

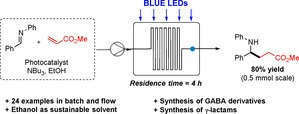
Abstract
Owing to their wide range of biological properties, γ‐aminobutyric acid derivatives (GABA) have been extensively studied and found noteworthy industrial applications. However, atom‐economical and efficient processes for their production are scarce and would greatly benefit from further investigations. Herein, we demonstrate that an iridium‐based photocatalyst promotes the direct reductive cross‐coupling of imines with olefins upon irradiation with visible light to give GABA derivatives in good yields and selectivities. We also stress the enabling triple role of tributylamine additive in this process, discuss the advantages of strategies based on proton‐coupled electron transfer (PCET) and demonstrate the scale‐up of this reaction in continuous flow.
Keywords: continuous-flow photocatalysis, heterocycles, imine umpolung, proton-coupled electron transfer, visible light photoredox catalysis
One amine–3 tasks: Gamma‐aminobutyric acid derivatives could easily be synthesized by the visible‐light photoredox‐catalyzed coupling of imines with olefins. The reaction works well for a range of substrates and tolerates many functional groups. A continuous‐flow strategy considerably improved the efficiency of this methodology.
Introduction
Over the last decade, the synthetic potential of visible light photoredox catalysis became more and more evident. Low loadings of catalysts, inexpensive light sources as well as mild reaction conditions contrast with most of the requirements for classical thermal or UV‐mediated radical reactions.1 Ruthenium‐ and iridium‐based photocatalysts1c, 1e with bipyridine and phenylpyridine ligands offer highly tunable and predictable reduction and oxidation potentials and therefore were applied by our group and others to a vast number of transformations. Interestingly, although a multitude of catalysts were designed for reductive quenching catalytic cycles, mostly aiming at a high excited‐state oxidation potential (E1/2 ox(M*/M−)=1.21 V vs. SCE for Ir[dF(CF3)ppy]2(dtbbpy)+, 1.45 V vs. SCE for Ru(bpz)3 2+),2 catalysts with high excited‐state reduction potential, used in oxidative quenching cycles, are virtually limited to Ir(ppy)3 (E1/2 red(M*/M+)=−1.73 V vs. SCE).3 Therefore, understanding the oxidation of ground‐state catalysts in reductive quenching cycles is of importance to widen the scope of accessible substrates.
The development of methods for the umpolung of carbonyl compounds has been of great interest over the past years, as it opens new retrosynthetic opportunities for the synthesis of complex molecules.4 Although umpolung of aldehydes and ketones has been extensively investigated,4 the number of efficient methods for umpolung of imines are limited.5 A now common strategy is the deprotonation of imines to form 2‐azaallyl anions.5 These intermediates can be used in organocatalyzed or metal‐catalyzed cross‐coupling processes, yielding alkylated or arylated umpolung products.6 A major shortcoming of this strategy is the need for strong bases and biased substrates to ensure regioselectivity in the functionalization of the 2‐azaallyl anion. Another strategy is the one‐electron reduction of an imine to give a free amino radical anion or a 3‐membered metallacycle depending on the reductant used.7 However, direct electron‐transfer is difficult due to the high reduction potentials of imines (E1/2 red=−1.98 V vs. SCE for N‐benzylideneaniline),8 and either specially designed substrates with lower reduction potentials or overstoichiometric amounts of highly reducing metals salts based on zirconium,7a samarium7b, 7c or titanium7d, 7e are required.
We previously showed that visible light induced reduction of imines to α‐amino radicals using photocatalysts with low ground state reduction potentials is possible by a strategic choice of reductive quencher.9, 10 Tributylamine acted as both electron‐donor for the catalyst and hydrogen‐bond donor for the substrate,9 permitting a proton‐coupled electron transfer11, 12 as key step. The resulting radical then underwent dimerization to give symmetrical diamines. Very recently, various groups reported photoredox‐catalyzed radical–radical cross‐coupling reactions between the persistent radical formed by reduction of an imine and a transient radical formed by oxidation, however the photoredox‐catalyzed reductive cross‐coupling of imines with olefins has been less investigated.13, 14 We therefore questioned the possibility of a general cross‐coupling reaction exploiting the α‐amino radical intermediate.
We hypothesized that such a cross‐coupling might be mechanistically similar to our previously reported coupling of α‐chloroamides with olefins (Scheme 1)15: upon excitation by visible light, the photocatalyst becomes highly oxidizing and is reduced by tributylamine to give the corresponding radical cation A, which rapidly undergoes a 1,2‐hydrogen shift to form the α‐ammonium radical BH+. This intermediate activates the imine 1 as C for a subsequent proton‐coupled electron transfer that regenerates the catalyst and gives the α‐amino radical D along with B. Cross‐coupling between these two nucleophilic radicals is disfavored in the presence of electron‐deficient olefins 2, onto which D readily adds in accordance with the polar effect.16 The resulting electrophilic radical E undergoes hydrogen atom transfer from B to give F and the expected saturated product 3.17 Thus, the additive plays here a triple role: reduction of the excited‐state catalyst, hydrogen‐bonding to render the reduction of the imine less endergonic, and finally hydrogen‐atom donation. Noticeably, such a reaction manifold avoids side‐reactions typically observed when using trialkylamines as reductive quenchers,18 such as reduction of D by HAT from tributylamine or B and radical addition of D onto F.
Scheme 1.
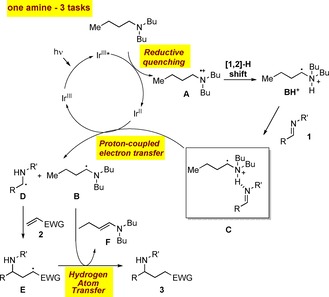
Proposed mechanism for the visible light photoredox‐catalyzed reductive coupling of imines with olefins ([1,2]‐H shift=1,2‐hydrogen shift).
Results and Discussion
Based on our previous studies, we started our investigation on the photoredox‐catalyzed reductive cross‐coupling of imines with olefins by irradiating a mixture of imine, tributylamine and methyl acrylate in DMF, using Ir[F(CF3)ppy]2(bpy)PF6 (4 a) or Ir(ppy)2(dtbbpy)PF6 (4 b) as photoredox catalyst (see Figure 1 for the structure of the catalysts). In contrast with many other N‐protected substrates, N‐benzylideneaniline gave little to no imino‐pinacol coupling and selectively coupled with the olefin in 43 and 59 % yield, respectively (Table S2, entries 1 and 3).
Figure 1.
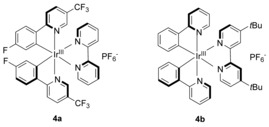
Iridium‐photocatalysts discussed in this work.
Further optimization showed that no additives other than tributylamine were needed, and that the use of ethanol as a sustainable alternative to DMF greatly enhanced the yield. Under the optimal conditions, N‐benzylideneaniline 1 a was dissolved in ethanol in the presence of 3 equivalents of methyl acrylate, 1.5 equivalents of tributylamine and 2 mol % of 4 a and irradiated at 450 nm with blue LEDs for 24 hours to give the expected product 3 a in 84 % yield (Table S2, entry 7). Slightly higher yields could be obtained by running the reaction for 24 additional hours (Table S2, entry 8). No conversion was observed in the absence of tributylamine or photocatalyst, in the dark, or under air atmosphere. Addition of catalytic amounts of bases completely inhibited the reaction, confirming a proton‐coupled electron transfer as the key step.19
Although these reaction conditions were satisfying from a batch photochemistry point of view, we wondered whether we could decrease the reaction time and the catalyst loading. We therefore investigated the outcome of the reaction under continuous flow conditions.20 Pleasingly, when a 5 mL flow microreactor (PTFE tubing, id=0.8 mm, l=10 m) was irradiated with blue LEDs, a six‐fold decrease in reaction time could be observed. Additionally, the catalyst loading could even be reduced from 2 to 1 mol %, without substantial loss in yield and selectivity.19 Performing a flow experiment on a 0.5 mmol scale using 1 mol % of 4 a and a residence time of 4 hours gave 3 a in 80 % yield, emphasizing once more the advantages of flow over batch, for scaling‐up photocatalyzed reactions (Scheme 2).21
Scheme 2.
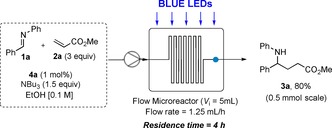
Photoredox‐catalyzed reductive coupling in continuous flow.
The integration of in‐line analytical devices into the flow stream of a synthetic methodology improves process automation and also reduces risks associated to handling and isolation of intermediates. In particular, IR flow cells have recently proved to be effective tools for reaction monitoring, mechanistic investigations as well as quantitative measurements in multistep processes and self‐optimization studies.22 To have a better understanding of the reaction kinetics, we next decided to monitor the reaction profile using a FlowIR instrument equipped with a DiComp ATR (diamond‐composite attenuated total reflection) probe. For in‐line analysis, we identified signals at ν=1633 cm−1 and ν=1607 cm−1 corresponding to starting imine 1 a and product 3 a, respectively. Monitoring the flow photoredox coupling of 1 a and 2 a under optimal conditions at different reaction times, substrate consumption and product formation could be observed in real‐time (Figure 2 and Figure S2). As depicted in Figure 3, the comparison between batch and continuous flow conditions clearly demonstrates that both processes have a similar reaction profile, but light irradiation in a flow microreactor ensures a faster transformation, which undergoes completion after only 4 hours.
Figure 2.
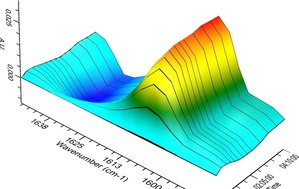
3D time‐resolved spectral data obtained from FlowIR in‐line analysis showing substrate consumption (peak at 1633 cm−1) and product formation (peak at 1607 cm−1).
Figure 3.
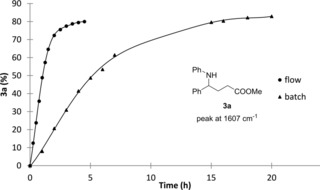
Comparison of batch and flow trend‐curves of 3 a formation obtained with FlowIR in‐line monitoring.
Once the best reaction conditions were identified for both batch and flow regimes, we evaluated the scope of the reaction (Scheme 3). First, we established that not only acrylates, but also various radical acceptors such as acrylonitrile, acrylamides as well as vinyl sulfones can be coupled with 1 a in good yields. Next, we showed that acrylates with side‐chains of various size and lipophilicity gave the corresponding products in very good yields. A free hydroxy group (3 h) as well as a trialkyl amine (3 i) were tolerated, without signs of oxidation under the reaction conditions.23, 24 A bromide‐containing acrylate derivative smoothly reacted to give the Br‐containing product 3 l, even though reduction of alkyl bromides by photoredox catalysis has been reported.25 As alkyl bromides and imines have similar reduction potentials,26 this example further demonstrates how our strategy involving reductive quenching followed by proton‐coupled electron transfer provides exquisite selectivity compared to alternative oxidative quenching strategies. Methacrylate and itaconate derivatives gave the expected products (3 j, 3 n, respectively) in good yields and selectivities, whereas using methyl 2‐acetamidoacrylate as acceptor gave the highly valuable γ‐aminobutyric amino acid derivative 3 m in high yield and moderate selectivity. Conjugated dienes were also competent substrates and use of methyl 4‐methylpentadien‐2,4‐oate as acceptor resulted in 3 o as the only product in good yield. Next, we decided to investigate the substrate scope regarding imines (Scheme 3).
Scheme 3.
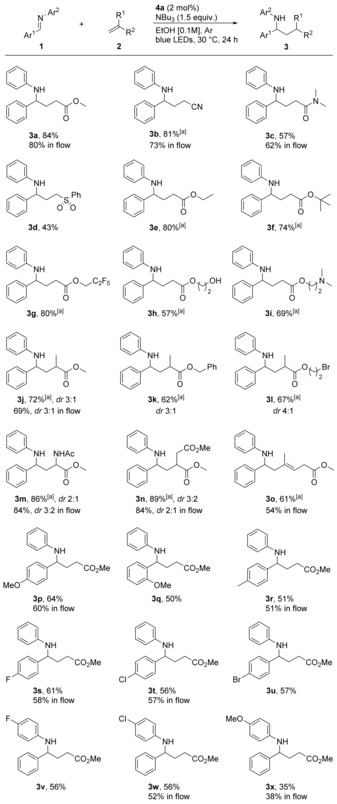
Reaction scope. Reaction conditions: imine 1 (0.2 mmol, 1 equiv), olefin 2 (0.6 mmol, 3 equiv), NBu3 (0.3 mmol, 1.5 equiv), Ir[F(CF3)ppy]2(bpy)PF6 4 a (2 mol %) in degassed EtOH (2 mL) under argon and blue LEDs (11 W rubber strip) for 24 hours, yields after purification. Reactions in flow performed as indicated in Scheme 2. [a] The reaction was performed with 5 equivalents of olefin.
While trying to broaden the scope of the nitrogen protecting group, we found that several imines underwent decomposition, pinacol coupling, reduction, or simply did not react at all under the standard reaction conditions. N‐arylimines however were robust and cleanly afforded the desired products in most cases. Electron‐rich as well as electron deficient groups were tolerated and, once again, no dehalogenation was observed (3 s–w). The synthetically valuable product 3 x was obtained in moderate yield.
Noticeably, for all examples tested in flow, the products were obtained in yields and selectivities comparable to the ones obtained in batch. The conversion of γ‐aminobutyric ester derivatives 3 a, 3 j, 3 m and 3 n to the corresponding γ‐lactams 5 proceeded almost quantitatively by heating the starting materials in toluene in the presence of catalytic amounts of para‐toluenesulfonic acid (Scheme 4 a).13b Additionally, our reaction was not only limited to reductive alkylation but could also be extended to reductive allylation of imines by using appropriate allyl sulfone derivatives as coupling partners (Scheme 4 b).27 Intramolecular conjugate addition of the products 6 a and 6 b would generate 2,4‐disubstituted pyrrolidines in one step.28
Scheme 4.
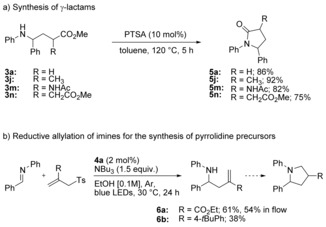
Application of our methodology to the synthesis of nitrogen‐containing heterocycles.
Conclusions
In conclusion, we developed a highly efficient reductive cross‐coupling reaction between imines and olefins using a visible light photoredox catalysis procedure. Tributylamine was shown to play a crucial role as it is involved in three distinct mechanistic steps. We also demonstrated how this reaction could be applied to the expedient and modular synthesis of heterocyclic building blocks, and how continuous flow technology would open opportunities for scale‐up and in‐line synthesis. Attempts to extend this reaction to other classes of substrates and to the synthesis of non‐racemic products are ongoing in our laboratories and will be reported in due course.
Experimental Section
Procedure for the visible light photoredox‐catalyzed reductive cross‐coupling of imines with olefins:
In an oven‐dried test tube equipped with a magnetic stir bar were placed Ir[F(CF3)ppy]2(bpy)PF6 4 a (3.9 mg, 0.004 mmol, 0.02 equiv), imine 1 (0.2 mmol, 1.0 equiv) and olefin 2 (1.0 mmol, 5.0 equiv, if solid). The tube was closed with a septum and purged three times with a sequence vacuum/argon before addition of degassed ethanol (2.0 mL), NBu3 (71 μL, 0.3 mmol, 1.5 equiv), and the corresponding olefin 2 (1.0 mmol, 5.0 equiv, if liquid). The tube was placed in a beaker wrapped with 11 W blue LEDs stripes and irradiated for 24 hours. After complete conversion, the reaction mixture was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (see Supporting Information for eluent used). In some cases, residual amounts of tributylamine were observed after column chromatography; they were removed by co‐evaporation in vacuo with isopropanol or water followed by toluene.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
R.P. thanks DAAD for a scholarship. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007‐2013)/ERC Grant Agreement no. 617044 (SunCatChem).
Q. Lefebvre, R. Porta, A. Millet, J. Jia, M. Rueping, Chem. Eur. J. 2020, 26, 1363.
References
- 1.
- 1a. Shi L., Xia W., Chem. Soc. Rev. 2012, 41, 7687; [DOI] [PubMed] [Google Scholar]
- 1b. Xi Y., Yi H., Wei A., Org. Biomol. Chem. 2013, 11, 2387; [DOI] [PubMed] [Google Scholar]
- 1c. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Beatty J. W., Stephenson C. R. J., Acc. Chem. Res. 2015, 48, 1474; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Teegardin K., Day J. I., Chan J., Weaver J., Org. Process Res. Dev. 2016, 20, 1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Lowry M. S., Goldsmith J. I., Slinker J. D., Rohl R., R. A. Pascal Jr. , Malliaras G. G., Bernhard S., Chem. Mater. 2005, 17, 5712; [Google Scholar]
- 2b. Crutchley R. J., Lever A. B. P., J. Am. Chem. Soc. 1980, 102, 7128. [Google Scholar]
- 3. Flamigni L., Barbieri A., Sabatini C., Ventura B., Barigelletti F., Top. Curr. Chem. 2007, 281, 143. [Google Scholar]
- 4.
- 4a. Corey E. J., Seebach D., Angew. Chem. Int. Ed. Engl. 1965, 4, 1075; [Google Scholar]; Angew. Chem. 1965, 77, 1134; [Google Scholar]
- 4b. Corey E. J., Seebach D., Angew. Chem. Int. Ed. Engl. 1965, 4, 1077; [Google Scholar]; Angew. Chem. 1965, 77, 1136; [Google Scholar]
- 4c. Stork G., Maldonado L., J. Am. Chem. Soc. 1971, 93, 5286; [Google Scholar]
- 4d. Seebach D., Angew. Chem. Int. Ed. Engl. 1979, 18, 239; [Google Scholar]; Angew. Chem. 1979, 91, 259; [Google Scholar]
- 4e. A. B. Smith III , Xian M., J. Am. Chem. Soc. 2006, 128, 66; [DOI] [PubMed] [Google Scholar]
- 4f. Fischer C., Smith S. W., Powell D. A., Fu G. C., J. Am. Chem. Soc. 2006, 128, 1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Finkbeiner P., Nachtsheim B., Nachr. Chem. 2015, 63, 1089; [Google Scholar]
- 5b. Tang S., Zhang Z., Sun J., Niu D., Chruma J. J., Chem. Rev. 2018, 118, 10393. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Zhu Y., Buchwald S. L., J. Am. Chem. Soc. 2014, 136, 4500; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Li M., Yücel B., Adrio J., Bellomo A., Walsh P. J., Chem. Sci. 2014, 5, 2383; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Matsumoto M., Harado M., Yamashita Y., Kobayashi S., Chem. Commun. 2014, 50, 13041; [DOI] [PubMed] [Google Scholar]
- 6d. Wu Y., Hu L., Li Z., Deng L., Nature 2015, 523, 445; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Cao C.-G., He B., Fu Z., Niu D., Org. Process Res. Dev. 2019, 23, 1758–1761. [Google Scholar]
- 7.
- 7a. Barluenga J., Sanz R., Fañanás F. J., J. Org. Chem. 1997, 62, 5953; [Google Scholar]
- 7b. Machrouhi F., Namy J.-L., Tetrahedron Lett. 1999, 40, 1315; [Google Scholar]
- 7c. Masson G., Py S., Vallée Y., Angew. Chem. Int. Ed. 2002, 41, 1772; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1850; [Google Scholar]
- 7d. Takahashi M., Micalizio G. C., J. Am. Chem. Soc. 2007, 129, 7514; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Takahashi M., Micalizio G. C., Chem. Commun. 2010, 46, 3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Root D. K., Smith W. H., J. Electrochem. Soc. 1982, 129, 1231. [Google Scholar]
- 9. Nakajima M., Fava E., Loescher S., Jiang Z., Rueping M., Angew. Chem. Int. Ed. 2015, 54, 8828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8952. [Google Scholar]
- 10.For Reviews on the use of α-aminoalkyl radicals in synthesis, see:
- 10a. Renaud P., Giraud L., Synthesis 1996, 913; [Google Scholar]
- 10b. Cossy J. in Radicals in Organic Synthesis, Vol. 1 (Eds.: P. Renaud, M. P. Sibi), Wiley-VCH, Weinheim, 2001, pp. 229–249; [Google Scholar]
- 10c. Aurrecoechea J. M., Suero R., ARKIVOC 2004, 14, 10; [Google Scholar]
- 10d. Yoon U. C., Mariano P. S., J. Photosci. 2003, 10, 89; [Google Scholar]
- 10e. Nakajima K., Miyake Y., Nishibayashi Y., Acc. Chem. Res. 2016, 49, 1946. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Reece S. Y., Hodgkiss J. M., Stubbe J., Nocera D. G., Philos. Trans. R. Soc. London Ser. B 2006, 361, 1351; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Weinberg D. R., Gagliardi C. J., Hull J. F., Murphy C. F., Kent C. A., Westlake B. C., Paul A., Ess D. H., McCafferty D. G., Meyer T. J., Chem. Rev. 2012, 112, 4016. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Knowles R., Yayla H., Synlett 2014, 2819; [Google Scholar]
- 12b. Gentry E. C., Knowles R. R., Acc. Chem. Res. 2016, 49, 1546; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Miller D. C., Tarantino K. T., Knowles R. R., Top. Curr. Chem. 2016, 374, 30; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Hoffmann N., Eur. J. Org. Chem. 2017, 1982. [Google Scholar]
- 13.For reductive coupling of imine derivatives with electron-deficient olefins using metals, see:
- 13a. Masson G., Cividino P., Py S., Vallée Y., Angew. Chem. Int. Ed. 2003, 42, 2265; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 2367; [Google Scholar]
- 13b. Yeh C.-H., Korivi R. P., Cheng C.-H., Angew. Chem. Int. Ed. 2008, 47, 4892; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4970; [Google Scholar]
- 13c.For photoredox-catalyzed radical cross-couplings involving imine derivatives, see: Hager D., MacMillan D. W. C., J. Am. Chem. Soc. 2014, 136, 16986; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Jeffrey J. L., Petronijević F. R., MacMillan D. W. C., J. Am. Chem. Soc. 2015, 137, 8404; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13e. Uraguchi D., Kinoshita N., Kizu T., Ooi T., J. Am. Chem. Soc. 2015, 137, 13768; [DOI] [PubMed] [Google Scholar]
- 13f. Fava E., Millet A., Nakajima M., Loescher S., Rueping M., Angew. Chem. Int. Ed. 2016, 55, 6776; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6888; [Google Scholar]
- 13g. Chen M., Zhao X., Yang C., Xia W., Org. Lett. 2017, 19, 3807. [DOI] [PubMed] [Google Scholar]
- 13h.For photoredox-catalyzed addition of imines onto vinyl sulfones or vinyl pyridines, see: Qi L., Chen Y., Angew. Chem. Int. Ed. 2016, 55, 13312; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13506; [Google Scholar]
- 13i. Lee K. N., Lei Z., Ngai M.-Y., J. Am. Chem. Soc. 2017, 139, 5003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13j. Cao K., Tan S. M., Lee R., Yang S., Jia H., Zhao X., Qiao B., Jiang Z., J. Am. Chem. Soc. 2019, 141, 5437. [DOI] [PubMed] [Google Scholar]
- 13k.For a photoredox-catalyzed three component reaction, see: Fuentes de Arriba A. L., Urbitsch F., Dixon D. J., Chem. Commun. 2016, 52, 14434. [DOI] [PubMed] [Google Scholar]
- 14.For a photoredox-catalyzed reaction between in situ-generated imines and dehydroalanine derivatives, see: Rossolini T., Leitch J. A., Grainger R., Dixon D. J., Org. Lett. 2018, 20, 6794. [DOI] [PubMed] [Google Scholar]
- 15. Nakajima M., Lefebvre Q., Rueping M., Chem. Commun. 2014, 50, 3619. [DOI] [PubMed] [Google Scholar]
- 16. Roberts B. P., Chem. Soc. Rev. 1999, 28, 25. [Google Scholar]
- 17.The formation of F was detected by 13C NMR analysis of the crude reaction mixture (resonance at δ C=97.0 ppm).
- 18. Furst L., Matsuura B. S., Narayanam J. M. R., Tucker J. W., Stephenson C. R. J., Org. Lett. 2010, 12, 3104. [DOI] [PubMed] [Google Scholar]
- 19.See Supporting Information for optimization studies and investigations about the reaction mechanism.
- 20.
- 20a. Atodiresei I., Vila C., Rueping M., ACS Catal. 2015, 5, 1972; [Google Scholar]
- 20b. Rueping M., Vila C., Bootwicha T., ACS Catal. 2013, 3, 1676; [Google Scholar]
- 20c. Chiroli V., Benaglia M., Cozzi F., Puglisi A., Annunziata R., Celentano G., Org. Lett. 2013, 15, 3590; [DOI] [PubMed] [Google Scholar]
- 20d. Puglisi A., Benaglia M., Chiroli V., Green Chem. 2013, 15, 1790; [Google Scholar]
- 20e. Rueping M., Bootwicha T., Sugiono E., Beilstein J. Org. Chem. 2012, 8, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Matsushita Y., Ichimura T., Ohba N., Kumada S., Sakeda K., Suzuki T., Tanibata H., Murata T., Pure Appl. Chem. 2007, 79, 1959; [Google Scholar]
- 21b. Coyle E. E., Oelgemöller M., Photochem. Photobiol. Sci. 2008, 7, 1313; [DOI] [PubMed] [Google Scholar]
- 21c. Oelgemöller M., Shvydkiv O., Molecules 2011, 16, 7522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21d. Oelgemöller M., Murata A., Med. Chem. News 2012, 4, 30; [Google Scholar]
- 21e. Knowles J. P., Elliott L. D., Booker-Milburn K. I., Beilstein J. Org. Chem. 2012, 8, 2025; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21f. Oelgemoeller M., Chem. Eng. Technol. 2012, 35, 1144; [Google Scholar]
- 21g. Shvydkiv O., Oelgemöller M., Microphotochemistry: Photochemical Synthesis in Microstructed Flow Reactors in CRC Handbook of Organic Photochemistry and Photobiology (Eds.: A. Griesbeck, M. Oelgemöller, F. Ghetti), CRC Press, Taylor & Francis Group, New York, 2012, Chapter 6, pp. 125; [Google Scholar]
- 21h. Tucker J. W., Zhang Y., Jamison T. F., Stephenson C. R. J., Angew. Chem. Int. Ed. 2012, 51, 4144; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4220; [Google Scholar]
- 21i. Amii H., Nagaki A., Yoshida J., Beilstein J. Org. Chem. 2013, 9, 2793; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21j. Su Y., Straathof N. J. W., Hessel V., Noel T., Chem. Eur. J. 2014, 20, 10562; [DOI] [PubMed] [Google Scholar]
- 21k. Lefebvre Q., Hoffmann N., Rueping M., Chem. Commun. 2016, 52, 2493. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Carter C. F., Lange H., Ley S. V., Baxendale I. R., Wittkamp B., Goode J. G., Gaunt N. L., Org. Process Res. Dev. 2010, 14, 393; [Google Scholar]
- 22b. Malet-Sanz L., Madrzak J., Ley S. V., Baxendale I. R., Org. Biomol. Chem. 2010, 8, 5324; [DOI] [PubMed] [Google Scholar]
- 22c. Lange H., Carter C. F., Hopkin M. D., Ley S. V., Chem. Sci. 2011, 2, 765; [Google Scholar]
- 22d. Fabry D. C., Sugiono E., Rueping M., React. Chem. Eng. 2016, 1, 129; [Google Scholar]
- 22e. Fabry D., Ho Y. A., Zapf R., Tremel W., Panthöfer M., Rueping M., Rehm T. H., Green Chem. 2017, 19, 1911; [Google Scholar]
- 22f. Cai Y., Tang Y., Fan L., Lefebvre Q., Hou H., Rueping M., ACS Catal. 2018, 8, 9471; [Google Scholar]
- 22g. Fabry D. C., Heddrich S., Sugiono E., Liauw M. A., Rueping M., React. Chem. Eng. 2019, 4, 1486. [Google Scholar]
- 23.
- 23a. Cano-Yelo H., Deronzier A., Tetrahedron Lett. 1984, 25, 5517; [Google Scholar]
- 23b. Hu J., Wang J., Nguyen T. H., Zheng N., Beilstein J. Org. Chem. 2013, 9, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rueping M., Vila C., Szadkowska A., Koenigs R., Fronert J., ACS Catal. 2012, 2, 2810. [Google Scholar]
- 25.
- 25a. Nguyen J. D., Tucker J. W., Konieczynska M. D., Stephenson C. R. J., J. Am. Chem. Soc. 2011, 133, 4160; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Wallentin C.-J., Nguyen J. D., Finkbeiner P., Stephenson C. R. J., J. Am. Chem. Soc. 2012, 134, 8875; [DOI] [PubMed] [Google Scholar]
- 25c. Narayanam J. M. R., Tucker J. W., Stephenson C. R. J., J. Am. Chem. Soc. 2009, 131, 8756. [DOI] [PubMed] [Google Scholar]
- 26. Lambert F. L., J. Org. Chem. 1966, 31, 4184. [Google Scholar]
- 27.
- 27a. Harvey I. W., Phillips E. D., Whitham G. H., J. Chem. Soc. Chem. Commun. 1990, 481; [Google Scholar]
- 27b. Larraufie M.-H., Pellet R., Fensterbank L., Goddard J.-P., Lacôte E., Malacria M., Ollivier C., Angew. Chem. Int. Ed. 2011, 50, 4463; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4555; [Google Scholar]
- 27c. Donck S., Baroudi A., Fensterbank L., Goddard J.-P., Ollivier C., Adv. Synth. Catal. 2013, 355, 1477; [Google Scholar]
- 27d. Baralle A., Fensterbank L., Goddard J.-P., Ollivier C., Chem. Eur. J. 2013, 19, 10809; [DOI] [PubMed] [Google Scholar]
- 27e. Zhang J., Li Y., Zhang F., Hu C., Chen Y., Angew. Chem. Int. Ed. 2016, 55, 1904; [Google Scholar]; Angew. Chem. 2016, 128, 1936. [Google Scholar]
- 28.
- 28a. Ichikawa J., Lapointe G., Iwai Y., Chem. Commun. 2007, 2698; [DOI] [PubMed] [Google Scholar]
- 28b. Pandey M. K., Disai A., Pandey A., Singh V. K., Tetrahedron Lett. 2005, 46, 5039. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary