

Abstract
Carbon dots (CDs) have been used for the first time as a sensitizer to initiate and activate free radical and controlled radical polymerization, respectively, based on an ATRP protocol with blue LEDs. Consideration of diverse heteroatom‐doped CDs indicated that N‐doped CDs could serve as an effective photocatalyst and photosensitizer in combination with LEDs emitting either at 405 nm or 470 nm. Free radical polymerization was initiated by combining the CDs with an iodonium or sulfonium salt in tri(propylene glycol) diacrylate. Polymerization of methyl methacrylate (MMA) by photo‐induced ATRP was achieved with CDs and ethyl α‐bromophenylacetate using CuII as catalyst in the ppm range. The polymers obtained showed temporal control, narrower dispersity ≲1.5, and chain‐end fidelity. The first‐order kinetics and ON/OFF experiments additionally gave evidence of the constant concentration of polymer radicals. No remarkable cytotoxic activity was observed for the CDs, underlining their biocompatibility.
Keywords: blue LEDs, carbon dots, cytotoxicity, free radical photopolymerization, photo-ATRP
On the dot: Carbon dots isolated from seaweed work well as a photocatalyst and sensitizer in living and free radical polymerization, while their low cytotoxic activity makes them interesting for medical applications.
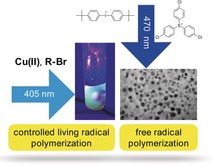
Photoinitiated polymerization with LEDs has received increased attention in many areas.1 Owing to specific regulatory requirements, academia and industry are replacing existing mercury‐based light sources with energy‐saving and environmentally friendly light sources such as LEDs.2 Interest has focused on UV LEDs and blue‐emitting LEDs owing to their use in 2D and 3D printing,3 coatings,4 and dentistry.5 Both free radical polymerization and controlled/living radical polymerization (CRP) have received increased interest in this field.1a The latter enables the synthesis of polymers with precise average molar masses, various compositions, and well‐defined structures. Numerous examples of photomediated CRP techniques have been successfully developed, including photo‐NMP,6 photo‐RAFT,1a, 1c, 1e, 7 and photo‐ATRP.1, 8 Regarding the latter, the in situ photolytic generation of CuI complexes from CuII under irradiation as an initiator for controlled polymerization facilitates CRP with a copper concentration in the ppm range.9 This has been successfully demonstrated with systems requiring UV,9d, 9e visible,9b, 9c or NIR9a light.
Some challenges still remain. Many photosensitizers and photocatalysts used for photo‐ATRP are based on synthetic materials that are accessible by synthetic routes with no sustainable feedback. Certainly, there is a demand for a sustainable photocatalyst or photosensitizer with negligible toxicological issues as well. From this point of view, materials derived from natural feedstocks would be highly suitable. Therefore, carbon dots (CDs) made from natural materials should present one feasible opportunity. This is the first report considering CDs as a photoinitiating system for both free radical and CRP based on ATRP with blue‐emitting LEDs. The manufacture of such materials from sodium alginate10 provides cheap photosensitive carbon dots at a cost (0.95 €/100 g CDs) that is competitive with the cost of current industrial photoinitiators based on petrochemicals (2 €/100 g TPO‐based photoinitiator; TPO=trimethylbenzoyl‐diphenyl‐phosphine oxide).
CDs have been known as novel fluorescent carbon nanomaterials exhibiting sizes below 10 nm.10 Photoluminescent CDs with ultrastable photoluminescence, easy preparation, and sustainable raw materials, have been employed in numerous areas including bioimaging,11 optoelectronics,12 and photocatalysis.13 CDs also demonstrate interesting electrochemical redox properties.14
Recently, CDs also received attention for photosensitized RAFT polymerization,15 which was the first report regarding their use in a photocontrolled radical polymerization. Exposure to visible light as well as sunlight resulted in low dispersities, generally around 1.1. On the other hand, to the best of our knowledge there has not been any report regarding the use of CDs either as a sensitizer for LED‐initiated free radical photopolymerization using iodonium salts or as a photocatalyst in photo‐ATRP protocols.
The aforementioned arguments regarding LED‐initiated polymerization have moved CDs into the focus of radiation curing. They might become a work horse for sensitizing photoinitiated radical polymerizations because of their photonic16 and electrochemical properties.14 They assume the function of a photosensitizer in combination with radical initiators such as onium salts.17 In particular, iodonium salts and sulfonium salts comprising low‐coordinating anions have received increased attention.17 According to the electrochemical properties reported previously,14 CDs may also assume a major function in photo‐ATRP protocols, as previously reported for metal‐free systems8 and CuII‐catalyzed systems, which required a heavy metal concentration in the ppm range.9 CuII‐catalyzed systems may serve as a general approach to synthesize tailor‐made polymers with a broader available range of LED sources.
Herein we use sodium alginate, a biopolymer obtained from brown seaweed, along with ethylenediamine to prepare carbon dots via a hydrothermal method. The synthetic details were reported previously.18, 19 Figure 1 shows a TEM image of the CDs synthesized. They exhibit a size <10 nm. Furthermore, X‐ray photoelectron spectroscopy (XPS) analysis showed that the N‐CDs contained Na (≈8 %), C (≈72 %), N (≈5 %), and O (≈13 %). High‐resolution scans on C, O, and N elements of N‐CDs exhibited signals for C=O, C−O, and O−Na, indicating that N‐CDs comprised carboxylic sodium moieties. Furthermore, the C−N−C signal of N‐CDs suggested that the hydrothermal treatment formed also amide moieties.16c Noteworthy, the CDs made from sodium alginate showed the best performance, while those synthesized from poly(acrylic) acid, phytic acid, or carboxylic sodium cellulose did not perform well for the purposes of this study, although they exhibit similar morphology (see Figure S1 in the Supporting Information).
Figure 1.
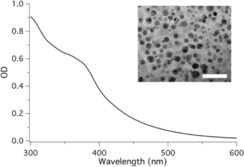
UV/Vis spectra of CDs in DMSO. Inset shows TEM images of CDs (scale bar=20 nm).
Photoexcited CDs made from sodium alginate facilitated free radical photopolymerization of tri(propylene glycol) diacrylate (TPGDA) in combination with onium salts serving as radical initiator in a photoinduced electron transfer protocol. TPGDA was the monomer of choice since it was compatible with photoinitiator components in previous investigations.17a Before the experiment, the biocompatibility of the CDs was investigated to ensure that no toxic product would be present in the photopolymerization studies. Up to a concentration of 400 μg mL−1 CDs did not show obvious inhibition of cell growth indicating their nice biocompatibility and green properties (see Figure S2 in the Supporting Information). The absorption of the CDs (size=5–10 nm, Figure 1 inset) covered the blue spectral range, facilitating the use of blue LEDs (Figure 1). The oxidation potential of 0.40 V (see Figure S3 in the Supporting Information) and the excitation energy of the CD at the wavelengths chosen facilitate the photolytic reduction of iodonium (1) and sulfonium (2) cations, which exhibit a reduction potential of −0.6 V and −1.1 V, respectively.17c This results in a negative free enthalpy for the photoinduced electron transfer in the case of both onium ions. This thermodynamic quantity indicates sufficient generation of initiating radicals in the sensitized free radical polymerization.2 The selected anions were aluminates (a)17b, 17c and bis(trifluoromethylsulfonyl) imides (b)17a, 17c to maintain the advantages of the initiator disclosed previously.17a These anions have received increasing attention as the counterions of onium salts because they do not generate HF, which can arise from the PF6 − anion under certain conditions17 (see refs.17a, 17c and references therein). Scheme 1 discloses the structures of the cations (1 and 2) and anions (a and b).
Scheme 1.
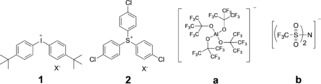
Structures of onium cations and anions used in this work.
Figure 2 shows the polymerization rate R p of TPGDA as determined by photo‐DSC (online determination of the heat flux by differential scanning calorimetry of polymerization initiated by light) employing a 405 nm LED. The results indicate a certain reactivity, as shown by the acceptable maximum for the polymerization rate, R p max. This does not differ significantly for the different salts though the molar concentration of 1 a was lower. The sulfonium salt 2 a exhibits less reactivity but the time to reach the maximum of R p max (t max) appears at a similar time frame, indicating similar efficiency. At R p max gelation and vitrification occur with equal efficiency, regardless of which salt served as the initiator.
Figure 2.
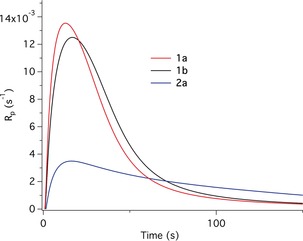
Polymerization as a function of exposure time obtained in the free radical polymerization of TPGDA using CDs with different onium salts in combination with a blue LED emitting at 405 nm (light intensity: 100 mW cm−2).
Replacement of the LED emitting at 405 nm by an LED exhibiting emission at 470 nm resulted in both a similar final conversion and R p max, although the t max was significantly longer and accompanied by a remarkable inhibition time (Table 1). It is remarkable that the reactivity (R p max) is similar, since the light source emitting at 470 nm exhibits significantly lower intensity, i.e. 0.5 mW cm−2. Such events usually scale with a power law, where the exponent of the light intensity ranges from 0.1 and 1 depending on the monomer, matrix binder, and conversion.20 Again, such systems might be of interest for applications requiring light exposure that excludes UV‐photons, for example in dentistry,5 because the comparable R p max observed at 405 nm and 470 nm at different intensities should result in similar cross‐linking density.
Table 1.
Blue LED (405 nm and 470 nm) initiated free radical polymerization of TPGDA with onium salts 1 a, 1 b, and 2 a with CDs.
|
Onium salt[a] |
R p max [s−1] |
t max [s] |
x ∞ |
λ exc [nm] |
|---|---|---|---|---|
|
1 a |
13.5 |
12.8 |
0.59 |
405 |
|
1 b |
12.5 |
16.8 |
0.64 |
405 |
|
2 a |
3.5 |
16.3 |
0.41 |
405 |
|
1 a |
12.1 |
597 (414)[a] |
0.64 |
470 |
[a] Inhibition time.
In the context of this contribution we extended our studies to living radical photopolymerizations based on ATRP. Protocols obtained for NIR irradiation were transferred to blue‐emitting LEDs.9a Scheme 2 proposes a possible reaction mechanism. Photoexcitation of the CD at 405 nm results in the population of its excited state CD*. Electron transfer between CD* and [CuIIL]Br2 results in reduction to the respective CuI complex, which initiates the living radical polymerization of alkyl bromides. The oxidized CD (CD+.) reacts with bromide in the photocatalytic cycle, resulting in regeneration of the CD. This is similar to the mechanism previously discussed for photo‐ATRP initiated by NIR exposure.9a
Scheme 2.
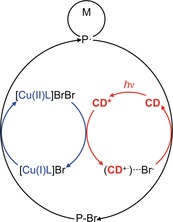
Proposed simplified mechanism of photoinduced ATRP using CDs as the photosensitizer, omitting chain termination.
The ability of CDs to mediate photoinduced ATRP was examined by irradiating solutions of CDs in DMSO with ethyl α‐bromophenylacetate (EBPA) as the alkyl halide source together with the CuBr2/tris(2‐pyridylmethyl)amine (TPMA) complex as the catalyst at 405 nm for the polymerization of methyl methacrylate (MMA) under reduced pressure under different polymerization conditions (Table 2). CDs showed initiation of living radical polymerization with acceptable conversions that could be explained by the favorable redox conditions to reduce CuII (vide supra). The polymers obtained with CDs showed narrower molecular weight characteristics (runs 1 and 2). To gain more insight into the polymerization mechanism, similar experiments were conducted in the absence of CDs and the CuII catalyst as shown by the respective runs 3 and 4. Run 4 proceeded via a typical free radical photopolymerization mechanism resulting in very high molecular weight and large dispersity. This demonstrates the necessity to operate with a catalytic amount of CuII in the ppm range in order to achieve living radical polymerization. However, no polymer was obtained in the absence of CDs. In addition, a longer irradiation time with a lower concentration of CDs (run 5) resulted in no polymer.
Table 2.
UV light‐induced ATRP of MMA using CDs under different experimental conditions (t=exposure time, L=TPMA; tris(2‐pyridylmethyl)amine)[a]
|
Run |
[MMA]/[RX]/[CD]/[CuBr2]/[L] |
t [h] |
x [b] [%] |
M n [c] [g mol−1] |
Đ [c] |
|---|---|---|---|---|---|
|
1 |
300:1:0.3:0.03:0.135 |
0.5 |
5.5 |
23 000 |
1.3 |
|
2 |
300:1:0.6:0.03:0.135 |
2.5 |
30 |
6000 |
1.2 |
|
3 |
300:1:0:0.03:0.135 |
1 |
– |
– |
– |
|
4 |
300:1:0.3:0:0.135 |
1 |
<1 |
40 0000 |
2.4 |
|
5 |
300:1:0.15:0.03:0.135 |
4 |
– |
– |
– |
[a] Polymerization experiments were performed at 405 nm at room temperature. [b] Conversion (x) determined gravimetrically. [c] Determined by gel permeation chromatography using PMMA standards.
Figure 3 depicts the GPC trace obtained for the photo‐ATRP after 30 min exposure. The higher dispersity Ð of 1.3 indicates the occurrence of chain‐terminating reactions. Longer exposure times resulted in an additional shoulder (compare Figure S4 in the Supporting Information) which was not evident at short irradiation times (Figure 3). These results were reproduced several times, resulting in GPC curves exhibiting a dispersity of 1.2 and M n of 6000 g mol−1 . (The 1H NMR spectrum of the polymer is shown in Figure S5 in the Supporting Information). More investigations would be necessary to discuss whether the heterogeneity of the CDs caused this change or whether chain termination became more important at higher conversion, resulting in the molecular weight distribution shown in Figure S4 in the Supporting Information.
Figure 3.
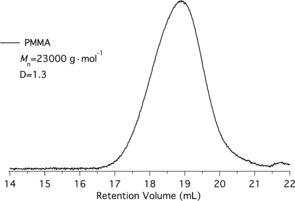
GPC trace of PMMA obtained after 30 min exposure. Conditions applied correspond to run 1 in Table 2.
Furthermore, light on–off experiments were performed under experimental conditions identical to those in Table 2. For this purpose, the polymerization mixture was exposed to repeated cycles of LED exposure, in which the sample was irradiated for 30 min and then kept in dark for 30 min. The results obtained demonstrated the polymerization its ultimate irradiation dependence. No polymerization occurred when the solutions were kept in the dark (Figure 4).
Figure 4.
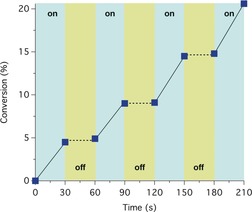
Monomer conversion (%) vs. time using CDs to determine the dependency on irradiation: light on (blue regions) light off (yellow regions) using the following concentration ratio: [MMA]0/[EBPA]0/[CuIIBr2]0/[TPMA]/[CDs]=300:1:0.03:0.135:0.3.
For further insight into the polymerization kinetics, several experiments were conducted to confirm the linear increase in the conversion during the irradiation time. A linear relationship between logarithm of monomer concentration ln([M]0/[M]) and time t starting at t=0 indicated first‐order kinetics with respect to the monomer in this system comprising CDs as photocatalyst while the concentration of radicals can be seen as constant21 (Figure 5).
Figure 5.
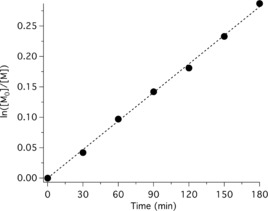
Kinetic plot of the polymerization system using CDs. ([MMA]0/[EBPA]0/[CuIIBr2]0/[TPMA]/[CDs]/=300:1:0.03:0.135:0.3).
Similar experiments were also performed without switching off the light, and the polymers obtained at each time interval were analyzed by GPC. GPC measurements indicated a molecular weight increase upon irradiation while the dispersities resided around 1.5 (Figure 6). Several factors can explain the higher dispersity. Chain termination may explain the higher dispersities obtained.22 Previous investigations carried out with up‐conversion nanoparticles also resulted in higher dispersities, and the heterogeneous nature of the photocatalyst was discussed as one possible reason.23 Nevertheless, this topic remains the subject of future experiments, which should give more detailed insight about the function of CDs as photocatalyst. Despite the fascinating properties of CDs, there are still many unanswered questions regarding their detailed structure and functionality.
Figure 6.
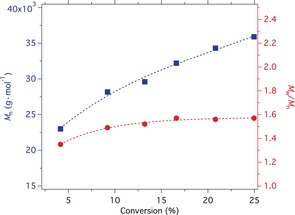
Molecular weight characteristics and dispersity of PMMA vs. irradiation time. ([MMA]0/[EBPA]0/[CuIIBr2]0/[TPMA]/[CDs]/=300:1:0.03:0.135:0.3).
Finally, a block copolymerization reaction confirmed the chain‐end functionality of this material. The polymer obtained under identical experimental conditions functioned also as macroinitiator. GPC showed shift of the maximum of the molecular weight of PMMA after restarting with styrene from 6000 g mol−1 for PMMA (run 2 in Table 2) to 12 000 g mol−1 for PMMA‐b‐PSt. Thus, the isolated block copolymer confirmed the chain‐end functionality. The appearing shoulder of the PMMA precursor (Figure S4) overlays with the shifted molecular weight of the block copolymer, resulting in the broad peak shown in Figure S6 in the Supporting Information. Furthermore, the Br‐CH2 signal appearing in the 1H NMR spectrum between 4–4.5 ppm also supported the expected end group in the styrene moiety of the block copolymer.
These examples complement the general picture of CDs with regard to their use as photocatalysts in particular for photo‐ATRP. Since these materials do not exhibit remarkable cytotoxic activity, they can be used as photosensitizers in free radical photopolymerization as shown in this first report. Medical applications will definitively benefit from these advantages. Additional work might focus on shifting the absorption to higher wavelengths, which would provide access to diode lasers emitting in the near‐infrared region. This would link such systems further technologies such as coatings, imaging, and printing based on these sources. Future work should focus on structural investigations and the improving the compatibility with traditional polymerizing systems. This will definitively lead to a deeper understanding of CDs in polymerizing systems.
Experimental Section
The supporting information summarizes information on materials used, synthesis, UV/Vis measurements of CDs, general procedure for photoinduced ATRP of MMA, block copolymerization experiment (PMMA‐b‐PSt), light on–off experiments, kinetic studies of the polymerization, instrumentation, gel permeation chromatography (GPC), NMR spectroscopy, UV/Visible spectroscopy, and photo‐DSC experiments.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
C.Z. acknowledges the National Natural Science Foundation of China (grant 31890774 and 31800494) and the Young Elite Scientists Sponsorship Program (CAST, grant 2018QNRC001). B.S. thanks the state of North Rhine‐Westphalia for funding the project REFUBELAS (grant 005‐1703‐0006, managed by PTJ). B.S. also acknowledges the Project D‐NL‐HIT carried out in the framework of the INTERREG‐Program Deutschland‐Nederland, which is cofinanced by the European Union, the MWIDE of NRW, the Ministerie van Economische Zaken en Klimaat, and the provinces of Limburg, Gelderland, Noord‐Brabant, and Overijssel.
C. Kütahya, P. Wang, S. Li, S. Liu, J. Li, Z. Chen, B. Strehmel, Angew. Chem. Int. Ed. 2020, 59, 3166.
Contributor Information
Prof. Dr. Zhijun Chen, Email: chenzhijun@nefu.edu.cn.
Prof. Dr. Bernd Strehmel, Email: bernd.strehmel@hs-niederrhein.de.
References
- 1.
- 1a. Dadashi-Silab S., Doran S., Yagci Y., Chem. Rev. 2016, 116, 10212–10275; [DOI] [PubMed] [Google Scholar]
- 1b. Seidler K., Griesser M., Kury M., Harikrishna R., Dorfinger P., Koch T., Svirkova A., Marchetti-Deschmann M., Stampfl J., Moszner N., Gorsche C., Liska R., Angew. Chem. Int. Ed. 2018, 57, 9165–9169; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9305–9310; [Google Scholar]
- 1c. Corrigan N., Yeow J., Judzewitsch P., Xu J., Boyer C., Angew. Chem. Int. Ed. 2019, 58, 5170–5189; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5224–5243; [Google Scholar]
- 1d. Quéméner F., Subervie D., Morlet-Savary F., Lalevée J., Lansalot M., Bourgeat-Lami E., Lacôte E., Angew. Chem. Int. Ed. 2018, 57, 957–961; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 969–973; [Google Scholar]
- 1e. Pan X., Tasdelen M. A., Laun J., Junkers T., Yagci Y., Matyjaszewski K., Prog. Polym. Sci. 2016, 62, 73–125. [Google Scholar]
- 2. Strehmel B., Schmitz C., Cremanns K., Göttert J., Chem. Eur. J. 2019, 25, 12855–12864. See also cited references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Ligon S. C., Liska R., Stampfl J., Gurr M., Mülhaupt R., Chem. Rev. 2017, 117, 10212–10290; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b.“Imaging Technology, 3. Imaging in Graphic Arts”: Baumann H., Hoffmann-Walbeck T., Wenning W., Lehmann H.-J., Simpson C. D., Mustroph H., Stebani U., Telser T., Weichmann A., Studenroth R., in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015, pp. 1–51. [Google Scholar]
- 4. Schmitz C., Pang Y., Gülz A., Gläser M., Horst J., Jäger M., Strehmel B., Angew. Chem. Int. Ed. 2019, 58, 4400–4404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4445–4450. see also cited references therein. [Google Scholar]
- 5.
- 5a. Palin W. M., Leprince J. G., Hadis M. A., Dent. Mater. 2018, 34, 695–710; [DOI] [PubMed] [Google Scholar]
- 5b. Ikemura K., Endo T., Dental Mater. J. 2010, 29, 481–501; [DOI] [PubMed] [Google Scholar]
- 5c. Stansbury J. W., Idacavage M. J., Dental Mater. 2016, 32, 54–64. [DOI] [PubMed] [Google Scholar]
- 6. Corrigan N., Xu J., Boyer C., Macromolecules 2016, 49, 3274–3285. [Google Scholar]
- 7. McKenzie T. G., Fu Q., Uchiyama M., Satoh K., Xu J., Boyer C., Kamigaito M., Qiao G. G., Adv. Sci. 2016, 3, 1500394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Discekici E. H., Anastasaki A., Read de Alaniz J., Hawker C. J., Macromolecules 2018, 51, 7421–7434; [Google Scholar]
- 8b. Ribelli T. G., Konkolewicz D., Bernhard S., Matyjaszewski K., J. Am. Chem. Soc. 2014, 136, 13303–13312. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Kütahya C., Schmitz C., Strehmel V., Yagci Y., Strehmel B., Angew. Chem. Int. Ed. 2018, 57, 7898–7902; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8025–8030; [Google Scholar]
- 9b. Pan X., Malhotra N., Simakova A., Wang Z., Konkolewicz D., Matyjaszewski K., J. Am. Chem. Soc. 2015, 137, 15430–15433; [DOI] [PubMed] [Google Scholar]
- 9c. Konkolewicz D., Schröder K., Buback J., Bernhard S., Matyjaszewski K., ACS Macro Lett. 2012, 1, 1219–1223; [DOI] [PubMed] [Google Scholar]
- 9d. Tasdelen M. A., Uygun M., Yagci Y., Macromol. Rapid Commun. 2011, 32, 58–62; [DOI] [PubMed] [Google Scholar]
- 9e. Tasdelen M. A., Uygun M., Yagci Y., Macromol. Chem. Phys. 2011, 212, 2036–2042. [Google Scholar]
- 10.
- 10a. Lim S. Y., Shen W., Gao Z., Chem. Soc. Rev. 2015, 44, 362–381; [DOI] [PubMed] [Google Scholar]
- 10b. Zhang X., Jiang M., Niu N., Chen Z., Li S., Liu S., Li J., ChemSusChem 2018, 11, 11–24. [DOI] [PubMed] [Google Scholar]
- 11. Du J., Xu N., Fan J., Sun W., Peng X., Small 2019, 15, 1805087. [DOI] [PubMed] [Google Scholar]
- 12. Choi H., Ko S.-J., Choi Y., Joo P., Kim T., Lee B. R., Jung J.-W., Choi H. J., Cha M., Jeong J.-R., Hwang I.-W., Song M. H., Kim B.-S., Kim J. Y., Nat. Photonics 2013, 7, 732. [Google Scholar]
- 13.
- 13a. Liu J., Liu Y., Liu N., Han Y., Zhang X., Huang H., Lifshitz Y., Lee S.-T., Zhong J., Kang Z., Science 2015, 347, 970. [DOI] [PubMed] [Google Scholar]
- 14. Karthikeyan R., Nelson D. J., Ajith A., John S. A., J. Electroanal. Chem. 2019, 848, 113297. [Google Scholar]
- 15. Jiang J., Ye G., Wang Z., Lu Y., Chen J., Matyjaszewski K., Angew. Chem. Int. Ed. 2018, 57, 12037–12042; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12213–12218. [Google Scholar]
- 16.
- 16a. Xu X., Ray R., Gu Y., Ploehn H. J., Gearheart L., Raker K., Scrivens W. A., J. Am. Chem. Soc. 2004, 126, 12736–12737; [DOI] [PubMed] [Google Scholar]
- 16b. Sun Y.-P., Zhou B., Lin Y., Wang W., Fernando K. A. S., Pathak P., Meziani M. J., Harruff B. A., Wang X., Wang H., Luo P. G., Yang H., Kose M. E., Chen B., Veca L. M., Xie S.-Y., J. Am. Chem. Soc. 2006, 128, 7756–7757; [DOI] [PubMed] [Google Scholar]
- 16c. Wang P., Liu C., Tang W., Ren S., Chen Z., Guo Y., Rostamian R., Zhao S., Li J., Liu S., Li S., ACS Appl. Mater. Interfaces 2019, 11, 19301–19307. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Brömme T., Oprych D., Horst J., Pinto P. S., Strehmel B., RSC Adv. 2015, 5, 69915–69924; [Google Scholar]
- 17b. Klikovits N., Knaack P., Bomze D., Krossing I., Liska R., Polym. Chem. 2017, 8, 4414–4421; [Google Scholar]
- 17c. Kocaarslan A., Kütahya C., Keil D., Yagci Y., Strehmel B., ChemPhotoChem 2019, 3, 1127–1132. [Google Scholar]
- 18.
- 18a. Hu B., Wang K., Wu L., Yu S.-H., Antonietti M., Titirici M.-M., Adv. Mater. 2010, 22, 813–828; [DOI] [PubMed] [Google Scholar]
- 18b. Wang J., Wang C.-F., Chen S., Angew. Chem. Int. Ed. 2012, 51, 9297–9301; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9431–9435; [Google Scholar]
- 18c. Wang Y., Hu A., J. Mater. Chem. C 2014, 2, 6921–6939. [Google Scholar]
- 19. Tao S., Lu S., Geng Y., Zhu S., Redfern S. A. T., Song Y., Feng T., Xu W., Yang B., Angew. Chem. Int. Ed. 2018, 57, 2393–2398; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2417–2422. [Google Scholar]
- 20. Timpe H. J., Strehmel B., Makromol. Chem. 1991, 192, 779–791. [Google Scholar]
- 21. Stenzel M. H., Barner-Kowollik C., Mater. Horiz. 2016, 3, 471–477. [Google Scholar]
- 22.“General Concepts and History of Living Radical Polymerization”: Matyjaszewski K., in Handbook of Radical Polymerization (Eds.: K. Matyjaszewski, T. P. Davis), Wiley, Hoboken, 2002, chapter 8. [Google Scholar]
- 23. Chen Z., Oprych D., Xie C., Kutahya C., Wu S., Strehmel B., ChemPhotoChem 2017, 1, 499–503. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary