

Abstract
Oxidative aromatic coupling occupies a fundamental place in the modern chemistry of aromatic compounds. It is a method of choice for the assembly of large and bewildering architectures. Considerable effort was also devoted to applications of the Scholl reaction for the synthesis of chiral biphenols and natural products. The ability to form biaryl linkages without any prefunctionalization provides an efficient pathway to many complex structures. Although the chemistry of this process is only now becoming fully understood, this reaction continues to both fascinate and challenge researchers. This is especially true for heterocoupling, that is, oxidative aromatic coupling with the chemoselective formation of a C−C bond between two different arenes. Analysis of the progress achieved in this field since 2013 reveals that many groups have contributed by pushing the boundary of structural possibilities, expanding into surface‐assisted (cyclo)dehydrogenation, and developing new reagents.
Keywords: biaryls, Lewis acids, nanographenes, oxidative coupling, Scholl reaction
Coupling up: The dehydrogenative coupling of arenes is an ultimate example of C−H activation. This Review highlights the most exciting examples of the Scholl reaction, ranging from the synthesis of biphenols through natural products to nanographenes and on‐surface dehydrogenation reported since 2013.
1. Introduction
Oxidative aromatic coupling and the Scholl reaction, both discovered more than 100 years ago,1, 2, 3 have not lost any attraction to organic chemists in the last decade, despite the appearance of many modern C−H activation procedures. These reactions are often the methods of choice for the synthesis of large π‐extended scaffolds as well as smaller benzenoid or heteroaromatic compounds possessing biaryl linkages (Scheme 1). The Scholl reaction is undoubtedly the most useful for the construction of nanographenes4, 5 and their heterocyclic analogues.6 Indeed, in some cases, more than 100 C−C bonds are formed in one synthetic operation to furnish truly amazing polycyclic aromatic hydrocarbons from suitable precursors. There is no other reaction that can duplicate this result for a discrete molecule (i.e. not a polymer). However, despite ubiquitous utilization, the ability to foresee when and how this reaction will occur is still lacking. Indeed, the love‐hate relationship between synthetic organic chemists and dehydrogenative coupling is strongly related to the fact that it works with stunning efficiency in some cases, and that it fails in many others. Although we presented a comprehensive overview on the century‐long history of the Scholl reaction in 2013,3 a significant amount of experimental data in various versions of oxidative aromatic coupling has been accumulated within the last seven years. At the same time, these studies are increasingly accompanied by DFT calculations, which often, post‐factum, prove that the unanticipated course of a reaction really was to be expected as far as energies of transition states are concerned. Unfortunately, because of space limitations, not all the interesting results could be included in this Review. The photocyclodehydrogenation of stilbenes and derivatives of o‐terphenyls,7, 8 although very interesting and often complementary to the Scholl reaction, will not be described here for this reason. This Review is comprised of four sections that deal with intermolecular oxidative aromatic coupling, the intramolecular Scholl reaction, on‐surface (cyclo)dehydrogenation, and an outlook summarizing progress from 2013 until April 2019.
Scheme 1.
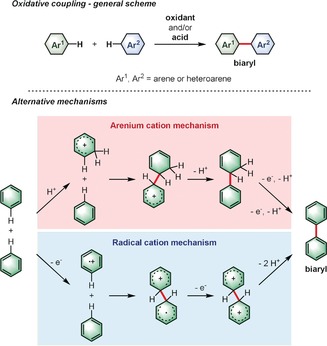
General scheme for the oxidative coupling of arenes through the two alternative, generally accepted, mechanisms using the formation of biphenyl as an example.
Scheme 1 depicts the general case of the oxidative coupling of arenes and explains the two generally accepted mechanisms: via arenium cation and radical cation intermediates.
2. Intermolecular Oxidative Aromatic Coupling
In the following section we will focus on key achievements in the field of intermolecular oxidative aromatic coupling that have appeared in the literature since our previous review was published in 2013. Occasional references to earlier work will be mentioned where appropriate.
2.1. Oxidative Homocoupling of Arenes and Heteroarenes
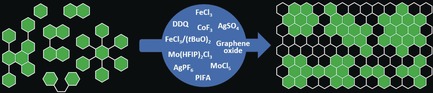
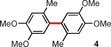
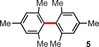
Among the numerous oxidants typically used for the construction of a new C−C bond in an oxidative manner, the most frequently applied is iron(III) chloride.9 New reagents are, however, continually proposed for homocoupling processes under oxidative conditions. Indeed, a graphene oxide (GO)/BF3⋅OEt2 system is able to promote oxidative C−H/C−H coupling between two electron‐rich arenes in a selective way.10 The utility of boron(III)‐based Lewis acids presumably relies on the activation of epoxy sites on a GO surface to form active radical species. EPR studies support a radical pathway for this transformation. An oxidative coupling process for simple 1‐ or 2‐substituted naphthalene derivatives can also proceed in the presence of AgSO4,11 albeit in moderate yields in most cases. Nevertheless, under these conditions, 1‐(trifluoromethyl)naphthalene gives 5,5′‐bis(trifluoromethyl)‐1,1′‐binaphthyl in 17 % yield, thus showing that the oxidizing power of silver(II)‐based systems can partially overcome the electronic limitations caused by an electron‐withdrawing CF3 group. The corresponding binaphthyls derived from 1‐nitro‐ and 1‐cyanonaphthalene are not formed under these conditions.
Molybdenum pentachloride is a selective, one‐electron oxidizing reagent frequently employed in many batch oxidative coupling processes,12 and more recently under continuous‐flow conditions.13 In most MoCl5‐mediated reactions of this type, undesired chlorination can be successfully avoided, as the C−C coupling is much faster than other side reactions. However, for highly electron‐rich substrates or when the coupling process proceeds slowly, chlorination becomes an issue.14 The application of MoCl5 together with a Lewis acid (usually TiCl4), which is believed to bind the coformed chloride anions, increases the overall efficiency of the C−C coupling,15 but this beneficial effect is not general. To solve this problem, the Waldvogel group designed16 the two dinuclear molybdenum(V) complexes 1 and 2 (Figure 1), which were prepared from MoCl5 and either 1,1,1,3,3,3‐hexafluoroisopropanol (HFIP) or 2,2,2‐trifluoroethanol (TFE) in 95 % and 97 % yield, respectively. According to electrochemical measurements and DFT calculations, only complex 1 has an oxidizing power comparable to that of MoCl5 [E p(MoCl5)=1.16 V; E p(1)=1.22 V, versus FcH/FcH+; FcH=ferrocene], while electron transfer from 3,4‐dimethoxytoluene to complex 2 should be strongly prevented because of the low potential value [E p(2)=0.31 V, versus FcH/FcH+] and the sterically congested coordination sphere. Problematic substrates were subjected to oxidative coupling reactions mediated by MoCl5, MoCl5/TiCl4, or complex 1 (Table 1). In all cases, the authors obtained considerably higher yields for reactions mediated by complex 1 compared with the other two systems and this fact was associated with such factors as the lower chlorine content of the reagent 1 (thus avoiding chlorination), the lower nucleophilicity of the HFIP ligands compared to chloride, and the considerably higher reaction rate of the C−C coupling compared with side processes induced by the higher oxidation potential of complex 1.
Figure 1.
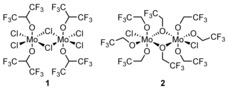
Structures of newly designed Mo‐based mediators for oxidative coupling.16
Table 1.
Comparison of the efficiencies of oxidative coupling reactions for different molybdenum(V)‐based systems.16
|
Compound |
Yield [%][a] |
|||
|---|---|---|---|---|
|
|
MoCl5 [c] |
MoCl5/TiCl4 [d] |
1 [e] |
|
|
|
|
47 |
41 |
60 |
|
|
|
67 |
44 |
82 |
|
|
|
34 |
<5[b] |
78 |
[a] Yields of isolated products. [b] Yield determined by GC. [c] 2.2 equiv of MoCl5. [d] 1.4–2.2 equiv of both MoCl5 and TiCl4. [e] 0.7–1.1 equiv of 1.
It is not only molybdenum(V) complexes that are useful mediators in the oxidative formation of new C−C bonds. An electrochemical system consisting of an active molybdenum anode, a graphite cathode, and HFIP as a mediator was found to be highly effective in the dehydrogenative homocoupling of simple electron‐rich arenes.17 The elaborated method tolerates a broad scope of arenes and is particularly important, especially for environmental reasons, as it does not generate a large amount of reagent waste.
An interesting discovery was made by Boyd and Sperry,18 who examined the influence of various oxidizing agents on the dimerization process of hemidendridine acetates of type 6 (Scheme 2). Under the influence of a wide range of typical oxidants [FeCl3⋅SiO2, Ag2O, Pb(OAc)4, K3[Fe(CN)6], PhI(OAc)2 (PIDA)] 6 decomposes, and no trace of a dimer is detected in the reaction mixture. Nevertheless, the 6,6′‐coupled, symmetrical dimer 8 is formed exclusively through the oxidative dimerization of 6 mediated by (tBuO)2 in toluene at 130 °C followed by protection of the hydroxy groups in 7. This is the result of an ortho‐ortho coupling, which is usually observed for phenols in the absence of steric hindrance19 or catalyst control.20 Completely different regioselectivity is observed if an indole precursor bears an iPrO substituent. Under Scholl‐type conditions, the best result was obtained with the MoCl5/TiCl4 system, thus 6 was successfully transformed into 4,4′‐bistryptamine 9 in 32 % yield. Besides 9, several other unsymmetrical products were isolated from the reaction mixture in a total yield of 14 %. In this case, the reaction outcome is undoubtedly governed by the bulky OiPr group, which enforces the dominant para–para coupling. Such a regioselectivity can also be achieved by blocking the ortho positions relative to the OH group, as was recently shown by Porco and co‐workers.21
Scheme 2.
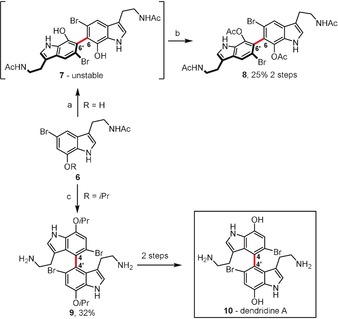
Reagents and conditions: a) (tBuO)2 (1.5 equiv), toluene, 130 °C; b) Ac2O, pyridine; c) MoCl5 (4 equiv), TiCl4 (4 equiv), 0 °C.
The oxidative dimerization of larger phenols represents one of the most convenient pathways toward complex 2,2′‐dihydroxybiaryl compounds. According to the Tsubaki group, butterfly‐shaped molecule 11 can be obtained by the CuII‐mediated dimerization of dinaphthofuran‐6‐ol (Figure 2).22 The dihedral angle between the two central naphthyl moieties can be altered by chemical modification of the central region; for example, acid‐catalyzed dehydration involving the central OH groups delivers an additional furan system, which in turn decreases the dihedral angle from 86.4° to 14.6°, as predicted by DFT methods. The protected 1,1′‐bi‐2‐pyrenol (12, Figure 2) can be prepared by the FeIII‐23 or CuII‐mediated24 oxidative dimerization of the protected precursor of 2‐pyrenol. After dimerization, diastereomers of type 12 can be effectively separated24 using conventional chromatography and then be deprotected to deliver optically pure 1,1′‐bi‐2‐pyrenols. Another complex phenol 13, based on the naphtho[2,1,8‐def]coumarin scaffold, was synthesized by employing a FeCl3/(tBuO)2 system.25 Numerous attempts to perform the second coupling process to generate a heterocyclic analogue of 2,4:9,11‐dinaphthoperylene were ineffective. In contrast, 3,6‐dibromo‐2,7‐dihydroxynaphthalene dimerizes selectively26 at C1 and C8 to form a mixture of dihydroxyperylenequinones, which after reduction and alkylation affords the octasubstituted perylene analogue 14.
Figure 2.
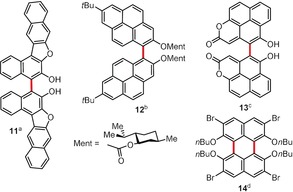
Reagents and conditions of oxidative dimerization: a) Cu(NO3)2⋅3 H2O (2 equiv), phenylethylamine, THF/MeOH, RT; b) FeCl3 (4 equiv), EtOH, reflux or Cu(BF4)2⋅n H2O (4 equiv), MeCN, reflux; c) FeCl3 (5 mol %), (tBuO)2 (2 equiv), DCE, 100 °C; d) 1. KMnO4 (0.25 equiv), MeOH/H2O, RT; 2. Na2S2O4, MeOH, RT; 3. BuBr, KOH, TBAB, dioxane, 80 °C. DCE=1,2‐dichloroethane, TBAB=tetrabutylammonium bromide.
Anilines, as a consequence of their electron‐rich character and thus lower oxidation potentials, should undergo oxidative aromatic coupling more easily than phenolic derivatives. Indeed, treatment of some secondary and tertiary anilines with p‐chloranil as an oxidizing agent27 produces dimerization products 15–19 (Scheme 3). From a structural point of view, the method tolerates a variety of functional groups (even those with electron‐withdrawing character) as well as substrates with sterically hindered reactive sites. In parallel, Kita and co‐workers proved28 that the dimerization of 1‐naphthylarylamines, 1‐naphthyldiarylamines, or 2‐naphthyldiphenylamine mediated by phenyliodine(III) bis(trifluoroacetate) (PIFA) takes place selectively at the naphthyl site of the molecule. Similar to the p‐chloranil‐mediated method, these conditions are also applicable when steric hindrance is present within a substrate molecule.
Scheme 3.
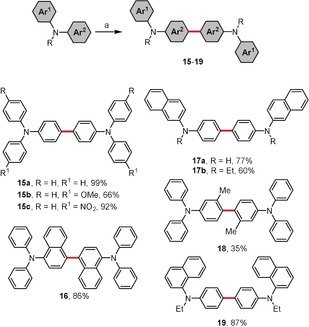
Reagents and conditions: a) p‐chloranil (2 equiv), MsOH/CH2Cl2, 0–25 °C. MsOH=methanesulfonic acid.
Scheme 4 depicts a catalytic approach for the oxidative homocoupling of anilines.29 In this case, a system comprised of rhodium adsorbed on carbon, air, and TFA as a solvent showed the highest efficiency. Biaryl amines 20–26 (including π‐expanded analogues 20 a–d) were obtained selectively in high to excellent yields (up to 99 %). Interestingly, when HFIP was used as a solvent, the authors obtained the carbazole‐based compound 21 as the sole product in 70 % yield. The oxidative cross‐coupling of aromatic amines was also reported by Pappo and co‐workers as well as the Shindo group.30, 31
Scheme 4.
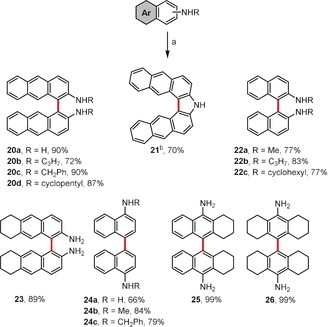
Reagents and conditions: a) Rh/C (5 mol %), TFA, air, RT; b) Rh/C (5 mol %), HFIP, air, RT. TFA=trifluoroacetic acid.
In 2017, Knölker and co‐workers investigated the reactivity of diarylamines of type 27 towards iron(III)‐based catalysts: hexadecafluorinated iron‐phthalocyanine (FeF16Pc) and iron(III) chloride hexahydrate (Scheme 5).32 By using FeF16Pc as a catalyst, 2,2′‐di(arylamino)biaryls 28, tetraarylhydrazines 29, and 5,6‐dihydrobenzo[c]cinnolines 30 can be obtained in reasonable yields. The selectivity is governed by the use of acidic or basic additives. Namely, in the presence of a FeF16Pc/MsOH system, diarylamines 27 give exclusively the 2,2′‐coupled products 28. By changing the character of the additive from acidic to basic, 27 undergoes N−H/N−H oxidative coupling to form the tetraarylhydrazines 29. Unexpectedly, when 27 was treated with a stoichiometric amount of FeCl3⋅6 H2O, spiro‐products 31 were obtained in reasonable yields.
Scheme 5.
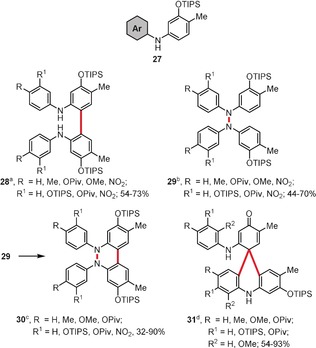
Reagents and conditions: a) [FeF16Pc] (1 mol %), MsOH, air, CH2Cl2, RT; b) [FeF16Pc] (5 mol %), NEt(iPr)2, air (1 atm), DCE, 60 °C; c) [FeF16Pc] (10 mol %), AcOH, air, CH2Cl2, RT; d) FeCl3⋅6 H2O (2.4 equiv), CH2Cl2, reflux, argon. Piv=pivaloyl, TIPS=triisopropylsilyl.
Recently, Venkatakrishnan and co‐workers33 found that, whereas carbazoles containing electron‐donating groups form 3,3′‐coupled products, substrates bearing electron‐withdrawing groups tend to form predominantly 1,1′‐coupled molecules. The dimerization of N‐phenylcarbazole can also be achieved using Eaton's reagent (composed of 7.5 wt % phosphorus pentoxide in methanesulfonic acid).34
The synthesis of large graphenic structures from relatively simple molecules is usually not a straightforward task. Among the many approaches leading to nanographenes, the benzene ring seems to be an ideal candidate for the bottom‐up synthesis of such structures.5, 6, 35, 36 Zarabin and co‐workers attempted37 the construction of nanographene sheets from benzene at a liquid–liquid interface. According to their strategy, a single benzene ring should undergo FeCl3‐mediated oxidative coupling to give oligo(paraphenylene) and/or poly(paraphenylene). The poly(paraphenylene) fractions should spontaneously migrate to the water–benzene interface, where the second polymerization step that creates lateral chains occurs. In the final step, a series of intramolecular oxidative coupling processes between adjacent phenyl rings within the branched polymer takes place. The proposed method allows the preparation of large graphene nanosheets with a surface area of about 800 nm2.
Recently, oxidized active carbon/oxygen,38 2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone (DDQ)/MsOH,39 and NOBF4/oxygen40 systems were found to be applicable for the construction of 2,2′‐ or 3,3′‐coupled motifs starting from benzofused heterocycles such as indole, benzothiophene, or benzofuran.
Short‐chain oligopyrroles constitute outstanding substrates for the formation of various pyrrolic macrocycles, which may vary significantly in their electronic properties and metal‐binding properties.41, 42, 43 Anand and co‐workers investigated the reactivity of benzo‐dipyrromethanes under oxidative conditions (Scheme 6),44 and they uncovered that the reaction outcome highly depends on the exact structure of the dipyrromethene substrate. The DDQ‐mediated oxidation of monobenzo‐dipyrromethanes 32 to the corresponding dipyrromethenes 33 (Scheme 6 A) followed by treatment with Cu(OAc)2 leads to cyclic trimers of type 34. If the same sequence was applied to 32 b in high dilution, a mixture of dimer 35 and trimer 34 b could be obtained. Subjecting 36 to the same reaction sequence (Scheme 6 B) resulted in cyclodimer 37 containing two fused seven‐membered rings in the central part of the molecule. Finally, the stepwise, DDQ/Cu(OAc)2‐mediated oxidation of dibenzo‐dipyrromethane 38 gives access to the unsymmetrical, acyclic dimer 39 (Scheme 6 C) with six‐ and seven‐membered rings at its core.
Scheme 6.
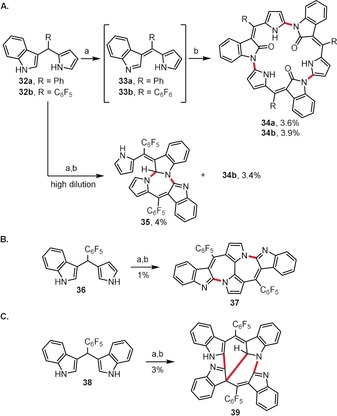
Reagents and conditions: a) DDQ (2.2 equiv), THF, RT; b) Cu(OAc)2 (1 equiv), THF, RT.
Oxidative homo‐ and cross‐coupling processes are key synthetic steps towards helical π‐conjugated oligopyrrins with a bipyrrole linkage (Scheme 7).45 A series of DDQ‐mediated oxidative couplings between the monoacylated bilane 40 and/or tripyrrane (41) lead to oligopyrrolic molecules 42–44 containing six, seven, or eight pyrrolic units, respectively. These oligopyrrolic derivatives readily undergo double complexation with copper(II) acetate to form bis(copper(II)) complexes, which distinctly differ in geometry. Complex 45 adopts a helical coil geometry, whereas complexes 46 and 47 exhibit S‐shaped open‐spiral‐type arrangements.
Scheme 7.
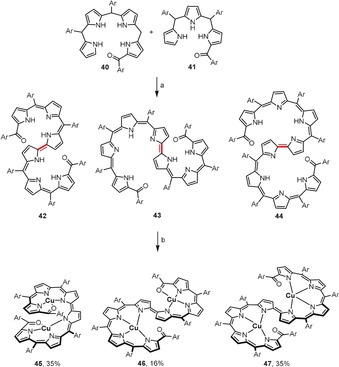
Reagents and conditions: a) DDQ (4 equiv), CH2Cl2, RT; b) Cu(OAc)2, CH2Cl2/MeOH, RT. Yields after two steps. Ar=pentafluorophenyl.
2.2. Oxidative Cross‐Coupling of Arenes and Heteroarenes
Cross‐coupling processes occurring between two different arenes could potentially afford three different products (or more, if a given substrate has two inequivalent reactive sites). To reduce the number of possible products, Pappo's model46 based on an interplay between global nucleophilicity and oxidation potentials may be considered. Another system consisting of a salen‐based catalyst (56) and atom‐economical oxygen as the terminal oxidant was found to be broadly effective in oxidative cross‐coupling reactions to give compounds 50–55 (Scheme 8).20 According to Kozlowski, the 2,6‐disubstituted phenol 48 should be applied as the substrate to carry out a selective cross‐coupling process and reduce the number of possible products.47 It was assumed that such a phenol (called Type I) should bind selectively at its para site to a less‐hindered site of a metal‐bound radical or radical cation of the second phenolic partner (49). Taking into account the regioselectivity of the method, substrates of type 49 were assigned as Type II (2,4‐, 3,4,5‐, 3,5‐, and 2,3,5‐substituted phenols) and Type III (2,5‐substituted phenols), as they provide para–ortho (p‐o) and para–para (p‐p) coupled products, respectively.
Scheme 8.
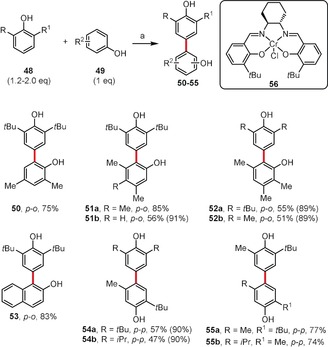
Reagents and conditions: a) 56 (5 mol %), O2, DCE, 50–85 °C. Yields in parentheses are based on recovered substrate.
Electrochemical oxidation is currently being considered as a green alternative for classical oxidative aromatic coupling, because such a method does not require a metal‐based catalyst or a stoichiometric amount of an oxidant to form the crucial intermediate—a radical cation. However, the selective generation of a new C−C bond between two different aryl moieties under either classical or electrochemical conditions is challenging due to a strong tendency to form homocoupled products.
Recently, Waldvogel and co‐workers proved that the use of HFIP as a mediator, combined with a boron‐doped diamond electrode (BDD) as the anode constitutes a powerful system for selective phenol–arene cross‐coupling under electrochemical conditions.48, 49, 50, 51, 52 The importance of this specific solvent and the source of selectivity can be attributed to the strongly different solvation of the individual coupling partners and, therefore, the decoupling of the oxidation potential from the nucleophilicity. The efficiency and selectivity of the cross‐coupling process can be improved when HFIP/methanol or HFIP/water mixtures are used as mediators.53, 54 For example, the electrochemical oxidation of N‐protected aniline derivatives using HFIP/methanol as a mediator and glassy carbon as the anode material leads to a variety of unsymmetrical 2,2′‐diaminobiaryls in moderate to good yields and excellent regioselectivity.55 Easily removable protecting groups including Boc, acetyl, benzoyl, pivaloyl, and trifluoroacetyl are fully compatible with the electrochemical environment; thus, after deprotection under mild conditions, this method provides expedient access to important structural motifs.
Recently, Waldvogel and co‐workers successfully elaborated an electrochemical alternative for the arylation of sulfur‐containing heterocycles. Thiophenes56 and benzothiophenes57 were effectively coupled with phenol derivatives to form biaryl systems with high selectivity and efficiency. Importantly, with 2‐ as well as 3‐substituted benzothiophenes they obtained a broad scope of products in yields up to 88 %. In the case of thiophenes, applying an excess of phenolic substrate (3 equiv) leads to diarylated products in high yields.
Hypervalent iodine(III)‐based compounds serve as excellent oxidizing agents in oxidative aromatic coupling processes,58, 59 and they are typically used in stoichiometric or excess amounts. The first organocatalytic version of an oxidative cross‐coupling reaction between sulfonylanilides and aromatic hydrocarbons was developed by the Kita group (Figure 3).60 During this process, the organocatalyst 57 based on a 2,2′‐diiodobiaryl scaffold is oxidized to its hypervalent iodine(III) analogue 58 by mCPBA. After the cross‐coupling reaction, the organocatalyst is then regenerated by the same means as its activation. The process is slower and a lower yield was obtained when 0.75 equiv of 58 was used instead of a catalytic amount of 57. A series of derivatives 59–62 containing naphthyl or phenanthryl substituents were obtained using the organocatalytic method, with yields of up to 99 %.
Figure 3.
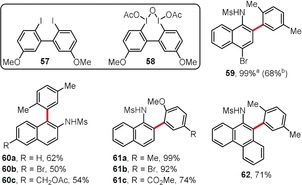
Reagents and conditions: a) 57 (5 mol %), mCPBA (1.5 equiv), TFE/CH2Cl2, RT; b) 58 (0.75 equiv), TFE/CH2Cl2, RT. mCPBA=meta‐chloroperbenzoic acid, TFE=2,2,2‐trifluoroethanol.
In their follow‐up publication, the Kita group explored simple iodobenzenes as potential organocatalysts in oxidative phenol–arene or phenol–phenol cross‐coupling reactions.61 A catalytic system consisting of 4‐iodoanisole (10 mol %), oxone (1.2 equiv), 18‐crown‐6, and CH3COOH in HFIP was shown to be the most efficient method. High regioselectivity and broad substrate scope represent indisputable advantages of this method. According to the studies of More and Jeganmohan,62 oxone mixed with the phase‐transfer catalyst Bu4N+ HSO3 − in acetic acid also constitutes a selective system for the formation of unsymmetrical biphenols; however, the yields obtained were only moderate.
Recently, Pappo and co‐workers made a significant contribution to this field by the discovery46, 63 that, depending on the solvent used during an oxidative coupling process, two different products (64 and 65) can be obtained starting from 2,6‐dimethoxyphenol (63; Scheme 9, top). In a HFIP solution, the initially formed radical [63−H]. is stabilized to a greater extent than in DCE, thus it reacts with the most nucleophilic site of phenol 63 and leads to 64 through a radical–anion coupling mechanism. In DCE, [63−H]. is poorly stabilized, and it undergoes a rapid recombination process through a radical–radical coupling mechanism. Based on this finding, the authors developed a predictive model based on the interplay between the theoretical global nucleophilicity (N) and the susceptibility to oxidation (derived from a direct comparison of the oxidation potentials measured in HFIP).46 According to this model, an efficient cross‐coupling process between phenols A and B can be performed as long as:
Scheme 9.
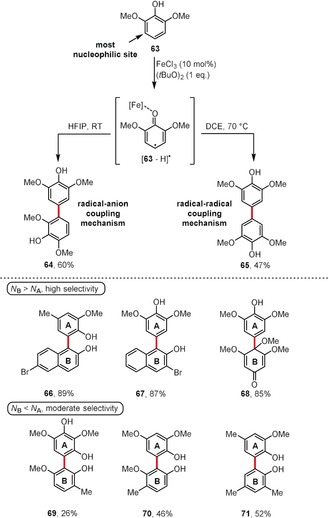
Pappo's model for selective oxidative cross‐coupling.63
one of the phenols (A) can be selectively oxidized to a corresponding phenoxyl radical (E ox A < E ox B) in the presence of the second phenolic partner B;
the second phenolic partner (B) exhibits a more pronounced nucleophilic character (that is when N B>N A or ΔN>0).
A set of unsymmetrically coupled biaryls 66–68 was selectively constructed using this model when ΔN>0 (Scheme 9, bottom). When ΔN<0, the selectivity of this process decreases, thus only moderate yields are achieved for cross‐coupling reactions (e.g. products 69–71). During the carbon–carbon bond‐forming step in the FeCl3/(tBuO)2/HFIP‐mediated process, both phenolic components are attached to the metal center. As an extension of the proposed model, the same group later discovered64 that a system consisting of an iron meso‐tetraphenylporphyrin chloride (Fe[TPP]Cl) complex and HFIP allows for the selective oxidative cross‐coupling of readily oxidized phenols with poorly nucleophilic phenolic partners (ΔN<0). It is believed that the high selectivity of the process results from the presence of a single available coordination site for phenol binding on the metal center in Fe[TPP]Cl.
The authors have demonstrated that the use of the iron(III)/(tBuO)2 couple in fluorinated solvents enables many valuable cross‐coupling products to be obtained, such as biaryls 72–73 and 75–76, as well as triaryl 74 (46 % yield; Figure 4).46, 63 Importantly, polycyclic aromatic hydrocarbons larger than naphthalene, such as anthracene, pyrene, and benzopyrene, are also reactive in this transformation and lead to 77–79. This study was recently further extended to include cross‐coupling catalyzed by CoII‐salen and CrIII‐salen complexes.65, 66
Figure 4.
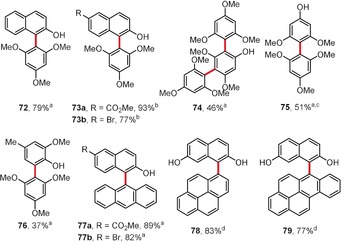
Reagents and conditions: a) FeCl3 (5–15 mol %), (tBuO)2 (1.5–2 equiv), HFIP, RT; b) FeCl3⋅6 H2O (5 mol %), (tBuO)2 (2 equiv), HFIP, RT; c) TFE was used as a solvent; d) 1‐phenyl‐2,2,2‐trifluoroethanol was used as a solvent at 40 °C.
As shown in Figure 4, the reaction of 3,5‐dimethoxyphenol and 1,3,5‐trimethoxybenzene (2.1 equiv) gives the bisarylated product 74 in reasonable yield. Intrigued by this result, the same authors evaluated the dilemma of polyarylation in depth (Scheme 10).67 According to their study, the first coupling reaction of the phenolic substrate 80 occurs at the available ortho/para position(s) relative to the hydroxy group. Then, they found that the subsequent reaction strongly depends on the character of the substituent R. If R=H, the next coupling process leads to ortho‐arylated derivative 82. In the case of R being a methyl group, the second/third arylation reaction exhibits meta selectivity, finally providing a compound with an exemplary substitution pattern of type 83. When R is a methoxy group, the third arylation reaction tends to proceed chemoselectively at the OH group to form a C−O coupling product 84. The EPR studies performed on the key persistent phenoxyl radicals clearly demonstrated that the coupling selectivity strongly correlates with the distribution of high spin density over the above‐mentioned radicals.
Scheme 10.
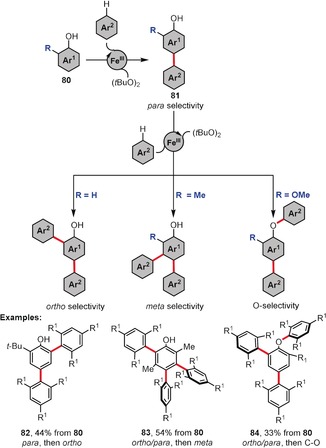
Catalyst and oxidant loadings: FeCl3 (15 mol %), (tBuO)2 (3–5 equiv). R1=OMe.
Polyarylated phenolic derivatives can also be assembled by the iron(III)‐catalyzed functionalization of 4‐aryl‐4‐methoxy‐2,5‐cyclohexadienones 85 (Scheme 11, top).68 Although this reaction is not an oxidative aromatic coupling, but rather a polar reaction of an electrophile 85 with arene nucleophiles, we decided to present it here for the sake of completeness of strategies leading to polyarylated phenols. Substrates of type 85 were prepared using the PIDA‐mediated oxidation of the corresponding 4‐arylphenols in the presence of MeOH.69, 70 Initially, compounds 85 are arylated at the α‐position relative to the keto group in the presence of a catalytic amount of iron(III) chloride, thereby leading to the 2,4‐diarylphenolic derivatives 86–90. However, for a substrate formed by the PIDA‐mediated oxidation of 4‐methyl‐1‐hydroxynaphthalene in MeOH they obtained a mixture of α‐ and β‐arylated products in yields of 76 and 20 %, respectively. Products of monoarylation, that is 86 a or 86 c, can be subsequently oxidized to 91 a,b and arylated again to give 2,4,6‐triarylphenolic molecules 92–93 (Scheme 11, bottom). One major drawback of the above method is that a significant excess of the arene (Ar2H up to 10 equiv) is required to achieve monoarylation.
Scheme 11.
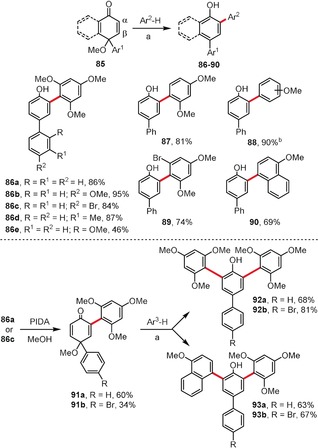
Reagents and conditions: a) FeCl3 (10 mol %), MeCN, RT; b) a mixture of regioisomers (ortho/para=1:2) was obtained.
It is well known that 9‐phenanthrenol undergoes an oxidative aromatic homocoupling reaction at C10 under the influence of CuII species.71, 72 Wang et al. found73 that 9‐methoxyphenanthrene (94) shows a similar reactivity towards 2‐methoxynaphthalene derivatives in the presence of (NH4)2S2O8, with C10/C1‐linked molecules 95 being formed with excellent regioselectivity (Scheme 12). Interestingly, when 2‐naphthylamine derivatives were employed in this cross‐coupling reaction, C3‐arylation products 96 were obtained in good to excellent yields. According to DFT calculations employed in this study, 94 and 2‐methoxynaphthalene cation radicals have the highest spin density distribution at positions C10 and C1, respectively, and they follow a radical coupling pathway leading to the C10/C1 linked product (Scheme 12, bottom). In contrast, 94 reacts with 2‐naphthylamine at the most electrophilic position (C3), because the oxidation potential of naphthylamine increases significantly upon protonation in acidic media, thereby preventing the effective formation of a radical cation.
Scheme 12.
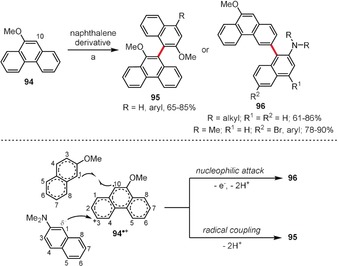
Reagents and conditions: a) (NH4)2S2O8 (6 equiv), TFA/CH2Cl2, 25 °C.
Kita and co‐workers successfully performed a series of PIFA‐mediated intermolecular oxidative coupling processes between pyrrole derivatives and 3,4‐ethylenedioxythiophene (Figure 5).74 The dimers of type 97 were synthesized in moderate to good yields with excellent regioselectivity. In the case of dimers containing an unprotected pyrrole unit, intramolecular hydrogen bonds enforce high coplanarity of both subunits, as proven by X‐ray crystallography, potentially allowing these dimers to serve as excellent substrates for efficient conducting polymers.
Figure 5.

Cross‐coupling conditions: PIFA (1 equiv), Me3SiBr (2 equiv), HFIP, RT.
2.3. Enantioselective Synthesis of Chiral Biaryls
Hindered rotation around the Caryl–Caryl bond, such as in binaphthyl or ortho‐substituted biphenyl derivatives, gives rise to axial chirality, which is present in many natural products75, 76, 77, 78 and is the basis of various asymmetric catalytic systems.79 Hence, it is not surprising that the oxidative coupling of arenes, being one of the simplest ways to synthesize biaryls, was quickly recognized as a potential method to access optically active products.47, 76, 78 Early successful reports were based on the use of copper complexes with chiral amines as catalysts for the enantioselective synthesis of BINOL derivatives under aerobic conditions (Scheme 13).80, 81, 82, 83 The enantioselectivity was significantly improved after mononuclear84, 85 and binuclear86, 87 vanadium complexes were introduced.88 Recently, Egami and Katsuki developed chiral iron(salan) complexes (Scheme 13), which afforded unsubstituted BINOL and 3,3′‐disubstitued derivatives with an enantiomeric excess of 64 % and up to 97 %, respectively.89 Progress in the asymmetric synthesis of biaryls has been briefly summarized in recent articles.47, 88, 90, 91 The most important recent contributions are presented below.
Scheme 13.
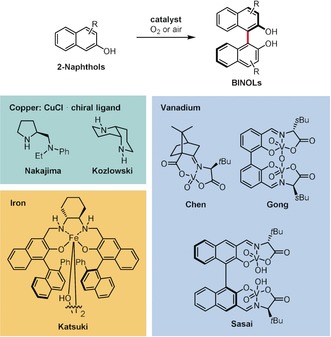
Early catalytic systems for the enantioselective synthesis of BINOL derivatives by the oxidative dimerization of 2‐naphthols.
Pappo and co‐workers developed a new chiral iron complex 99 bearing three BINOL‐phosphate ligands which turned out to be a very efficient catalyst for the enantioselective homo‐ and cross‐coupling of 2‐naphthols.92 Scheme 14 presents selected examples of BINOLs (98 a–f) prepared with good ee values using as little as 2.5 mol % of catalyst 99 in a mixture of DCE and HFIP as a solvent and with di‐tert‐butyl peroxide (1 equiv) as a terminal oxidant. Importantly, the authors have also found that enantiopure BINOLs undergo racemization in the presence of FeIII and CuII salts, which may reduce the enantiomeric excess of the products from the oxidative coupling. In the case of cross‐coupling, the reaction yields were significantly lower due to the formation of homocoupled side products. Nevertheless, this is one of only a few examples of enantioselective oxidative cross‐coupling reactions to date.
Scheme 14.
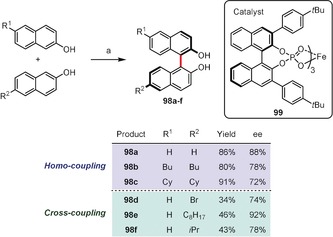
Reaction conditions: a) 99 (2.5 mol %), (tBuO)2 (1 equiv), DCE/HFIP 1:1, RT.
Dinuclear vanadium catalysts are among the most efficient examples for the enantioselective syntheses of BINOLs,86, 87, 88, 93 useful even in the dimerization of PAH‐based phenols.94 However, recent studies have revealed that chiral mononuclear vanadium(V) catalysts are also useful. Takizawa, Oh, and co‐workers found that 4‐substituted 2‐naphthols undergo oxidation with oxygen in the presence of mononuclear catalyst 101 or dinuclear 102 to give either R or S enantiomers, respectively, of BINOLs 100 in good yields and high ee values (Scheme 15).95 The mononuclear catalyst 101 had to be used in higher loadings (5 mol %) and resulted in slightly lower ee values.
Scheme 15.
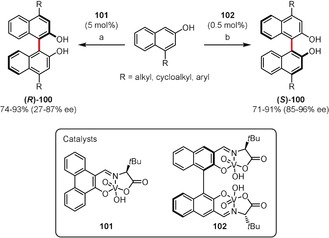
Reaction conditions: a) 101 (5 mol %), O2, CCl4, RT; b) 102 (0.5 mol %), O2, CH2Cl2, RT.
Compound 107, also a mononuclear vanadium complex, was found by Takizawa, Sasai, and co‐workers to catalyze the enantioselective formation of oxa[9]helicenes 106 in one step from 2‐hydroxybenzo[c]phenanthrenes 103 (Scheme 16).96 In this transformation, 107 acts simultaneously as both a redox catalyst (enantioselective aerobic oxidative coupling) and Lewis acid (metal‐mediated intramolecular cyclization) catalyst. The authors proposed that this process may proceed via intermediates 104 or 105, although these could not be detected in the reaction mixture. This method tolerates a broad scope of substrates, giving (M)‐oxa[9]helicenes with ee values up to 94 %.
Scheme 16.
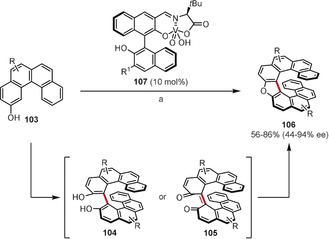
Reaction conditions: a) O2, CCl4, 50 °C. R=H, alkyl, aryl, Br; R1=3,5‐Ph2C6H3.
Although numerous chiral catalysts have been developed for the asymmetric synthesis of BINOLs by oxidative coupling, the enantioselective preparation of chiral biphenyl derivatives has remained challenging until very recently. Kozlowski and co‐workers addressed this issue and performed an extensive catalyst optimization study using dinuclear vanadium catalysts as a starting point.91, 97 It turned out that nitro‐substituted mononuclear vanadium complex 109 actually provided the best results, especially in the presence of acetic acid or lithium chloride as additives (Scheme 17). A series of biphenols of type 108 were obtained in good yields and moderate ee values. N‐Benzyl‐2‐hydroxycarbazoles also reacted under similar conditions to give the corresponding 1,1′‐coupled dimers enantioselectively. Kozlowski and co‐workers utilized the new catalyst in the total synthesis of chaetoglobin, where a key chiral biaryl intermediate was prepared by atroposelective oxidative dimerization of phenols catalyzed by 109.98
Scheme 17.
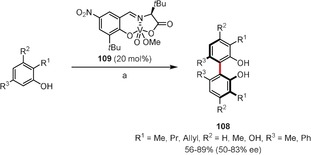
Reaction conditions: a) 109 (20 mol %), AcOH (6.25 equiv) or LiCl, O2, toluene, 0 °C or 25 °C.
Very recently, Takizawa, Sasai, and co‐workers developed a dinuclear vanadium complex that mediated the oxidative homocoupling of various 5‐arylresorcinols to furnish the corresponding biaryls with up to 98 % ee.99
2.4. Metal‐Catalyzed and Metal‐Mediated Intermolecular Oxidative Coupling of Electron‐Poor Arenes
In terms of both the mechanism and the scope, the metal‐catalyzed intermolecular dehydrogenative coupling of electron‐poor arenes differs significantly from classic oxidative aromatic coupling. However, we have decided to include an overview of this synthetic methodology, with an emphasis on copper and rhodium catalysis as well as a few selected examples of palladium‐mediated reactions which have been more generally reviewed recently.100, 101, 102, 103, 104
2.4.1. Copper
In 2013, Bolm and co‐workers reported interesting copper‐mediated oxidative coupling reactions of 1,3,4‐oxadiazoles and benzoxazoles with pentafluorobenzene and its analogues (Figure 6).105 Compounds 110–112 were obtained in yields of 15–68 %.
Figure 6.
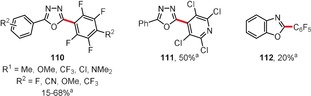
Reaction conditions: a) Heterocycle (1 equiv), halogenated arene (5–30 equiv), CuBr (1 equiv), 1,10‐phenantroline (1 equiv), tBuOLi, O2, CH3CN, RT.
Miura and co‐workers realized the copper‐mediated regioselective dehydrogenative biaryl coupling of naphthylamides and 1,3‐azoles using copper(II) acetate in the presence of pivalic acid.106 As a representative example, the reaction of 113 with benzoxazole leading to 114 is given in Scheme 18. Deuterium labeling experiments suggest that the metalation takes place at the benzoxazole first, with the species formed then undergoing a directed C−H activation at the naphthalene derivative.
Scheme 18.
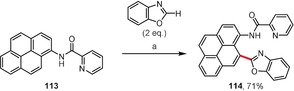
Reaction conditions: a) Cu(OAc)2 (3 equiv), PivOH (1 equiv), mesitylene, 165 °C.
2.4.2. Rhodium
In the last few years some interesting reports on rhodium‐catalyzed dehydrogenative arene coupling reactions have appeared, often within the context of directed C−H activation. You, Lan, and co‐workers reported the chelation‐assisted rhodium(III)‐catalyzed oxidative C−H/C−H cross‐coupling of indoles and pyrroles with heteroarenes preferentially at the C2‐position (Figure 7).107 Their strategy allows the selective coupling between an electron‐rich heteroarene with a directing group and both electron‐rich and electron‐deficient heteroarenes. For example, 2‐pyrimidyl‐protected indoles or 2‐methylpyrrole react with electron‐poor heterocycles in the presence of a [Cp*RhCl2]2/AgSbF6 catalyst system to afford coupling products 115–120. Closely related reactions were published by Kambe and co‐workers at almost the same time.108 More recently, the procedure has been extended to the synthesis of mono‐ and bisarylated phenols.109
Figure 7.
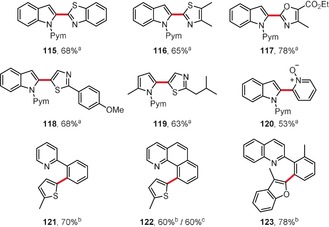
Reaction conditions: a) indole (1 equiv), heteroarene (3 equiv), [Cp*RhCl2]2 (2.5 mol %), AgSbF6 (10 mol %), Ag2CO3 (2 equiv), PivOH (2 equiv), DMF, 150 °C; b) pyridine derivative (1 equiv), heteroarene (3 equiv), [Cp*RhCl2]2 (5 mol %), AgSbF6 (20 mol %), Cu(OAc)2 (3 equiv), DCE, 140 °C; c) pyridine derivative (1 equiv), heteroarene (3 equiv), [Ru(p‐cymene)Cl2]2 (5 mol %), KPF6 (20 mol %), Cu(OAc)2⋅H2O (2 equiv), DCE, 140 °C. Cp*=C5Me5, PivOH=pivalic acid, Pym=2‐pyrimidyl.
In the context of directed C−H activation, You and co‐workers also reported the rhodium‐ or ruthenium‐catalyzed oxidative C−H/C−H cross‐coupling between various heterocycles and 2‐aryl‐substituted pyridines or quinolines in the presence of [Cp*RhCl2]2 or [{Ru(p‐cymene)Cl2}2] as catalysts.110 Three exemplary products 121–123 are presented in Figure 7. Similar reactions were reported with oxime ethers as substrates.111 Later the same group reported the use of the Wilkinson catalyst for the ortho‐heteroarylation of pivaloyl anilides.112
Li and co‐workers developed rhodium(I) catalysts for the regiospecific homodehydrogenative coupling of aromatic carboxylic acids in water (Scheme 19).113, 114 The dimerization of variously substituted benzoic acids upon reaction with the chloro(norbornadiene)rhodium(I) dimer as a catalyst and manganese dioxide or sodium chlorite as the oxidant gave coupling products of type 124 in good yields. In a subsequent report, the authors showed that cross‐dehydrogenative coupling is also possible under similar reaction conditions if one of the carboxylic acids is electron‐rich.115
Scheme 19.
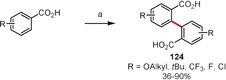
Reaction conditions: a) [Rh(nbd)Cl]2 (5 mol %), MnO2 (3 equiv), H2O, air, 150 °C.
2.4.3. Palladium
Most of the reported metal‐catalyzed or ‐mediated C−H/C−H dehydrogenative coupling reactions involve palladium as the catalyst metal. There are some reports on palladium‐catalyzed arylations by coupling an arene with another bearing an ortho directing group.100, 101, 102, 103, 104 In this context, Song, You, and co‐workers reported reactions of anilide derivatives with arenes, which were catalyzed by palladium(II) acetate in the presence of ammonium peroxodisulfate as the oxidant and TFA as the solvent (Figure 8).116 Cross‐coupled products such as 125–127 were obtained in yields ranging from 31 % to 95 %. A closely related reaction, namely the palladium(II)‐catalyzed dehydrogenative coupling of arenes with indolines at C7, has also been reported (products 128; Figure 8).117
Figure 8.
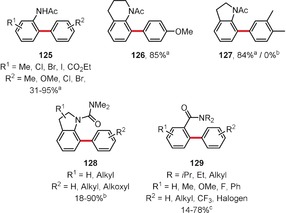
Reaction conditions: a) anilide (1 equiv), arene (20 equiv), Pd(OAc)2 (10 mol %), (NH4)2S2O8 (2 equiv), TFA (20 equiv), RT; b) Pd(OAc)2 (10 mol %), Cu(OAc)2 (1 equiv) or O2, TFA, arene as solvent, 50 °C; c) PdCl2 (10 mol %), AgOTf (20 mol %), NaOTf (20 mol %), K2S2O8 (2 equiv), DMA (2 equiv), arene as solvent, 80 °C. DMA=N,N‐dimethylacetamide.
Guan and co‐workers showed that biaryls of type 129 can be synthesized by coupling tertiary benzamides with arenes by utilizing the in situ generated palladium(II) triflate as the catalyst and dipotassium peroxodisulfate as the oxidant (Figure 8).118
Homo‐ and cross‐dehydrogenative coupling reactions yielding biaryls were also reported by Zhang and Rao, who used HFIP as the solvent and sodium periodate/dipotassium peroxodisulfate as oxidants.119 Homodimers of types 130–132 as well as unsymmetric biaryls (e.g. 133–135) were obtained in good yields (Figure 9).
Figure 9.
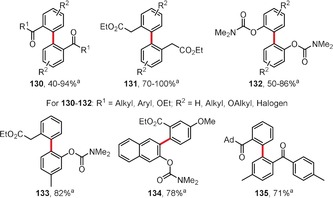
Reaction conditions: a) Pd(OAc)2 (7.5 mol %), NaIO4 (0.6 equiv), K2S2O8 (0.5 equiv), TfOH, HFIP, 25–70 °C. TfOH=trifluoromethanesulfonic acid.
In an interesting mechanochemical approach, Xu and co‐workers showed that the rates of dehydrogenative coupling reactions can significantly increase when ball milling is used instead of conventional heating. Long reaction times of up to 24 h for conventional reactions were reduced to 1 h, without the need for external heating.120
Although the dehydrogenative coupling reactions mentioned so far involve benzene derivatives that result in biphenyl‐derived products, there are also reports on the coupling of heterocycles. Yu and co‐workers reported an interesting coupling reaction of pyridines to afford 3,3′‐bipyridyl derivatives (e.g. 136) in addition to a minor amount of the 2,3′‐isomer.121 The reaction is catalyzed by palladium(II) acetate in the presence of 1,10‐phenanthroline and gives the homocoupling products with turnover numbers (TONs) of up to 10.3 (Scheme 20). Cross‐coupling reactions of pyridines with benzene derivatives to give 2‐phenylpyridines 137 were also reported.
Scheme 20.
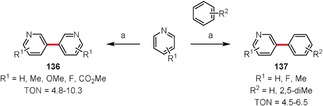
Reaction conditions: a) Pd(OAc)2 (10 mol %), 1,10‐phenanthroline (10 mol %), Ag2CO3 (2 equiv), K2CO3 (2 equiv), pyridine, 130 °C.
3. Intramolecular Oxidative Aromatic Coupling
Although numerous impressive examples have been known for a long time, only in recent years has the methodology for the intramolecular oxidative coupling of arenes reached its synthetic potential and become a well‐established tool used in a variety of applications. The growing importance of research in such fields as organic electronics, nanotechnology, and bioimaging, has led to an increased demand for simple and effective methods for the creation of intramolecular Caryl–Caryl bonds. Often simply (but not entirely correctly) referred to as the “Scholl reaction”, the intramolecular oxidative coupling of arenes is a versatile and widely used method for the synthesis of various polycyclic aromatic hydrocarbons (PAHs), including so‐called nanographenes, as well as expansions/planarizations of π‐conjugated systems and for the preparation of strained, curved, or twisted systems. For space reasons, the application of cyclodehydrogenation in the synthesis of small‐molecule targets is presented in the Supporting Information.
3.1. Large Planar PAHs, Expanded Acenes, and Nanographenes
Perhaps there is no better way to demonstrate the potential of intramolecular oxidative coupling methods than in the syntheses of large polycyclic aromatic hydrocarbons and nanographenes, where multiple (sometimes hundreds or even thousands) Caryl–Caryl bonds are formed in a single operation. It is easy to imagine that a similar goal would be impossible to achieve using alternative methods of Caryl–Caryl coupling, since it would require the presence of numerous activating units (e.g. halides, boronic acids/esters, etc.) strategically positioned over the precursor molecules.
One of the most important examples is hexa‐peri‐hexabenzocoronene (HBC, 138, R=H; Figure 10). Although HBC and its derivatives had been obtained previously, the yields were typically very low, and it was not until Müllen and co‐workers developed the CuII/AlCl3 or FeCl3‐promoted oxidation of hexaphenylbenzenes that HBC derivatives could be obtained in good yields.36 The latter conditions in particular, where FeCl3 is dissolved in nitromethane prior to its addition to a solution of the precursor in dichloromethane, provide the best yields for the broadest scope of HBCs (138; Figure 10),35, 122, 123 which opened the door to the synthesis of large PAHs and nanographenes.
Figure 10.
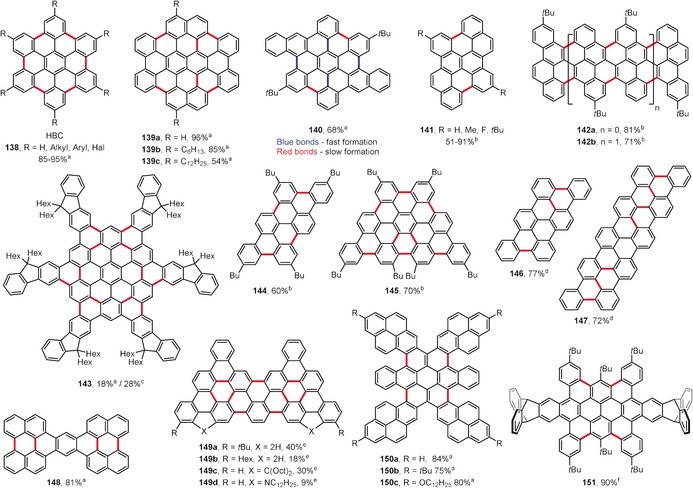
Selected examples of PAHs obtained by intramolecular oxidative coupling. Cyclization conditions: a) FeCl3, CH2Cl2, CH3NO2, RT; b) DDQ, TfOH, CH2Cl2, 0 °C; c) DDQ, MsOH, CH2Cl2, 0 °C; d) FeCl3, CH2Cl2, 0 °C (146) or RT (147); e) FeCl3, CH2Cl2, 4 Å MS, 0 °C; f) FeCl3, DDQ, CH2Cl2, RT.
Figure 10 depicts some recently reported large PAHs that were synthesized under intramolecular oxidative coupling conditions. Tetrabenzocircumpyrene derivatives 139 a–c have been synthesized in good yields using FeCl3 as an oxidant in a CH2Cl2/CH3NO2 mixture.124
Compound 140 is an extended hexabenzocoronene that was obtained by Dichtel and co‐workers.125 The authors observed that the formation of the first four bonds is very fast (Figure 10, blue bonds), thereby providing an isolable partly fused, twisted intermediate, which after prolonged exposure to the FeCl3 oxidant furnishes the final HBC derivative in good yield.
In addition to FeCl3, DDQ in the presence of Brønsted acids (Rathore conditions) is also frequently used in the synthesis of PAH molecules by oxidative cyclization. Compounds 141–145 are examples of PAHs synthesized using DDQ as an oxidant in the presence of triflic or methanesulfonic acid (Figure 10).126, 127, 128, 129 In the case of the hexafluorenyl‐fused HBC 143, DDQ/MsOH proved to be more efficient than FeCl3 (28 % versus 18 % yield).128 As a consequence of the presence of twelve hexyl chains pointing out of the PAH plane, the aggregation of 143 in solution is restricted and it shows good solubility in common organic solvents.
Murakami, Itami, and co‐workers have recently developed an interesting method for the palladium‐catalyzed annulative dimerization of monochlorinated oligo‐para‐phenylenes to afford substituted triphenylenes.130 The products contain (oligo)phenylene residues in a parallel orientation. Two of these triphenylene derivatives have been subjected to oxidative cyclization conditions with FeCl3 in dichloromethane to give the corresponding products 146 and 147 in good yields (Figure 10). The analogous oxidative coupling of phenylene residues was not possible when two linear oligophenylene moieties were connected by only one covalent bond, thus indicating that the triphenylene core is necessary to hold the residues in a parallel orientation to enable the reaction to proceed efficiently. However, the analogous reaction was reported to be successful for a singly bonded tri‐para‐phenylene dimer when the subunits were end‐capped with octyloxy substituents,131 thereby furnishing the tetraoctyloxy derivative of 147 in good yield. A similar approach towards triphenylene derivatives, but employing oligo‐para‐phenylene iodides instead of chlorides, was independently developed by Shi and co‐workers; the products also underwent oxidative planarization upon treatment with FeCl3.132
Tao and co‐workers have utilized the FeCl3/CH3NO2/CH2Cl2 system for oxidative cyclization reactions in the synthesis of numerous extended planar and curved PAHs.133, 134, 135, 136 Tetranaphthopentacene 148 is one of many interesting examples prepared by this research group (Figure 10).135
Roberts, Krische, and co‐workers have exploited ruthenium‐catalyzed diol‐diene benzannulation for the construction of various polyphenylene‐type PAH precursors.137 Treatment of zigzag‐type oligophenylene precursors with FeCl3 in dichloromethane in the presence of 4 Å molecular sieves led to the formation of nine new Caryl–Caryl bonds and afforded PAHs 149 a–d in moderate yields (9–40 %; Figure 10). The presence of nitrogen bridges in 149 d significantly decreased the reaction efficiency.
Müllen, Chen, and co‐workers have reported the synthesis of a series of tetrapyrene‐fused benzocoronenes 150 a–c (Figure 10).138 All compounds were obtained in very good yields by the FeCl3‐mediated oxidation of the corresponding tetrapyrenylpentacene‐quinodimethanes.
Mastalerz and co‐workers reported the synthesis of triptycene‐flanked tetrabenzoovalene 151, which was obtained in 90 % yield upon oxidation with a FeCl3/DDQ mixture (Figure 10).139 As a result of the large steric hindrance, the whole system is contorted and easily loses the two central tert‐butyl substituents under acidic conditions.
In addition to the structures shown in Figure 10, many other PAHs have been synthesized using the oxidative coupling method140, 141, 142, 143, 144, 145, 146, 147 which, due to space limitations, cannot be included in this Review.
Wei and co‐workers developed a convenient method for the synthesis of threefold symmetrical nanographenes 153 and 154 by reacting 1,3,5‐tribenzylbenzene derivatives 152 with aromatic aldehydes in the presence of FeCl3 as a catalyst/oxidant and acetic anhydride as a dehydrant in a CH2Cl2/CH3NO2 mixture (Scheme 21).148 The reaction is a combination of Friedel–Crafts‐type substitution (between formyl groups of aldehydes and aryl rings), dehydrogenative aromatization, and intramolecular oxidative coupling and produces the nanographenes 153 and 154 a–c in good yields.
Scheme 21.
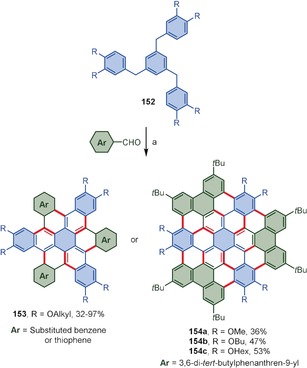
Reaction conditions: a) FeCl3 (10 mol %), Ac2O, CH2Cl2, CH3NO2, RT, then excess FeCl3.
The in situ 1,2‐aryl shift often changes the course of the oxidative aromatic coupling. Indeed, 1,2‐aryl shifts during the course of oxidative aromatic coupling reactions have been regularly encountered over the last few years.149, 150 A highly interesting example was published by Müllen and co‐workers during attempts to synthesize tetrabenzo[a,cd,j,lm]perylene 156 by the Scholl reaction of 6,7,13,14‐tetraphenylbenzo[k]tetraphene 155 (Scheme 22).151 Instead of 156, compound 157 was obtained as a result of two 1,2‐aryl shifts. Müllen and co‐workers showed that a radical cation mechanism is much more likely for the first 1,2‐aryl shift. In the following study, the authors utilized the discovered rearrangement in the synthesis of fused dibenzo[a,m]rubicene, a bowl‐shaped subunit of C70 fullerene.152
Scheme 22.
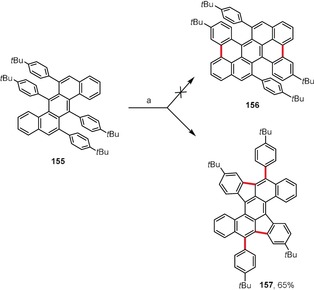
Exemplary dehydrogenative coupling accompanied by a double 1,2‐aryl shift. Reaction conditions: a) DDQ (5 equiv), TfOH, CH2Cl2, 0 °C.
Another example of this trend has been presented by Hartley and co‐workers, who have shown that terphenyl 158 undergoes single oxidative coupling and a 1,2‐aryl shift to afford 160 and not hexacycle 159 (Scheme 23).153
Scheme 23.
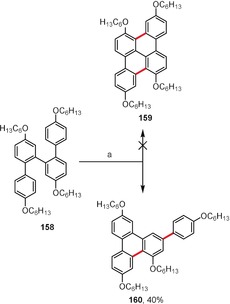
Reagents and conditions: a) FeCl3 (7.3 equiv), CH3NO2, CH2Cl2, RT.
Under oxidative coupling conditions (FeCl3 or DDQ), acenes bearing aryl groups attached to the central benzene rings generally cyclize to form new five‐membered rings.142, 144 Scheme 24 depicts an especially interesting example, where oxidation of symmetric tetracene 161 led to the unsymmetric product 162 containing two new rings of different sizes: five‐ and six‐membered.154
Scheme 24.
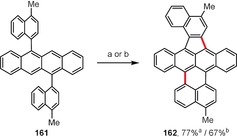
Reagents and conditions: a) FeCl3 (16 equiv), CH2Cl2, CH3NO2, RT; b) DDQ (2.4 equiv), TfOH, CH2Cl2, RT.
The bottom‐up synthesis of carbon nanoribbons is yet another impressive demonstration of the prowess of intramolecular oxidative coupling. Belonging to the nanographene class, carbon nanoribbons are ribbon‐ or tape‐like graphene fragments which are highly interesting because of their promising optoelectronic and semiconducting properties. Their syntheses have been extensively reviewed recently.5, 155, 156 Figure 11 presents the structures of the two exemplary carbon nanoribbons 163 157 and 164 158 obtained in excellent yields by Feng, Müllen, and co‐workers from the corresponding polymeric precursors using FeCl3 as an oxidant in a CH2Cl2/CH3NO2 mixture. The precursor for nanoribbon 163 was obtained by Diels–Alder polymerization159 of an appropriate acetylene‐functionalized tetraarylcyclopentadienone monomer. The weight‐average molecular weight (M w) of the resulting polymer reached 640 kg mol−1. FeCl3‐promoted planarization of the polymer furnished the nanoribbon with a cove‐type edge structure and width of 0.7–1.1 nm.157 The poly‐para‐phenylene scaffold of the precursor for nanoribbon 164 was constructed by Suzuki polymerization.158 After treatment with FeCl3, the necklace‐like nanoribbon 164 was obtained in high yield.
Figure 11.
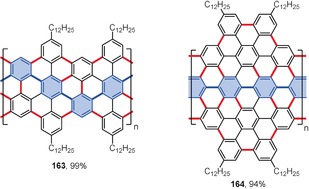
Examples of graphene nanoribbons. Cyclization conditions: FeCl3 (excess), CH2Cl2, CH3NO2, RT. The benzene rings constituting the main polyphenylene polymer chain are marked in blue.
Recent reports show that large PAHs, such as HBC, triangular C60H42, or even the hexagonal graphene plate C222H150, can be successfully synthesized from the corresponding branched polyphenylene precursors in a mechanochemical fashion by grinding with FeCl3 in a planetary ball mill.160
3.2. Dyes and Heterocyclic Polyarenes
The intramolecular oxidative coupling of arenes typically results in a connection of two aromatic rings in a coplanar orientation, which elongates the conjugated system of π‐electrons and usually strongly affects the photophysical properties of the obtained polycyclic aromatic products. Hence, it is not surprising that this method has been widely used for π‐expansion in various organic chromophores and fluorophores.
Coumarins are bright organic fluorophores with multiple applications in fluorescence imaging.161 Figure 12 presents the structures of π‐expanded coumarins obtained by the Scholl reaction or by intramolecular oxidative coupling. The pentacene‐based coumarin dimer 165 162 and its regioisomer 166 163 can both be efficiently synthesized from the corresponding precursors through an FeCl3‐promoted intramolecular oxidative coupling in the presence of a catalytic amount of BF3⋅Et2O. Compound 165 can also be obtained by a photochemical cyclization; however, the yield of the transformation was considerably lower than the oxidation with FeCl3.162
Figure 12.
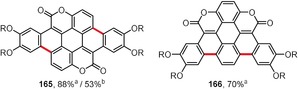
Conditions: a) FeCl3 (10 equiv), cat. BF3⋅Et2O, CH2Cl2, RT; b) hν (365 nm), air, THF, RT. R=Hexyl.
Pyrrolo[3,2‐b]pyrrole is the most electron‐rich among the known neutral 10π‐aromatic systems.164 This heterocycle recently gained more attention because of the discovery of an efficient one‐step, multicomponent synthesis of 1,2,4,5‐tetraarylpyrro[3,2‐b]pyrrole dyes with strong fluorescence. By using this method, 2,5‐di(biaryl)‐substituted derivatives 167 have been obtained from the reaction of ortho‐arylbenzaldehyes with 4‐alkylanilines and diacetyl (Scheme 25).165 These products readily react with FeCl3 to give the series of π‐expanded pyrrolopyrroles 168 a–e. Gryko and co‐workers devoted much effort to the synthesis of the bowl‐shaped pyrrolopyrrole 171, which would have an inverse Stone‐Thrower‐Wales166, 167 topology (Scheme 25). To meet this goal, compounds 169 and 170 were prepared as precursors for 171, with FeCl3 used to efficiently close the six‐membered and seven‐membered rings, respectively.168, 169 Unfortunately, all attempts to convert these precursors into 171 by means of classic organic synthesis failed. Finally, however, compound 171 was generated on an Au(111) surface by thermal annealing of the precursor 170 under ultrahigh vacuum (see Section 4).169
Scheme 25.
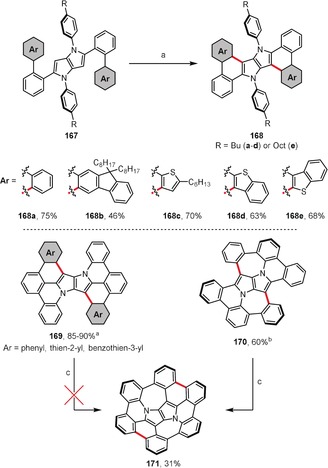
FeCl3‐mediated π‐expansions of pyrrolo[3,2‐b]pyrroles. Cyclization conditions: a) FeCl3 (20 equiv), CH2Cl2, CH3NO2, RT; b) FeCl3 (20 equiv), DCE, CH3NO2, 80 °C; c) Au(111), 320 °C.
Gryko and co‐workers revealed the first case of a 1,2‐aryl shift for aromatic heterocycles (Scheme 26).170 Tetraarylpyrrolo[3,2‐b]pyrrole (TAPP) 172 possessing [1,1′‐biphenyl]‐2‐yl substituents attached to the pyrrolic nitrogen atoms undergoes selective double dehydrogenative cyclization accompanied by a twofold 1,2‐aryl shift under oxidative aromatic coupling conditions. As a result, instead of the expected product 174 possessing two seven‐membered rings, dye 173 is formed.
Scheme 26.
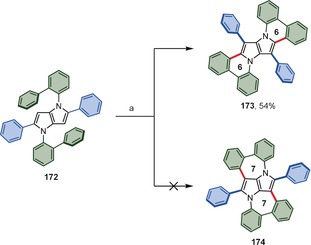
Reagents and conditions: a) FeCl3 (20 equiv), DCE, CH3NO2, 80 °C.
Treatment of bulky tetrabromo‐substituted tetraarylpyrrolo[3,2‐b]pyrrole 175 with FeCl3 gave the spirocyclic cationic fluorene derivative 176 instead of the expected product with new six‐membered rings (Scheme 27).171 Computational studies revealed that, in the case of 175, the formation of the spiro‐five‐membered ring is preferred in terms of both the transition‐state energy and the relative energy of the product. Steric hindrance is undoubtedly the main reason for the different reactivity of compound 175 compared to its analogues that lack additional substituents on the peripheral benzene rings (e.g. 167; see Scheme 25). In a broader context, these two studies have shown that a C−C bond can form at an already occupied carbon atom if the highest electron density is located there.
Scheme 27.
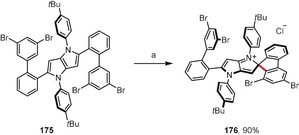
Intramolecular oxidative coupling of pyrrolo[3,2‐b]pyrroles. Reagents and conditions: a) FeCl3 (20 equiv), CH2Cl2, CH3NO2, RT.
Intramolecular oxidative coupling has also been used as a convenient procedure for the ring fusion of perylene diimides (PDI),172, 173, 174 as well as for π‐expansions of BODIPY and aza‐BODIPY‐based dyes,175, 176, 177, 178, 179 which has led to significant bathochromic shifts of the absorption and emission maxima. A library of π‐expanded imidazoles has been prepared by employing a PIFA/BF3⋅Et2O system for the oxidative cyclization of variously substituted 1,2,4,5‐tetraarylimidazole derivatives.180
As a consequence of their high electron density, pyrrole‐based compounds are often cumbersome substrates for reactions involving strong oxidants and acids.181 However, the efficient intramolecular coupling of pyrrole rings can be achieved under carefully optimized conditions.182 A prominent example was provided in 2007 by Müllen and co‐workers, who synthesized the rim‐fused hexapyrrolylbenzene 177 (Figure 13).183 The electron‐rich nature of pyrrole was evident by the fact that the FeCl3‐mediated cyclization initially led to an overoxidized dicationic form of 177, which could be reduced to the neutral 177 with hydrazine. More recently, the authors have extended this synthetic method to include analogues of 177 with one, two, or three pyrrole moieties replaced by benzene units (e.g. 178).184
Figure 13.
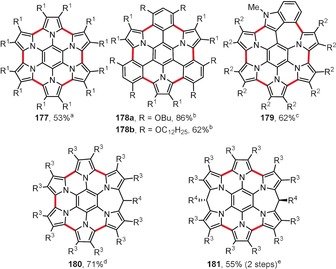
Hexapyrrolocoronene analogues and related compounds. Cyclization conditions: a) FeCl3 (36 equiv), CH2Cl2, CH3NO2, RT, then N2H4; b) FeCl3 (38 equiv), CH2Cl2, CH3NO2, RT; c) BAHA (12 equiv), Et2O/THF, RT, then Zn amalgam; d) DDQ (6 equiv), CH2Cl2, RT, then N2H4; e) i. DDQ (5 equiv), CH2Cl2, RT, then HBF4/H2O, ii. NaBH4, THF. R1=4‐(trifluoromethyl)phenyl, R2=4‐chlorophenyl, R3=4‐butoxyphenyl, R4=4‐nitrophenyl.
Stępień and co‐workers have synthesized an analogue of 177 with one pyrrole ring replaced by an indole unit (179; Figure 13).185 The cyclization was accomplished using tris(4‐bromophenyl)ammoniumyl hexachloroantimonate (BAHA, also known as “Magic Blue”), a radical cation oxidant. Similar to 177, the oxidation leads to an overoxidized aromatic dication of 179 (62 % yield), which after reduction with zinc amalgam affords the neutral 179 quantitatively. The same research group also reported the DDQ‐mediated synthesis of compounds 180 and 181, expanded derivatives of 177 containing one or two methylene bridges in the outer hexapyrrole rim.186 Another spectacular case of multiple intramolecular oxidative aromatic coupling involving eight pyrrole units to form a twisted aza‐PAH containing two unusual N‐doped heptagons was revealed by Uno and co‐workers.187
Osuka and co‐workers reported yet another example of an efficient intramolecular oxidative coupling of pyrrole rings (Figure 14). Tetrapyrrole[8]circulene analogue 182 was prepared by employing a so‐called “fold‐in” strategy,188 which is a method developed earlier by Stępień and co‐workers to construct strained macrocycles by contraction of the larger, less‐strained precursors.189, 190 Here, a macrocycle consisting of four pyrrole rings connected by ortho‐phenylene moieties was oxidatively contracted to 182 in excellent yield by the action of DDQ and scandium(III) triflate in boiling toluene. Products 183–185 were prepared in a similar way.191 Depending on the conditions and oxidant used, the bis(indolyl)helicene 183 or the closed, nonplanar circulene analogue 184 can be obtained in good yields from the same precursor. The eight‐membered ring in the latter case readily forms under the same conditions as those used for the synthesis of circulene 182, whereas the preparation of its open form 183 requires the use of much milder conditions (PIFA in CH2Cl2 at −78 °C). The efficiency of the DDQ/Sc(OTf)3 system for pyrrole–pyrrole intramolecular oxidative coupling has also been demonstrated by the synthesis of paracyclophane 186, which contains a segregated donor‐acceptor‐donor system with stacked tetrafluorobenzene rings as acceptors.192
Figure 14.
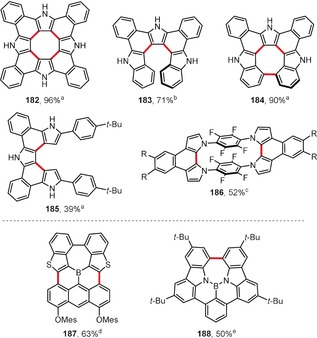
A selection of pyrrole‐based and boron‐doped heterocyclic polycyclic arenes obtained by oxidative cyclization. Reaction conditions: a) DDQ (5 equiv), Sc(OTf)3 (5.6 equiv), toluene, reflux; b) PIFA (2.5 equiv), CH2Cl2, −78 °C; c) DDQ (2 equiv), Sc(OTf)3 (7 equiv), CH2Cl2, RT; d) FeCl3 (8 equiv), CH2Cl2, CH3NO2, RT; e) DDQ (2.5 equiv), TfOH, CH2Cl2, 0 °C.
Boron‐doped polycyclic arenes are gaining more and more attention because of their promising properties and use in organic electronics, light‐emissive materials, and fluorescent sensors.193, 194 Yamaguchi and co‐workers have synthesized the first planarized, fully conjugated triarylborane 187 using FeCl3‐mediated cyclization, which led to the formation of two new bonds between the benzothienyl substituents and an anthracene moiety (Figure 14).195 The same group has also used FeCl3 as an oxidant in the synthesis of a larger PAH containing two boron atoms.196
The polycyclic arene 188 doped with a nitrogen‐boron‐nitrogen unit has been synthesized by using DDQ in the presence of triflic acid to efficiently close the seven‐membered ring (Figure 14).197
Many π‐extended porphyrins have been synthesized by inter‐ or intramolecular oxidative coupling; however, they will not be described here due to space restrictions and the fact that this subject has been reviewed recently.198, 199
As reported by Tanaka, Kim, Osuka, and co‐workers, similar to porphyrin analogues, meso‐meso‐linked corrole dimers 189 a–c bearing various aryl substituents can also be oxidatively planarized to the corresponding triply linked products 190 a–c (Scheme 28).200 The reaction was accomplished using DDQ in boiling chloroform and required high dilution of the starting materials to minimize unwanted intermolecular coupling reactions. The cyclization was accompanied by overoxidation, thereby leading to the loss of one NH hydrogen atom in each corrole macrocycle, which as a consequence became non‐aromatic. Similar overoxidation also occurred in the case of the doubly linked corrole dimer 191, which was synthesized under similar conditions.201 The aromaticity of the corrole ring in both types of dimers, 190 and 191, can be restored by reduction with NaBH4; however, they slowly reconvert back to the stable non‐aromatic forms in air.
Scheme 28.
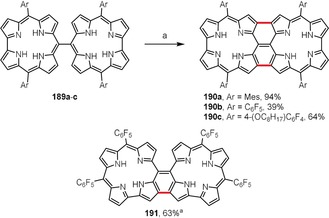
Synthesis of fused corrole dimers. Reaction conditions: a) DDQ (3–3.5 equiv), CHCl3, reflux.
3.3. Curved, Twisted, and Strained Structures
Nonplanar, curved, and twisted polycyclic aromatic architectures have attracted considerable attention because of their unusual geometries, strained structures, and interesting photophysical and electrochemical properties, which are significantly different from those of their planar analogues.202 The synthesis of curved PAHs remains challenging because of the large strain. Nevertheless, recent years have seen a proliferation of curved or twisted PAHs synthesized by the intramolecular oxidative coupling method.
Planar PAHs naturally consist mainly of fused six‐membered benzene rings. The introduction of rings of different sizes usually induces curvature to the system. Hence, the presence of five‐membered rings usually leads to bowl‐shaped compounds with positive curvature (e.g. corannulene and C60). In contrast, larger seven‐ and eight‐membered rings lead to saddle‐shaped systems with negative curvatures (e.g. [7]circulene). Figure 15 presents the structures of saddle‐shaped PAHs synthesized under intramolecular oxidative coupling conditions.
Figure 15.
Saddle‐shaped PAHs. Cyclization conditions: a) Cu(OTf)2 (24 equiv), AlCl3 (24 equiv), CS2, 30 °C; b) FeCl3 (16 equiv), CH2Cl2, −20 °C; c) DDQ (1–4 equiv/bond), TfOH, CH2Cl2, 0 °C (193, 195 a,b, 196 b,c) or RT (194); d) FeCl3 (excess), CH3NO2, CH2Cl2, RT.
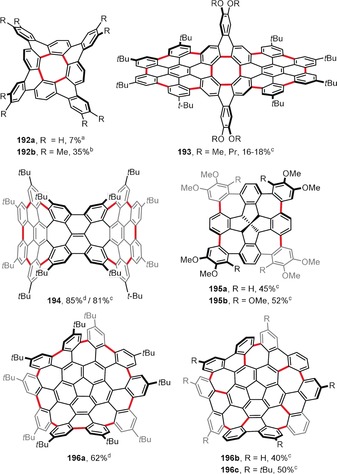
In 2013, Sakamoto and Suzuki reported an elegant synthesis of tetrabenzo[8]circulenes 192 a and 192 b by the oxidation of the corresponding macrocyclic octaphenylene precursors with Cu(OTf)2/AlCl3 and FeCl3, respectively (Figure 15).203 This was the second successful reported synthesis of an [8]circulene derivative; the first report preceded it only by a few weeks.204 In Section 3.2 we described the synthesis of a tetrapyrrole analogue of 192, compound 182, which was obtained by a similar “fold‐in” strategy, but using DDQ/Sc(OTf)3 as an oxidant.188 In contrast to 182, the products 192 a,b are saddle‐shaped, as evidenced by X‐ray crystallography. By using the same approach, Miao and co‐workers recently synthesized a much larger derivative of [8]circulene, a saddle‐shaped PAH 193 flanked by two fused HBC moieties (Figure 15).205 DDQ in TfOH/CH2Cl2 enabled the formation of all required 14 Caryl–Caryl bonds in one step, thereby providing 193 in 16–18 % yield. An alternative method to construct the tetrabenzo[8]circulene scaffold starts with tetraphenyl‐substituted tetra‐ortho‐phenylenes which already contain a central eight‐membered ring. These phenylenes are treated with DDQ/TfOH to effect the oxidative closure of four external benzene rings.206 This approach is efficient (47–72 % yield) for the synthesis of variously substituted derivatives of tetrabenzo[8]circulene that can undergo further functionalization.
The dipleiadiene‐embedded PAH‐saddle 194 contains two seven‐membered rings embedded within two fused [7]circulene moieties (Figure 15). Its synthesis has been realized by Miao and co‐workers in high yields using FeCl3 and DDQ as oxidants.207 The same group also synthesized other heptagon‐containing saddle‐shaped PAHs using intramolecular oxidative coupling in key steps.208, 209
Kuck, Chow, and co‐workers have utilized the DDQ/TfOH oxidative system for the synthesis of molecules 195 a and 195 b bearing tetracoordinated carbon atom cores within saddle‐shaped o,p,o,p,o,p,o,p‐cyclooctaphenylene belts (Figure 15).210 Despite large strain, both products were obtained in relatively good yields of about 50 %. The authors have also recently reported the synthesis of enantiopure isomers of 195 b.211
An oxidative closure of five seven‐membered rings around a corannulene core yields the large asymmetric saddle‐shaped PAHs 196 a–c, as reported by Itami, Scott, and co‐workers (Figure 15).212, 213, 214 The authors demonstrated two different oxidative coupling strategies to construct the curved skeleton of these compounds. Oxidation of deca(4‐tert‐butylphenyl)corannulene with FeCl3 afforded the product 196 a in 62 % yield,212 whereas penta(biphenyl)‐substituted corannulenes yielded the corresponding curved PAHs 196 b and 196 c upon treatment with DDQ/TfOH (40 and 50 % yield, respectively).212, 213, 214 The ease of closure of the seven‐membered rings is noteworthy, since an analogous reaction failed for the model compound [6]helicene.212 Further functionalization of 196 c by C−H borylation and introduction of polar groups by Suzuki coupling provided a water‐soluble warped nanographene which could be used to promote photoinduced cell death in living HeLa cells.214 The authors have also reported an analogue of 196 with five benzene rings replaced by thiophene rings.215
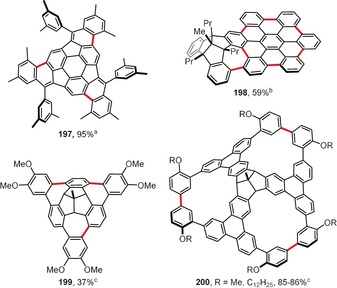
Figure 16 presents four examples of bowl‐shaped PAHs synthesized using intramolecular oxidative coupling in the final step. Compound 197 is a derivative of hemifullerene (C60 half), which was obtained in excellent yield and regioselectivity by Amaya, Ito, and Hirao through DDQ/Sc(OTf)3‐promoted oxidative cyclization of the precursor derived from the condensation of sumanene with three molecules of benzophenone.216 Mughal and Kuck synthesized the tribenzotriquinacene‐HBC hybrid 198 using the classical Müllen oxidative coupling conditions by employing copper(II) triflate/aluminum chloride in CS2 (Figure 16).217 The product contains one seven‐membered ring formed between the HBC and one of the benzene rings in the triquinacene core. Attempts to synthesize a C 3‐symmetric analogue of 198 containing three HBC moieties failed.218
Figure 16.
Bowl‐shaped compounds. Cyclization conditions: a) DDQ (6 equiv), Sc(OTf)3, CH2Cl2, RT; b) Cu(OTf)2 (42 equiv), AlCl3 (43 equiv), CS2, 45 °C; c) DDQ (4.5–5 equiv), TfOH, CH2Cl2, 0 °C.
Kuck, Chow, and co‐workers have, however, reported the syntheses of other bowl‐shaped derivatives of tribenzotriquinacene bearing threefold symmetry: compounds 199 219, 220 and 200 221 (Figure 16). Triquinacene 199 was obtained in 37 % yield from an unsymmetric triaryltribenzotriquinacene precursor by oxidative closure of three seven‐membered rings upon treatment with DDQ/TfOH. The product 199 contains an o,p,o,p,o,p‐cyclohexaphenylene belt at the rim of the bowl. Even more interesting is the synthesis of derivative 200, a rare example of oxidative macrocyclization, which was achieved using similar conditions as for 199, however, in a much higher yield (ca. 85 %; Figure 16). The high efficiency of the threefold macrocyclization can be attributed to the rigid framework of the tris(triphenyleno)triquinacene system, which spatially prearranges the alkoxyphenyl groups for intramolecular oxidative coupling.
Smith and Lucas have reported the synthesis of interesting tetraarylene‐bridged cavitands based on resorcin[4]arene using FeCl3 or DDQ as the oxidants.222
Helicenes have fired researchers’ imagination ever since the first synthesis and resolution of [6]helicene in 1956.223 The synthesis of helicenes has long been dominated by iodine‐catalyzed photocyclizations of stilbene derivatives.224, 225 Numerous examples published in recent years clearly show that the intramolecular oxidative coupling can be no worse, or even superior, for the construction of strained helicene scaffolds, especially for products containing multiple helicene fragments.
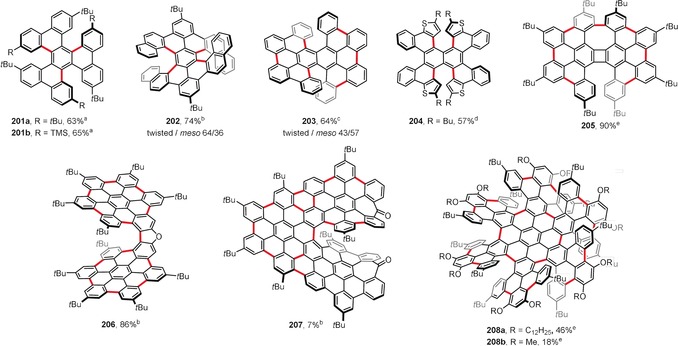
The pioneering synthesis of dibenzo[5]helicene by Durola and co‐workers using FeCl3 as the oxidant indicated that intramolecular oxidative coupling can be a very efficient method for the preparation of helicenes.226 By using this strategy the authors have synthesized in high yields the tris[5]helicenes 201 a 226 and 201 b 227 (Figure 17), as well as many other distorted PAHs with embedded helicene moieties.228 Many reports of oxidatively synthesized helicenes soon followed—selected examples are shown in Figure 17.
Figure 17.
Helicenes. Cyclization conditions: a) FeCl3 (20 equiv), CH2Cl2, CH3NO2, RT; b) DDQ (1.0–1.2 equiv/bond), TfOH, CH2Cl2, 0 °C; c) DDQ (7 equiv), DCE, TfOH, 0 °C; d) MoCl5 (10 equiv), CH2Cl2, 4 Å MS, 0 °C; e) DDQ (1.1–2.0 equiv/bond), MsOH, CH2Cl2, 0 °C. For all products obtained as mixtures of enantiomers/diastereoisomers, the structure of one selected isomer is shown.
Müllen, Narita, and co‐workers reported that, instead of the expected perihexacene derivative, the reaction of the tetranaphthyl‐substituted precursor with DDQ/TfOH produced the double [7]helicene 202 in 74 % yield as a mixture of diastereoisomers (a pair of twisted enantiomers and a meso form, Figure 17).229 Such a regioselectivity of the cyclization is impressive, since [7]helicene moieties are even more strained than [5]helicenes. Recently, an analogue of 202 with two [6]helicene parts has also been described by using oxidative coupling in the final step.230 Moreover, Miao and co‐workers described a similar double helicene with a PAH skeleton regioisomeric to 202.231
Itami and co‐workers synthesized a double [6]helicene 203 by the reaction of tetra(biphenyl)‐substituted naphthalene with DDQ/TfOH in dichloroethane (Figure 17).232 Similar to 202, the product was also obtained as a mixture of three diastereoisomers. An analogous reaction of a thiophene‐based precursor with MoCl5 in dichloromethane over 4 Å molecular sieves afforded the not fully cyclized tetrahelicene 204 instead of the expected double helicene.233 The formation of 204 was kinetically controlled; upon heating, the propeller‐like arrangement of its thiophene‐containing wings underwent quantitative isomerization to the less‐strained diastereoisomer with the wings alternately tilted “up” and “down” relative to the central naphthalene plane. Repeating the reaction with MoCl5 at room temperature without the addition of molecular sieves led to the formation of the two remaining Caryl−Caryl bonds, thereby providing an analogue of 203 (double helicene) containing four chlorine atoms as a consequence of the MoCl5‐mediated chlorination.234
Octa(4‐tert‐butylphenyl)biphenylene undergoes efficient oxidation with DDQ/MsOH to form the distorted PAH 205, which is based on a [7]helicene moiety with one four‐membered ring (Figure 17).235 Interestingly, one twisted eight‐membered ring was also formed in this reaction.
The remaining three compounds presented in Figure 17 (206–208) were all reported in 2018 and belong to the class known as “superhelicenes”, a term coined for unusually large helical PAHs. Here the synthetic power of intramolecular oxidative coupling is fully unleashed, as almost all members of this class are prepared using the oxidative coupling method.
Compound 206 was synthesized by Jux and co‐workers using DDQ/TfOH to oxidatively close 13 new Caryl–Caryl bonds in one operation (Figure 17).236 206 consists of two penta(tert‐butyl‐HBC) units fused to a central furan core, hence, forming an oxa[7]helicene derivative. Replacing DDQ by FeCl3 as an oxidant leads to the non‐fully cyclized bis‐HBC ether (lacking the furan ring) which, however, readily cyclizes to 206 upon exposure to light.
Campaña and co‐workers synthesized the superhelicene 207 in 7 % yield by employing DDQ/TfOH as an oxidant (Figure 17).237 The authors have also reported the synthesis of other large PAH‐helicenes and superhelicenes by oxidative coupling.238, 239
Compounds 208 a, 208 b,240 and larger analogues241 reported by Wang and co‐workers are amongst the most impressive examples of curved PAH derivatives prepared by oxidative coupling (Figure 17). These propeller‐shaped hexapole [7]helicenes based on a HBC core were obtained by oxidation of the corresponding dendrimer‐like oligophenylene precursor with DDQ/MsOH, which resulted in the formation of 18 new Caryl–Caryl bonds in one step. The enantiomers of 208 a were separated by HPLC on a chiral phase and the helicity of the fractions was elucidated from direct observation with a STM microscope.
Figure 18 presents helicene‐like compounds based on thiophene and pyrrole. Bu using FeCl3 as an oxidant, Aida and co‐workers synthesized propeller‐shaped molecules containing multiple thiophene rings, that is, 209 and its two C 3‐symmetric regioisomers.242
Figure 18.
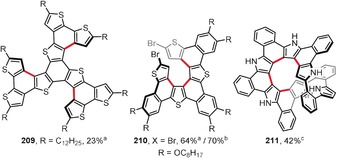
Heterocyclic helicene‐like compounds. Cyclization conditions: a) FeCl3 (6.6–12 equiv), CH2Cl2, CH3NO2, 0 °C; b) DDQ (4.5 equiv), BF3⋅Et2O, CH2Cl2, 0 °C; c) DDQ (5 equiv), Sc(OTf)3, toluene, reflux.
Heterohelicene 210 was synthesized by Dehaen and co‐workers as a demonstrative application of the oxidative coupling conditions they had optimized for the synthesis of dithienyltriphenylene analogues.243 Cyclization using DDQ or FeCl3 was found to be much faster than photocyclization (1.5–2 h versus 72 h for completion).
As an extension of the synthesis of the [8]circulene analogue 182,188 Osuka and co‐workers performed a similar reaction for a hexameric analogue, which afforded the non‐fully cyclized product 211 (Figure 18).244 Its structure contains a pentaaza[9]helicene moiety with its ends connected through a 2,5‐diphenylpyrrole bridge. An analogous reaction for a macrocyclic hexathiophene counterpart led to a product composed of [9]‐ and [5]helicene segments.
The Müllen245, 246 and Jasti247 research groups tried to apply intramolecular oxidative coupling in the synthesis of segments of carbon nanotubes (so‐called nanobelts); however, their attempts failed because of strain‐relieving rearrangements. Only very recently, Chi, Miao, and co‐workers succeeded in the synthesis of the armchair and chiral carbon nanobelts 212 and 213, respectively, using FeCl3 or DDQ/Sc(OTf)3 as the oxidants (Figure 19).248 This impressive achievement is a crowning demonstration of the capabilities of the oxidative coupling method. The same group has recently reported the preparation of a thiophene‐based nanoring using FeCl3 as the oxidant.249
Figure 19.
Cyclization conditions: a) FeCl3 (100 equiv), CH2Cl2, −20 °C; b) DDQ (22 equiv), Sc(OTf)3, toluene, reflux. R=OPr.
4. Surface‐Assisted (Cyclo)Dehydrogenation (CDH)
In contrast to a typical oxidative aromatic coupling reaction, a surface‐assisted CDH process can be considered a greener alternative because this method generally produces a minimal amount of by‐products and it does not require any solvent. An exemplary process of this type is the formation of tribenzo[a,g,m]coronene (215) from its molecular precursor 214 (Scheme 29). Research into the assembly of carbon‐rich materials by an on‐surface method was initiated by the synthesis of fullerene C60 and its triaza‐analogue,250 hexabenzocoronene (138, R=H),251 and other aromatic hemispheres252 from compounds 138. Since then, many challenging syntheses of carbon‐based nanostructures have been realized by employing this approach, including single‐chirality (that is, having one single chirality index (n,m) indicating the length and the direction of the circumventing vector) single‐walled carbon nanotubes,253 the longest to‐date acenes254 and periacenes,255 PAHs with embedded nonhexagonal rings,256 and carbon‐rich molecules with embedded nitrogen atoms.169, 250, 257 An indisputable advantage of this method is the clear preference of the arene rings to undergo exclusively the desired cyclization that leads to the extended nanographenes and results in low‐defect assemblies. Surface‐assisted synthesis has been elegantly summarized in recent literature,258, 259, 260, 261 which is why we will only briefly describe the most important contributions to this field in the following section.
Scheme 29.
The concept of a surface‐assisted CDH process toward carbon‐rich materials.262 Reagents and conditions: a) Cu(111), ca. 230 °C.
As a consequence of the excellent physicochemical properties of graphene, a great amount of effort has been made by researchers to prepare atomically precise graphene nanoribbons (GNRs). The general concept for the rational synthesis of such assemblies employing a CDH reaction usually includes two steps: 1) an on‐surface polymerization of a halogenated small‐molecule precursor by a homolytic cleavage of the carbon–halogen bond after evaporation onto a clean noble metal surface under an ultrahigh vacuum and 2) a CDH sequence at elevated temperature, which yields the fully conjugated nanoribbon structure. Following this concept, many atomically precise GNRs have been prepared from the corresponding molecular precursors (Scheme 30; GNR 217,263 219,264 221,265 223,266 225,267 227,268 229,269 and 231 127). The physicochemical properties of such GNRs may by altered not only by changing the size of the assemblies (Scheme 30 A), but also by doping with heteroatoms (Scheme 30 B) or changing the edge structure of the GNRs (Scheme 30 C).
Scheme 30.
Selected examples for the on‐surface synthesis of atomically precise graphene nanoribbons (GNRs) from small‐molecule precursors.
As has been proved many times, the CDH process occurs typically between two arene rings in proximity. Fasel, Müllen, and co‐workers have found that, during the CDH process, methyl groups that are placed in proximity to aryl rings undergo oxidative cyclization with neighboring aromatic units.270 Specifically, the molecular precursor 232 undergoes polymerization followed by a series of CDH processes to afford graphene nanoribbon 234 with a zigzag edge topology (Scheme 31 A). This concept was further extended to dibromodimethylterphenyl 235 (Scheme 31 B),271 where the polymerization/surface‐assisted oxidative ring‐closure sequence between a methyl group and the neighboring aryl moiety gives rise to indenofluorene‐based polymers 237 and 238. This strategy could eventually be employed in the preparation of graphene nanoribbons containing five‐membered rings, because such a modification at specific positions can allow for the fine‐tuning of electronic properties. According to the same strategy, ultra‐low‐gap open‐shell molecules, such as peri‐tetracene (240 from 239), a benzenoid graphene fragment with a zigzag edge topology, and dibenzo[a,m]dicyclohepta[bcde,nopq]rubicene (242 from 241), a nonbenzenoid nonalternant structural isomer of peri‐tetracene with two embedded azulene units, were synthesized272 by surface‐assisted CDH (Scheme 31 C). Spin‐polarized DFT calculations revealed that both compounds should exhibit an open‐shell singlet ground state, thus making them promising candidates for spintronic applications.
Scheme 31.
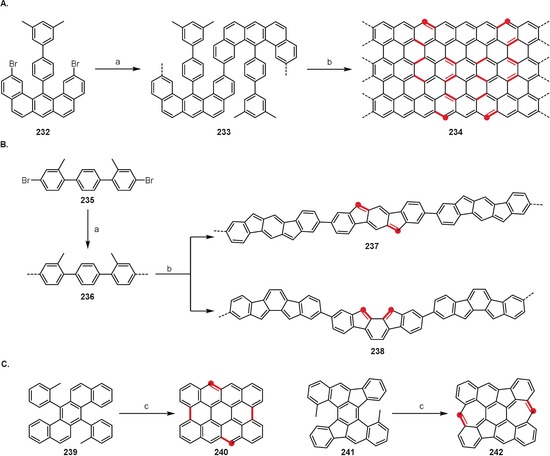
Reagents and conditions: a) Au(111), 200 °C; b) Au(111), 350 °C; c) Au(111), 300 °C. Red dots denote carbon atoms originating from methyl groups.
The power of the CDH reaction has been recently highlighted by Fasel, Müllen, and co‐workers (Scheme 32).273 A typical DDQ‐mediated oxidative aromatic coupling performed on 243 leads to the propeller‐shaped molecule 244. Although a classical oxidative aromatic coupling method was unsuccessful, the CDH process was accomplished on a Au(111) surface at elevated temperature, thus giving the nonplanar porous nanographene analogue 245.
Scheme 32.
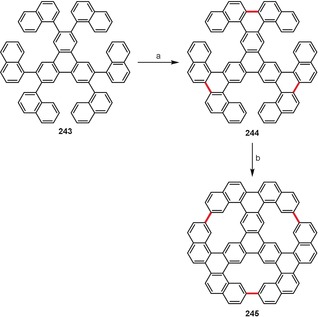
Reagents and conditions: a) DDQ (3.6 equiv), CH2Cl2/CF3SO3H (9:1), 0→25 °C, 75 %; b) Au(111), 350 °C.
Despite all the advantages described above, the CDH method still has some limitations. Many functional groups are not stable to the elevated temperatures required and, thus, the possibility of derivatizing the structures prepared by CDH is rather limited. Moreover, the synthesized nanographenic structures may also be unstable under CDH conditions. For practical applications, large amounts of a given material should ideally be obtainable in a relatively short amount of time; however, the CDH method does not yet fulfill this criterion.
5. Summary and Outlook
From small‐molecule natural products and drugs, through fluorescent dyes and polycyclic aromatic hydrocarbons to nanographenes, curved arenes, and nanobelts—the wealth of structures that can be synthesized by the oxidative coupling of arenes is tremendous. Enormous efforts by many researchers, from both theoretical and experimental standpoints, has led to an improved understanding of oxidative coupling and provided definitive evidence for the two main mechanisms: via arenium and radical cation intermediates. Slowly, predominantly thanks to the studies by the groups of Waldvogel, Müllen, and Pappo, we are approaching the point where it is possible to predict the reaction outcome before the reactants are even put into the flask; however, a lot of research is still required to reach this goal.
Simultaneously to the mechanistic studies, significant progress has been made in the development of new reagents and conditions, in particular those enabling the more efficient and more selective intermolecular oxidative homo‐ and cross‐coupling of arenes. The most interesting of these include MoCl3[OCH(CF3)2]2, (tBuO)2, Rh/C, and AgSO4. However, in many cases of cross‐coupling a large excess of one arene is still necessary to achieve good yields of heterobiaryls. The synthetic possibilities have also been vastly expanded by the broader application of electrochemical methods.
In the meantime, the intramolecular oxidative coupling of arenes has revealed its full synthetic potential, which can be concluded from the vast number of different structures that have been synthesized in recent years. Compared to the intermolecular version, the regioselectivity of intramolecular oxidative coupling is much easier to predict and plan. Moreover, many of the structures that are accessible by intramolecular oxidative coupling, in particular large planar and curved PAHs, would be nearly impossible to synthesize using other methods for forming biaryl bonds because of the required prefunctionalization. Although a variety of valuable reactants are being developed for oxidative coupling, intramolecular oxidative coupling is dominated by two oxidants: FeCl3 and DDQ/acid (Rathore conditions). These two reagents have been used by the Müllen, Itami, Miao, and Wu groups for the preparation of the most spectacular PAH molecules to date, including nanographenes, superhelicenes, and nanobelts. It is also worth emphasizing that some very strained products, such as helicenes, bowls and saddles, can be synthesized using this method. Indeed, in contrast to earlier assumptions, the Scholl reaction is capable of proceeding even if ring closure is accompanied by the introduction of significant steric strain that leads to nonplanar molecules.274, 275 Progress has been particularly impressive in the case of surface‐assisted dehydrogenation, which has enabled the synthesis of nanographenes of unprecedented size by scanning tunneling microscopy.169, 276
Despite all the impressive progress summarized herein, methods for the oxidative coupling of arenes still have some limitations, which will no doubt be the focus of future studies.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Marek Grzybowski obtained his PhD from the Institute of Organic Chemistry of the Polish Academy of Sciences in 2014 under the supervision of Prof. D. T. Gryko. He then carried out postdoctoral research under a JSPS fellowship in the group of Prof. S. Yamaguchi at Nagoya University. Currently he is an assistant professor at the Institute of Organic Chemistry of the Polish Academy of Sciences. His research focuses on the development of photostable fluorophores for bioimaging and the synthesis of extended π‐systems and strained polycyclic arenes.

Biographical Information
Bartłomiej Sadowski was born in 1990 in Nowy Dwór Mazowiecki, Poland. He completed his MSc in 2014 at the Warsaw University of Technology. Subsequently, he received his PhD from the Institute of Organic Chemistry of the Polish Academy of Sciences under the guidance of Prof. D. T. Gryko. His research interests focus on the synthesis of functional aromatic molecules, notably dipyrrolonaphthyridinediones.

Biographical Information
Holger Butenschön obtained his PhD with A. de Meijere in 1983. After a postdoctoral year with Prof. K. P. C. Vollhardt at the University of California at Berkeley, he carried out independent research at the Max‐Planck‐Institute for Coal Research in Mülheim an der Ruhr, Germany. After habilitation in organic chemistry at the University of Hamburg in 1991, he was a Heisenberg fellow (DFG) at the University of Wuppertal. Since 1993 he has been a professor of organic chemistry at Leibniz University Hannover and was a visiting professor at Kyushu University in 1999 as well as at the University of California at Berkeley in 2008.

Biographical Information
Daniel T. Gryko obtained his PhD from the Institute of Organic Chemistry of the Polish Academy of Sciences in 1997, under the supervision of Prof. J. Jurczak. After postdoctoral research with Prof. J. Lindsey at North Carolina State University, he started his independent career in Poland. Recently, he became Director of the Institute of Organic Chemistry of the Polish Academy of Sciences. He received the Society of Porphyrins and Phthalocyanines Young Investigator Award in 2008 and Foundation for Polish Science Award in 2017.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The work was financially supported by the Foundation for Polish Science (TEAM POIR.04.04.00‐00‐3CF4/16‐00) and the Polish National Science Centre (Grants PRELUDIUM 2016/23/N/ST5/00054 and SONATA 2018/31/D/ST5/00432).
M. Grzybowski, B. Sadowski, H. Butenschön, D. T. Gryko, Angew. Chem. Int. Ed. 2020, 59, 2998.
Dedicated to Professor Armin de Meijere on the occasion of his 80th birthday
Contributor Information
Prof. Dr. Holger Butenschön, Email: holger.butenschoen@mbox.oci.uni-hannover.de.
Prof. Dr. Daniel T. Gryko, Email: dtgryko@icho.edu.pl.
References
- 1. Griefsmayer V., Justus Liebigs Ann. Chem. 1871, 160, 40–56. [Google Scholar]
- 2. Scholl R., Mansfeld J., Ber. Dtsch. Chem. Ges. 1910, 43, 1734–1746. [Google Scholar]
- 3. Grzybowski M., Skonieczny K., Butenschön H., Gryko D. T., Angew. Chem. Int. Ed. 2013, 52, 9900–9930; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10084–10115. [Google Scholar]
- 4. Ito H., Segawa Y., Murakami K., Itami K., J. Am. Chem. Soc. 2019, 141, 3–10. [DOI] [PubMed] [Google Scholar]
- 5. Narita A., Wang X. Y., Feng X., Müllen K., Chem. Soc. Rev. 2015, 44, 6616–6643. [DOI] [PubMed] [Google Scholar]
- 6. Stępień M., Gońka E., Żyła M., Sprutta N., Chem. Rev. 2017, 117, 3479–3716. [DOI] [PubMed] [Google Scholar]
- 7. Mallory F. B., Mallory C. W., in Org. React., Wiley, Hoboken, 1984, pp. 1–456. [Google Scholar]
- 8. Jørgensen K. B., Molecules 2010, 15, 4334–4358.20657445 [Google Scholar]
- 9. Sarhan A. A. O., Bolm C., Chem. Soc. Rev. 2009, 38, 2730–2744. [DOI] [PubMed] [Google Scholar]
- 10. Morioku K., Morimoto N., Takeuchi Y., Nishina Y., Sci. Rep. 2016, 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Budniak A. K., Masny M., Prezelj K., Grzeszkiewicz M., Gawraczyński J., Dobrzycki Ł., Cyrański M. K., Koźmiński W., Mazej Z., Fijałkowski K. J., et al., New J. Chem. 2017, 41, 10742–10749. [Google Scholar]
- 12. Waldvogel S. R., Trosien S., Chem. Commun. 2012, 48, 9109–9119. [DOI] [PubMed] [Google Scholar]
- 13. Beil S. B., Uecker I., Franzmann P., Müller T., Waldvogel S. R., Org. Lett. 2018, 20, 4107–4110. [DOI] [PubMed] [Google Scholar]
- 14. Kovacic P., Lange R. M., J. Org. Chem. 1965, 30, 4251–4254. [Google Scholar]
- 15. Kramer B., Fröhlich R., Waldvogel S. R., Eur. J. Org. Chem. 2003, 3549–3554. [Google Scholar]
- 16. Schubert M., Leppin J., Wehming K., Schollmeyer D., Heinze K., Waldvogel S. R., Angew. Chem. Int. Ed. 2014, 53, 2494–2497; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2527–2530. [Google Scholar]
- 17. Beil S. B., Müller T., Sillart S. B., Franzmann P., Bomm A., Holtkamp M., Karst U., Schade W., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 2450–2454; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2475–2479. [Google Scholar]
- 18. Boyd E. M., Sperry J., Org. Lett. 2015, 17, 1344–1346. [DOI] [PubMed] [Google Scholar]
- 19. Armstrong D. R., Breckenridge R. J., Cameron C., Nonhebel D. C., Pauson P. L., Perkins P. G., Tetrahedron Lett. 1983, 24, 1071–1074. [Google Scholar]
- 20. Lee Y. E., Cao T., Torruellas C., Kozlowski M. C., J. Am. Chem. Soc. 2014, 136, 6782–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu X., Iwata T., Scharf A., Qin T., Reichl K. D., Porco J. A., J. Am. Chem. Soc. 2018, 140, 5969–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakanishi K., Sasamori T., Kuramochi K., Tokitoh N., Kawabata T., Tsubaki K., J. Org. Chem. 2014, 79, 2625–2631. [DOI] [PubMed] [Google Scholar]
- 23. Hassan K., Yamashita K., Hirabayashi K., Shimizu T., Nakabayashi K., Imai Y., Matsumoto T., Yamano A., Sugiura K., Chem. Lett. 2015, 44, 1607–1609. [Google Scholar]
- 24. Rajbangshi S., Sugiura K. I., Synthesis 2017, 49, 3145–3148. [Google Scholar]
- 25. Nazir R., Stasyuk A. J., Gryko D. T., J. Org. Chem. 2016, 81, 11104–11114. [DOI] [PubMed] [Google Scholar]
- 26. Li Y., Hong Y., Guo J., Huang X., Wei H., Zhou J., Qiu T., Wu J., Zeng Z., Org. Lett. 2017, 19, 5094–5097. [DOI] [PubMed] [Google Scholar]
- 27. Maddala S., Mallick S., Venkatakrishnan P., J. Org. Chem. 2017, 82, 8958–8972. [DOI] [PubMed] [Google Scholar]
- 28. Morimoto K., Koseki D., Dohi T., Kita Y., Synlett 2017, 28, 2941–2945. [Google Scholar]
- 29. Matsumoto K., Dougomori K., Tachikawa S., Ishii T., Shindo M., Org. Lett. 2014, 16, 4754–4757. [DOI] [PubMed] [Google Scholar]
- 30. Forkosh H., Vershinin V., Reiss H., Pappo D., Org. Lett. 2018, 20, 2459–2463. [DOI] [PubMed] [Google Scholar]
- 31. Matsumoto K., Yoshida M., Shindo M., Angew. Chem. Int. Ed. 2016, 55, 5272–5276; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5358–5362. [Google Scholar]
- 32. Fritsche R. F., Theumer G., Kataeva O., Knölker H. J., Angew. Chem. Int. Ed. 2017, 56, 549–553; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 564–568. [Google Scholar]
- 33. Mallick S., Maddala S., Kollimalayan K., Venkatakrishnan P., J. Org. Chem. 2019, 84, 73–93. [DOI] [PubMed] [Google Scholar]
- 34. Yuan J., Jin L., Chen R., Tang X., Xie X., Tang Y., Huang W., New J. Chem. 2018, 42, 14704–14708. [Google Scholar]
- 35. Müller M., Kübel C., Müllen K., Chem. Eur. J. 1998, 4, 2099–2109. [Google Scholar]
- 36. Wu J., Pisula W., Müllen K., Chem. Rev. 2007, 107, 718–747. [DOI] [PubMed] [Google Scholar]
- 37. Salvatierra R. V., Souza V. H. R., Matos C. F., Oliveira M. M., Zarbin A. J. G., Carbon 2015, 93, 924–932. [Google Scholar]
- 38. Wirtanen T., Mäkelä M. K., Sarfraz J., Ihalainen P., Hietala S., Melchionna M., Helaja J., Adv. Synth. Catal. 2015, 357, 3718–3726. [Google Scholar]
- 39. Wirtanen T., Muuronen M., Hurmalainen J., Tuononen H. M., Nieger M., Helaja J., Org. Chem. Front. 2016, 3, 1738–1745. [Google Scholar]
- 40. Bering L., Paulussen F. M., Antonchick A. P., Org. Lett. 2018, 20, 1978–1981. [DOI] [PubMed] [Google Scholar]
- 41. Seidel D., Lynch V., Sessler J. L., Angew. Chem. Int. Ed. 2002, 41, 1422–1425; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1480–1483. [Google Scholar]
- 42. Köhler T., Seidel D., Lynch V., Arp F. O., Ou Z., Kadish K. M., Sessler J. L., J. Am. Chem. Soc. 2003, 125, 6872–6873. [DOI] [PubMed] [Google Scholar]
- 43. Gadekar S. C., Reddy B. K., Anand V. G., Angew. Chem. Int. Ed. 2013, 52, 7164–7167; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7305–7308. [Google Scholar]
- 44. Gadekar S. C., Reddy B. K., Panchal S. P., Anand V. G., Chem. Commun. 2016, 52, 4565–4568. [DOI] [PubMed] [Google Scholar]
- 45. Zhang K., Savage M., Li X., Jiang Y., Ishida M., Mitsuno K., Karasawa S., Kato T., Zhu W., Yang S., et al., Chem. Commun. 2016, 52, 5148–5151. [DOI] [PubMed] [Google Scholar]
- 46. Libman A., Shalit H., Vainer Y., Narute S., Kozuch S., Pappo D., J. Am. Chem. Soc. 2015, 137, 11453–11460. [DOI] [PubMed] [Google Scholar]
- 47. Kozlowski M. C., Acc. Chem. Res. 2017, 50, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kirste A., Schnakenburg G., Stecker F., Fischer A., Waldvogel S. R., Angew. Chem. Int. Ed. 2010, 49, 971–975; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 983–987. [Google Scholar]
- 49. Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2014, 53, 5210–5213; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5311–5314. [Google Scholar]
- 50. Wiebe A., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 11801–11805; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11979–11983. [Google Scholar]
- 51. Waldvogel S. R., Lips S., ChemElectroChem 2018, 6, 1649–1660. [Google Scholar]
- 52. Möhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149. [Google Scholar]
- 53. Kirste A., Elsler B., Schnakenburg G., Waldvogel S. R., J. Am. Chem. Soc. 2012, 134, 3571–3576. [DOI] [PubMed] [Google Scholar]
- 54. Elsler B., Wiebe A., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Chem. Eur. J. 2015, 21, 12321–12325. [DOI] [PubMed] [Google Scholar]
- 55. Schulz L., Enders M., Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2017, 56, 4877–4881; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4955–4959. [Google Scholar]
- 56. Wiebe A., Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2017, 56, 14727–14731; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14920–14925. [Google Scholar]
- 57. Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 13325–13329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13509–13513. [Google Scholar]
- 58. Morimoto K., Dohi T., Kita Y., Synlett 2017, 28, 1680–1694. [Google Scholar]
- 59. Morimoto K., Sakamoto K., Ohnishi Y., Miyamoto T., Ito M., Dohi T., Kita Y., Chem. Eur. J. 2013, 19, 8726–8731. [DOI] [PubMed] [Google Scholar]
- 60. Ito M., Kubo H., Itani I., Morimoto K., Dohi T., Kita Y., J. Am. Chem. Soc. 2013, 135, 14078–14081. [DOI] [PubMed] [Google Scholar]
- 61. Morimoto K., Sakamoto K., Ohshika T., Dohi T., Kita Y., Angew. Chem. Int. Ed. 2016, 55, 3652–3656; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3716–3720. [Google Scholar]
- 62. More N. Y., Jeganmohan M., Org. Lett. 2015, 17, 3042–3045. [DOI] [PubMed] [Google Scholar]
- 63. Gaster E., Vainer Y., Regev A., Narute S., Sudheendran K., Werbeloff A., Shalit H., Pappo D., Angew. Chem. Int. Ed. 2015, 54, 4198–4202; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4272–4276. [Google Scholar]
- 64. Shalit H., Libman A., Pappo D., J. Am. Chem. Soc. 2017, 139, 13404–13413. [DOI] [PubMed] [Google Scholar]
- 65. Reiss H., Shalit H., Vershinn V., More N. Y., Forckosh H., Pappo D., J. Org. Chem. 2019, 84, 7950–7960. [DOI] [PubMed] [Google Scholar]
- 66. Nieves-Quinones Y., Paniak T. J., Lee Y. E., Kim S. M., Tcyrulnikov S., Kozlowski M. C., J. Am. Chem. Soc. 2019, 141, 10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dyadyuk A., Sudheendran K., Vainer Y., Vershinin V., Shames A. I., Pappo D., Org. Lett. 2016, 18, 4324–4327. [DOI] [PubMed] [Google Scholar]
- 68. Sawama Y., Masuda M., Nakatani R., Yokoyama H., Monguchi Y., Dohi T., Kita Y., Sajiki H., Adv. Synth. Catal. 2016, 358, 3683–3687. [Google Scholar]
- 69. Tamura Y., Yakura T., ichi Haruta J., Kita Y., J. Org. Chem. 1987, 52, 3927–3930. [Google Scholar]
- 70. Mitchell A. S., Russell R. A., Tetrahedron 1997, 53, 4387–4410. [Google Scholar]
- 71. Norman D. W., Carraz C. A., Hyett D. J., Pringle P. G., Sweeney J. B., Orpen A. G., Phetmung H., Wingad R. L., J. Am. Chem. Soc. 2008, 130, 6840–6847. [DOI] [PubMed] [Google Scholar]
- 72. Aydin J., Kumar K. S., Sayah M. J., Wallner O. A., Szabó K. J., J. Org. Chem. 2007, 72, 4689–4697. [DOI] [PubMed] [Google Scholar]
- 73. Wang T., Ma H., Zhang S., Li Z. J., Zhang M., Li F., Sun F., Xiang J., Ke M., Wang Q., Org. Lett. 2018, 20, 3591–3595. [DOI] [PubMed] [Google Scholar]
- 74. Morimoto K., Nakamura A., Dohi T., Kita Y., Eur. J. Org. Chem. 2016, 4294–4297. [Google Scholar]
- 75. Bringmann G., Walter R., Weirich R., Angew. Chem. Int. Ed. Engl. 1990, 29, 977–991; [Google Scholar]; Angew. Chem. 1990, 102, 1006–1019. [Google Scholar]
- 76. Kozlowski M. C., Morgan B. J., Linton E. C., Chem. Soc. Rev. 2009, 38, 3193–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bringmann G., Gulder T., Gulder T. A. M., Breuning M., Chem. Rev. 2011, 111, 563–639. [DOI] [PubMed] [Google Scholar]
- 78. Wencel-Delord J., Panossian A., Leroux F. R., Colobert F., Chem. Soc. Rev. 2015, 44, 3418–3430. [DOI] [PubMed] [Google Scholar]
- 79. Brunel J. M., Chem. Rev. 2005, 105, 857–898. [DOI] [PubMed] [Google Scholar]
- 80. Nakajima M., Miyoshi I., Kanayama K., Hashimoto S. I., Noji M., Koga K., J. Org. Chem. 1999, 64, 2264–2271. [Google Scholar]
- 81. Li X., Yang J., Kozlowski M. C., Org. Lett. 2001, 3, 1137–1140. [DOI] [PubMed] [Google Scholar]
- 82. Kim K. H., Lee D. W., Lee Y. S., Ko D. H., Ha D. C., Tetrahedron 2004, 60, 9037–9042. [Google Scholar]
- 83. Allen S. E., Walvoord R. R., Padilla-Salinas R., Kozlowski M. C., Chem. Rev. 2013, 113, 6234–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Barhate N. B., Chen C. T., Org. Lett. 2002, 4, 2529–2532. [DOI] [PubMed] [Google Scholar]
- 85. Chu C. Y., Uang B. J., Tetrahedron: Asymmetry 2003, 14, 53–55. [Google Scholar]
- 86. Guo Q. X., Wu Z. J., Bin Luo Z., Liu Q. Z., Ye J. L., Luo S. W., Cun L. F., Gong L. Z., J. Am. Chem. Soc. 2007, 129, 13927–13938. [DOI] [PubMed] [Google Scholar]
- 87. Takizawa S., Katayama T., Somei H., Asano Y., Yoshida T., Kameyama C., Rajesh D., Onitsuka K., Suzuki T., Mikami M., et al., Tetrahedron 2008, 64, 3361–3371. [Google Scholar]
- 88. Takizawa S., Gröger H., Sasai H., Chem. Eur. J. 2015, 21, 8992–8997. [DOI] [PubMed] [Google Scholar]
- 89. Egami H., Katsuki T., J. Am. Chem. Soc. 2009, 131, 6082–6083. [DOI] [PubMed] [Google Scholar]
- 90. Shalit H., Dyadyuk A., Pappo D., J. Org. Chem. 2019, 84, 1677–1686. [DOI] [PubMed] [Google Scholar]
- 91. Kang H., Herling M. R., Niederer K. A., Lee Y. E., Vasu Govardhana Reddy P., Dey S., Allen S. E., Sung P., Hewitt K., Torruellas C., et al., J. Org. Chem. 2018, 83, 14362–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Narute S., Parnes R., Toste F. D., Pappo D., J. Am. Chem. Soc. 2016, 138, 16553–16560. [DOI] [PubMed] [Google Scholar]
- 93. Sako M., Takizawa S., Yoshida Y., Sasai H., Tetrahedron: Asymmetry 2015, 26, 613–616. [Google Scholar]
- 94. Takizawa S., Kodera J., Yoshida Y., Sako M., Breukers S., Enders D., Sasai H., Tetrahedron 2014, 70, 1786–1793. [Google Scholar]
- 95. Kim H. Y., Takizawa S., Sasai H., Oh K., Org. Lett. 2017, 19, 3867–3870. [DOI] [PubMed] [Google Scholar]
- 96. Sako M., Takeuchi Y., Tsujihara T., Kodera J., Kawano T., Takizawa S., Sasai H., J. Am. Chem. Soc. 2016, 138, 11481–11484. [DOI] [PubMed] [Google Scholar]
- 97. Kang H., Lee Y. E., Reddy P. V. G., Dey S., Allen S. E., Niederer K. A., Sung P., Hewitt K., Torruellas C., Herling M. R., et al., Org. Lett. 2017, 19, 5505–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kang H., Torruellas C., Liu J., Kozlowski M. C., Org. Lett. 2018, 20, 5554–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sako M., Aoki T., Zumbrägel N., Schober L., Gröger H., Takizawa S., Sasai H., J. Org. Chem. 2019, 84, 1580–1587. [DOI] [PubMed] [Google Scholar]
- 100. Wu Y., Wang J., Mao F., Kwong F. Y., Chem. Asian J. 2014, 9, 26–47. [DOI] [PubMed] [Google Scholar]
- 101. Hussain I., Singh T., Adv. Synth. Catal. 2014, 356, 1661–1696. [Google Scholar]
- 102. Liu C., Yuan J., Gao M., Tang S., Li W., Shi R., Lei A., Chem. Rev. 2015, 115, 12138–12204. [DOI] [PubMed] [Google Scholar]
- 103. Yang Y., Lan J., You J., Chem. Rev. 2017, 117, 8787–8863. [DOI] [PubMed] [Google Scholar]
- 104. Zhang Y. F., Shi Z. J., Acc. Chem. Res. 2019, 52, 161–169. [DOI] [PubMed] [Google Scholar]
- 105. Zou L. H., Mottweiler J., Priebbenow D. L., Wang J., Stubenrauch J. A., Bolm C., Chem. Eur. J. 2013, 19, 3302–3305. [DOI] [PubMed] [Google Scholar]
- 106. Odani R., Hirano K., Satoh T., Miura M., J. Org. Chem. 2013, 78, 11045–11052. [DOI] [PubMed] [Google Scholar]
- 107. Qin X., Liu H., Qin D., Wu Q., You J., Zhao D., Guo Q., Huang X., Lan J., Chem. Sci. 2013, 4, 1964–1969. [Google Scholar]
- 108. Reddy V. P., Qiu R., Iwasaki T., Kambe N., Org. Lett. 2013, 15, 1290–1293. [DOI] [PubMed] [Google Scholar]
- 109. Wu Q., Chen Y., Yan D., Zhang M., Lu Y., Sun W. Y., Zhao J., Chem. Sci. 2017, 8, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Dong J., Long Z., Song F., Wu N., Guo Q., Lan J., You J., Angew. Chem. Int. Ed. 2013, 52, 580–584; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 608–612. [Google Scholar]
- 111. Qin D., Wang J., Qin X., Wang C., Gao G., You J., Chem. Commun. 2015, 51, 6190–6193. [DOI] [PubMed] [Google Scholar]
- 112. Huang Y., Wu D., Huang J., Guo Q., Li J., You J., Angew. Chem. Int. Ed. 2014, 53, 12158–12162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12354–12358. [Google Scholar]
- 113. Gong H., Zeng H., Zhou F., Li C. J., Angew. Chem. Int. Ed. 2015, 54, 5718–5721; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5810–5813. [Google Scholar]
- 114. Liu Y., Zhu Y., Li C., Front. Chem. Sci. Eng. 2018, 12, 3–8. [Google Scholar]
- 115. Zhang H., Deng G. J., Li S., Li C. J., Gong H., RSC Adv. 2016, 6, 91617–91620. [Google Scholar]
- 116. Yang F., Song F., Li W., Lan J., You J., RSC Adv. 2013, 3, 9649–9652. [Google Scholar]
- 117. Jiao L. Y., Smirnov P., Oestreich M., Org. Lett. 2014, 16, 6020–6023. [DOI] [PubMed] [Google Scholar]
- 118. Yang Z., Qiu F. C., Gao J., Li Z. W., Guan B. T., Org. Lett. 2015, 17, 4316–4319. [DOI] [PubMed] [Google Scholar]
- 119. Zhang C., Rao Y., Org. Lett. 2015, 17, 4456–4459. [DOI] [PubMed] [Google Scholar]
- 120. Lou S. J., Mao Y. J., Xu D. Q., He J. Q., Chen Q., Xu Z. Y., ACS Catal. 2016, 6, 3890–3894. [Google Scholar]
- 121. Gao G. L., Xia W., Jain P., Yu J. Q., Org. Lett. 2016, 18, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wehmeier M., Epsch R., Brand J. D., Kübel C., Müllen K., Rabe J. P., Ito S., Chem. Eur. J. 2000, 6, 4327–4342. [DOI] [PubMed] [Google Scholar]
- 123. Fechtenkötter A., Saalwächter K., Harbison M. A., Müllen K., Spiess H. W., Angew. Chem. Int. Ed. 1999, 38, 3039–3042; [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 3224–3228. [Google Scholar]
- 124. Dorel R., Manzano C., Grisolia M., Soe W. H., Joachim C., Echavarren A. M., Chem. Commun. 2015, 51, 6932–6935. [DOI] [PubMed] [Google Scholar]
- 125. Arslan H., Uribe-Romo F. J., Smith B. J., Dichtel W. R., Chem. Sci. 2013, 4, 3973–3978. [Google Scholar]
- 126. Wu T. L., Kuo C. H., Lin B. C., Tao Y. T., Hsu C. P., Liu R. S., J. Mater. Chem. C 2015, 3, 7583–7588. [Google Scholar]
- 127. Liu J., Li B. W., Tan Y. Z., Giannakopoulos A., Sanchez-Sanchez C., Beljonne D., Ruffieux P., Fasel R., Feng X., Müllen K., J. Am. Chem. Soc. 2015, 137, 6097–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Navale T. S., Ivanov M. V., Hossain M. M., Rathore R., Angew. Chem. Int. Ed. 2018, 57, 790–794; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 798–802. [Google Scholar]
- 129. Li G., Yoon K. Y., Zhong X., Zhu X., Dong G., Chem. Eur. J. 2016, 22, 9116–9120. [DOI] [PubMed] [Google Scholar]
- 130. Koga Y., Kaneda T., Saito Y., Murakami K., Itami K., Science 2018, 359, 435–439. [DOI] [PubMed] [Google Scholar]
- 131. Sen C. P., Valiyaveettil S., Chem. Eur. J. 2017, 23, 1686–1693. [DOI] [PubMed] [Google Scholar]
- 132. Zhu C., Wang D., Wang D., Zhao Y., Sun W. Y., Shi Z., Angew. Chem. Int. Ed. 2018, 57, 8848–8853; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8986–8991. [Google Scholar]
- 133. Kumar S., Ho M. T., Tao Y. T., Org. Lett. 2016, 18, 200–203. [DOI] [PubMed] [Google Scholar]
- 134. Tao Y. T., Pola S., Kumar S., Islam M. M., J. Org. Chem. 2017, 82, 8067–8071. [DOI] [PubMed] [Google Scholar]
- 135. Kumar S., Huang D. C., Venkateswarlu S., Tao Y. T., J. Org. Chem. 2018, 83, 11614–11622. [DOI] [PubMed] [Google Scholar]
- 136. Kumar S., Tao Y. T., Org. Lett. 2018, 20, 2320–2323. [DOI] [PubMed] [Google Scholar]
- 137. Kasun Z. A., Sato H., Nie J., Mori Y., Bender J. A., Roberts S. T., Krische M. J., Chem. Sci. 2018, 9, 7866–7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Chen Y., Marszalek T., Fritz T., Baumgarten M., Wagner M., Pisula W., Chen L., Müllen K., Chem. Commun. 2017, 53, 8474–8477. [DOI] [PubMed] [Google Scholar]
- 139. Baumgärtner K., Meza Chincha A. L., Dreuw A., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2016, 55, 15594–15598; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15823–15827. [Google Scholar]
- 140. Zhao K. Q., Jing M., An L. L., Du J. Q., Wang Y. H., Hu P., Wang B. Q., Monobe H., Heinrich B., Donnio B., J. Mater. Chem. C 2017, 5, 669–682. [Google Scholar]
- 141. Tokoro Y., Oishi A., Fukuzawa S.-ichi, Chem. Eur. J. 2016, 22, 13908–13915. [DOI] [PubMed] [Google Scholar]
- 142. Naibi Lakshminarayana A., Chang J., Luo J., Zheng B., Huang K. W., Chi C., Chem. Commun. 2015, 51, 3604–3607. [DOI] [PubMed] [Google Scholar]
- 143. Kawamura M., Tsurumaki E., Toyota S., Synthesis 2018, 50, 134–138. [Google Scholar]
- 144. Purvis L. J., Gu X., Ghosh S., Zhang Z., Cramer C. J., Douglas C. J., J. Org. Chem. 2018, 83, 1828–1841. [DOI] [PubMed] [Google Scholar]
- 145. Lungerich D., Hitzenberger J. F., Donaubauer W., Drewello T., Jux N., Chem. Eur. J. 2016, 22, 16697. [DOI] [PubMed] [Google Scholar]
- 146. Zeng W., Phan H., Herng T. S., Gopalakrishna T. Y., Aratani N., Zeng Z., Yamada H., Ding J., Wu J., Chem 2017, 2, 81–92. [Google Scholar]
- 147. Hensel T., Andersen N. N., Plesner M., Pittelkow M., Synlett 2016, 27, 498–525. [Google Scholar]
- 148. Zhang Q., Peng H., Zhang G., Lu Q., Chang J., Dong Y., Shi X., Wei J., J. Am. Chem. Soc. 2014, 136, 5057–5064. [DOI] [PubMed] [Google Scholar]
- 149. Zhang X., Xu Z., Si W., Oniwa K., Bao M., Yamamoto Y., Jin T., Nat. Commun. 2017, 8, 15073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Nobusue S., Fujita K., Tobe Y., Org. Lett. 2017, 19, 3227–3230. [DOI] [PubMed] [Google Scholar]
- 151. Liu J., Narita A., Osella S., Zhang W., Schollmeyer D., Beljonne D., Feng X., Müllen K., J. Am. Chem. Soc. 2016, 138, 2602–2608. [DOI] [PubMed] [Google Scholar]
- 152. Liu J., Osella S., Ma J., Berger R., Beljonne D., Schollmeyer D., Feng X., Müllen K., J. Am. Chem. Soc. 2016, 138, 8364–8367. [DOI] [PubMed] [Google Scholar]
- 153. He J., Mathew S., Kinney Z. J., Warrell R. M., Molina J. S., Hartley C. S., Chem. Commun. 2015, 51, 7245–7248. [DOI] [PubMed] [Google Scholar]
- 154. Chaolumen, Murata M., Wakamiya A., Murata Y., Angew. Chem. Int. Ed. 2017, 56, 5082–5086; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5164–5168. [Google Scholar]
- 155. Shekhirev M., Sinitskii A., Phys. Sci. Rev. 2017, 2, 1–21. [Google Scholar]
- 156. Narita A., Chen Z., Chen Q., Müllen K., Chem. Sci. 2019, 10, 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Narita A., Feng X., Hernandez Y., Jensen S. A., Bonn M., Yang H., Verzhbitskiy I. A., Casiraghi C., Hansen M. R., Koch A. H. R., et al., Nat. Chem. 2014, 6, 126–132. [DOI] [PubMed] [Google Scholar]
- 158. Schwab M. G., Narita A., Osella S., Hu Y., Maghsoumi A., Mavrinsky A., Pisula W., Castiglioni C., Tommasini M., Beljonne D., et al., Chem. Asian J. 2015, 10, 2134–2138. [DOI] [PubMed] [Google Scholar]
- 159. Hou I. C. Y., Hu Y., Narita A., Müllen K., Polym. J. 2018, 50, 3–20. [Google Scholar]
- 160. Grätz S., Beyer D., Tkachova V., Hellmann S., Berger R., Feng X., Borchardt L., Chem. Commun. 2018, 54, 5307–5310. [DOI] [PubMed] [Google Scholar]
- 161. Tasior M., Kim D., Singha S., Krzeszewski M., Ahn K. H., Gryko D. T., J. Mater. Chem. C 2015, 3, 1421–1446. [Google Scholar]
- 162. Węcławski M. K., Tasior M., Hammann T., Cywiński P. J., Gryko D. T., Chem. Commun. 2014, 50, 9105–9108. [DOI] [PubMed] [Google Scholar]
- 163. Węcławski M. K., Deperasińska I., Banasiewicz M., Young D. C., Leniak A., Gryko D. T., Chem. Asian J. 2019, 14, 1763–1770. [DOI] [PubMed] [Google Scholar]
- 164. Krzeszewski M., Gryko D., Gryko D. T., Acc. Chem. Res. 2017, 50, 2334–2345. [DOI] [PubMed] [Google Scholar]
- 165. Krzeszewski M., Gryko D. T., J. Org. Chem. 2015, 80, 2893–2899. [DOI] [PubMed] [Google Scholar]
- 166. Stone A. J., Wales D. J., Chem. Phys. Lett. 1986, 128, 501–503. [Google Scholar]
- 167. Lusk M. T., Wu D. T., Carr L. D., Phys. Rev. B 2010, 81, 155444. [Google Scholar]
- 168. Krzeszewski M., Kodama T., Espinoza E. M., Vullev V. I., Kubo T., Gryko D. T., Chem. Eur. J. 2016, 22, 16478–16488. [DOI] [PubMed] [Google Scholar]
- 169. Mishra S., Krzeszewski M., Pignedoli C. A., Ruffieux P., Fasel R., Gryko D. T., Nat. Commun. 2018, 9, 1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Krzeszewski M., Sahara K., Poronik Y. M., Kubo T., Gryko D. T., Org. Lett. 2018, 20, 1517–1520. [DOI] [PubMed] [Google Scholar]
- 171. Krzeszewski M., Świder P., Dobrzycki Ł., Cyrański M. K., Danikiewicz W., Gryko D. T., Chem. Commun. 2016, 52, 11539–11542. [DOI] [PubMed] [Google Scholar]
- 172. Yang T., Pu J., Zhang J., Wang W., J. Org. Chem. 2013, 78, 4857–4866. [DOI] [PubMed] [Google Scholar]
- 173. Zhong H., Wu C. H., Li C. Z., Carpenter J., Chueh C. C., Chen J. Y., Ade H., Jen A. K. Y., Adv. Mater. 2016, 28, 951–958. [DOI] [PubMed] [Google Scholar]
- 174. Hartnett P. E., Ramakrishna Matte H. S. S., Eastham N. D., Jackson N. E., Wu Y., Chen L. X., Ratner M. A., Chang R. P. H., Hersam M. C., Wasielewski M. R., et al., Chem. Sci. 2016, 7, 3543–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Yu C., Jiao L., Li T., Wu Q., Miao W., Wang J., Wei Y., Mu X., Hao E., Chem. Commun. 2015, 51, 16852–16855. [DOI] [PubMed] [Google Scholar]
- 176. Heyer E., Retailleau P., Ziessel R., Org. Lett. 2014, 16, 2330–2333. [DOI] [PubMed] [Google Scholar]
- 177. Sheng W., Zheng Y. Q., Wu Q., Wu Y., Yu C., Jiao L., Hao E., Wang J. Y., Pei J., Org. Lett. 2017, 19, 2893–2896. [DOI] [PubMed] [Google Scholar]
- 178. Cui J., Sheng W., Wu Q., Yu C., Hao E., Bobadova-Parvanova P., Storer M., Asiri A. M., Marwani H. M., Jiao L., Chem. Asian J. 2017, 12, 2486–2493. [DOI] [PubMed] [Google Scholar]
- 179. Sheng W., Wu Y., Yu C., Bobadova-Parvanova P., Hao E., Jiao L., Org. Lett. 2018, 20, 2620–2623. [DOI] [PubMed] [Google Scholar]
- 180. Skonieczny K., Jaźwiński J., Gryko D. T., Synthesis 2017, 49, 4651–4662. [Google Scholar]
- 181. Ono T., Xu N., Koga D., Ideo T., Sugimoto M., Hisaeda Y., RSC Adv. 2018, 8, 39269–39273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Firmansyah D., Hong S. J., Dutta R., He Q., Bae J., Jo H., Kim H., Ok K. M., Lynch V. M., Byon H. R., et al., Chem. Eur. J. 2019, 25, 3525–3531. [DOI] [PubMed] [Google Scholar]
- 183. Takase M., Enkelmann V., Sebastiani D., Baumgarten M., Müllen K., Angew. Chem. Int. Ed. 2007, 46, 5524–5527; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5620–5623. [Google Scholar]
- 184. Takase M., Narita T., Fujita W., Asano M. S., Nishinaga T., Benten H., Yoza K., Müllen K., J. Am. Chem. Soc. 2013, 135, 8031–8040. [DOI] [PubMed] [Google Scholar]
- 185. Zyła M., Gońka E., Chmielewski P. J., Cybińska J., Stępień M., Chem. Sci. 2016, 7, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Gońka E., Chmielewski P. J., Lis T., Stępień M., J. Am. Chem. Soc. 2014, 136, 16399–16410. [DOI] [PubMed] [Google Scholar]
- 187. Oki K., Takase M., Mori S., Shiotari A., Sugimoto Y., Ohara K., Okujima T., Uno H., J. Am. Chem. Soc. 2018, 140, 10430–10434. [DOI] [PubMed] [Google Scholar]
- 188. Chen F., Hong Y. S., Shimizu S., Kim D., Tanaka T., Osuka A., Angew. Chem. Int. Ed. 2015, 54, 10639–10642; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10785–10788. [Google Scholar]
- 189. Myśliwiec D., Stępień M., Angew. Chem. Int. Ed. 2013, 52, 1713–1717; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1757–1761. [Google Scholar]
- 190. Kondratowicz M., Myśliwiec D., Lis T., Stępień M., Chem. Eur. J. 2014, 20, 14981–14985. [DOI] [PubMed] [Google Scholar]
- 191. Chen F., Tanaka T., Mori T., Osuka A., Chem. Eur. J. 2018, 24, 7489–7497. [DOI] [PubMed] [Google Scholar]
- 192. Takase M., Inabe A., Sugawara Y., Fujita W., Nishinaga T., Nomura K., Org. Lett. 2013, 15, 3202–3205. [DOI] [PubMed] [Google Scholar]
- 193. Ji L., Griesbeck S., Marder T. B., Chem. Sci. 2017, 8, 846–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Hirai M., Tanaka N., Sakai M., Yamaguchi S., Chem. Rev. 2019, 119, 8291–8331. [DOI] [PubMed] [Google Scholar]
- 195. Saito S., Matsuo K., Yamaguchi S., J. Am. Chem. Soc. 2012, 134, 9130–9133. [DOI] [PubMed] [Google Scholar]
- 196. Dou C., Saito S., Matsuo K., Hisaki I., Yamaguchi S., Angew. Chem. Int. Ed. 2012, 51, 12206–12210; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12372–12376. [Google Scholar]
- 197. Yang D.-T., Nakamura T., He Z., Wang X., Wakamiya A., Peng T., Wang S., Org. Lett. 2018, 20, 6741–6745. [DOI] [PubMed] [Google Scholar]
- 198. Lewtak J. P., Gryko D. T., Chem. Commun. 2012, 48, 10069–10086. [DOI] [PubMed] [Google Scholar]
- 199. Tanaka T., Osuka A., Chem. Eur. J. 2018, 24, 17188–17200. [DOI] [PubMed] [Google Scholar]
- 200. Ooi S., Tanaka T., Park K. H., Kim D., Osuka A., Angew. Chem. Int. Ed. 2016, 55, 6535–6539; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6645–6649. [Google Scholar]
- 201. Ooi S., Tanaka T., Park K. H., Lee S., Kim D., Osuka A., Angew. Chem. Int. Ed. 2015, 54, 3107–3111; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3150–3154. [Google Scholar]
- 202. Majewski M. A., Stępień M., Angew. Chem. Int. Ed. 2019, 58, 86–116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 90–122. [Google Scholar]
- 203. Sakamoto Y., Suzuki T., J. Am. Chem. Soc. 2013, 135, 14074–14077. [DOI] [PubMed] [Google Scholar]
- 204. Feng C. N., Kuo M. Y., Wu Y. T., Angew. Chem. Int. Ed. 2013, 52, 7791–7794; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7945–7948. [Google Scholar]
- 205. Cheung K. Y., Chan C. K., Liu Z., Miao Q., Angew. Chem. Int. Ed. 2017, 56, 9003–9007; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9131–9135. [Google Scholar]
- 206. Miller R. W., Averill S. E., Van Wyck S. J., Whalley A. C., J. Org. Chem. 2016, 81, 12001–12005. [DOI] [PubMed] [Google Scholar]
- 207. Pun S. H., Chan C. K., Luo J., Liu Z., Miao Q., Angew. Chem. Int. Ed. 2018, 57, 1581–1586; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1597–1602. [Google Scholar]
- 208. Cheung K. Y., Xu X., Miao Q., J. Am. Chem. Soc. 2015, 137, 3910–3914. [DOI] [PubMed] [Google Scholar]
- 209. Luo J., Xu X., Mao R., Miao Q., J. Am. Chem. Soc. 2012, 134, 13796–13803. [DOI] [PubMed] [Google Scholar]
- 210. Wong W. S., Ng C. F., Kuck D., Chow H. F., Angew. Chem. Int. Ed. 2017, 56, 12356–12360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12528–12532. [Google Scholar]
- 211. Wong W. S., Tse H. W., Cheung E., Kuck D., Chow H. F., J. Org. Chem. 2019, 84, 869–878. [DOI] [PubMed] [Google Scholar]
- 212. Kawasumi K., Zhang Q., Segawa Y., Scott L. T., Itami K., Nat. Chem. 2013, 5, 739–744. [DOI] [PubMed] [Google Scholar]
- 213. Kato K., Segawa Y., Scott L. T., Itami K., Chem. Asian J. 2015, 10, 1635–1639. [DOI] [PubMed] [Google Scholar]
- 214. Lin H. A., Sato Y., Segawa Y., Nishihara T., Sugimoto N., Scott L. T., Higashiyama T., Itami K., Angew. Chem. Int. Ed. 2018, 57, 2874–2878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2924–2928. [Google Scholar]
- 215. Lin H.-A., Kato K., Segawa Y., Scott L. T., Itami K., Chem. Sci. 2019, 10, 2326–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Amaya T., Ito T., Hirao T., Angew. Chem. Int. Ed. 2015, 54, 5483–5487; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5573–5577. [Google Scholar]
- 217. Mughal E. U., Kuck D., Chem. Commun. 2012, 48, 8880–8882. [DOI] [PubMed] [Google Scholar]
- 218. Mughal E. U., Neumann B., Stammler H. G., Kuck D., Eur. J. Org. Chem. 2014, 7469–7480. [Google Scholar]
- 219. Ip H. W., Ng C. F., Chow H. F., Kuck D., J. Am. Chem. Soc. 2016, 138, 13778–13781. [DOI] [PubMed] [Google Scholar]
- 220. Ip H. W., Chow H. F., Kuck D., Org. Chem. Front. 2017, 4, 817–822. [Google Scholar]
- 221. He L., Ng C. F., Li Y., Liu Z., Kuck D., Chow H. F., Angew. Chem. Int. Ed. 2018, 57, 13635–13639; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13823–13827. [Google Scholar]
- 222. Smith J. N., Lucas N. T., Chem. Commun. 2018, 54, 4716–4719. [DOI] [PubMed] [Google Scholar]
- 223. Newman M. S., Lednicer D., J. Am. Chem. Soc. 1956, 78, 4765–4770. [Google Scholar]
- 224. Shen Y., Chen C. F., Chem. Rev. 2012, 112, 1463–1535. [DOI] [PubMed] [Google Scholar]
- 225. Gingras M., Chem. Soc. Rev. 2013, 42, 968–1006. [DOI] [PubMed] [Google Scholar]
- 226. Pradhan A., Dechambenoit P., Bock H., Durola F., Angew. Chem. Int. Ed. 2011, 50, 12582–12585; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12790–12793. [Google Scholar]
- 227. Pradhan A., Dechambenoit P., Bock H., Durola F., J. Org. Chem. 2013, 78, 2266–2274. [DOI] [PubMed] [Google Scholar]
- 228. Pradhan A., Dechambenoit P., Bock H., Durola F., Chem. Eur. J. 2016, 22, 18227–18235. [DOI] [PubMed] [Google Scholar]
- 229. Hu Y., Wang X.-Y., Peng P.-X., Wang X.-C., Cao X.-Y., Feng X., Müllen K., Narita A., Angew. Chem. Int. Ed. 2017, 56, 3374–3378; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3423–3427. [Google Scholar]
- 230. Yamano R., Shibata Y., Tanaka K., Chem. Eur. J. 2018, 24, 6364–6370. [DOI] [PubMed] [Google Scholar]
- 231. Yang Y., Yuan L., Shan B., Liu Z., Miao Q., Chem. Eur. J. 2016, 22, 18620–18627. [DOI] [PubMed] [Google Scholar]
- 232. Fujikawa T., Segawa Y., Itami K., J. Am. Chem. Soc. 2015, 137, 7763–7768. [DOI] [PubMed] [Google Scholar]
- 233. Fujikawa T., Segawa Y., Itami K., J. Am. Chem. Soc. 2016, 138, 3587–3595. [DOI] [PubMed] [Google Scholar]
- 234. Fujikawa T., Mitoma N., Wakamiya A., Saeki A., Segawa Y., Itami K., Org. Biomol. Chem. 2017, 15, 4697–4703. [DOI] [PubMed] [Google Scholar]
- 235. Schlütter F., Nishiuchi T., Enkelmann V., Müllen K., Angew. Chem. Int. Ed. 2014, 53, 1538–1542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1564–1568. [Google Scholar]
- 236. Reger D., Haines P., Heinemann F. W., Guldi D. M., Jux N., Angew. Chem. Int. Ed. 2018, 57, 5938–5942; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6044–6049. [Google Scholar]
- 237. Cruz C. M., Castro-Fernández S., Maçôas E., Cuerva J. M., Campaña A. G., Angew. Chem. Int. Ed. 2018, 57, 14782–14786; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14998–15002. [Google Scholar]
- 238. Márquez I. R., Fuentes N., Cruz C. M., Puente-Muñoz V., Sotorrios L., Marcos M. L., Choquesillo-Lazarte D., Biel B., Crovetto L., Gómez-Bengoa E., et al., Chem. Sci. 2017, 8, 1068–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Cruz C. M., Márquez I. R., Mariz I. F. A., Blanco V., Sánchez-Sánchez C., Sobrado J. M., Martín-Gago J. A., Cuerva J. M., Maçôas E., Campaña A. G., Chem. Sci. 2018, 9, 3917–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Zhu Y., Xia Z., Cai Z., Yuan Z., Jiang N., Li T., Wang Y., Guo X., Li Z., Ma S., et al., J. Am. Chem. Soc. 2018, 140, 4222–4226. [DOI] [PubMed] [Google Scholar]
- 241. Zhu Y., Guo X., Li Y., Wang J., J. Am. Chem. Soc. 2019, 141, 5511–5517. [DOI] [PubMed] [Google Scholar]
- 242. Xiao Q., Sakurai T., Fukino T., Akaike K., Honsho Y., Saeki A., Seki S., Kato K., Takata M., Aida T., J. Am. Chem. Soc. 2013, 135, 18268–18271. [DOI] [PubMed] [Google Scholar]
- 243. Waghray D., De Vet C., Karypidou K., Dehaen W., J. Org. Chem. 2013, 78, 11147–11154. [DOI] [PubMed] [Google Scholar]
- 244. Chen F., Tanaka T., Hong Y. S., Mori T., Kim D., Osuka A., Angew. Chem. Int. Ed. 2017, 56, 14688–14693; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14880–14885. [Google Scholar]
- 245. Golling F. E., Quernheim M., Wagner M., Nishiuchi T., Müllen K., Angew. Chem. Int. Ed. 2014, 53, 1525–1528; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1551–1554. [Google Scholar]
- 246. Golling F. E., Osella S., Quernheim M., Wagner M., Beljonne D., Müllen K., Chem. Sci. 2015, 6, 7072–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Sisto T. J., Zakharov L. N., White B. M., Jasti R., Chem. Sci. 2016, 7, 3681–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Cheung K. Y., Gui S., Deng C., Liang H., Xia Z., Liu Z., Chi L., Miao Q., Chem 2019, 5, 838–847. [Google Scholar]
- 249. Yang Y., Chu M., Miao Q., Org. Lett. 2018, 20, 4259–4262. [DOI] [PubMed] [Google Scholar]
- 250. Otero G., Biddau G., Sánchez-Sánchez C., Caillard R., López M. F., Rogero C., Palomares F. J., Cabello N., Basanta M. A., Ortega J., et al., Nature 2008, 454, 865–868. [DOI] [PubMed] [Google Scholar]
- 251. Beernink G., Gunia M., Dötz F., Öström H., Weiss K., Müllen K., Wöll C., ChemPhysChem 2001, 2, 317–320. [DOI] [PubMed] [Google Scholar]
- 252. Rim K. T., Siaj M., Xiao S., Myers M., Carpentier V. D., Liu L., Su C., Steigerwald M. L., Hybertsen M. S., McBreen P. H., et al., Angew. Chem. Int. Ed. 2007, 46, 7891–7895; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8037–8041. [Google Scholar]
- 253. Sanchez-Valencia J. R., Dienel T., Gröning O., Shorubalko I., Mueller A., Jansen M., Amsharov K., Ruffieux P., Fasel R., Nature 2014, 512, 61–64. [DOI] [PubMed] [Google Scholar]
- 254. Krüger J., García F., Eisenhut F., Skidin D., Alonso J. M., Guitián E., Pérez D., Cuniberti G., Moresco F., Peña D., Angew. Chem. Int. Ed. 2017, 56, 11945–11948; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12107–12110. [Google Scholar]
- 255. Rogers C., Chen C., Pedramrazi Z., Omrani A. A., Tsai H. Z., Jung H. S., Lin S., Crommie M. F., Fischer F. R., Angew. Chem. Int. Ed. 2015, 54, 15143–15146; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15358–15361. [Google Scholar]
- 256. Hieulle J., Carbonell-Sanromà E., Vilas-Varela M., Garcia-Lekue A., Guitián E., Peña D., Pascual J. I., Nano Lett. 2018, 18, 418–423. [DOI] [PubMed] [Google Scholar]
- 257. Pinardi A. L., Martínez J. I., Jančařík A., Stará I. G., Starý I., López M. F., Méndez J., Martín-Gago J. Á., Chem. Commun. 2014, 50, 1555–1557. [DOI] [PubMed] [Google Scholar]
- 258. On-Surface Synthesis (Ed.: A. Gourdon), Springer, Cham, 2016. [Google Scholar]
- 259. Fischer F. R., in Adv. Polym. Sci., Springer, Cham, 2017, pp. 33–65. [Google Scholar]
- 260. Talirz L., Ruffieux P., Fasel R., Adv. Mater. 2016, 28, 6222–6231. [DOI] [PubMed] [Google Scholar]
- 261. Wang T., Zhu J., Surf. Sci. Rep. 2019, 74, 97–140. [Google Scholar]
- 262. Treier M., Pignedoli C. A., Laino T., Rieger R., Müllen K., Passerone D., Fasel R., Nat. Chem. 2011, 3, 61–67. [DOI] [PubMed] [Google Scholar]
- 263. Basagni A., Sedona F., Pignedoli C. A., Cattelan M., Nicolas L., Casarin M., Sambi M., J. Am. Chem. Soc. 2015, 137, 1802–1808. [DOI] [PubMed] [Google Scholar]
- 264. Cai J., Ruffieux P., Jaafar R., Bieri M., Braun T., Blankenburg S., Muoth M., Seitsonen A. P., Saleh M., Feng X., et al., Nature 2010, 466, 470–473. [DOI] [PubMed] [Google Scholar]
- 265. Chen Z., Wang H. I., Teyssandier J., Mali K. S., Dumslaff T., Ivanov I., Zhang W., Ruffieux P., Fasel R., Räder H. J., et al., J. Am. Chem. Soc. 2017, 139, 3635–3638. [DOI] [PubMed] [Google Scholar]
- 266. Kawai S., Saito S., Osumi S., Yamaguchi S., Foster A. S., Spijker P., Meyer E., Nat. Commun. 2015, 6, 8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Wang X. Y., Urgel J. I., Barin G. B., Eimre K., Di Giovannantonio M., Milani A., Tommasini M., Pignedoli C. A., Ruffieux P., Feng X., et al., J. Am. Chem. Soc. 2018, 140, 9104–9107. [DOI] [PubMed] [Google Scholar]
- 268. Bronner C., Stremlau S., Gille M., Brauße F., Haase A., Hecht S., Tegeder P., Angew. Chem. Int. Ed. 2013, 52, 4422–4425; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4518–4521. [Google Scholar]
- 269. Rizzo D. J., Veber G., Cao T., Bronner C., Chen T., Zhao F., Rodriguez H., Louie S. G., Crommie M. F., Fischer F. R., Nature 2018, 560, 204–208. [DOI] [PubMed] [Google Scholar]
- 270. Ruffieux P., Wang S., Yang B., Sanchez-Sanchez C., Liu J., Dienel T., Talirz L., Shinde P., Pignedoli C. A., Passerone D., et al., Nature 2016, 531, 489–492. [DOI] [PubMed] [Google Scholar]
- 271. Di Giovannantonio M., Urgel J. I., Beser U., Yakutovich A. V., Wilhelm J., Pignedoli C. A., Ruffieux P., Narita A., Müllen K., Fasel R., J. Am. Chem. Soc. 2018, 140, 3532–3536. [DOI] [PubMed] [Google Scholar]
- 272. Mishra S., Lohr T. G., Pignedoli C. A., Liu J., Berger R., Urgel J. I., Müllen K., Feng X., Ruffieux P., Fasel R., ACS Nano 2018, 12, 11917–11927. [DOI] [PubMed] [Google Scholar]
- 273. Xu K., Urgel J. I., Eimre K., Di Giovannantonio M., Keerthi A., Komber H., Wang S., Narita A., Berger R., Ruffieux P., et al., J. Am. Chem. Soc. 2019, 141, 7726–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Pun S. H., Wang Y., Chu M., Chan C. K., Li Y., Liu Z., Miao Q., J. Am. Chem. Soc. 2019, 141, 9680–9686. [DOI] [PubMed] [Google Scholar]
- 275. Navakouski M., Zhylitskaya H., Chmielewski P. J., Lis T., Cybińska J., Stępień M., Angew. Chem. Int. Ed. 2019, 58, 4929–4933; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4983–4987. [Google Scholar]
- 276. Urgel J. I., Di Giovannantonio M., Segawa Y., Ruffieux P., Scott L. T., Pignedoli C. A., Itami K., Fasel R., J. Am. Chem. Soc. 2019, 141, 13158–13164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary