
- 6.
- 6a. Pawlicki M., Collins H. A., Denning R. G., Anderson H. L., Angew. Chem. Int. Ed. 2009, 48, 3244–3266; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3292–3316; [Google Scholar]
- 6b. Mayer D. C., Manzi A., Medishetty R., Winkler B., Schneider C., Kieslich G., Pöthig A., Feldmann J., Fischer R. A., J. Am. Chem. Soc. 2019, 141, 11594–11602. [DOI] [PubMed] [Google Scholar]
- 7. Parker C. A., Hatchard C. G., Proc. R. Soc. London Ser. A 1962, 269, 574–584. [Google Scholar]
- 8. Singh-Rachford T. N., Castellano F. N., Coord. Chem. Rev. 2010, 254, 2560–2573. [Google Scholar]
- 9. Gray V., Moth-Poulsen K., Albinsson B., Abrahamsson M., Coord. Chem. Rev. 2018, 362, 54–71. [Google Scholar]
- 10. Huang L., Kakadiaris E., Vaneckova T., Huang K., Vaculovicova M., Han G., Biomaterials 2019, 201, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng Y. Y., Khoury T., Clady R. G. C. R., Tayebjee M. J. Y., Ekins-Daukes N. J., Crossley M. J., Schmidt T. W., Phys. Chem. Chem. Phys. 2010, 12, 66–71. [DOI] [PubMed] [Google Scholar]
- 12. Kouno H., Sasaki Y., Yanai N., Kimizuka N., Chem. Eur. J. 2019, 25, 6124–6130. [DOI] [PubMed] [Google Scholar]
- 13. Baluschev S., Katta K., Avlasevich Y., Landfester K., Mater. Horiz. 2016, 3, 478–486. [Google Scholar]
- 14. Filatov M. A., Baluschev S., Landfester K., Chem. Soc. Rev. 2016, 45, 4668–4689. [DOI] [PubMed] [Google Scholar]
- 15. Turshatov A., Busko D., Baluschev S., Miteva T., Landfester K., New J. Phys. 2011, 13, 083035. [DOI] [PubMed] [Google Scholar]
- 16. Duan P., Yanai N., Kimizuka N., J. Am. Chem. Soc. 2013, 135, 19056–19059. [DOI] [PubMed] [Google Scholar]
- 17. Kwon O. S., Song H. S., Conde J., Kim H.-I., Artzi N., Kim J.-H., ACS Nano 2016, 10, 1512–1521. [DOI] [PubMed] [Google Scholar]
- 18. Askes S. H. C., Pomp W., Hopkins S. L., Kros A., Wu S., Schmidt T., Bonnet S., Small 2016, 12, 5579–5590. [DOI] [PubMed] [Google Scholar]
- 19. Liu Q., Yang T., Feng W., Li F., J. Am. Chem. Soc. 2012, 134, 5390–5397. [DOI] [PubMed] [Google Scholar]
- 20. Baluschev S., Yakutkin V., Wegner G., Minch B., Miteva T., Nelles G., Yasuda A., J. Appl. Phys. 2007, 101, 023101. [DOI] [PubMed] [Google Scholar]
- 21. Kamada K., Sakagami Y., Mizokuro T., Fujiwara Y., Kobayashi K., Narushima K., Hiratad S., Vachad M., Mater. Horiz. 2017, 4, 83–87. [Google Scholar]
- 22. Hosoyamada M., Yanai N., Ogawa T., Kimizuka N., Chem. Eur. J. 2016, 22, 2060–2067. [DOI] [PubMed] [Google Scholar]
- 23. Son H.-J., Jin S., Patwardhan S., Wezenberg S. J., Jeong N. C., So M., Wilmer C. E., Sarjeant A. A., Schatz G. C., Snurr R. Q., Farha O. K., Wiederrecht G. P., Hupp J. T., J. Am. Chem. Soc. 2013, 135, 862–869. [DOI] [PubMed] [Google Scholar]
- 24. Rowe J. M., Zhu J., Soderstrom E. M., Xu W., Yakovenko A., Morris A. J., Chem. Commun. 2018, 54, 7798–7801. [DOI] [PubMed] [Google Scholar]
- 25. Oldenburg M., Turshatov A., Busko D., Wollgarten S., Adams M., Baroni N., Welle A., Redel E., Wöll C., Richards B. S., Howard I. A., Adv. Mater. 2016, 28, 8477–8482. [DOI] [PubMed] [Google Scholar]
- 26. Park J., Xu M., Li F., Zhou H.-C., J. Am. Chem. Soc. 2018, 140, 5493–5499. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Ahmad S., Liu J., Gong C., Zhao J., Sun L., ACS Appl. Energy Mater. 2018, 1, 249–253; [Google Scholar]
- 27b. Meinardi F., Ballabio M., Yanai N., Kimizuka N., Bianchi A., Mauri M., Simonutti R., Ronchi A., Campione M., Monguzzi A., Nano Lett. 2019, 19, 2169–2177. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Morris W., Volosskiy B., Demir S., Gándara F., McGrier P. L., Furukawa H., Cascio D., Stoddart J. F., Yaghi O. M., Inorg. Chem. 2012, 51, 6443–6445; [DOI] [PubMed] [Google Scholar]
- 28b. Feng D., Gu Z.-Y., Li J.-R., Jiang H.-L., Wei Z., Zhou H.-C., Angew. Chem. Int. Ed. 2012, 51, 10307–10310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10453–10456. [Google Scholar]
- 29. Deibert B. J., Li J., Chem. Commun. 2014, 50, 9636–9639. [DOI] [PubMed] [Google Scholar]
- 30. Liu Y., Howarth A. J., Hupp J. T., Farha O. K., Angew. Chem. Int. Ed. 2015, 54, 9001–9005; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9129–9133. [Google Scholar]
- 31. Li P., Klet R. C., Moon S.-Y., Wang T. C., Deria P., Peters A. W., Klahr B. M., Park H.-J., Al-Juaid S. S., Hupp J. T., Farha O. K., Chem. Commun. 2015, 51, 10925–10928. [DOI] [PubMed] [Google Scholar]
- 32. Deria P., Gómez-Gualdrón D. A., Hod I., Snurr R. Q., Hupp J. T., Farha O. K., J. Am. Chem. Soc. 2016, 138, 14449–14457. [DOI] [PubMed] [Google Scholar]
- 33. He T., Ni B., Xu X., Li H., Lin H., Yuan W., Luo J., Hu W., Wang X., ACS Appl. Mater. Interfaces 2017, 9, 22732–22738. [DOI] [PubMed] [Google Scholar]
- 34. Park K. C., Seo C., Gupta G., Kim J., Lee C. Y., ACS Appl. Mater. Interfaces 2017, 9, 38670–38677. [DOI] [PubMed] [Google Scholar]
- 35. Li H., Cao X., Zhang X., Yu Q., Zhao Z., Niu X., Sun X., Liu Y., Ma L., Li Z., RSC Adv. 2017, 7, 16273–16281. [Google Scholar]
- 36. Liu W., Wang Y.-M., Li Y.-H., Cai X.-J., Yin X.-B., He X.-W., Zhang Y.-K., Small 2017, 13, 1603459. [Google Scholar]
- 37. Bai Y., Dou Y., Xie L.-H., Rutledge W., Li J.-R., Zhou H.-C., Chem. Soc. Rev. 2016, 45, 2327–2367. [DOI] [PubMed] [Google Scholar]
- 38. Tannaci J. F., Noji M., McBee J. L., Tilley T. D., J. Org. Chem. 2008, 73, 7895–7900. [DOI] [PubMed] [Google Scholar]
- 39. Serevičius T., Komskis R., Adomėnas P., Adomėnienė O., Jankauskas V., Gruodis A., Kazlauskasa K., Juršėnasa S., Phys. Chem. Chem. Phys. 2014, 16, 7089–7101. [DOI] [PubMed] [Google Scholar]
- 40. Kaupp G., Liebigs Ann. Chem. 1977, 254–275. [Google Scholar]
- 41. Al-Hazmi M. H., Choi Y., Apblett A. W., Adv. Phys. Chem. 2014, 429751. [Google Scholar]
- 42. Matsuzawa H., Tsuda M., Minami H., Iwahashi M., Food Nutr. Sci. 2013, 4, 25–32. [Google Scholar]
- 43. Sun D., Adiyala P. R., Yim S.-J., Kim D.-P., Angew. Chem. Int. Ed. 2019, 58, 7405–7409; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7483–7487. [Google Scholar]
- 44. Senge M. O., MacGowan S. A., O'Brien J. M., Chem. Commun. 2015, 51, 17031–17063. [DOI] [PubMed] [Google Scholar]
- 45. McCusker C. E., Castellano F. N., Top. Curr. Chem. 2016, 374, 175–199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary