

Abstract
Herein, ferumoxytol (Fer) capped antiprogrammed cell death-ligand 1 (PD-L1) antibodies (aPD-L1) loaded ultralarge pore mesoporous silica nanoparticles (Fer-ICB-UPMSNPs) are formulated for a sequential magnetic resonance (MR) image guided local immunotherapy after cabazitaxel (Cbz) chemotherapy for the treatment of prostate cancer (PC). The highly porous framework of UPMSNP provides a large capacity for aPD-L1. Fer capping of the pores extends the period of aPD-L1 release and provides MR visibility of the aPD-L1 loaded UPMSNP. As-chosen Cbz chemotherapy prior to the local immunotherapy induces strong immunogenic cell death, dendritic cell maturation, and upregulation of PD-L1 of tumor cells. Finally, tumor growth inhibition of sequential MR image-guided local delivery of Fer-ICB-UPMSNPs and a tumor specific adoptive immune reaction are demonstrated in the pretreated Tramp C1 PC mouse model with Cbz chemotherapy. The tumor suppression is superior to those obtained with systemic ICB treatment after Cbz, only Fer-ICB-UPMSNP or only Cbz. As a proof-of concept, MR image-guided local ICB immunotherapy using Fer-ICB-UPMSNPs after chemotherapy suggests a new perspective of translational local immunotherapy for patients who are treated with standard chemotherapies.
Keywords: cabazitaxel, chemo-immunotherapy, image-guided cancer immunotherapy, prostate cancer, ultralarge pore mesoporous silica nanoparticles
1. Introduction
Since Dr. William Coley developed the earliest form of immunotherapy using bacterial toxins in 1908, immune-cancer therapies that harness the power of our own immune system to treat cancers have become one of most promising cancer therapeutic approaches. In the past few years, it has been revealed that standard cancer therapies (chemotherapy, radiation, and local ablation) can induce immunogenic stress or death of tumors cells as well as generate a large number of tumor antigens.[1] Many clinical trials of combination immunotherapies with various chemotherapies or other conventional therapies are developed. Over 700 clinical trials[2] are testing the efficacy of combination immunotherapy in cancer patients who have been already treated with standard chemotherapies. A large set of data demonstrates that a pretreatment with standard chemotherapeutics mediates antitumor effect of immunotherapies by inducing immunogenic cell death (ICD) and tumor-suppressive immunity.[3–8] The combinational immune-chemotherapy is now becoming a part of the standard of care.[9–12] Various immune related compartments, such as immune suppressive cytokines, regulatory T cell (Treg), immunosuppressive myeloid cells, immune checkpoint and so on, are involving with the maintenance of immunosuppression. For the last decades, immune checkpoint blockades (ICBs) including cytotoxic T-lymphocyte associated protein 4, programmed cell death protein 1 (PD-1), programmed cell death-ligand 1 (PD-L1), Tim-3, and indolamine 2,3-dioxygenase showed their capability to manage the immune status.[13] Importantly, PD-1/PD-L1 axis demonstrated the most significant increasement of tumor patients as cancer immunotherapy.[14] Nevertheless, low delivery efficiency and systemic exposure of immune therapeutic molecules such as ICB have limited therapeutic responses in a large portion of patients.[15,16]
Prostate cancer (PC) is one of the most common cancer and a leading cause of death.[17] Relapse occurs in about 30% of cases and resistance to chemohormonal therapy in the meta-static setting remains a prominent cause of morbidity and mortality.[18–20] A recent clinical trial testing the sequential ICB immunotherapy after a standard taxane-family chemotherapy demonstrated somewhat encouraging results compared to monotherapies but resulted in dismal therapeutic responses and long-term survival.[21,22] Novel new approaches are critically needed to extend the curative potential of these synergistic combinations to a broader range of patients.[23–29]
We propose anti-PD-L1 antibody (aPD-L1) ICB loaded multifunctional carriers that can be delivered with magnetic resonance (MR) image guidance after a standard taxane-based chemotherapy for the treatment of PC (Scheme 1). Here, ultralarge pore mesoporous silica nanoparticles (UPMSNPs) providing excellent loading capacity were synthesized and aPD-L1 was loaded into the pores. Food and Drug Administration (FDA) approved magnetic resonance imaging (MRI) visible iron oxide ferumoxytol (Fer, brand name: Feraheme) was employed for capping the UPMSNP pores loaded with aPD-L1 and MR imaging guidance. ICB loading procedure and the amount of Fer in UPMSNPs were optimized for sequential MR image-guided local ICB delivery after cabazitaxel (Cbz) chemotherapy. The immunotherapeutic efficacy of sequential MR image guided Fer-capped aPD-L1 loaded UPMSNPs (Fer-ICB-UPMSNPs) delivery after Cbz treatment was investigated with in vitro and in vivo Tramp C1 PC mice model.
Scheme 1.
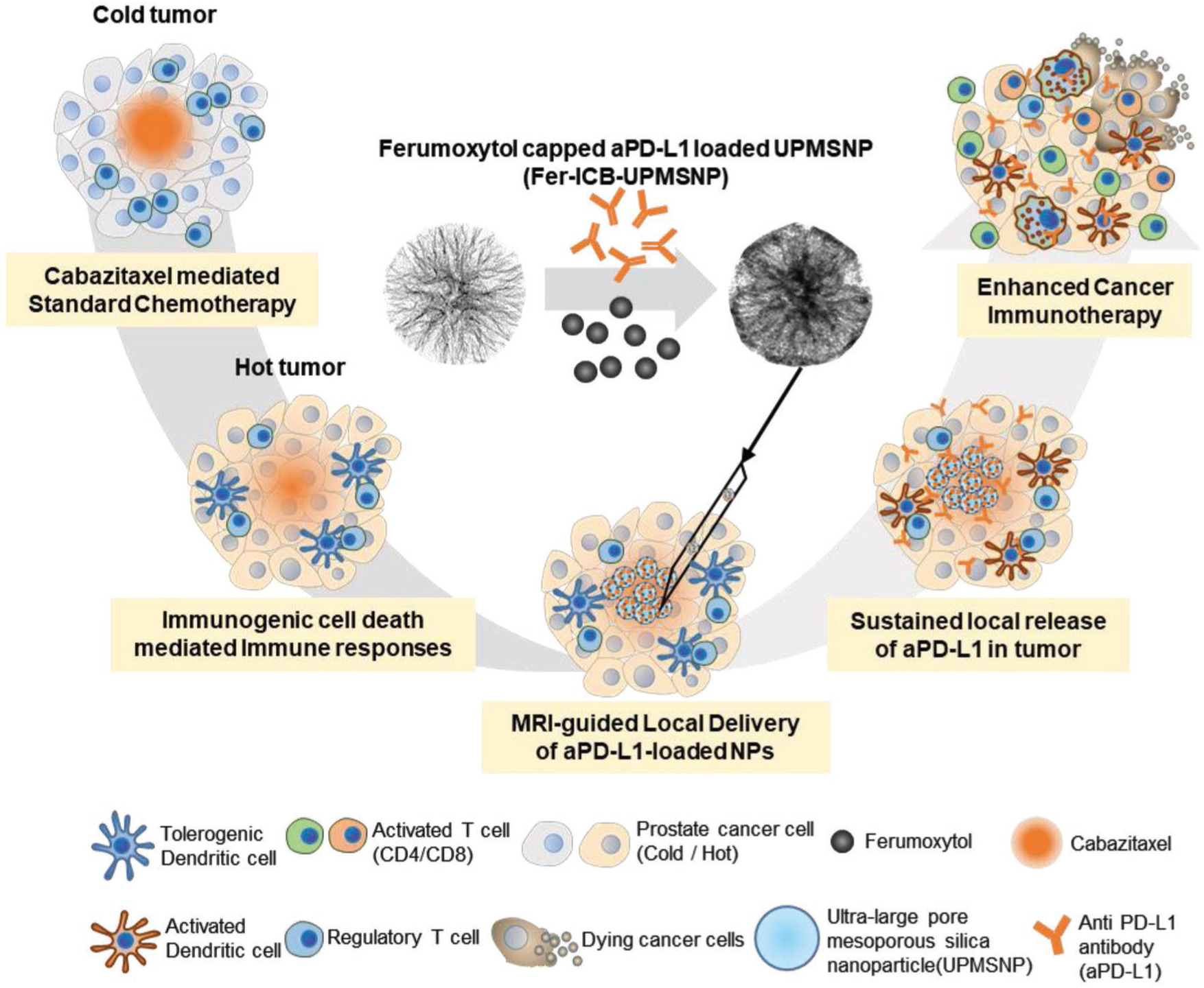
Schematic illustration of sequential local aPD-L1 ICB immunotherapy after chemotherapy. Intratumorally administered Cbz induces immunogenic tumor status and induces immunogenic cell death (ICD) to recruit immune cells. MRI-visible Fer-ICB-UPMSNPs are injected into the central tumor region via MR image guidance. Sustained release of ICB from UPMSNP inhibits the immunosuppressive tumor microenvironment and activates the tumor specific dendritic cells (DC). Activated antigen presenting cells educate the tumor specific naïve T cells and cytotoxic T cells are infiltrated to the tumor for the regression
2. Results and Discussion
2.1. Characterization of UPMSNP and Optimization of aPD-L1 Loading
To formulate Fer-ICB-UPMSNP carriers (Figure 1A), amine terminated UPMSNPs were synthesized by the procedures as previously reported.[30–32] The synthesized bare UPMSNPs showed well-defined ultralarge porous structure and monodispersed with a mean size of 300 nm (Figure 1B). Large pores between 14.6 and 25 nm in size were measured and showed a high surface area of 1155.174 m2 g−1 (Figure 1C and Table S1, Supporting Information). Such a large pore size was enough to load aPD-L1 (8 nm hydrodynamic size).[33,34] Loading capacity of aPD-L1 in UPMSNP was measured to be ≈88 μg per 1 mg of UPMSNP (Figure 1D). The loading efficiency of aPD-L1 was varied with initial aPD-L1 feeding amounts. The feeding of 62.5 μg or lower amount of aPD-L1 to 1 mg of UPMSNP showed over 95.7% loading efficiency but the loading efficiency was decreased to 18.9% with increasing the initial aPD-L1 feeding up to 500 μg (aPD-L1) mg−1 (UPMSNP) (Figure 1D).
Figure 1.
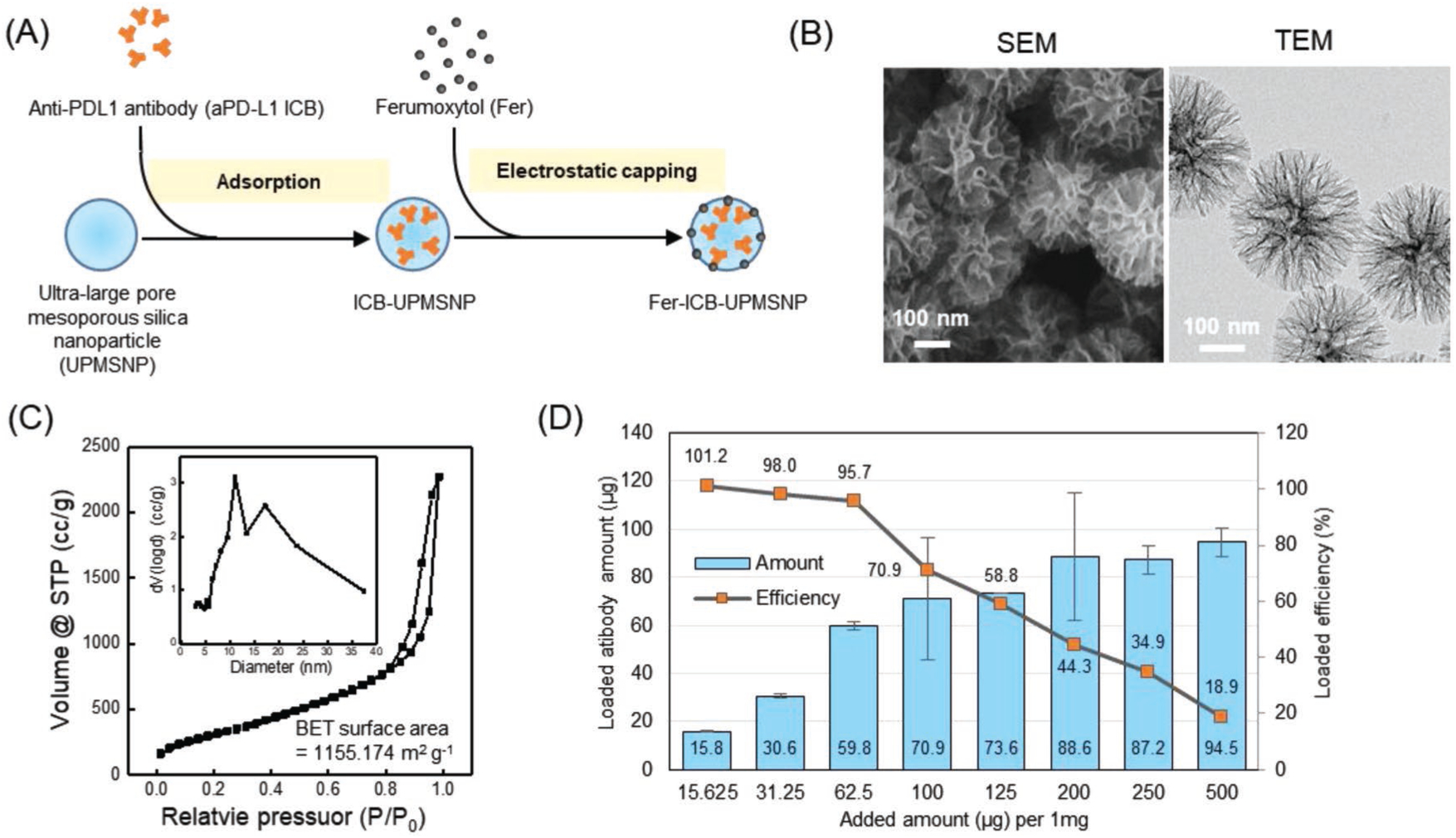
Characterization of Fer capped aPD-L1 loaded UPMSNP. A) Schematic illustration of aPD-L1 ICB loading and Fer capping in UPMSNP. B) Scanning electron microscope (SEM) and TEM images of synthesized UPMSNP. C) Nitrogen adsorption–desorption isotherms and the pore size distributions of UPMSNP. D) aPD-L1 ICB loading-amount and loading-efficiency of UPMSNP.
2.2. Formulation of MRI-Visible Fer Capped ICB-UPMSNP
Although UPMSNP demonstrated a higher loading capacity, the release rate of loaded aPD-L1 could be too fast to deliver ICB in local tumor regions. To prevent fast and burst release of the aPD-L1 loaded into UPMSNP pores, Fer was added to block the pores of ICB loaded UPMSNPs (ICB-UPMSNP). Negatively charged Fer (ζ-potential: −24.4 mV, average hydrodynamic size: 30 nm) with carboxyl methyl groups was electrostatically bound with positively charged ICB-UPMSNP (ζ-potential, +10.2 mV) (Figure 2A). Initial zeta-potential of amine-terminated UPMSNP (ζ-potential, +28.5 mV) was decreased to +10.2 mV after aPD-L1 loading. The addition of strong negative surface charged Fer (ζ-potential, −26.8 mV) to ICB-UPMSNP reversed the positive charge of ICB-UPMSNP to a negative charge (ζ-potential, −12.6 mV). Transmission electron microscope (TEM) analysis of Fer-ICB-UPMSNP confirmed the Fer attachment to the surface of UPMSNP. Corona-like formation of Fer outside of ICB-UPMSNP was significantly different with bare UPMSNP (Figure 2B). Up to ≈320–400 μg of Fer could be attached to the surface of 1 mg of UPMSNP (Figure S1A, Supporting Information) and ≈220 μg of Fer was attached after ICB loading in UPMSNP formulation (Figure S1B, Supporting Information). Fer attachment on the surface of UPMSNP did not affect the ICB loading inside of pores (Figure 1D and Figure S1C, Supporting Information). The strong electrostatic coupling[35] between Fer and UPMSNP could be accommodative for the sustained release of aPD-L1 with a steric hindrance effect.[36,37] In vitro ICB release study, attached Fer capping UPMSNP pores significantly sustained the release of aPD-L1 compared to only ICB-UPMSNP sample (Figure 2C). Fer-ICB-UPMSNP released only 25% of loaded aPD-L1 for 72 h while ICB-UPMSNP released 83% of loaded aPD-L1 antibodies (Figure 2C). Our proposed image guided local delivery of the Fer-ICB-UPMSNP will allow superior tumor-targeted ICB activity compared to the intravenously injected. As demonstrated excellent clinical MRI T2 contrast properties of the Fer, Fer-ICB-UPMSNP also exhibited strong MR T2 contrast effect in a phantom study (Figure 2D). The MRI visibility of Fer-ICB-UPMSNP would be critical to optimize catheter directed local infusion procedure and localize ICB delivery into tumors.
Figure 2.
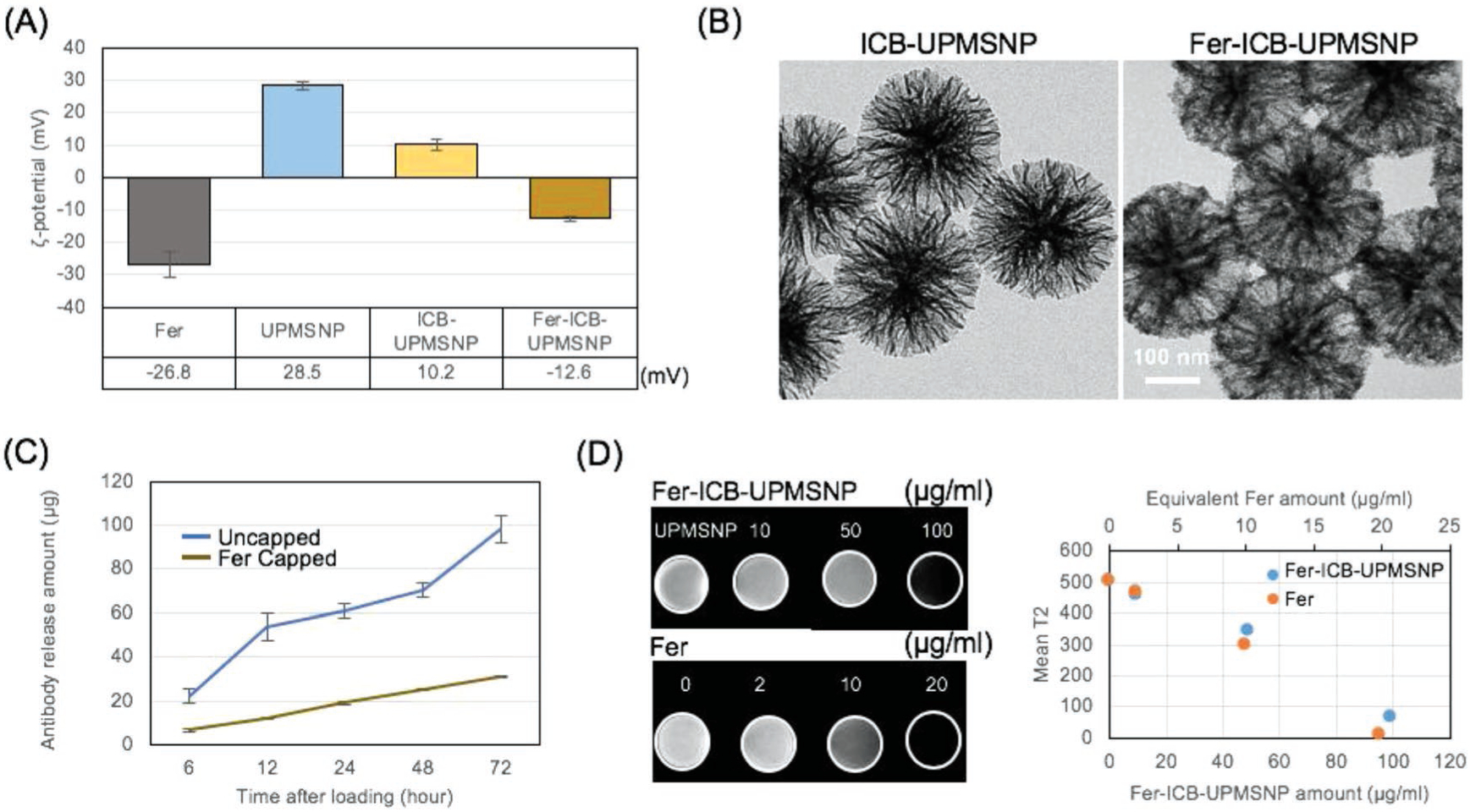
Formulation of Fer capped ICB-UPMSNP. A) ζ-potential of particles is measured by dynamic light scattering. The carboxyl residue decorating Fer and the amine residue on the UPMSNPs showed −24.4 mV and +28.5 mV, respectively. ICB-UPMSNP (+10.2 mV) can be capped with Fer. Fer capped ICB-UPMSNP had −12.6 mV. B) TEM images of ICB-UPMSNP and Fer-ICB-UPMSNP after 7 d. C) Fer capping after ICB loading to UPMSNPs resulted in the sustained release of antibodies compared to uncapped ICB-UPMSNPs. Without Fer capping of UPMSNP completely releases ICB within 72 h. D) T2 weighted MRI and T2 signal changes with increased concentrations of Fer and Fer-ICB-UPMSNP. Samples were dispersed into the 1% agarose to obtain the phantom T2W images with 7T MRI.
2.3. ICD and PD-L1 Expression after Cbz Chemotherapy
Demonstrated effective ICB loading and sustained release along with MRI contrast effect of the formulated Fer-ICB-UPMSNP will be effective for the proposed sequential MR image-guided local ICB immunotherapy after a standard chemotherapy. To achieve the synergistic combinations, ICD induction of chemotherapy prior to ICB immunotherapy is critical for the synergistic anticancer effect of the proposed combination. Because nonimmunogenic tumor microenvironment (TME) and low PD-L1 expression on cancer cells are very common in various solid tumors, such as prostate cancers, hepatocellular carcinoma, and nonsmall-cell lung carcinoma, the confirmation of ICD inducing chemotherapies among standard chemotherapies would be critical to enhance the sequential ICB immunotherapeutic outcomes.[15,16] Here, tubulin-binding Cbz, one of the most promising anticancer agents for the treatment of PC,[38] was chosen as a chemotherapy for the sequential immunotherapy of PC. As the first step, ICD induction of Cbz was confirmed with in vitro characterization of calreticulin (CRT) exposure and high mobility group box 1 protein (HMGB1) (Figure 3A). CRT expression from the surface of dying cells provides an “eat-me” signal for dendritic cell (DC) phagocytosis. HMGB1 is one of immunostimulatory signals that induce the DC migration and maturation.[6] When Tramp C1 was treated with 0.5 × 10−6 m of Cbz for 18 h, mean fluorescent intensities of CRT and HMGB1 expression were nearly doubled compared to nontreated control (Figure S2, Supporting Information). Flow cytometry analysis revealed that 91.0% of cell population shifted to ICD stages (positive CRT and HMGB1), which was greater than 23.4% of phosphate buffered saline (PBS) treated cells (Figure 3B). Significantly increased actual HMGB1 secretion after Cbz treatment was also confirmed with enzyme linked immunosorbent assay (ELISA) (Figure 3C). Further, the ICD induction after the Cbz treatment directly promotes the tumor antigen uptake of immature DCs and DC maturation, resulting in the consequent tumor-specific T cell differentiation and proliferation. To confirm those sequences of adaptive immune responses by the Cbz mediated ICD induction, in vivo tumor-specific DC activation and maturation following by the Cbz treatment (1 μg/50 μL) were also characterized with B7 maturation markers (CD80 and CD86) in Tramp C1 PC mice model. CD80+ and CD86+ expression significantly increased about three times higher compared to that of the nontreated control group at 7 d-post Cbz treatment (Figure 3D). Furthermore, these DCs secreted proinflammatory cytokines, tumor necrosis factor alpha (TNF-α), IL12p70 and the tolerogenic cytokine, IL-10 (Figure 3E).[39] Taken results together, Cbz mediated ICD induces immunogenic DC maturation for the adaptive immune responses. Subsequently, the activated and maturated DCs by the Cbz treatment directly augmented tumor specific T cell proliferation (Figure 3F).
Figure 3.
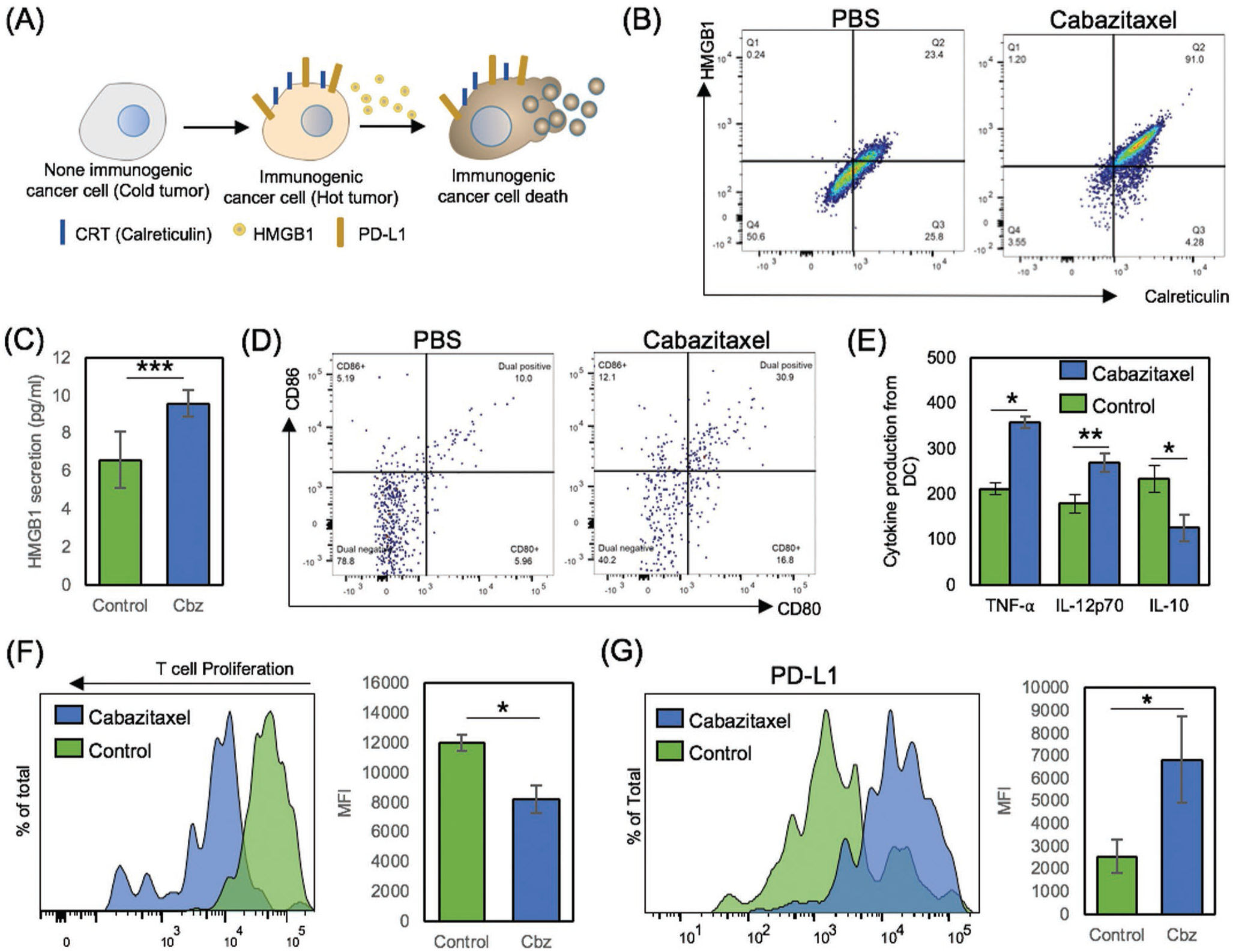
Immunogenic cell death induction (ICD) of Tramp C1 and followed adaptive immune responses with PD-L1 overexpression. A) Schematic illustration of ICD and following CRT, HMGB1, and PD-L1 expression. B,C) Validation of ICD after Cbz treatment. 1 × 106 Tramp C1 cells were cultured with 0.5 × 10−6 m of Cbz for 18 h. CRT and HMGB1, immunogenic cell death markers were analyzed by ELISA and flow cytometry. D) In vivo DC maturation test after Cbz treatment. Tramp C1 prostate tumor bearing mice were treated with PBS as negative control and Cbz only for ICD induction. Splenocytes were isolated and prepared with CD11c as DC marker, CD80, and CD86 as DC maturation marker. Data were obtained and analyzed from CD11c+ gate by flow cytometry. E) Production of TNF-α, IL-12p70 and IL-10 from ICD induced DCs after ex vivo restimulation. Cytokine profiles were determined by ELISA. F) Tumor specific T cell activation and proliferation. Tramp C1 cells were cultured with or without 0.5 × 10−6 m of Cbz for 24 h. Then, DCs were added to uptake ICD induced Tramp C1 cells. After 4 h, CMFDA labeled immunized T cells from donor mice were cocultured with primed DCs. After 5 d of cultivation, Tramp C1 specific T cell proliferation was observed and analyzed by flow cytometry. G) Cbz treatment dependent PD-L1 expression change was investigated after 18 h (PBS, 0.5 × 10−6 m of Cbz).
However, most of these ICD and immune responses are actually concluded in anergy state because of immune suppressive mechanisms including PD-L1 overexpression in vivo TME. Our data also demonstrated a clear overexpression of PD-L1 after the Cbz treatment (Figure 3G).[40] Ironically, the increased expression of PD-L1 in Tramp C1 presents the chance of inducing tumor specific adaptive immune responses when aPD-L1 ICB mediated immunotherapy is effectively applied.[41–43] aPD-L1 ICB immunotherapy also can interfere the PD-1/PD-L1 axis between DC and T cells, as shown in synergistic DC maturation and T cell activation of aPD-L1 treated groups (Figure S3, Supporting Information).[44] The local delivery of aPD-L1 with Fer-ICB-UPMSNP to the PD-L1 expressing tumors will effectively block the PD-1/PD-L1 axis (as demonstrated in vitro fluorescent images in Figure S4, Supporting Information) during the favorable immunogenic TME induced by Cbz pretreatment.
2.4. Sequential MR Image-Guided Delivery of Fer-ICB-UPMSNP after Cbz Chemotherapy and Analysis of the Tumor Infiltrated Lymphocytes
As-shown the sustained aPD-L1 release in Fer-ICB-UPMSNP would be ideal for the extended period of ICB treatment in the local tumors. However, the targeting efficiency of nanocarriers with the conventional intravenous injection are about 5% in maximum due to biological barriers and TME.[45] Intravenously injected nanocarriers also may lose the loaded therapeutics during the blood circulation. Image-guided intratumoral (IT) local administration may overcome the limitation of intravenously injected nanocarriers and potentially increase the therapeutic efficacy of our sequential ICB immunotherapy using Fer-ICB-UPMSNP.[46] Hence, MR image guided local delivery of Fer-ICB-UPMSNP after the ICD induction of Cbz chemotherapy was tested as a proof-of-concept in the Tramp C1 PC mice model. Autochthonous Tramp C1 prostate tumor model is suitable for evaluating the sequential chemo-immunotherapies. Tramp C1 to male C57/BL6 is the well characterized syngeneic animal model to study the prostate tumor progression for preclinical therapeutic establishment.[47] Suppressed PD-L1 expression and enhanced Treg population are also well representing human immune-responses.[48,49] After the confirmation of 5 mm sized Tramp C1 tumor generation after 12–14 weeks, five experimental groups of only PBS (Group 1), only Cbz treatment (Group 2), only Fer-ICB-UPMSNP (Group 3), sequential Fer-ICB-UPMSNP after Cbz (Group 4), and sequential systemic ICB treatment after Cbz (Group 5) were prepared and the immune therapeutic efficacies of each groups were compared. MR image guided IT infusion was successfully performed using an MR compatible 24 gauge (G) catheter with MRI T2 images (Figure 4A). MR T2 baseline scans provided the structural orientation of tumor that allowed for the placement of the catheter into the central tumor region. During the infusion of Fer-ICB-UPMSNP, MR contrast effect of Fer-ICB-UPMSNP permitted the monitoring of the intratumoral distribution of Fer-ICB-UPMSNP and the adjustment of catheter position for uniform distribution of samples in a specific central tumor region (Figure 4A). To evaluate the tumor specific immune responses in each group, the T cell subset composition from the harvested tumor-infiltrating lymphocytes (TILs) was analyzed by flow cytometry at 14 d[50]-post infusion. Sequential Fer-ICB-UPMSNP after Cbz (Group 4) showed a significantly increased total number of TILs and both CD8+ and CD4+ T cells compared to other groups (Figure 4B,C and Figure S5, Supporting Information). In contrast, sequential systemic ICB treatment after Cbz (Group 5) showed only limited T-cell infiltration as shown in Groups 1, 2, and 3. The percentage of CD8+ T cells in the tumor after the sequential Fer-ICB-MSNP after Cbz (Group 4) was approximately twofold higher than Group 5. When it is compared with Groups 1, 2, and 3, fivefold higher CD8+ cells were measured (Figure 4D). Importantly, Tregs are essential for peripheral immune tolerance in response to the activation of effector T cells (Teffs). During a tumor development, Tregs in TME expresses significant amounts of PD-1 and PD-L1 for an active suppression of local immunity.[51] Therefore, the ratio and population of the Treg versus Teff in TME are the crucial components for evaluating the effectiveness of immunotherapies. Here, the sequential local aPD-L1 immunotherapy after Cbz (Group 4) decreased the number of Treg and increased the number of Teff in TME.[52] Cbz treatment itself The sustained aPD-L1 release in the tumor region induced only 2.60% of Treg population in CD4+ T cells (Treg/CD4+ = 0.1)), that were significantly different with the systemic ICB treatment (Group 5) exhibiting 12.4% of Treg population in CD4+ T cells (Treg/CD4+ = 0.68) (Figure 4E and Figure S6, Supporting Information). The production of IL-10 tolerogenic cytokine by the Cbz treatment (Figure 3E) and local aPD-L1 treatment synergistically depleted more pre-existing Tregs in tumor. The ratio of CD8+ and Treg in Group 4 was 12, that was much higher than 5 of Group 5 (Figure 4F). Taken together, the sustained aPD-L1 local release from sequentially delivered Fer-ICB-UPMSNP after Cbz (Group 4) significantly increased infiltration of CD8+ cytotoxic T cells to the tumor sites and decreased number of Tregs. Consequently, the treatment group with a single session of sequential MR image guided Fer-ICB-UPMSNPs infusion after Cbz chemotherapy (Group 4) showed clear tumor rejection efficiency compared to the systemic ICB injection after Cbz (Group 5) within 14 d (Figure 4G). These results clearly indicated that our sequential MR image guided local delivery of Fer-ICB-UPMSNP in the tumor (Group 4) enhanced the therapeutic efficacy of the sequential aPD-L1 immunotherapy after immunogenic chemotherapy. As considering future translational feature of nanocarriers, biodegradable UPMSNP[53,54] can be considered for the safe exclusion of nanocarriers minimizing any potential toxic effect in long term.
Figure 4.
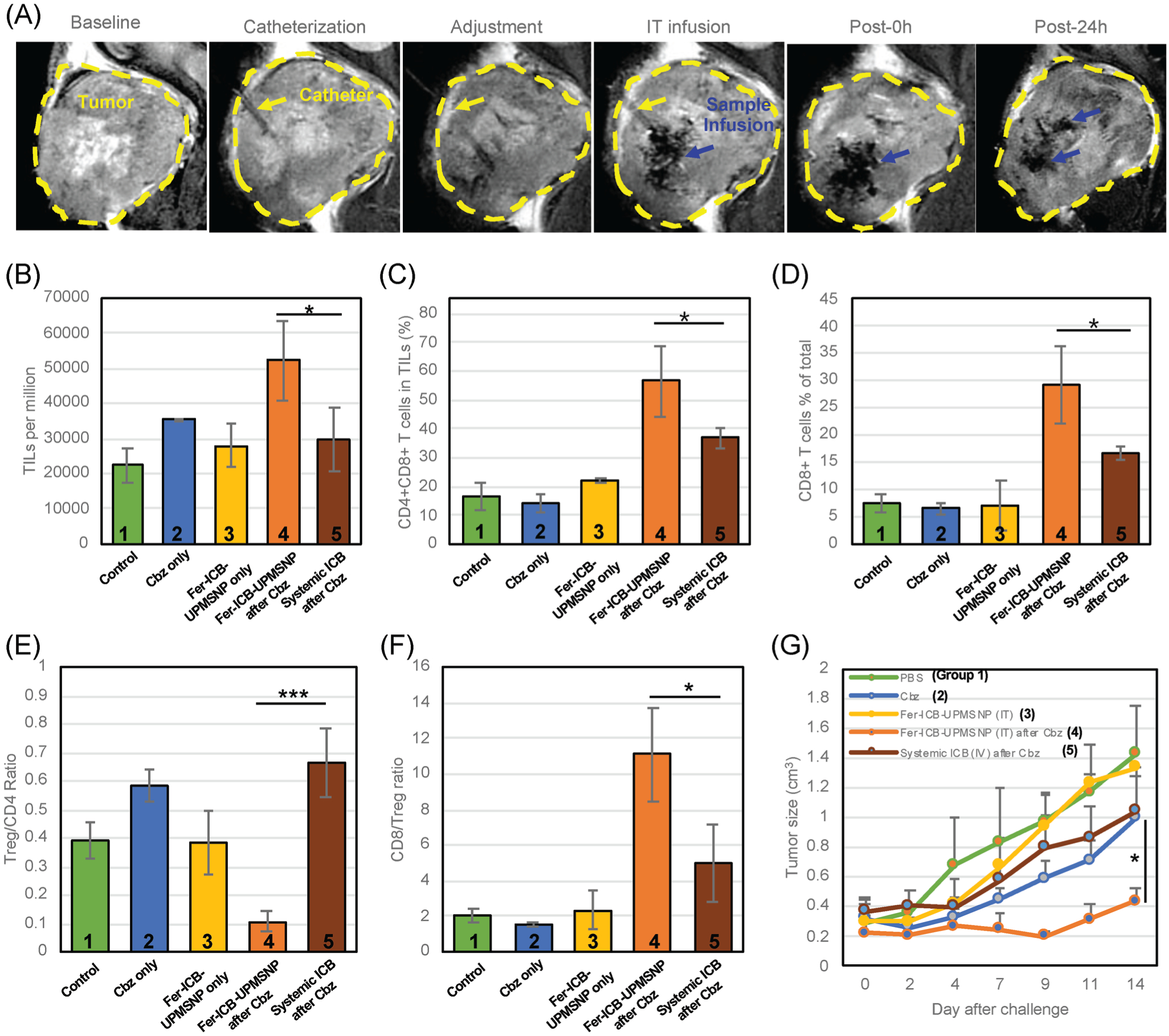
MR image guided Fer-ICB-UPMSNP delivery after Cbz treatment to Tramp C1 prostate tumor and analysis of the tumor infiltrated lymphocytes. A) T2 weighted MR images during intratumoral (IT) infusion of Fer-ICB-UPMSNP. Yellow dash indicates the tumor formation. B) TIL infiltration per 1 million cells from homogenized tumors after 14 d of ICB administration. C) CD4+CD8+ T cell percentages were gated from CD3 common T cell marker in TIL population. D) CD8+ T cell accumulation in total TILs after 2 weeks of each treatment. The infiltration of CD8+ T cells showed significant changes in the local tumor area. E) Treg/CD4 ratio analysis from TILs. Even tumor accumulation of CD4 T cell was significantly higher in a group of Fer-ICB-UPMSNP after Cbz than other groups but Treg population in total CD4 T cell was decreased with the localized ICB treatment. F) CD8/Treg ratio analysis from TILs. Because of significant decrease of Tregs in CD4 T cell population, CD8/Treg T cell ratio relatively increased. G) Tramp C1 tumor growth after the treatment in each group. (Group 1: only PBS, Group 2: only Cbz treatment, Group 3: only Fer-ICB-UPMSNP, Group 4: sequential Fer-ICB-UPMSNP after Cbz, Group 5: sequential systemic ICB treatment after Cbz) (*p < 0.05).
3. Conclusion
Various immune checkpoint deficient cold tumors including prostate cancer showed dismal prognosis with their low immunogenicity in clinical trials. Recently sequential ICB immunotherapies after chemotherapy showed a promising potential. However, optimizing synergistic combination and local delivery of effective ICB dose are required for effectively activating antitumor immune responses. In this study, MRI visible aPD-L1 loaded carriers (Fer-ICB-UPMSNPs) were formulated for a sequential MR image guided local immunotherapy after Cbz chemotherapy. Ultralarge pores and high surface area in UPMSNP provided a large capacity of aPD-L1 ICB. Fer capping of the UPMSNP pores successfully sustained the release of aPD-L1. Cbz chemotherapy clearly induced the ICD, DC maturation, tumor specific T cell development, and upregulation of PD-L1 of cancer cells. In vivo studies using PC mice model demonstrated that sequential MR image guided local delivery of Fer-ICB-UPMSNPs after the Cbz treatment effectively activated T cell infiltration and decreased Treg insisting a tumor specific adoptive immune reaction. Finally, the tumor growth was significantly suppressed with the sequential local delivery of Fer-ICB-UPMSNP after Cbz treatment. It was superior to the obtained with systemic ICB treatment after Cbz at the same total dose. Our approach of MR image guided local immunotherapy using multifunctional UPMSNP suggests a new perspective of translational local immunotherapy for patients who have been treated with standard chemotherapies.
4. Experimental Section
Synthesis of MSNP:
Mesoporous silica nanoparticles (UPMSNP) were prepared via anionic and cationic surfactant micelle complex.[30–32] Triethanolamine was dissolved in deionized water in a 100 mL three-neck flask. After raising the temperature to 80 °C, cetyltrimethylammonium chloride (CTAC) and NaSal were added into the solution, and then tetraethyl orthosilicate was added. (The molar ratio between CTAC and anionic surfactant differ from the desired pore size.) After further stirring at 80 °C for the reaction times, the reaction solution was collected by centrifugation. The reaction solution was washed with ethanol three times. The synthesized UPMSNPs were extracted at 50 °C for 2 h to remove the residual surfactants from the UPMSNPs. The morphology of UPMSNPs was observed using a emission scanning electron microscope (Nova Nano 200, Field Electron and Ion Company (FEI), USA) and transmission electron microscope (Talos F200X, FEI, USA). The Fourier-transform infrared (FTIR) spectra data were obtained by Nicolet iS10 FTIR spectrophotometer (Thermo scientific, USA). For amine functionalization on the surface of the UPMSNPs, UPMSNPs dispersed into toluene were mixed with 2% w/v aminopropyltriethoxysilane. The mixture solution was stirred in a water bath for 5 h at 80 °C. After the reaction, the solution was centrifuged and washed with methanol three times.
Anti PD-L1 Antibody Loading Test:
Antimouse PD-L1 Rat polyclonal antibody (LifeSpan Bioscience, USA) was used. 0 to 1 mg of antibodies was diluted in 300 μL of PBS buffer, then cocultured with various size of 1 mg of UPMSNP (300 nm) for 18 h at room temperature (RT) with continuous invert. After incubation, the sample was centrifuged (8000 rpm, 10 min), the supernatant was transferred to other tubes and the protein concentration was measured by bicinchoninic acid analysis (Pierce scientific, USA).
Ferumoxytol Capping of ICB Loaded UPMSNP:
1 mg of ICB loaded UPMSNP was cultured with 0 to 1 mg of Fer in 300 μL of PBS buffer with a repetitive inverting at RT for 4 h. Then, the samples were centrifuged (8000 rpm, 10 min) and the supernatants were collected to measure the concentration of residual ferumoxytol through QuantiChrom Iron Assay Kit (Bioassay, USA). Next, 590 nm wavelength absorbance is obtained by Cytation3 spectrophotometer (BioTek, USA). Based on the standard curves, each antibody loading amount and Fer loading amount was calculated by a linear fitting equation.
Phantom Imaging of Fer-ICB-MSNPs:
Imaging phantoms were prepared in 1% agar with 0 to 100 μg mL−1 of Fer-ICB-MSNPs or only Fer. Samples were homogenously suspended in 0.3 mL of 1% agarose gel. Additionally, sample mixtures were laid on the prebuffered layer of 0.5 mL of 1% agarose gel in microcentrifuge tubes for MR phantom images. MRI studies were performed using a Bruker 7.0T ClinScan (Bruker BioSpin, Ettlingen, Germany) high-field small animal MRI system with a commercial mouse coil (Bruker Biospin).
Induction of Immunogenic Cell Death:
To validate ICD induction by cabazitaxel (Apexbio Technology, USA), 1 × 106 cells of Tramp C1 cells per 100 μL of complete media (followed American type culture collection guidance) were cultured with 0.5 × 10−6 m of Cbz diluted complete media for 18 h. After the incubation, cells were fixed and permeabilized (Becton Dickinson (BD), cytofix/Cytoperm, USA) to stain the intracellular HMGB1 and CRT (BD). The amount of ICD population was analyzed by Flow cytometry (BD, LSRII Fortessa, USA).
In Vivo DC Maturation:
After 7 d of each treatment, mouse spleens were cut into small pieces and treated with 400 collagenase D (Roche, Switzerland) in 5 mL of Hank’s balanced salt solution (Thermofisher, USA), and subsequently digested with 37 °C, 30 min. After treatment with 100 μL of 0.5 M ethylenediaminetetraacetic acid for an additional 5 min, CD11c positive DCs were positively sorted by magnetic-activated cell sorting (Miltenyi Biotech, Germany) from single-cell suspensions of splenocytes. CD80 and DC86, DC maturation markers (CD11c-APC, CD80-FITC, CD86-PE purchased from BD, USA) were stained for 4 °C, 30 min and analyzed by Flow cytometry (BD, LSRII Fortessa, USA).
HMGB1 and DC Cytokine ELISA after ICD Induction:
To quantify cytokines after ICD induction, each ICD induced tumor supernatants and DC-conditioned supernatants were collected after restimulation with lipopolysaccharide (LPS, 100 ng mL−1) for 18 h. The concentration of HMGB-1 (Lifespan bioscience, USA), TNF-α, IL-12p70, and IL-10 was measured by ELISA (eBioscience, USA) with manufacturer’s protocol.
In Vitro T Cell Proliferation Assay:
To obtain the Tramp C1 specific T cells, 100 Gy irradiated 1 × 106 Tramp C1 cells were subcutaneously injected to 6–7 weeks female mice with 100 ng of LPS as an adjuvant. Immunization was repeated 2 weeks interval. After three times of injection, mice were sacrificed and both CD3 positive T cells and CD11c positive DCs were enriched by magnetic separation. DCs were primed with each treatment in the presence of 100 ng mL−1 concentration of LPS. After 3 h of culture, 5-chloromethylfluorescein diacetate (CMFDA) dye (BD, USA) labeled T cells were cocultured and the proliferation was tracked for 5 d. The decreased amount of CMFDA fluorescence intensity corresponding to T cell proliferation was analyzed by Flow cytometry (BD, LSRII Fortessa, USA).
Image-Guided Fer-ICB-MSNP Delivery:
Animal experiments were performed in accordance with protocols approved by Institutional Animal Care and Use Committee (IACUC) of Northwestern University. Tramp C1 tumor bearing mice were anesthetized with an isoflurane induction dose (mixture of 5% isoflurane and oxygen at 3 L min−1). MR image-guided intratumoral injection of Fer-ICB-MSNPs was performed with MR T2 scanning and 24G polyurethane catheter (Terumo Medical Co., Somerset, USA). After injection, the guide needle was removed, and the location of catheter was adjusted by post-MR T2 scanning. The catheter was filled with 100 μg of Fer-ICB-MSNPs in 50 μL of PBS and injected very slowly. Biodistribution of samples was observed by MRI immediately after injection and 24 h later.
T Cell Characterization:
To test the immune cell distribution from chemo-immunotherapy, 5 mm sized tumor bearing mice were intratumorally injected with 1 μg of Cbz in 50 μL of PBS via 30 G insulin syringes (BD). After 72 h, 100 μg of Fer-ICB-MSNPs in 50 μL of PBS were injected to tumor via MR image guidance. Tumor sizes are recorded by 2 d interval for 2 weeks. Mice were euthanized and TILs from homogenized tumor cells were stained with indicated antibodies (CD3-PE, CD4-PEcy7, CD8-FITC, CD25-APCcy7 and FoxP3-PECF594 purchased from BD, USA) for the analysis with flow cytometry (BD, LSRII Fortessa, USA).
Immunohistochemistry of Infiltrated T Lymphocytes into Prostate Cancer:
Histology samples were embedded with the optimal cutting temperature compounds (Fisher scientific, USA) and freezed with dry ice immediately. CD3-AF488 antibodies (eBioscience, USA) were permeabilized and stained into the slide of cryosection. Immunofluorescent images were obtained by confocal microscopy (A1R spectral, Nikon, Japan).
Statistical Analysis:
Analysis results were presented as mean ± standard deviation, representing at least three independent experiments. Nonparametric Mann–Whitney U-test was performed with EXCEL (Microsoft). Statistical significance was set at *p < 0.05; **p < 0.01; ***p < 0.001, as indicated in the figure legends.
Supplementary Material
Acknowledgements
This work was supported by grants R01CA218659 and R01EB026207 from the National Cancer Institute and National Institute of Biomedical Imaging and Bioengineering. This work was also supported by the Center for Translational Imaging and Mouse Histology and Phenotyping Laboratory at Northwestern University.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/smll.201904378.
Contributor Information
Bongseo Choi, Department of Radiology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA.
Huijin Jung, Molecular Recognition Research Center, Korea Institute of Science and Technology (KIST), Division of Nano and Information Technology, KIST School, Korea University of Science and Technology (UST), Seoul 02792, South Korea.
Joonseok Lee, Molecular Recognition Research Center, Korea Institute of Science and Technology (KIST), Division of Nano and Information Technology, KIST School, Korea University of Science and Technology (UST), Seoul 02792, South Korea.
Dong-Hyun Kim, Department of Radiology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA; Robert H. Lurie Comprehensive Cancer Center, Chicago, IL 60611, USA; Department of Biomedical Engineering, McCormick School of Engineering, Evanston, IL 60208, USA.
References
- [1].Binnewies M, Roberts EW, Krummel MF, Nat. Med 2018, 24, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Riley RS, June CH, Mitchell MJ, Nat. Rev. Drug Discovery 2019, 18, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ladoire S, Penault-Llorca F, Kroemer G, Autophagy 2015, 11, 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Topalian SL, Taube JM, Pardoll DM, Nat. Rev. Cancer 2016, 16, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dosset M, Vargas TR, Apetoh L, Oncoimmunology 2018, 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lu JQ, Liu XS, Nel AE, ACS Nano 2018, 12, 11041. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [7].Hanahan D, Weinberg RA, Cell 2011, 144, 646. [DOI] [PubMed] [Google Scholar]
- [8].Schumacher TN, Schreiber RD, Science 2015, 348, 69. [DOI] [PubMed] [Google Scholar]
- [9].Cook AM, Lesterhuis WJ, Lake RA, Curr. Opin. Immunol 2016, 39, 23. [DOI] [PubMed] [Google Scholar]
- [10].Morrissey KM, Yuraszeck TM, Kasichayanula S, Clin. Transl. Sci 2016, 9, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Miller JFAP, Sadelain M, Cancer Cell 2015, 27, 439. [DOI] [PubMed] [Google Scholar]
- [12].Mueller KL, Science 2015, 348, 54. [DOI] [PubMed] [Google Scholar]
- [13].Pitt JM, Vetizou M, Zitvogel L, Immunity 2016, 44, 1255. [DOI] [PubMed] [Google Scholar]
- [14].Sun C, Mezzadra R, Schumacher TN, Immunity 2018, 48, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martin AM, Nirschl TR, Drake CG, Prostate Cancer Prostatic Dis. 2015, 18, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang PP, Ma YY, Yang Y, Cancer Sci. 2016, 107, 1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huggins C, Hodges CV, J. Urol 2002, 168, 9. [DOI] [PubMed] [Google Scholar]
- [18].Crawford ED, Eisenberger MA, Goodman PJ, Engl N. J. Med 1989, 321, 419. [DOI] [PubMed] [Google Scholar]
- [19].Eisenberger MA, Blumenstein BA, Lowe BA, Engl N. J. Med 1998, 339, 1036. [DOI] [PubMed] [Google Scholar]
- [20].Calcagno F, Nguyen T, Thiery-Vuillemin A, Clin. Med. Insights: Oncol 2013, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sharma P, Narayan V, Fizazi K, Genitourin. Cancers Symp 2019, 4, 8. [Google Scholar]
- [22].Gao JJ, Ward JF, Sharma P, Nat. Med 2017, 23, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rudzki JD, Memo 2018, 11, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Powles T, Duran I, Ravaud A, Lancet 2018, 391, 748. [DOI] [PubMed] [Google Scholar]
- [25].Rittmeyer A, Barlesi F, von Pawel J, O. A. K. S. Group, Lancet 2017, 389, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fehrenbacher L, Spira A, Mazieres J, Group PS, Lancet 2016, 387, 1837. [DOI] [PubMed] [Google Scholar]
- [27].Chen Q, Wang C, Gu Z, Adv. Healthcare Mater 2018, 7, e1800424. [DOI] [PubMed] [Google Scholar]
- [28].Chen Q, Wang C, Gu Z, Nat. Nanotechnol 2019, 14, 89. [DOI] [PubMed] [Google Scholar]
- [29].Wang C, Wang J, Gu Z, Sci. Transl. Med 2018, 10. [DOI] [PubMed] [Google Scholar]
- [30].Polshettiwar V, Cha D, Basset JM, Angew. Chem., Int. Ed. Engl 2010, 49, 9652. [DOI] [PubMed] [Google Scholar]
- [31].Moon DS, Lee JK, Langmuir 2012, 28, 12341. [DOI] [PubMed] [Google Scholar]
- [32].Yang YN, Bernardi S, Yu CZ, Chem. Mater 2016, 28, 704. [Google Scholar]
- [33].Kwon D, Cha BG, Kim J, Nano Lett. 2017, 17, 2747. [DOI] [PubMed] [Google Scholar]
- [34].Lu Y, Yang YN, Yu CZ, Biomaterials 2018, 175, 82. [DOI] [PubMed] [Google Scholar]
- [35].Stace AJ, Bichoutskaia E, Soft Matter 2012, 8, 6210. [Google Scholar]
- [36].Vallet-Regi M, Izquierdo-Barba I, Colilla M, Philos. Trans. R. Soc., A 2012, 370, 1400. [DOI] [PubMed] [Google Scholar]
- [37].Xu C, Lei C, Yu C, Front. Chem 2019, 7, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tsao CK, Cutting E, Oh WK, Ther. Adv. Urol 2014, 6, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maldonado RA, von Andrian UH, Adv. Immunol 2010, 108, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brown JS, Sundar R, Lopez J, Br. J. Cancer 2018, 118, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Burotto M, Singh N, Madan RA, Front. Oncol 2014, 4, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Galluzzi L, Senovilla L, Kroemer G, Nat. Rev. Drug Discovery 2012, 11, 215. [DOI] [PubMed] [Google Scholar]
- [43].Kepp O, Galluzzi L, Kroemer G, Cancer Metastasis Rev. 2011, 30, 61. [DOI] [PubMed] [Google Scholar]
- [44].Versteven M, Van den Bergh JMJ, Lion E, Front. Immunol 2018, 9, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wilhelm S, Tavares AJ, Chan WCW, Nat. Rev. Mater 2016, 1, 16014. [Google Scholar]
- [46].Kim DH, J. Imaging 2018, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Foster BA, Gingrich JR, Greenberg NM, Cancer Res. 1997, 57, 3325. [PubMed] [Google Scholar]
- [48].Wu CT, Chen WC, Chen MF, Cancers 2019, 11, 20. [Google Scholar]
- [49].Ngiow SF, von Scheidt B, Smyth MJ, Cancer Res. 2011, 71, 3540. [DOI] [PubMed] [Google Scholar]
- [50].Pulendran B, Nat. Rev. Immunol 2009, 9, 741. [DOI] [PubMed] [Google Scholar]
- [51].Sakaguchi S, Yamaguchi T, Ono M, Cell 2008, 133, 775. [DOI] [PubMed] [Google Scholar]
- [52].Kamada T, Togashi Y, Nishikawa H, Proc. Natl. Acad. Sci. U. S. A 2019, 116, 9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kong M, Tang JM, Zhang ZP, Theranostics 2017, 7, 3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Park JH, Gu L, Sailor MJ, Nat. Mater 2009, 8, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.