

Abstract
Dietary exposure to aflatoxin B1 (AFB1) is a significant contributor to the incidence of hepatocellular carcinomas globally. AFB1 exposure leads to the formation of AFB1-N7-guanine (AFB1-N7-Gua) and two diastereomers of the imidazole ring-opened 8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxyaflatoxin B1 (AFB1-FapyGua) in DNA. These adducts lead to G → T transversion mutations with the ring-opened adduct being more mutagenic than the cationic species. Accurate measurement of these three adducts as biomarkers in DNA and urine will help identify dietary exposure to AFB1 as a risk factor in the development of hepatocellular carcinoma worldwide. Herein, we report an improved methodology for the measurement of AFB1-N7-Gua and the two diastereomers of AFB1-FapyGua using liquid chromatography-tandem mass spectrometry with isotope dilution. We measured the levels of these compounds in liver DNA of six control mice and six AFB1-treated mice. Levels varying from 1.5 to 45 lesions/106 DNA bases in AFB1-treated mice were detected depending on the compound and animal. No background levels of these adducts were detected in control mice. We also tested whether the AFB1 treatment caused oxidatively induced DNA base damage using gas chromatography-tandem mass spectrometry with isotope dilution. Although background levels of several pyrimidine- and purine-derived lesions were detected, no increases in these levels were found upon AFB1 treatment of mice. On the other hand, significantly increased levels of (5′R)- and (5′S)-8,5′-cyclo-2′-deoxyadenosines were observed in liver DNA of AFB1-treated mice. The impact of this work is expected to achieve the accurate measurement of three AFB1-DNA adducts and oxidatively induced DNA lesions as biomarkers of AFB1 exposure as germane to investigations designed for the prevention of aflatoxin-related hepatocellular carcinomas and for determining the effects of genetic deficiencies in human populations.
Graphical Abstract
INTRODUCTION
Hepatocellular carcinomas (HCCs) are a major cause of worldwide cancer deaths affecting upward of 800,000 people each year.1–4 The incidence of these cancers is concentrated in economically developing regions including Eastern and Southeastern Asia, Central America, and sub-Saharan Africa, in which there are dietary exposures to aflatoxin from the molds Aspergillus flavus and Aspergillus parasiticus that frequently contaminate staple grains and nuts. The relationship of the prevalence of these molds in staple food products and cancer is supported by epidemiological studies where the highest levels of aflatoxin contamination of food products lead to a large shift in not only the age of onset of HCCs, but also in the incidence rate.5 Although the aflatoxins constitute a family of closely related structures, aflatoxin B1 (AFB1) has been shown to be the most potent hepatocarcinogen.6–8 Following ingestion, AFB1 is activated primarily in the liver by microsomal cytochrome P450 enzymes to produce AFB1-exo-8,9-epoxide, which forms a covalently bound cationic adduct at N7 of guanine in DNA, that is, 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 (AFB1-N7-Gua)9–13 (Figure 1). Under physiological conditions or by acid treatment, the glycosidic bond of AFB1-N7-Gua in DNA is hydrolyzed, releasing AFB1-N7-Gua.14 Moreover, AFB1-N7-Gua in DNA readily undergoes imidazole ring opening at physiological pH, due to the positive charge on the imidazole ring, leading to the formation of the highly persistent 8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihy-dropyrimid-5-yl-formamido)-9-hydroxyaflatoxin B1 (AFB1-FapyGua) in DNA (Figure 1).13–15 AFB1-FapyGua in DNA exists in the α- and β-anomers of its 2′-deoxynucleoside, that is, α-AFB1-FapydG (major) and β-AFB1-FapydG (minor), which are distinguished by the orientation of the glycosidic bond relative to the 2′-deoxyribose moiety.13
Figure 1.

Mechanism of formation in DNA of AFB1-N7-Gua, cis-AFB1-FapyGua and trans-AFB1-FapyGua.
Under acidic conditions, AFB1-N7-Gua and AFB1-FapyGua are released from DNA by hydrolysis of the glycosidic bond, with the latter existing in the cis-form (minor) and the transform (major) (Figure 1).12,13 Replication of aflatoxin-modified DNAs generates a distinct mutational signature, dominated by G → T transversions with a small amount of G → A transitions (≈12%),6,12,16–20 underscoring a causal link between these adducts and genetic alterations in HCCs. In primate cells, AFB1-FapyGua has been shown to be at least 6-fold more mutagenic than AFB1-N7-Gua with dominating G → T transversions in both mutagenic spectra.12,21,22 In terms of the anomeric forms of its 2′-deoxynudeoside, β-AFB1FapydG is a major contributor to mutagenesis, while α-AFB1-FapydG strongly blocks replication.12
There is evidence that both AFB1-N7-Gua and AFB1-FapyGua are repaired by nucleotide excision repair (NER).23,24 This is supported by the fact that AFB1N7-Gua accumulates in NER-deficient XPA cells when compared to controls and that Xpa−/− mice are more susceptible to HCC than wild-type mice.23,25 The role of base excision repair (BER) in the repair of AFB1-N7-Gua and AFB1-FapyGua has also been investigated. The data suggested that AFB1-FapyGua may be a substrate of E. coli Fpg.26 However, another study using Fpg-deficient E. coli cells did not support the role of Fpg in the repair of AFB1-FapyGua.23,25 Recently, it has been hypothesized that DNA glycosylase NEIL1 may play a major role in the repair of AFB1-FapyGua.27 Indeed, increased accumulation of AFB1-FapyGua has been observed in Neil1−/− mice when compared to wild-type mice.27 Furthermore, Neil1−/− mice were significantly more susceptible to AFB1-induced HCCs than wild-type mice. These data unequivocally implicated NEIL1 as the major DNA glycosylase in BER for the repair of AFB1-FapyGua.
In addition to the formation of AFB1-N7-Gua and AFB1-FapyGua, cells exposed to AFB1 have been shown to form free radicals and other oxygen-derived species.28,29 Moreover, oxidatively induced DNA damage has been investigated in both animals and humans, and the formation of 8-hydroxy-2′-deoxyguanosine (8-OH-dG) [the nucleoside form of 8-hydroxyguanine (8-OH-Gua)] in DNA has been observed.30–33 However, no other oxidatively induced DNA base damage has been reported. 8-OH-Gua is known to cause G → T transversions,34,35 potentially contributing to this type of mutations caused by AFB1 treatment. A recent study showed an increase in the BER activity and OGG1 expression in lung extracts of mice following acute exposure to AFB1; however, no increase in BER activity was observed in liver extracts.36 Taken together, these findings suggest that AFB1-induced oxidative stress may be an important mechanism, contributing to the mutagenicity and carcinogenicity of AFB1 in vivo. On the other hand, some other studies showed that chronic low-dose exposure to AFB1 did not affect BER and OGG1 expression in lungs or livers of heterozygous p53 knockout mice.37 Furthermore, the OGG1 status had no significant effect on AFB1-induced oxidatively damaged DNA or carcinogenicity.38
Here, we report on the development of an improved methodology using liquid chromatography-tandem mass spectrometry (LC-MS/MS) with isotope dilution for the simultaneous measurement of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua in liver DNA of AFB1-treated mice. Furthermore, we present the evidence for oxidatively induced DNA damage in livers of mice upon AFB1 treatment.
EXPERIMENTAL PROCEDURES
Ethics Statement
The breeding and care of the Neil1−/− C57BL/6J mice were in accordance with the protocols approved by the Animal Care and Use Committee of Oregon Health & Science University, Portland, Oregon (Protocol number IS00002316-A967). Mice were euthanized by CO2 inhalation, followed by cervical dislocation. All efforts were made to minimize any discomfort to the mice, in accordance with approved animal care protocols.
Exposure of Mice to AFB1
To obtain experimental mice for AFB1 exposure that would maximize AFB1-induced DNA adducts, breeding pairs were set up using Neil1−/− mice that have been backcrossed into the C57Bl6 background through 17 generations. Neil1−/− mice were chosen for these investigations because, in the absence of NEIL1-initiated BER of AFB1-FapyGua, this lesion accumulates to ≈3-fold greater levels than observed in mice that are wild-type for NEIL1.27 A total of 6 mice (5 days old) were injected intraperitoneally with 7.5 mg/kg of AFB1 in dimethyl sulfoxide (DMSO) using 100 μL Hamilton syringe with 30X1/2 needle. The volume of injected solution was 10 μL per mouse. In addition to AFB1-injected mice, 6 control 5 day old mice were each injected with 10 μL of DMSO. Animals were euthanized by CO2 affixation, followed by cervical dislocation 48 h post-injection. Livers were isolated, and DNA extracted using a previously described method.15 No phenol was used to avoid artificial oxidation of DNA bases during DNA isolation. Furthermore, isolated DNA was washed twice in 70% ethanol and stored at −80 °C under 100% ethanol until use to avoid oxidation of DNA bases.
Preparation of AFB1-N7-Gua and AFB1-FapyGua and Their 15N5-Labeled Analogues
AFB1N7-Gua was prepared by reacting calf thymus DNA dissolved in water (1 mg/mL) with AFB1-exo-8,9-epoxide in acetone, as described previously.14,39,40 After 10 min, the residual acetone was removed by nitrogen, and 2 volumes of ice-cold ethanol were added to precipitate the DNA. The resulting mixture was centrifuged at 10,000 g for 10 min to precipitate the DNA. The pellet was hydrolyzed with 0.1 mol/L HCl by heating at 99 °C for 10 min, followed by neutralization with 1 mol/L ammonium formate. AFB1-N7-Gua was isolated and purified by high-performance liquid chromatography (HPLC) as described.40 Purified aliquots of the AFB1-N7-Gua were concentrated under nitrogen and quantified by absorbance at 360 nm with the extinction coefficient of 18,000 L mol−1 cm−1. AFB1N7-Gua-15N5 was similarly prepared using a uniformly 15N-labeled DNA isolated from algae grown in a pure 15N-environment.40 Further characterization of the purified standards was performed by LC-MS/MS. AFB1-FapyGua and AFB1-FapyGua-15N5 were prepared from AFB1-N7-Gua and AFB1-N7-Gua-15N5, respectively, by treatment with 0.1 mol/L NaOH for 10 min. The solution was readjusted to pH 5 prior to further purification as outlined above. Purified AFB1-FapyGua and AFB1-FapyGua-15N5 were quantified by absorbance at 362 nm with the extinction coefficient of 18,000 L mol−1 cm−1. Each compound contained both cis- and trans-isomers in a ratio of 1 to 3.
Measurement of AFB1-Adducts by Liquid Chromatography-Tandem Mass Spectrometry
Ethanol was removed from the DNA samples in a SpeedVac under vacuum. Dried DNA samples were dissolved in water overnight at 4 °C. The quality and quantity of DNA in water were determined through absorption spectrophotometry between 200 and 350 nm. The absorbance at 260 nm was used to measure the DNA concentration (absorbance of 1 corresponds to 50 μg of DNA/mL). Subsequently, three replicates of 50 μg aliquots of DNA samples from each mouse were dried in a SpeedVac under vacuum. For the measurement of the AFB1-adducts, 1.8 pmol of AFB1N7-Gua-15N5, and 0.6 pmol of AFB1-FapyGua-15N5 as internal standards were added to each dried DNA sample. The samples were dissolved in 100 μL of 0.1 mol/L HCl in Teflon-capped glass vials and then heated at 95 °C for 1 h. After cooling, the samples were frozen in liquid nitrogen and lyophilized for 18 h. The dried samples were dissolved in 100 μL of water prior to LC-MS/MS analysis.
Hydrolyzed DNA samples were analyzed by LC-MS/MS using a Thermo-Scientific Finnigan TSQ Quantum Ultra AM triple quadrupole MS/MS system with an installed heated electrospray-ionization source. Analysis of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua was performed using a Zorbax Extend C18 narrow-bore LC column (2.1 mm × 100 mm, 1.8 μm particle size) (Agilent Technologies, Wilmington, DE) with an attached Agilent Eclipse XDB-C8 guard column (2.1 mm × 12.5 mm, 5 μm particle size). In all instances, the autosampler and column temperature were kept at 5 and 40 °C, respectively. Mobile phase A was a mixture of water (98%) and acetonitrile (2%), and mobile phase B was acetonitrile, both containing 0.1% formic acid (v/v). A gradient analysis of 4% of B/min starting from 100% A was used with a flow rate of 0.3 mL/min. After 10 min, B was increased to 90% in 0.5 min and kept at this level for 5 min and then another 15 min at 8% to equilibrate the column. The total analysis time was 25 min. The following MS/MS parameters were used for all measurements: spray voltage = 3.5 kV; tube lens offsets = 89 V for Q1 and Q3; vaporizer temperature = 250 °C; capillary temperature = 340 °C; sheath gas (nitrogen) pressure = 50 (arbitrary units); auxiliary gas (nitrogen) pressure = 30 (arbitrary units); collision gas (argon) pressure = 6.67 × 10−5 Pa (5 mTorr). Selected reaction monitoring (SRM) data were acquired in the positive ionization mode at a mass range of m/z 100 to m/z 1500 with scan width m/z 2.000 and scan time 0.10 s.
The limit of quantification (LOQ) (at least 10-fold over the background level) of both AFB1-N7-Gua and AFB1-FapyGua was determined to be 0.036 fmol injected on column. The recovery of AFB1-N7-Gua and AFB1-FapyGua from the column was determined by injecting a mixture of these compounds on column and measuring the peak areas of their mass spectral responses. Subsequently, a similar injection was made, and the effluents were collected. The collected effluents were then analyzed, and their mass spectral responses were determined. These measurements were repeated 6 times. The recovery of both AFB1-N7-Gua and AFB1-FapyGua was found to be 80 ± 5%. The within-day reproducibility was determined by analyzing the triplicates of mixtures of AFB1-N7-Gua and AFB1-FapyGua 3 times per day with 4 h increments. This experiment was repeated over a 3 day period to determine the between-day reproducibility. On the basis of the first triplicate analyses of the first day, both the within-day reproducibility and the between-day reproducibility were 100%.
Measurement of R-cdA and S-cdA by Liquid Chromatography-Tandem Mass Spectrometry
Internal standards (5′R)-8,5′-cyclo-2′-deoxyadenosine-15N5 (R-cdA-15N5) and (5′S)-8,5′-cyclo-2′-deoxyadenosine-15N5 (S-cdA-15N5) were synthesized and isolated as described.41,42 Aliquots of R-cdA-15N5 and S-cdA-15N5 were added to each of the 50 μg DNA samples. Subsequently, the samples were dissolved in 50 μL of 10 mmol/L Tris-HCl buffer (pH 7.5) containing 45 mmol/L ZnCl2, supplemented with 2.5 μL of 1 mol/L sodium acetate (final pH 6.0), and then hydrolyzed with nuclease P1, snake venom phosphodiesterase, and alkaline phosphatase as described previously.43 After hydrolysis, the samples were filtered using ultrafiltration membranes with a molecular mass cutoff of 3 kDa by centrifugation at 12,000 g for 30 min. Separations were performed using a Zorbax SB-Aq rapid resolution narrow-bore LC column (2.1 mm × 150 mm, 3.5 μm particle size) (Agilent Technologies, Wilmington, DE) with an attached Agilent Eclipse XDB-C8 guard column (2.1 mm × 12.5 mm, 5 μm particle size). Mobile phase A was a mixture of water (98%) and acetonitrile (2%), and mobile phase B was acetonitrile, both containing 0.1% formic acid (v/v). A gradient analysis of 3% of B/min starting from 100% A was used with a flow rate of 0.5 mL/min. After 10 min, B was increased to 95% in 0.1 min and kept at this level for 5 min and then another 15 min at 100% A to equilibrate the column. The total analysis time was 30 min. The conditions of the MS/MS system were as previously described.44 The SRM mass transitions were m/z 250 → m/z 164 for R-cdA and S-cdA and m/z 255 → m/z 169 for R-cdA-15N5 and S-cdA15N5.
Measurement of Oxidatively Induced DNA Bases by Gas Chromatography-Tandem Mass Spectrometry
Another set of DNA samples isolated from livers of AFB1-treated mice was used to identify and quantify 5-hydroxy-5-methylhydantoin (5-OH-5-MeHyd), 5-hydroxycytosine (5-OH-Cyt), thymine glycol (ThyGly), 4,6-diamino-5-formamidopyrimidine (FapyAde), 8-hydroxyadenine (8-OH-Ade), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua), and 8-OH-Gua. DNA samples (50 μg each) were supplemented with the aliquots of the stable isotope-labeled analogues of these compounds, that is, 5-OH-5-MeHyd-13C,15N2, 5-OH-Cyt-13C,15N2, ThyGly-2H4, FapyAde-13C,15N2, FapyGua-13C,15N2, 8-OH-Ade-13C,15N2 and 8-OH-Gua-15N5 as internal standards, which are a part of the National Institute of Standards and Technology (NIST) Standard Reference Material 2396 Oxidative DNA Damage Mass Spectrometry Standards (for details see refs 45 and 46). The samples were dried in a SpeedVac under vacuum and then dissolved in 50 μL of an incubation buffer consisting of 50 mmol/L phosphate buffer (pH 7.4), 100 mmol/L KCl, 1 mmol/L EDTA, and 0.1 mmol/L dithiothreitol. Subsequently, they were incubated with 2 μg of E. coli Fpg protein and 2 μg of E. coli Nth protein at 37 °C for 1 h to release the modified DNA bases from DNA. This procedure has been shown to quantitatively release the modified bases from DNA at the levels observed with acidic hydrolysis.47,48 An aliquot of 100 μL ethanol was added to precipitate DNA. After centrifugation, the supernatant fractions were separated, lyophilized, trimethylsilylated,49 and then analyzed by GC-MS/MS using multiple reaction monitoring as described.43
Statistical Analysis
The statistical analysis of the data was performed using the GraphPad Prism 6 software (La Jolla, CA, USA) and unpaired, two-tailed nonparametric Mann-Whitney test with Gaussian approximation and confidence level of 99%.
RESULTS
The aim of this work was to develop an improved LC-MS/MS methodology with isotope dilution for the measurement of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua in vivo and to demonstrate the applicability of this methodology to the in vivo measurement of these DNA adducts. We also investigated whether AFB1 treatment leads to the in vivo formation of oxidatively induced DNA lesions. Figure 2 illustrates the fragmentation patterns of AFB1-N7-Gua and AFB1-FapyGua and their 15N5-labeled analogues as previously described.9,40,50 The cleavage of the bond between Gua and AFB1 moieties of AFB1-N7-Gua gives rise to the m/z 152 and m/z 329 product ions. AFB1-N7-Gua-15N5 yields the m/z 157 and m/z 329 product ions, because the Gua moiety contains five 15N atoms, whereas there is no labeling on the AFB1 moiety. The loss of H2O from AFB1-FapyGua and AFB1-FapyGua-15N5 yields the m/z 480 and m/z 485 product ions, respectively. Further loss of CO from these ions results in the formation of the m/z 452 and m/z 457 ions, respectively.
Figure 2.

Fragmentation mechanisms of AFB1-N7-Gua and AFB1-FapyGua and their 15N5-labeled analogues, leading to product ions. Protonated molecular ions (MH+), product ions, and mass transitions are shown.
To achieve the highest measurement sensitivity, the optimum collision energies were determined for the two mass transitions of AFB1-N7-Gua and AFB1-FapyGua. The collision energies were varied from 5 to 50 V in 5 V increments. Supporting Information Figure 1 illustrates the measured intensity versus the collision energy for the two mass transitions of each compound. The collision energy for the m/z 480 → m/z 152 transition of AFB1-N7-Gua reached a plateau after 20 V, whereas 20 V was the optimum collision energy for the m/z 480 → m/z 329 transition (Supporting Information Figure 1A). The former mass transition had a greater intensity than the latter. Hence, the m/z 480 → m/z 152 transition would provide a greater sensitivity for the measurement of AFB1-N7-Gua. However, the simultaneous monitoring of the m/z 480 → m/z 329 transition would serve as validation. The m/z 498 → m/z 452 transition of both cis-AFB1-FapyGua and trans-AFB1-FapyGua had a maximum collision energy of 20 V, whereas 15 V provided the highest intensity for the m/z 498 → m/z 480 transition of both compounds (Supporting Information Figure 1B). The m/z 498 → m/z 452 transition was almost 2-fold more intense than the m/z 498 → m/z 480 transition, meaning that the former would provide greater sensitivity than the latter for the measurement of both cis- and trans-isomers of AFB1-FapyGua. However, the simultaneous monitoring of both transitions would validate the identification. Supporting Information Figure 2 illustrates the ion-current profiles of both mass transitions of the standards AFB1-N7-Gua and cis- and trans-isomers of AFB1-FapyGua. A baseline separation and excellent peak shapes of the signals of all three compounds were achieved.
The LOQ with a signal-to-noise ratio of 10 for both AFB1-N7-Gua and AFB1-FapyGua was determined to be 0.036 fmol injected on column. This LOQ is very similar to the value published previously [0.02 pg on-column (0.042 fmol on-column)],40 with 0.036 fmol corresponding to approximately 2.2 lesions/1010 DNA bases. Thus, with the LOQ being 0.036 fmol, at least 2.2 lesions/1010 DNA bases can be detected with the methodology described in this work. We used 50 μg of DNA for all analyses. However, as the mass spectral responses (Figure 3) along with the very low LOQ indicate, lower amounts of DNA could be used to facilitate the measurement of AFB1-N7-Gua and AFB1-FapyGua. It should also be pointed out that 0.1 mol/L HCl (100 μL per 50 μg of DNA) at 95 °C for 1 h was used for the hydrolysis of DNA, which may not quantitatively release AFB1-N7-Gua and AFB1-FapyGua from DNA. Considering the weak nature of the glycosidic bond and the fact that these hydrolysis conditions were previously used in the literature,14,27 a strong acid such as HCl at the concentration used in this work is expected to quantitatively remove these adducts from DNA.
Figure 3.

Ion-current profiles of the mass transitions of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua and their 15N5-labeled analogues recorded during the LC-MS/MS analysis of a liver DNA sample from a control mouse (A) and an AFB1-treated mouse (B). For each of the three adducts, two mass transitions shown in Figure 2 were used. The arrows in (A) show the elution positions of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua and indicate their absence in liver DNA of control mice.
The recovery of both AFB1-N7-Gua and AFB1-FapyGua from the column was found to be 80 ± 5%. The within-day reproducibility and the between-day reproducibility varied from 95% to 115%. To test the accuracy, the ratios of the peak areas of the MS/MS response versus the ratios of the unlabeled and labeled forms of AFB1-N7-Gua and AFB1-FapyGua were generated. The amount of their labeled analogues was kept constant. Within the amount ratios ranging from 0.1 to 20.0, a linear response was observed with the coefficient of determination R2 = 0.9985.
Measurement of AFB1-N7-Gua and cis- and trans-Isomers of AFB1-FapyGua in Liver DNA of Mice
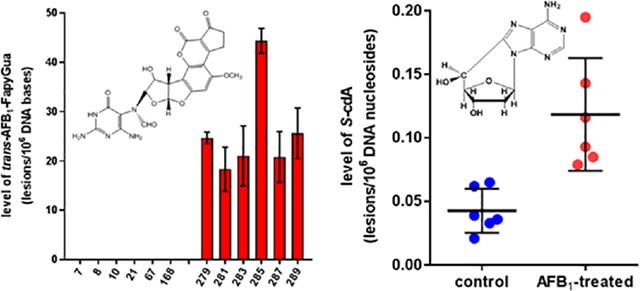
The developed methodology was applied to identify and quantify AFB1-N7-Gua and cis- and trans-isomers of AFB1-FapyGua in DNA of livers of control mice and AFB1-treated mice. In each set, six mice were used, and DNA was isolated from the livers of untreated mice and AFB1-treated mice. The experiment was performed in a double-blind fashion. The codes of the DNA samples were not known until after the LC-MS/MS analysis. Three DNA samples were isolated from each liver. Hydrolyzed DNA samples were analyzed by SRM using corresponding mass transitions of the analytes. As an example of the measurements, the data in Figure 3 illustrate the ion-current profiles of each of the two mass transitions of AFB1-N7-Gua, AFB1-N7-Gua-15N5, AFB1-FapyGua, and AFB1-FapyGua-15N5 as recorded during the analysis of DNA samples from a control mouse and an AFB1-treated mouse. The arrows in Figure 3A show the absence of the signals of the mass transitions of both AFB1-N7-Gua and AFB1-FapyGua in contrast to the signals of their 15N5-labeled analogues, indicating neither of these two adducts were detected in liver DNA of control mice. In contrast, the data in Figure 3B show the discernible signals of the two mass transitions of both AFB1-N7-Gua and AFB1-FapyGua, unequivocally identifying them in liver DNA of AFB1-treated mice. The quantification of AFB1-N7-Gua and cis- and trans-isomers of AFB1-FapyGua is shown in Figure 4. DNA samples with the numbers 1–6 were isolated from control mice, whereas those with the numbers 7–12 were from AFB1-treated mice. As was mentioned above, no AFB1-N7-Gua, cis-AFB1-FapyGua, or trans-AFB1-FapyGua was detected in control samples. In DNA samples isolated from AFB1-treated mice, these adducts were readily detectable. The level of AFB1-N7-Gua in liver DNA of treated mice varied from 15 to 25 lesions/106 DNA bases (Figure 4A). One animal had a significantly greater level of AFB1-N7-Gua than the other animals. The level of cis-AFB1-FapyGua varied among the animals from 1.5 to 6 lesions/106 DNA bases (Figure 4B). trans-AFB1-FapyGua had significantly greater levels than cis-AFB1-FapyGua, varying from 20 to 45 lesions/106 DNA bases (Figure 4C).
Figure 4.

Levels of AFB1-N7-Gua (A), cis-AFB1-FapyGua (B), and trans-AFB1-FapyGua (C) in liver DNA of control mice (1–6) and AFB1-treated mice (7–12). Uncertainties are standard deviations (n = 3).
Measurement of Oxidatively Induced DNA Base Lesions
Seven modified DNA bases were identified and quantified: 5-OH-5-MeHyd, 5-OH-Cyt, ThyGly, FapyAde, 8-OH-Ade, FapyGua, and 8-OH-Gua. However, the DNA samples collected 48 h post-AFB1 injection showed no increase in the levels of these modified DNA bases in their liver DNA when compared to their levels in liver DNA of control mice. Supporting Information Figure 3 shows the levels of FapyGua and 8-OH-Gua in control and AFB1-treated mice, with no statistically significant differences between the two groups. The levels of the DNA lesions varied between animals.
We also tested whether the AFB1 treatment would cause the formation of 8,5′-cyclo-2′-deoxynucleosides in mouse liver. Supporting Information Figure 4 shows the ion-current profiles of the mass transitions of R-cdA and S-cdA and their 15N5-labeled analogues recorded during the LC-MS/MS analysis of a liver DNA sample from an AFB1-treated mouse. The quantification of R-cdA and S-cdA in control and AFB1-treated mice is shown Figure 5. When compared to control mice, a statistically significant increase in the levels of these modified nucleosides was observed in liver DNA of AFB1-treated mice.
Figure 5.

Levels of R-cdA (A) and S-cdA (B) in liver DNA of control mice and AFB1-treated mice. Uncertainties are standard deviations.
DISCUSSION
To better understand the effect of aflatoxin toxicity on humans and the development of HCCs, robust assays are essential for the accurate measurement of aflatoxin adducts in DNA and urine of humans. Such assays would also help understand cellular repair of aflatoxin adducts and, consequently, the effect of genetic variants and polymorphisms in DNA repair mechanisms on the occurrence of HCCs. Various analytical methodologies have been developed for the measurement of aflatoxin adducts in biological samples with different sensitivities, advantages, and drawbacks.9,11,51–54 Most of these assays lack the detection of AFB1-FapyGua diastereomers and the positive identification of analytes with insufficient sensitivity and accuracy of quantification. Mass spectrometric assays and stable isotope-labeled standards have been developed and applied in the past for the measurement of aflatoxin-DNA adducts in urine and DNA.27,40,50 Oxidatively induced DNA damage caused by aflatoxin treatment has also been investigated. However, this was limited to the measurement of 8-OH-dG only by various methodologies, which resulted in conflicting conclusions.30–33 No other DNA lesions were measured.
In this work, we developed an improved methodology for the simultaneous measurement in vivo of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua by LC-MS/MS with isotope dilution. This paper marks the first time that an isotopically labeled internal standard for the AFB1-FapyGua adducts has been utilized in a quantitative study. These 15N-labeled standards have not been reported in prior publications from our laboratory or any other laboratory. Furthermore, proper LC separations of AFB1-N7-Gua and AFB1-FapyGua adducts and their isotopically labeled analogues were demonstrated for the first time in this work. LC-MS/MS conditions provided a baseline separation of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua from one another with excellent peak profiles. Two mass transitions of each compound were used to validate the identification. We also determined, for the first time, the optimal collision energy for each compound and mass transition to achieve the highest sensitivity. No background levels of AFB1-N7-Gua, cis-AFB1-FapyGua, and trans-AFB1-FapyGua were detected in control mice. However, these adducts were readily identified and quantified in liver DNA of mice treated with AFB1. Detected levels varied from 1 lesion/106 DNA bases to 45 lesions/106 DNA bases depending on the lesion and the animal. The limit of quantification (at least 10-fold over the background level) was 0.036 fmol on-column for all three AFB1 adducts. This is also reported, for the first time, in this work. Moreover, the recovery of AFB1-N7-Gua and AFB1-FapyGua from the LC column was determined to be ≈80%. In this context, it should be emphasized that there are many factors that can affect the operational measurement of molecules such as DNA adducts. All DNA adducts measured in this study had isotopically labeled internal standards to control for individual recovery. Thus, the data reported in this manuscript are normalized to the recovery of the individual isotopically labeled standard. In future applications of this technology, the sample amount needed for an analysis will be largely driven by the level of environmental exposure which can vary a lot across populations. These factors will then have to be accounted for to ensure the measurement of these adducts can be achieved. In our experience that has included many experimental studies and human epidemiological investigations, this is the critical factor for measuring DNA adducts. The within-day reproducibility and the between-day reproducibility with triplicate analysis did not significantly differ between measurements.
We also determined whether oxidatively induced DNA damage occurs in liver DNA of mice upon AFB1 treatment. No significant elevation of the background levels of DNA base lesions was observed in liver DNA of AFB1-treated mice when compared to control mice. In the case of each lesion, some animals had high levels compared to the others, possibly indicating the individual susceptibility to AFB1 treatment. In contrast, we found greater levels of R-cdA and S-cdA in liver DNA of AFB1-treated mice than in control mice. This was surprising because both types of lesions are formed by the attack of hydroxyl radical on DNA. However, there is a stark difference between the mechanisms of formation of DNA base lesions and 8,5′-cyclo-2′-deoxynucleosides. It is not clear whether the difference between the formation mechanisms plays a role in the detection of higher levels of R-cdA and S-cdA in AFB1-treated mice than in control mice. Additionally, the DNA repair mechanisms that are responsible for repair of these DNA lesions are different, with NER responsible for the removal of the R-cdA and S-cdA, while the ring-fragmented and ring-saturated bases are repaired by BER. Since DNAs were harvested 48 h post-AFB1 injection, it is possible that the differential kinetics of BER and NER could account for these differences. On the other hand, the high background levels of DNA base lesions in control mice may prevent the detection of minor increases in the levels of these DNA lesions upon AFB1 treatment of mice.
AFB1-N7-Gua and AFB1-FapyGua cause G → T transversions with a small amount of G → A transitions as discussed above. Mutagenic effects of 8,5′-cyclo-2′-deoxynucleosides have been extensively investigated. S-cdA blocks both RNA transcription and some DNA polymerases, causing transcriptional mutagenesis, and A → T transversions and A → G transitions.55–60 R-cdA is expected to have a similar effect. Since G → T transversions are the main mutations caused by AFB1, it is not known whether A → T transversions and A → G transitions play a significant role in the mutagenicity of AFB1. However, since the in vivo levels of AFB1-N7-Gua and AFB1-FapyGua are significantly greater than those of R-cdA and S-cdA, deciphering the relative importance of the R-cdA and S-cdA adducts may be biologically challenging. Future mutagenicity studies may shed light on the role of 8,5′-cyclo-2′-deoxynucleosides on the mutagenic effect of AFB1.
Cellular repair of AFB1-N7-Gua and AFB1-FapyGua and genetic variants that increase carcinogenic susceptibility, both from in utero exposures and dietary ingestion throughout an individual’s lifespan, are poorly understood. A recent study presented the evidence that, in a murine model, DNA glycosylase NEIL1-initiated BER was several-fold more critical in the removal of AFB1-FapyGua from DNA.27 The steady-state level of this lesion was found to be greater in liver DNA of Neil1−/− mice than those in wild-type mice. Increased susceptibility to AFB1-induced HCCs in Neil1−/− mice was also observed. However, no distinction between the two diastereomers of AFB1-FapyGua was reported. Polymorphic variants of NEIL1 such as G83D-NEIL1 and C136R-NEIL1, which are devoid of DNA glycosylase activity, exist in humans.61,62 It is possible that humans carrying such inactive NEIL1 variants may be more prone to AFB1-induced HCCs than those with wild-type NEIL1.
The robust methodology presented in the present work will help positively identify and accurately quantify three AFB1-DNA adducts as biomarkers of AFB1 exposure and HCCs in a variety of biological samples including urine. This may also contribute to the elucidation of cellular repair mechanisms of these adducts including DNA repair enzymes and to the understanding of the role of genetic deficiencies in human population that increase susceptibility to AFB1-associated carcinogenesis.
Supplementary Material
Figure S1, determination of the optimum collision energies of AFB1-N7-Gua (A) and cis-AFB1-FapyGua and trans-AFB1-FapyGua (B) for two mass transitions of each compound. Figure S2, LC-MS/MS measurement of the standards. Ion-current profiles of the two mass transitions of each compound are shown. Figure S3, ion-current profiles of the mass transitions of R-cdA and S-cdA and their 15N5-labeled analogues recorded during the LC-MS/MS analysis of a liver DNA sample from an AFB1-treated mouse. Figure S4, levels of FapyGua (A) and 8-OH-Gua (B) in liver DNA of control mice (1–6) and AFB1-treated mice (7–12). Uncertainties are standard deviations (n = 3) (PDF)
ACKNOWLEDGMENTS
Certain commercial equipment or materials are identified in this paper in order to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials or equipment identified are necessarily the best available for the purpose. O.E. was supported in part by a grant from The Scientific and Technology Research Council of Turkey (TÜITAK) with the reference number of B.14.2.TBT.0.06.01-21514107-115.02-25615. This work was supported in part by NIH R56 ES027632 (RSL) and R01 ES029357 (Michael P. Stone PI, Vanderbilt University, Subaward to R.S.L.).
ABBREVIATIONS
- HCCs
hepatocellular carcinomas
- AFB1
aflatoxin B1
- AFB1-N7-Gua
8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1
- AFB1-FapyGua
8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxyaflatoxin B1
- dG
2′-deoxyguanosine
- BER
base excision repair
- 8-OH-dG
8-hydroxy-2′-deoxyguanosine
- 8-OH-Gua
8-hydroxyguanine
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- R-cdA
R-8,5′-cyclo-2′-deoxyadenosine
- S-cdA
S-8,5′-cyclo-2′-deoxyadenosine
- GC-MS/MS
gas chromatography-tandem mass spectrometry
- 5-OH-5-MeHyd
5-hydroxy-5-methylhydantoin
- 5-OH-Cyt
5-hydroxycytosine
- ThyGly
thymine glycol
- FapyAde
4,6-diamino-5-formamidopyrimidine
- FapyGua
2,6-diamino-4-hydroxy-5-formamidopyrimidine
- 8-OH-Ade
8-hydroxyadenine
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.8b00202.
REFERENCES
- (1).Blonski W, Kotlyar DS, and Forde KA (2010) Non-viral causes of hepatocellular carcinoma. World J. Gastroenterol 16, 3603–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bray F, Ren JS, Masuyer E, and Ferlay J (2013) Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int. J. Cancer 132, 1133–1145. [DOI] [PubMed] [Google Scholar]
- (3).Chitapanarux T, and Phornphutkul K (2015) Risk Factors for the Development of Hepatocellular Carcinoma in Thailand. J. Clin. Transl. Hepatol 3, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, and Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–386. [DOI] [PubMed] [Google Scholar]
- (5).Kensler TW, Roebuck BD, Wogan GN, and Groopman JD (2011) Aflatoxin: a 50-year odyssey of mechanistic and translational toxicology. Toxicol. Sci 120, S28–S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ayres JL, Lee DJ, Wales JH, and Sinnhuber RO (1971) Aflatoxin structure and hepatocarcinogenicity in rainbow trout (Salmo gairdneri). J. Natl. Cancer Inst 46, 561–564. [PubMed] [Google Scholar]
- (7).Wogan GN, Edwards GS, and Newberne PM (1971) Structure-activity relationships in toxicity and carcinogenicity of aflatoxins and analogs. Cancer Res. 31, 1936–1942. [PubMed] [Google Scholar]
- (8).Wogan GN, Kensler TW, and Groopman JD (2012) Present and future directions of translational research on aflatoxin and hepatocellular carcinoma. A review. Food Addit. Contam., Part A 29, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Essigmann JM, Croy RG, Nadzan AM, Busby WF Jr., Reinhold VN, Buchi G, and Wogan GN (1977) Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc. Natl. Acad. Sci. U. S. A 74, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Martin CN, and Garner RC (1977) Aflatoxin B -oxide generated by chemical or enzymic oxidation of aflatoxin B1 causes guanine substitution in nucleic acids. Nature 267, 863–865. [DOI] [PubMed] [Google Scholar]
- (11).Croy RG, Essigmann JM, Reinhold VN, and Wogan GN (1978) Identification of the principal aflatoxin B1-DNA adduct formed in vivo in rat liver. Proc. Natl. Acad. Sci. U. S. A 75, 1745–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, and Essigmann JM (2002) The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A 99, 6655–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Brown KL, Deng JZ, Iyer RS, Iyer LG, Voehler MW, Stone MP, Harris CM, and Harris TM (2006) Unraveling the aflatoxin-FAPY conundrum: structural basis for differential replicative processing of isomeric forms of the formamidopyrimidine-type DNA adduct of aflatoxin B1. J. Am. Chem. Soc 128, 15188–15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Groopman JD, Croy RG, and Wogan GN (1981) In vitro reactions of aflatoxin B1-adducted DNA. Proc. Natl. Acad. Sci. U. S. A 78, 5445–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Croy RG, and Wogan GN (1981) Temporal patterns of covalent DNA adducts in rat liver after single and multiple doses of aflatoxin B1. Cancer Res. 41, 197–203. [PubMed] [Google Scholar]
- (16).McMahon G, Hanson L, Lee JJ, and Wogan GN (1986) Identification of an activated c-Ki-ras oncogene in rat liver tumors induced by aflatoxin B1. Proc. Natl. Acad. Sci. U. S. A 83, 9418–9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, and Vogelstein B (1987) Prevalence of ras gene mutations in human colorectal cancers. Nature 327, 293–297. [DOI] [PubMed] [Google Scholar]
- (18).Bressac B, Kew M, Wands J, and Ozturk M (1991) Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 350, 429–431. [DOI] [PubMed] [Google Scholar]
- (19).Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, and Harris CC (1991) Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 350, 427–428. [DOI] [PubMed] [Google Scholar]
- (20).Bailey EA, Iyer RS, Stone MP, Harris TM, and Essigmann JM (1996) Mutational properties of the primary aflatoxin B1-DNA adduct. Proc. Natl. Acad. Sci. U. S. A 93, 1535–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lin YC, Li L, Makarova AV, Burgers PM, Stone MP, and Lloyd RS (2014) Error-prone replication bypass of the primary aflatoxin B1 DNA adduct, AFB1-N7-Gua. J. Biol. Chem 289, 18497–18506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lin YC, Li L, Makarova AV, Burgers PM, Stone MP, and Lloyd RS (2014) Molecular basis of aflatoxin-induced mutagenesis-role of the aflatoxin B1-formamidopyrimidine adduct. Carcinogenesis 35, 1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Alekseyev YO, Hamm ML, and Essigmann JM (2004) Aflatoxin B1 formamidopyrimidine adducts are preferentially repaired by the nucleotide excision repair pathway in vivo. Carcinogenesis 25, 1045–1051. [DOI] [PubMed] [Google Scholar]
- (24).Mulder IA, Khmelinskii A, Dzyubachyk O, de Jong S, Wermer MJH, Hoehn M, Lelieveldt BPF, and van den Maagdenberg A (2017) MRI Mouse Brain Data of Ischemic Lesion after Transient Middle Cerebral Artery Occlusion. Front Neuroinform 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Leadon SA, Tyrrell RM, and Cerutti PA (1981) Excision repair of aflatoxin B1-DNA adducts in human fibroblasts. Cancer Res. 41, 5125–5129. [PubMed] [Google Scholar]
- (26).Beranek DT, Weis CC, Evans FE, Chetsanga CJ, and Kadlubar FF (1983) Identification of N5-methyl-N5-formyl-2,5,6-triamino-4-hydroxypyrimidine as a major adduct in rat liver DNA after treatment with the carcinogens, N,N-dimethylnitrosamine or 1,2-dimethylhydrazine. Biochem. Biophys. Res. Commun 110, 625–631. [DOI] [PubMed] [Google Scholar]
- (27).Vartanian V, Minko IG, Chawanthayatham S, Egner PA, Lin YC, Earley LF, Makar R, Eng JR, Camp MT, Li L, Stone MP, Lasarev MR, Groopman JD, Croy RG, Essigmann JM, McCullough AK, and Lloyd RS (2017) NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proc. Natl. Acad. Sci. U. S. A 114, 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kodama M, Inoue F, and Akao M (1990) Enzymatic and non-enzymatic formation of free radicals from aflatoxin B1. Free Radical Res. Commun 10, 137–142. [DOI] [PubMed] [Google Scholar]
- (29).Towner RA, Qian SY, Kadiiska MB, and Mason RP (2003) In vivo identification of aflatoxin-induced free radicals in rat bile. Free Radical Biol. Med 35, 1330–1340. [DOI] [PubMed] [Google Scholar]
- (30).Shen HM, Ong CN, Lee BL, and Shi CY (1995) Aflatoxin B1-induced 8-hydroxydeoxyguanosine formation in rat hepatic DNA. Carcinogenesis 16, 419–422. [DOI] [PubMed] [Google Scholar]
- (31).Shen HM, Ong CN, and Shi CY (1995) Involvement of reactive oxygen species in aflatoxin B1-induced cell injury in cultured rat hepatocytes. Toxicology 99, 115–123. [DOI] [PubMed] [Google Scholar]
- (32).Barraud L, Douki T, Guerret S, Chevallier M, Jamard C, Trepo C, Wild CP, Cadet J, and Cova L (2001) The role of duck hepatitis B virus and aflatoxin B1 in the induction of oxidative stress in the liver. Cancer Detect. Prev 25, 192–201. [PubMed] [Google Scholar]
- (33).Guindon KA, Bedard LL, and Massey TE (2007) Elevation of 8-hydroxydeoxyguanosine in DNA from isolated mouse lung cells following in vivo treatment with aflatoxin B(1). Toxicol. Sci 98, 57–62. [DOI] [PubMed] [Google Scholar]
- (34).Kuchino Y, Mori F, Kasai H, Inoue H, Iwai S, Miura K, Ohtsuka E, and Nishimura S (1987) Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature 327, 77–79. [DOI] [PubMed] [Google Scholar]
- (35).Wood ML, Dizdaroglu M, Gajewski E, and Essigmann JM (1990) Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry 29, 7024–7032. [DOI] [PubMed] [Google Scholar]
- (36).Guindon-Kezis KA, Mulder JE, and Massey TE (2014) In vivo treatment with aflatoxin B1 increases DNA oxidation, base excision repair activity and 8-oxoguanine DNA glycosylase 1 levels in mouse lung. Toxicology 321, 21–26. [DOI] [PubMed] [Google Scholar]
- (37).Mulder JE, Bondy GS, Mehta R, and Massey TE (2015) The impact of chronic Aflatoxin B1 exposure and p53 genotype on base excision repair in mouse lung and liver. Mutat. Res., Fundam. Mol. Mech. Mutagen 773, 63–68. [DOI] [PubMed] [Google Scholar]
- (38).Mulder JE, Turner PV, and Massey TE (2015) Effect of 8-oxoguanine glycosylase deficiency on aflatoxin B1 tumourigenicity in mice. Mutagenesis 30, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Baertschi SW, Raney KD, Stone MP, and Harris TM (1988) Preparation of the 8,9-Epoxide of the Mycotoxin Aflatoxin-B1 - the Ultimate Carcinogenic Species. J. Am. Chem. Soc 110, 7929–7931. [Google Scholar]
- (40).Egner PA, Groopman JD, Wang JS, Kensler TW, and Friesen MD (2006) Quantification of aflatoxin-B1-N7-guanine in human urine by high-performance liquid chromatography and isotope dilution tandem mass spectrometry. Chem. Res. Toxicol 19, 1191–1195. [DOI] [PubMed] [Google Scholar]
- (41).Birincioglu M, Jaruga P, Chowdhury G, Rodriguez H, Dizdaroglu M, and Gates KS (2003) DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). J. Am. Chem. Soc 125, 11607–11615. [DOI] [PubMed] [Google Scholar]
- (42).Jaruga P, Birincioglu M, Rodriguez H, and Dizdaroglu M (2002) Mass spectrometric assays for the tandem lesion 8,5′-cyclo-2’-deoxyguanosine in mammalian DNA. Biochemistry 41, 3703–3711. [DOI] [PubMed] [Google Scholar]
- (43).Jaruga P, Coskun E, Kimbrough K, Jacob A, Johnson WE, and Dizdaroglu M (2017) Biomarkers of oxidatively induced DNA damage in dreissenid mussels: A genotoxicity assessment tool for the Laurentian Great Lakes. Environ. Toxicol 32, 2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jaruga P, Xiao Y, Nelson BC, and Dizdaroglu M (2009) Measurement of (5′R)- and (5′S)-8,5′-cyclo-2’-deoxyadenosines in DNA in vivo by liquid chromatography/isotope dilution tandem mass spectrometry. Biochem. Biophys. Res. Commun 386, 656–660. [DOI] [PubMed] [Google Scholar]
- (45).NIST Standard Reference Materials. http://www.nist.gov/srm/index.cfm.
- (46).SRM 2396 - Oxidative DNA Damage Mass Spectrometry Standards. https://www-s.nist.gov/srmors/view_detail.cfm?srm=2396.
- (47).Jaruga P, Speina E, Gackowski D, Tudek B, and Olinski R (2000) Endogenous oxidative DNA base modifications analysed with repair enzymes and GC/MS technique. Nucleic Acids Res. 28, No. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rodriguez H, Jurado J, Laval J, and Dizdaroglu M (2000) Comparison of the levels of 8-hydroxyguanine in DNA as measured by gas chromatography mass spectrometry following hydrolysis of DNA by Escherichia coli Fpg protein or formic acid. Nucleic Acids Res. 28, No. e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jaruga P, Kirkali G, and Dizdaroglu M (2008) Measurement of formamidopyrimidines in DNA. Free Radical Biol. Med 45, 1601–1609. [DOI] [PubMed] [Google Scholar]
- (50).Walton M, Egner P, Scholl PF, Walker J, Kensler TW, and Groopman JD (2001) Liquid chromatography electrospray-mass spectrometry of urinary aflatoxin biomarkers: characterization and application to dosimetry and chemoprevention in rats. Chem. Res. Toxicol 14, 919–926. [DOI] [PubMed] [Google Scholar]
- (51).Groopman JD, Donahue PR, Zhu JQ, Chen JS, and Wogan GN (1985) Aflatoxin metabolism in humans: detection of metabolites and nucleic acid adducts in urine by affinity chromatography. Proc. Natl. Acad. Sci. U. S. A 82, 6492–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Shepherd MJ, and Gilbert J (1984) An investigation of HPLC post-column iodination conditions for the enhancement of aflatoxin B1 fluorescence. Food Addit. Contam 1, 325–335. [DOI] [PubMed] [Google Scholar]
- (53).Harris CC, LaVeck G, Groopman J, Wilson VL, and Mann D (1986) Measurement of aflatoxin B1, its metabolites, and DNA adducts by synchronous fluorescence spectrophotometry. Cancer Res. 46, 3249–3253. [PubMed] [Google Scholar]
- (54).Wang JS, Shen X, He X, Zhu YR, Zhang BC, Wang JB, Qian GS, Kuang SY, Zarba A, Egner PA, Jacobson LP, Munoz A, Helzlsouer KJ, Groopman JD, and Kensler TW (1999) Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J. Natl. Cancer Inst 91, 347–354. [DOI] [PubMed] [Google Scholar]
- (55).Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, and Lindahl T (2000) Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. U. S. A 97, 3832–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, and Robbins JH (2000) The oxidative DNA lesion 8,5′-(S)-cyclo-2’-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem 275, 22355–22362. [DOI] [PubMed] [Google Scholar]
- (57).Marietta C, and Brooks PJ (2007) Transcriptional bypass of bulky DNA lesions causes new mutant RNA transcripts in human cells. EMBO Rep. 8, 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Pednekar V, Weerasooriya S, Jasti VP, and Basu AK (2014) Mutagenicity and genotoxicity of (5′S)-8,5′-cyclo-2’-deoxy-adenosine in Escherichia coli and replication of (5′S)-8,5′-cyclopurine-2’-deoxynucleosides in vitro by DNA polymerase IV, exo-free Klenow fragment, and Dpo4. Chem. Res. Toxicol 27, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).You C, Dai X, Yuan B, Wang J, Wang J, Brooks PJ, Niedernhofer LJ, and Wang Y (2012) A quantitative assay for assessing the effects of DNA lesions on transcription. Nat. Chem. Biol 8, 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).You C, Swanson AL, Dai X, Yuan B, Wang J, and Wang Y (2013) J. Biol. Chem 288, 28548–28556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Roy LM, Jaruga P, Wood TG, McCullough AK, Dizdaroglu M, and Lloyd RS (2007) Human polymorphic variants of the NEIL1 DNA glycosylase. J. Biol. Chem 282, 15790–15798. [DOI] [PubMed] [Google Scholar]
- (62).Prakash A, Carroll BL, Sweasy JB, Wallace SS, and Doublie S (2014) Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: activity, structure, and the effect of editing. DNA Repair 14, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, determination of the optimum collision energies of AFB1-N7-Gua (A) and cis-AFB1-FapyGua and trans-AFB1-FapyGua (B) for two mass transitions of each compound. Figure S2, LC-MS/MS measurement of the standards. Ion-current profiles of the two mass transitions of each compound are shown. Figure S3, ion-current profiles of the mass transitions of R-cdA and S-cdA and their 15N5-labeled analogues recorded during the LC-MS/MS analysis of a liver DNA sample from an AFB1-treated mouse. Figure S4, levels of FapyGua (A) and 8-OH-Gua (B) in liver DNA of control mice (1–6) and AFB1-treated mice (7–12). Uncertainties are standard deviations (n = 3) (PDF)