

Abstract
The all-cis stereoisomers of tetrasubstituted azetidine-2-carboxylic acids and derivatives that possess three chiral centers have been prepared in high yield and stereocontrol from silyl-protected Z-γ-substituted enoldiazoacetates and imido-sulfur ylides by asymmetric [3+1]-cycloaddition using chiral sabox copper(I) catalysis followed by Pd/C catalytic hydrogenation. Hydrogenation of the chiral p-methoxybenzyl azetine-2-carboxylates occurs with both hydrogen addition to the C=C bond and hydrogenolysis of the ester
Keywords: azetidines, azetines, copper catalysis, cycloaddition, hydrogenation
Four-membered ring heterocyclic compounds are of increasing importance and interest, not only because of the biological activities of β-lactams[1] but, also, due to expanding interest in unsaturated four-membered ring azetines and their applications.[2] These relatively high energy compounds have not been widely explored because of limited synthetic methods for their formation and because their well-recognized ability to undergo four-electron electrocyclic ring-opening to 1-aza-1,3-butadienes.[3] Available methods for 2-azetine synthesis include those from already-formed four-membered ring heterocycles (3-methoxyazetidine derivatives and 3-azetidinones),[4] vinyldiazo compounds via aziridination/ring expansion reactions,[5] as well as from benzyne via [2+2]-cyclo-addition[6] or allenyl imides with imines[7] (Scheme 1a,b). However, none of the available methods are suitable for the synthesis of chiral 2-azetines.
Scheme 1.

Synthetic methods for construction of azetine ring.
Chiral azetidine-2-carboxylic acid, the first known example of naturally occurring azetidines,[8] and their derivatives (potential products from reduction of 2-azetine-carboxylates) are structural units of several natural products[9] and the thrombin inhibitor melagatran[10] (Figure 1). Various methods have been used for their preparation,[11] but all of them are multistep syntheses from chiral reactants that provide access only to mono- or disubstituted azetidine-2-carboxylic acids and their derivatives. Herein, we report a robust methodology for the highly enantioselective synthesis of 2-azetine-carboxylates catalyzed by copper(I) with chiral sabox ligand and their stereoselective hydrogenation to form a single stereoisomer of tetrasubstituted azetidine-2-carboxylate derivatives (Scheme 1c).
Figure 1.

Natural products and a pharmaceutical containing the structural unit of chiral azetidine-2-carboxylates or their reduced forms.
In previous research from our laboratory we reported that copper(I)-catalyzed [3+1]-cycloaddition of silyl group protected enoldiazoacetates with α-acyl sulfur ylides was effective in forming 2-silyloxy-1-cyclobutene carboxylates.[12] This transformation was the first example in which the dipolar intermediate underwent nucleophilic displacement of a leaving group (R2S) to achieve product formation in a metal carbene [3+n]-cycloaddition reaction.[13]
The high yields and enantioselectivities achieved in these reactions prompted us to investigate whether [3+1]-cyclo-addition could also occur with “nitrene” donor species. However, N-acyl-imido sulfur ylides,[14] used in place of the N-acylsulfur ylides, with the same catalysts and under the same conditions, were unreactive even at elevated temperatures due to a lack of reactivity of the imido ylide. Aryl-, sulfonyl-, and acyl-azides were, similarly, unable to undergo cycloaddition with metal carbenes formed from enoldiazoacetates and, instead, formed imine products.[15] However, use of N-arylimido sulfur ylides (S,S-disubstituted N-arylsulfilimines)[16] allowed cycloaddition to proceed smoothly at room temperature. Only copper(I) catalysis was effective for this transformation, and Cu(MeCN)4PF6 gave the highest product yield [Eq. (1)]. Dichloromethane was the preferred solvent, and diphenylsulfur ylides gave higher product yields than their dimethyl or methylphenyl analogues. Reactions were performed at room temperature to avoid electroreversion of the azetine.[3] Moreover, [3+1]-cycloaddition occurred with the triisopropylsilyl (TIPS)- but not with the tert-butyldimethylsilyl (TBS)-protected enoldiazoacetate.
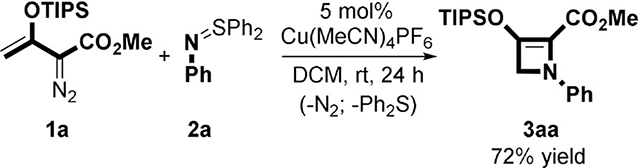 |
(1) |
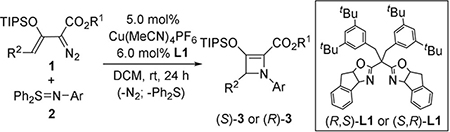
To produce azetines that possess a chiral center, γ-substituents on the enoldiazoacetate were introduced. Because [3+1]-cycloaddition with N-arylsulfilimines occurred with the Z-but not the E-enoldiazoacetate isomer, a new methodology for the nearly exclusive synthesis of the Z-geometrical isomer was developed.[17] A series of TIPS-protected γ-substituted Z- enoldiazoacetates (Z:E > 20:1) was prepared, and we identified the optimal chiral sabox ligand L1 that provided highest yield and % ee of azetine in the reaction of γ-methyl-substituted enoldiazoacetate 1b with N-(p-chlorophenyl)imido diphenyl sulfur ylide 2b.[18] Competing reactions that included 1,4-hydrogen transfer from the intermediate metallo-vinylcarbene to produce conjugated diene products were minor. Although azetine 3bb was obtained in good yield (82%), enantioselectivity was only moderate (75% ee). To improve enantiocontrol of the cycloaddition we varied substituents at the γ-position of enoldiazoacetates and studied the effects of the carboxylate ester group and substituents in the aromatic ring of N-arylimido sulfur ylides 2 (Table 1).
Table 1:
Substrate scope of donor–acceptor azetines obtained via CuI-catalyzed [3+1]-cycloaddition of enoldiazoacetates 1 and N-arylimido sulfur ylides 2.
 | ||||||
|---|---|---|---|---|---|---|
| Entry[a] | R1 | R2 | Ar | 3 | Yield [%][b] | ee [%][c] |
| 1 | Me | Me | 4-Cl-C6H4 | 3 bb | 82 | 75 |
| 2 | Me | Me | 3-F-C6H4 | 3 bc | 83 | 90 |
| 3 | Me | Et | C6H5 | 3 ca | 86 | 85 |
| 4 | Me | Et | 4-Cl-C6H4 | 3 cb | 92 | 90 |
| 5 | Me | Et | 3-Cl-C6H4 | 3 cd | 89 | 91 |
| 6 | Me | Et | 3,5-diCl-C6H3 | 3 ce | 80 | 82 |
| 7 | Me | Et | 4-Br-C6H4 | 3 cf | 88 | 89 |
| 8 | Me | Et | 4-F-C6H4 | 3 cg | 84 | 88 |
| 9 | Me | Et | 3-F-C6H4 | 3 cc | 87 | 92 |
| 10 | Me | Et | 2-F-C6H4 | 3 ch | 86 | 88 |
| 11 | Me | Et | 4-CF3-C6H4 | 3 ci | 87 | 91 |
| 12 | Me | Et | 3-OMe-C6H4 | 3 cj | 80 | 77 |
| 13 | Me | Et | 3-Me-C6H4 | 3 ck | 85 | 74 |
| 14 | Me | Et | 4-Me-C6H4 | 3 cl | 76 | 70 |
| 15 | Me | Et | 4-Et-C6H4 | 3 cm | 78 | 80 |
| 16[d] | Me | octyl | 3-F-C6H4 | 3 dc | 78 | 95 |
| 17[d] | Me | Bn | 4-Cl-C6H4 | 3 hb | 73 | 90 |
| 18[d] | Me | i-Pr | 4-Cl-C6H4 | 3 ib | 70 | 92 |
| 19 | PMB | Me | 3-F-C6H4 | 3 ec | 92 | 90 |
| 20[d] | PMB | Et | C6H5 | 3 fa | 83 | 88 |
| 21 | PMB | Et | 3-F-C6H4 | 3 fc | 94 | 99 |
| 22 | PMB | Et | 4-Cl-C6H4 | 3 fb | 95 | 99 |
| 23 | Bn | Et | 4-Cl-C6H4 | 3 jb | 87 | 92 |
| 24 | pCF3-Bn | Et | 4-Cl-C6H4 | 3 kb | 73 | 87 |
| 25 | PMB | Et | 2,4-diF-C6H3 | 3 fn | 83 | 87 |
| 26[d] | PMB | Et | 3-Me-C6H4 | 3 fk | 82 | 86 |
| 27[d] | PMB | Et | 4-CN-C6H4 | 3 fo | 75 | 96 |
| 28[d] | PMB | Et | 4-CO2Et-C6H4 | 3 fp | 68 | 82 |
| 29[d] | PMB | Et | 4-NO2-C6H4 | 3 fq | 66 | 93 |
| 30 | PMB | Et | 4-F-C6H4 | 3 fg | 88 | 99 |
| 31 | PMB | Et | 2-F-C6H4 | 3 fh | 76 | 90 |
| 32[d] | PMB | Et | 4-Et-C6H4 | 3 fm | 76 | 84 |
| 33[d] | PMB | octyl | 3-F-C6H4 | 3 gc[e] | 87 | 99 |
Reactions were carried out on a 0.20 mmol scale of N-arylsulfilimine 2 and 0.24 mmol of Z-enoldiazoacetate 1 with 5 mol% of Cu(MeCN)4PF6 and 6 mol% of (S,R)-L1 or (R,S)-L1 in 4.0 mL of DCM at room temperature.
Isolated yields after flash-chromatography are reported (average of two runs).
Enantiomeric excess was determined using a Daicel Chiralpak AD-H or a Chiralcel OD-H chiral column.
Reaction time was 72 h.
For additional examples of the synthesis of azetines 3, see: Table S2, Supporting Information.
Analysis of substituent effects revealed the influence of the three variable substitution patterns on reactivity and enantioselectivity for [3+1]-cycloaddition as a function of either or both electronic and steric factors. Increasing the size of the aliphatic chain at the γ-position of enoldiazoacetate 1 led to an improvement in yield and enantiocontrol of the reaction [82% yield, 75% ee (Me, 3bb) to 92% yield, 90% ee (Et, 3cb); entries 1 and 4 in Table 1]. However, introduction of octyl, benzyl and isopropyl substituents resulted in reduced reactivity (70–78% yield after 3 days, entries 16–18), but with excellent enantioselectivity (90–95% ee). These results suggest a significant steric effect by the γ-substituent on enantiocontrol for cycloaddition, and the Et substituent at the γ-position was chosen for further studies (diazo compound 1c). Only modest variations in yields and enantioselectivities were observed for the CuI-catalyzed reaction of 1c with a series of sulfilimines 2. The introduction of halogen and CF3 substituents to the aromatic ring of 2 at the ortho or para positions (entries 4, 7, 8, 10, 11) had virtually no influence on product yields (82–92%) or their ee values (86–91%) compared to the results obtained with unsubstituted sulfilimine 2a (3ca, 86% yield, 85% ee). However, with para-alkyl substituents (entries 14 and 15) diminished enantiocontrol (70–80% ee) and yields (76–78%) of the [3+1]-cycloaddition products were obtained. Moreover, changing the position of the substituent on the aromatic ring (para-, meta-, ortho-) of 2 (entries 5, 9, 12, 13) did not provide much difference in %ee or yield of 3, which suggests minor electronic or steric influence by arylsulfilimine substituents. Optimization of the carboxylate group revealed that 4-methoxybenzyl (PMB) enoldiazo esters 1e-g exhibit exceptionally high enantiocontrol (entries 21, 22, 30, 33) compared to their methyl ester analogues, although reaction times were extended to 3 days to achieve full conversions. Employment of benzyl or 4-CF3-benzyl enoldiazo esters led to decreases in % ee compared to the PMB ester (entries 23, 24). Sulfilimines 2 that possess strong EWGs (nitro, cyano, CO2Et) produced the corresponding azetines 3 in lower yields (66–75%, entries 27–29) but with excellent enantioselectivities (up to 96% ee).
A proposed mechanism for [3+1]-cycloaddition of enoldiazoacetates with imido-sulfur ylides, consistent with our previous report on carbonyl-sulfur ylides[12] (Scheme 2) includes a CuI-catalyzed generation of Z-metallo-enolcarbene A that, depending on reaction conditions, can form the corresponding stable donor–acceptor cyclopropene B, which serves as a resting state for Z-metallo-enolcarbene A,[13] or undergo an irreversible proto-decupration to produce diene C. The major reaction pathway proceeds by nucleophilic addition of imido-sulfur ylide 2 onto the electrophilic vinylogous carbon of Z-metallo-enolcarbene A to generate vinyl-copper-carbene intermediate D. Displacement of the diphenyl sulfide, a good leaving group, forms cyclic intermediate E, which upon dissociation of the copper catalyst produces donor–acceptor azetine 3. The facility of N-aryl-imidosulfur ylides for this cycloaddition compared to N-acylimidosulfur ylides does not appear to be due to enhanced electron donation from the aryl group because of the limited influence by substituents on product yields and selectivities but, instead, on the inherently low nucleophilicity of the imido nitrogen of acylimidosulfur ylides.
Scheme 2.

Proposed mechanism for copper(I)-catalyzed [3+1]-cyclo-addition of silyl-protected γ-substituted Z-enoldiazoacetates with N-arylsulfilimines and possible competing reactions.
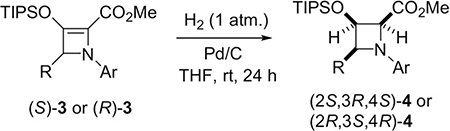
Removal of the silyl-protective group of these chiral azetine structures produces a β-ketoester that is inherently unstable to nucleophilic ring opening.[19] We anticipated that classic (Pd/C)-catalyzed cis-hydrogenation of the double bond of the highly reactive, strained donor–acceptor azetines 3 would retain the four-membered ring, but also lead to strain release and provide easy access to chiral tetrasubstituted 2-azetidine-carboxylates that possess three chiral centers. Indeed, we observed very efficient hydrogenation of methyl azetine-2-carboxylates 3 to azetidine-2-carboxylates 4 at room temperature under 1 atm. of H2 using only 2 wt.% of palladium metal (Table 2).
Table 2:
Substrate scope of azetidine-2-carboxylates 4 obtained via palladium-catalyzed hydrogenation of donor–acceptor azetines 3.
 | ||||||
|---|---|---|---|---|---|---|
| Entry[a] | R | Ar | 4 | Yield [%][b] | dr[c] | ee [%][d] |
| 1 | H | C6H5 | 4 aa | 93 | N/A | racemic |
| 2 | Me | 3-F-C6H4 | 4 bc | 96 | > 20:1 | 90 |
| 3 | Et | C6H5 | 4 ca | 95 | > 20:1 | 85 |
| 4 | Et | 4-Cl-C6H4 | 4 cb | 92 | > 20:1 | 90 |
| 5 | Et | 3-Cl-C6H4 | 4 cd | 90 | > 20:1 | 91 |
| 6 | Et | 4-F-C6H4 | 4 cg | 92 | > 20:1 | 88 |
| 7 | Et | 3-F-C6H4 | 4 cc | 96 | > 20:1 | 92 |
| 8 | Et | 2-F-C6H4 | 4 ch | 97 | 7:1 | 88 |
| 9 | Et | 4-CF3-C6H4 | 4 ci | 96 | 10:1 | 91 |
| 10 | Et | 3-OMe-C6H4 | 4 cj | 94 | > 20:1 | 77 |
| 11 | Et | 3-Me-C6H4 | 4 ck | 95 | > 20:1 | 74 |
| 12 | Et | 4-Me-C6H4 | 4 cl | 97 | > 20:1 | 70 |
| 13 | Et | 4-Et-C6H4 | 4 cm | 93 | > 20:1 | 80 |
| 14 | octyl | 3-F-C6H4 | 4 dc | 95 | > 20:1 | 95 |
Reactions were carried out on a 0.25 mmol scale of azetine-2-carboxylate 3 in 4.0 mL of THF at room temperature with Pd on activated charcoal (2 wt. % of Pd metal) under H2 (1 atm) for 24 h.
Isolated yields after flash-chromatography are reported.
Determined from the 1H NMR spectra of purified compounds.
Enantiomeric excesses were determined using a Daicel Chiralpak AD-H and Chiralcel OD-H chiral columns.
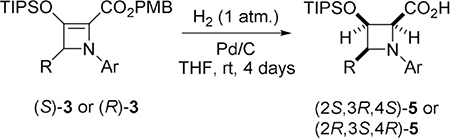
What is particularly striking, cis-hydrogenation occurs from the site opposite to substituent R at the 4-position of azetidine ring with exceptional diastereocontrol (dr > 20:1). A noticeable decrease in diastereocontrol is only observed with fluorinated azetidines 4ch (dr = 7:1) and 4ci (dr = 10:1), however all azetidine-2-carboxylates 4 are obtained in excellent yields (> 90%) and with complete retention of enantiopurity (confirmed by comparison of HPLC traces for racemic and chiral 3ca and 4ca). As anticipated, methyl azetidine-2-carboxylate without a substituent at the 4-position (4aa, R = H) is obtained as a racemic mixture of cis-hydrogenation products, and this is also confirmed by chiral HPLC analysis. Hydrogenation of PMB azetine-2-carboxylates 3 performed under the same reaction conditions as their methyl analogs led to mixtures of PMB azetidine-2-carboxylates 4 and azetidine-2-carboxylic acids 5—products of hydrogenolysis of the PMB group (Table 3) obtained in 24 h. The reaction time (4 days) that provided complete conversion to azetidine-2-carboxylic acids 5 as the sole product in very high yields (87–95%) was determined, and exceptional diastereoselectivities with dr > 20:1 (Table 3) were obtained. The structure and absolute configuration of azetidine-2-carboxylic acid 5ec obtained using sabox ligand (R,S)-L1, was established by X-ray diffraction crystallographic analysis as 2S,3R,4S- (Figure 2 represents the monomeric structure of dimeric 5ec).[20]
Table 3.
Substrate scope of azetidine-2-carboxylic acids 5 obtained via palladium-catalyzed hydrogenation of donor–acceptor azetines 3.
 | ||||||
|---|---|---|---|---|---|---|
| Entry[a] | R | Ar | 5 | Yield [%][b] | dr[c] | ee [%][d] |
| 1 | Et | 4-Me-C6H4 | 5 fl | 88 | > 20:1 | 80 |
| 2 | Et | C6H5 | 5 fa | 94 | > 20:1 | 88 |
| 3 | Et | 4-CN-C6H4 | 5 fo | 93 | > 20:1 | 96 |
| 4[e] | Et | 4-CF3-C6H4 | 4 fi | 86 | > 20:1 | 97 |
| 5 | Et | 3-Me-C6H4 | 5 fk | 93 | > 20:1 | 86 |
| 6 | Et | 3-OMe-C6H4 | 5 fj | 90 | > 20:1 | 88 |
| 7 | Et | 4-Et-C6H4 | 5 fm | 87 | > 20:1 | 84 |
| 8 | Et | 2,4-diF-C6H3 | 5 fn | 89 | > 20:1 | 87 |
| 9 | Et | 4-F-C6H4 | 5 fg | 95 | > 20:1 | 99 |
| 10 | Et | 3-F-C6H4 | 5 fc | 92 | > 20:1 | 99 |
| 11 | Et | 2-F-C6H4 | 5 fh | 92 | > 20:1 | 90 |
| 12 | Me | 3-F-C6H4 | 5 ec | 95 | > 20:1 | 90 |
| 13 | octyl | 3-F-C6H4 | 5 gc | 95 | > 20:1 | 99 |
Reactions were carried out on a 0.25 mmol scale of azetine-2-carboxylate 3 in 4.0 mL of THF at room temperature with Pd on activated charcoal (2 wt. % of Pd metal) under H2 (1 atm) for 4 days.
Isolated yields after flash-chromatography are reported.
Determined from the 1H NMR spectra of purified compounds.
Enantiomeric excess is reported based on the ee values of corresponding azetines 3 and confirmed by HPLC analyses.
PMB-ester was isolated.
Figure 2.

ORTEP diagram of the X-ray crystal structure of (2S,3R,4S)-1-(3-fluorophenyl)-4-methyl-3-[(triisopropylsilyl)oxy]azetidine-2-carboxylic acid 5ec obtained using (R,S)-L1.
Cleavage of the PMB group in hydrogenation of 4-CF3 substituted azetine-2-carboxylate 3 fi was not observed even after 4 days, and PMB azetidine-2-carboxylate 4 fi was isolated in high yield (86%, entry 4). With the exception of 3 fi, variation of the carboxylate ester group in 3 provided formation of either methyl azetidine-2-carboxylates 4 (Table 2) or azetidine-2-carboxylic acids 5 (Table 3), and both were formed in excellent yields and with exceptional stereocontrol.
Diol or polyol products from reduction of azetine-2-carboxylates or -carboxylic acids form derivatives that are a common structural unit in penaresidins A and B (Figure 1) and other naturally occurring or synthetic azetidine iminosugars—a class of compounds with potent glycosidase inhibitory activities also suitable for incorporation into peptides.[11d, 21] Chiral azetidine-carboxylates 4 or -carboxylic acids 5 appear to be suitable substrates for the synthesis of azetidine iminosugar derivatives. We have successfully demonstrated reduction/TIPS removal of azetidine 5 fa using lithium aluminum hydride to produce chiral azetidine-diol 6 in near quantitative yield (Scheme 3). Further functionalization of 6 with excess of p-toluenesulfonyl chloride and triethylamine afforded monotosyl-protected product 7 at the primary hydroxyl group exclusively in 88% isolated yield at room temperature.
Scheme 3.

Functionalization of chiral azetidine-2-carboxylic acids. Synthesis of chiral azetidine iminosugar derivatives.
In summary, we have established a new methodology for the highly enantioselective synthesis of tetrasubstituted azetines via asymmetric [3+1]-cycloaddition of γ-substituted enoldiazo-acetates with imido-sulfur ylides using chiral copper(I)-sabox catalysis. This reaction is remarkably versatile with TIPS-protected enoldiazo compounds and N-arylsulfilimines, affording a broad spectrum of 2-azetine derivatives in high yields and with good to excellent enantiocontrol. The azetine-2-carboxylates provide convenient access to tetrasubstituted azetidine-2-carboxylates and their carboxylic acids that possess three chiral centers, with exceptional diastereocontrol and retention of enantiopurity from the original 2-azetine using Pd-catalyzed hydrogenation. The reduction of azetidine-2-carboxylic acids results in the formation of chiral primary-secondary diols—azetidine iminosugar derivatives suitable for further functionalization. Other enantioselective transformations of strained and highly reactive 2-azetines and applications of [3+1]-cycloaddition methodology are currently in progress in our laboratory.
Supplementary Material
Acknowledgements
We acknowledge the U.S. National Science Foundation (CHE-1763168) for funding this research. The acquisition of a NMR spectrometer used in this research was supported by a grant from the U.S. National Science Foundation (CHE-1625963). K.D. acknowledges the support from the National Natural Science Foundation of China and Jiangsu province (21602148). We thank M. Venkateswaran and J. Vidal for their synthesis of starting materials.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/anie.201909929.
References
- [1] a).Fisher JF, Meroueh SO, Mobashery S, Chem. Rev. 2005, 105, 395–424; [DOI] [PubMed] [Google Scholar]; b) Brandi A, Cicchi S, Cordero FM, Chem. Rev 2008, 108, 3988–4035. [DOI] [PubMed] [Google Scholar]
- [2] a).Rousseau G, Robin S in Four-membered Heterocycles: Structure and Reactivity in Modern Heterocyclic Chemistry, 1st ed (Eds.: Alvarez-Builla J, Vaquero JJ, Barluenga J), Wiley-VCH, Weinheim, 2011, pp. 163–268; [Google Scholar]; b) Barluenga J, Gómez A, Santamaría J, Tomás M, Angew. Chem. Int. Ed. 2010, 49, 1306–1308; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1328–1330. [Google Scholar]
- [3] a).Shindoh N, Kitaura K, Takemoto Y, Takasu K, J. Am. Chem. Soc. 2011, 133, 8470–8473; [DOI] [PubMed] [Google Scholar]; b) Lopez SA, Houk KN, J. Org. Chem. 2014, 79, 6189–6195. [DOI] [PubMed] [Google Scholar]
- [4] a).Hodgson DM, Pearson CI, Kazmi M, Org. Lett. 2014, 16, 856–859; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baumann AN, Eisold M, Music A, Haas G, Kiw YM, Didier D, Org. Lett. 2017, 19, 5681–5684; [DOI] [PubMed] [Google Scholar]; c) Antermite D, Degennaro L, Luisi R, Org. Biomol. Chem. 2017, 15, 34–50. [DOI] [PubMed] [Google Scholar]
- [5].Barluenga J, Riesgo L, Lonzi G, Tomás M, López LA, Chem. Eur J. 2012, 18, 9221–9224. [DOI] [PubMed] [Google Scholar]
- [6].Singal KK, Kaur J, Synth. Commun. 2001, 31, 2809–2815. [Google Scholar]
- [7].Pang S, Yang X, Cao Z-H, Zhang Y-L, Zhao Y, Huang Y-Y, ACS Catal. 2018, 8, 5193–5199. [Google Scholar]
- [8].Fowden L, Nature 1955, 176, 347–348. [Google Scholar]
- [9] a).Matsuura F, Hamada Y, Shioiri T, Tetrahedron 1994, 50, 265–274; [Google Scholar]; b) Ma JF, Shinada T, Matsuda C, Nomoto K, J. Biol. Chem. 1995, 270, 16549–16554; [DOI] [PubMed] [Google Scholar]; c) Suzuki M, Takahashi M, Tsukamoto T, Watanabe S, Matsuhashi S, Yazaki J, Kishimoto N, Kikuchi S, Nakanishi H, Mori S, Nishizawa NK, Plant J. 2006, 48, 85–97; [DOI] [PubMed] [Google Scholar]; d) Stephan UW, Scholz G, Physiol. Plant. 1993, 88, 522–529; [Google Scholar]; e) Takahashi M, Terada Y, Nakai I, Nakanishi H, Yoshimura E, Mori S, Nishizawa NK, Plant Cell 2003, 15, 1263–1280; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kobayashi J, Cheng J-F, Ishibashi M, Wälchli MR, Yamamura S, Ohizumi Y, J. Chem. Soc. Perkin Trans. 1 1991, 1135–1137; [Google Scholar]; g) Knapp S, Dong Y, Tetrahedron Lett. 1997, 38, 3813–3816; [Google Scholar]; h) Fujiwara T, Hashimoto K, Umeda M, Murayama S, Ohno Y, Liu B, Nambu H, Yakura T, Tetrahedron 2018, 74, 4578–4591. [Google Scholar]
- [10] a).Evans HC, Perry CM, Faulas D, Drugs 2004, 64, 649–678; [DOI] [PubMed] [Google Scholar]; b) Brighton TA, Med. J. Aust. 2004, 181, 432–437; [DOI] [PubMed] [Google Scholar]; c) County F, Evano G, Org. Prep. Proced. Int. 2006, 38, 427–465. [Google Scholar]
- [11] a).Wessig P, Schwarz J, Helv. Chim. Acta 1998, 81, 1803–1814; [Google Scholar]; b) Couty F, Evano G, Vargas-Sanchez M, Bouzas G, J. Org. Chem. 2005, 70, 9028–9031; [DOI] [PubMed] [Google Scholar]; c) He G, Zhao Y, Zhang S, Lu C, Chen G, J. Am. Chem. Soc. 2012, 134, 3–6; [DOI] [PubMed] [Google Scholar]; d) Glawar AFG, Jenkinson SF, Thompson AL, Nakagawa S, Kato A, Butters TD, Fleet GWJ, ChemMedChem 2013, 8, 658–666; [DOI] [PubMed] [Google Scholar]; e) Mehra V, Lumb I, Anand A, Kumar V, RSC Adv. 2017, 7, 45763–45783. [Google Scholar]
- [12].Deng Y, Massey LA, Zavalij PY, Doyle MP, Angew. Chem. Int. Ed. 2017, 56, 7479–7483; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7587–7591. [Google Scholar]
- [13].a) For recent reviews on [3+n]-cycloaddition, see: Cheng Q-Q, Deng Y, Lankelma M, Doyle MP, Chem. Soc. Rev. 2017, 46, 5425–5443; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Marichev KO, Doyle MP, Org. Biomol Chem. 2019, 17, 4183–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14] a).Yoshimura T, Omata T, J. Org. Chem. 1976, 41, 1728–1733; [Google Scholar]; b) Bizet V, Buglioni L, Bolm C, Angew. Chem. Int. Ed. 2014, 53, 5639–5642; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5745–5748; [Google Scholar]; c) Hayashi R, Shimizu A, Song Y, Ashikari Y, Nokami T, Yoshida J.-i., Chem. Eur. J. 2017, 23, 61–64. [DOI] [PubMed] [Google Scholar]
- [15].Mandler MD, Truong PM, Zavalij PY, Doyle MP, Org. Lett. 2014, 16, 740–743. [DOI] [PubMed] [Google Scholar]
- [16] a).Gilchrist TL, Moody CJ, Chem. Rev. 1977, 77, 409–435; [Google Scholar]; b) García Ruano JL, Cid MB, Martín-Castro AM, Alemán J, Sci. Synth. 2007, 39, 245–390; [Google Scholar]; c) Tian X, Song L, Rudolph M, Rominger F, Oeser T, Hashmi ASK, Angew. Chem. Int. Ed. 2019, 58, 3589–3593; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3627–3631. [Google Scholar]
- [17].Dong K, Marichev KO, Xu X, Doyle MP, Synlett 2019, 30, 1457–1461. [Google Scholar]
- [18]. For ligand screening, see: Supporting Information. [Google Scholar]
- [19].Moyer MP, Feldman PL, Rappoport H, J. Org. Chem. 1985, 50, 5223–5230. [Google Scholar]
- [20]. For crystallographic report, see: Supporting Information. [Google Scholar]
- [21] a).Martínez RF, Fleet GWJ, Tetrahedron: Asymmetry 2014, 25, 373–380; [Google Scholar]; b) Lawande PP, Sontakke VA, Nair RJ, Khan A, Sabharwal SG, Shinde VS, Tetrahedron 2015, 71,5085–5090; [Google Scholar]; c) Lawande PP, Sontakke VA, Kumbhar NM, Bhagwat TR, Ghosh S, Shinde VS, Bioorg. Med. Chem. Lett. 2017, 27, 5291–5295; [DOI] [PubMed] [Google Scholar]; d) Gavale KS, Chavan SR, Khan A, Joshi R, Dhavale DD, Org. Biomol Chem. 2015, 13, 6634–6646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.