

Abstract
Upadacitinib plasma concentrations, efficacy, and safety data from 216 subjects with moderate‐to‐severe active Crohn's disease (CD) from the 16‐week induction period of the CELEST study were analyzed to characterize upadacitinib exposure–response relationships in CD. Subjects in CELEST received either placebo or upadacitinib (3, 6, 12, 24 mg b.i.d. or 24 mg q.d.). Exposure–response models were developed and utilized to simulate efficacy of induction doses of the immediate‐release (IR) and extended‐release (ER) formulations. Upadacitinib exposures associated with 18–24 mg b.i.d. (IR formulation) or 45–60 mg q.d. (ER formulation) are estimated to have greater efficacy during 12‐week induction in patients with CD compared with lower doses. No exposure–response relations were observed with decreases in hemoglobin or lymphocytes at week 16 or with herpes zoster infections, pneumonia, or serious infections during 16 weeks of treatment in this study. These analyses informed the selection of upadacitinib induction dose for phase III studies in CD.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Upadacitinib is an oral Janus kinase 1 inhibitor that was evaluated in a phase II study in patients with moderately‐to‐severely active Crohn's disease (CD; CELEST study) using immediate‐release formulation.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The relationships between upadacitinib plasma exposures and efficacy as well as safety were characterized in patients with CD during the induction period of CELEST. The models were used to predict efficacy for upadacitinib extended‐release (ER) regimens.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Upadacitinib plasma exposures associated with doses of 45 to 60 mg q.d. of the ER formulation are predicted to have greater efficacy during the induction period in subjects with CD compared with lower doses. No trends for exposure–response relationships were observed for the different safety end points evaluated.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Using model‐based approaches, this work characterizes the exposure–response relationships for upadacitinib efficacy and safety, supports the benefit/risk assessment in patients with CD, and sheds light on some of the analyses that informed the phase III dose selection and trial designs.
Crohn's disease (CD) is a chronic, progressive, inflammatory disease of the gastrointestinal tract that manifests as a spectrum of clinical and pathological complications with negative impact on quality of life.1 Current treatment strategies are aimed at symptomatic improvement and endoscopic healing of the intestinal mucosa, the latter of which is associated with improved long‐term outcomes.2, 3 Although currently available treatments, including corticosteroids, immunosuppressants, and biologics, reduce inflammation and ameliorate symptoms, some patients either fail to respond or do not achieve a sustained response.4 Patients who do not respond to medical treatment may ultimately require surgery,5 which, like current medical therapies, is not curative, although encouragingly, the number of patients requiring surgery has begun to decline.6
The inflammatory processes that underlie CD are believed to result in part from an imbalance between pro‐inflammatory and anti‐inflammatory cytokines, several of which signal via Janus kinase (JAK) pathways in the mucosal immune system.7 The JAKs are a family of four intracellular tyrosine kinases (JAK1, JAK2, JAK3, and tyrosine kinase 2) that play central roles in innate and adaptive immunity.8 Inhibition specifically of JAK1 blocks the signaling of several pro‐inflammatory cytokines (e.g., interleukin (IL)‐2, IL‐6, IL‐7, and IL‐15, among others) that seem to play a role in the pathogenesis of CD.9, 10
Upadacitinib is an oral, selective, inhibitor of JAK1 that is being developed for the treatment of CD11, 12 and several other autoimmune inflammatory diseases.14, 15 Results from a recent phase II study (CELEST, NCT02365649) with upadacitinib immediate‐release (IR) formulation administered primarily b.i.d. demonstrated favorable clinical and endoscopic efficacy after 16‐week induction treatment19 and continued improvements after 36‐week maintenance treatment20 in patients with moderate‐to‐severe active CD who had an inadequate response or intolerance to immunomodulators or antitumor necrosis factor (TNF) therapy.
After oral administration of upadacitinib IR formulation, upadacitinib maximum plasma concentration levels are reached within 1–2 hours followed by decline in plasma concentrations in a bi‐exponential manner.21, 22 The functional half‐life of upadacitinib from the IR formulation is 3–4 hours, which necessitated evaluation of b.i.d. administered regimens in the phase II CELEST study. The 24 mg q.d. treatment arm, an exploratory dose added to evaluate the potential of q.d. administration with the IR formulation, provides similar upadacitinib average plasma concentration during a dosing interval (Cave), but lower minimum plasma concentration (Cmin) compared with the 12 mg b.i.d. dose. In order to enhance compliance and provide a more convenient q.d. dosing regimen for patients while keeping comparable Cmin values to the b.i.d. administration of the IR formulation, upadacitinib extended‐release (ER) tablet formulation was developed and is being used in the ongoing phase III studies in CD.11, 12, 13, 23 The analyses reported herein were conducted to characterize the exposure–response relationships for upadacitinib efficacy and safety in CD using data from the CELEST study induction period. The exposure–response models were used to guide phase III dose selection by estimating the efficacy of different regimens of the IR formulation used in the phase II CELEST study and the ER formulation prior to its use in phase III studies.
Results
Available pharmacokinetic, efficacy, and safety data from a total of 216 of the 220 subjects enrolled in the CELEST study were included in the exposure–response analyses who received placebo, 3, 6, 12, or 24 mg b.i.d. or 24 mg q.d. using the IR formulation. Four subjects (of the total 220 subjects enrolled in CELEST) were randomized to active treatment but did not have pharmacokinetic measurements due to early study discontinuation and, hence, were excluded from all analyses. At baseline, the mean (SD) age of subjects included in analyses was 40.8 years (12.9 years), Crohn's Disease Activity Index (CDAI) score was 300 (59.9), and the Simple Endoscopic Score for Crohn's Disease (SES‐CD) score was 14.9 (8.4). In the CELEST study, 96% of patients had an inadequate response or intolerance to ≥ 1 TNF antagonist. No difference was observed in baseline characteristics among placebo and the different upadacitinib dosing groups. Patient demographics, disease characteristics, and disposition in the CELEST study are shown in Table 1.
Table 1.
Demographics and baseline characteristics of subjects included in exposure–response analyses
| Characteristics |
Placebo (N = 37) |
3 mg b.i.d. (N = 38) |
6 mg b.i.d. (N = 36) |
12 mg b.i.d. (N = 36) |
24 mg b.i.d. (N = 35) |
24 mg q.d. (N = 34) |
All subjects (N = 216) |
|
|---|---|---|---|---|---|---|---|---|
| Sex, n (%) | Male | 13 (35%) | 20 (53%) | 16 (44%) | 19 (53%) | 10 (29%) | 15 (44%) | 93 (43%) |
| Female | 24 (65%) | 18 (47%) | 20 (56%) | 17 (47%) | 25 (71%) | 19 (56%) | 123 (57%) | |
| Race, n (%) | White | 33 (89%) | 31 (82%) | 31 (86%) | 31 (86%) | 30 (86%) | 29 (85%) | 185 (86%) |
| Black | 1 (3%) | 1 (3%) | 1 (3%) | 3 (8%) | 3 (9%) | 3 (9%) | 12 (6%) | |
| Hispanic | 3 (8%) | 6 (16%) | 2 (6%) | 2 (6%) | 2 (6%) | 15 (7%) | ||
| Asian | 2 (6%) | 1 (3%) | 3 (1%) | |||||
| Other | 1 (3%) | 1 (0%) | ||||||
| Age (years) | Mean (SD) | 40.5 (12.1) | 39.2 (13.9) | 40.8 (13.5) | 40.8 (15.2) | 43.1 (9.64) | 40.4 (12.7) | 40.8 (12.9) |
| Weight (kg) | Mean (SD) | 77.3 (21.5) | 71.5 (13.5) | 75.5 (19.5) | 76.7 (18.5) | 71.2 (19.6) | 82.4 (26.1) | 75.7 (20.1) |
| CDAI at baseline | Mean (SD) | 288 (60.0) | 296 (59.7) | 308 (55.1) | 305 (60.0) | 291 (68.8) | 313 (54.4) | 300 (59.9) |
| SES‐CD at baseline | Mean (SD) | 15.8 (8.6) | 14.6 (8.9) | 16.3 (9.0) | 15.6 (9.4) | 14.0 (7.1) | 13.2 (7.5) | 14.9 (8.4) |
CD, Crohn's disease; CDAI, Crohn's disease Activity Index; SES‐CD, Simple Endoscopic Score for Crohn's disease.
Upadacitinib pharmacokinetics in subjects with CD
A population pharmacokinetic analysis was conducted including subjects with CD, rheumatoid arthritis (RA), and healthy subjects, as an extension of the previously conducted analysis in healthy subjects and subjects with RA.22 Upadacitinib pharmacokinetics were adequately characterized using a two‐compartment model with a linear elimination process. Upadacitinib absorption was described by a first‐order absorption process and absorption lag time for the IR formulation. Overviews of studies and subject demographics included in the pharmacokinetic analyses are presented in Table S1 and Table S2 , respectively. Summary of the parameter estimates from the population pharmacokinetics model and the differences between this model and a previously published model in healthy subjects and subjects with RA22 are presented in Table S3 . Upadacitinib apparent oral clearance (CL/F) in subjects with CD was estimated to be comparable to that in healthy subjects and slightly higher compared with subjects with RA, resulting in ~ 20% lower upadacitinib Cave in subjects with CD than in subjects with RA for the same dose. Intersubject variability in upadacitinib CL/F and apparent volume of distribution of the central compartment (Vc/F) were estimated to be 41% and 29%, respectively, in subjects with CD. Other covariates already included in the model based on the previous analysis21, 24 were renal impairment and sex on CL/F, as well as bodyweight and sex on Vc/F. Women were estimated to have 15% lower CL/F and 25% lower Vc/F compared with men. A subject with creatinine clearance of 50 mL/minute was estimated to have 28% higher Cave compared with a subject with normal renal function (creatinine clearance of 110 mL/minute) and women were estimated to have 18% higher upadacitinib Cave compared with men. Given the modest effect of these covariates on upadacitinib plasma exposures (< 30% difference in upadacitinib Cave among different covariate groups), their effect on upadacitinib exposure is not considered clinically relevant, as described previously.22 Summary of upadacitinib model‐estimated plasma exposures for the IR formulation regimens evaluated in CELEST is presented in Table 2. Upadacitinib dose was not a covariate on any model parameter supporting dose proportionality of upadacitinib plasma exposures across the evaluated dose range.
Table 2.
Upadacitinib model predicted (median (5th to 95th percentiles)) plasma exposures during a dosing interval at steady‐state for IR regimens evaluated in CELEST (based on the empirical Bayesian individual estimates) and for ER regimens (simulated)
| Plasma concentration | Upadacitinib IR regimen | Upadacitinib ER regimen | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
3 mg b.i.d. IR |
6 mg b.i.d. IR |
12 mg b.i.d. IR |
24 mg b.i.d. IR |
24 mg q.d. IR |
7.5 mg q.d. ER |
15 mg q.d. ER |
30 mg q.d. ER |
45 mg q.d. ER |
60 mg q.d. ER |
|
| Cave, ng/mLa | 6.9 (4.6–14.1) | 14.9 (7.6–25.2) | 26.7 (8.5–49.6) | 55.1 (29.8–91.8) | 25.1 (13.6–92.0) | 6.4 (3.1–13.1) | 12.9 (6.3–26.1) | 25.8 (12.5–52.3) | 38.7 (18.8–78.4) | 51.6 (25.1–104.6) |
| Cmax, ng/mLb | 21.8 (13.1–29.3) | 40.2 (21.7–61.6) | 80.7 (25.3–124) | 176 (106–265) | 161 (88.7–255) | 17.3 (7.5–40.1) | 34.7 (14.9–80.3) | 69.3 (29.9–160.5) | 104.0 (44.8–240.8) | 138.6 (59.7–321.1) |
| Cmin, ng/mLb | 1.2 (0.4–6.6) | 2.6 (0.2–7.5) | 4.9 (0.2–25.2) | 7.8 (1.8–20.7) | 1.7 (0.3–34.2) | 1.4 (0.3–5.6) | 2.9 (0.6–11.2) | 5.7 (1.3–22.3) | 8.6 (1.9–33.5) | 11.4 (2.5–44.7) |
Cave, average plasma concentration; Cmax, maximum plasma concentration; Cmin, minimum plasma concentration; ER, extended‐release; IR, immediate‐release.
Steady‐state Cave was calculated using the formula Cave = Dose[µg]/(CL/F[L/hour]*dosing interval [hour]) using individually estimated clearance for IR regimens from patients with Crohn's disease in the CELEST study and simulated clearance in patients with Crohn's disease for ER regimens.
Cmin and Cmax were derived from individually estimated pharmacokinetic profiles for IR regimens in patients with Crohn's disease in the CELEST study and from steady‐state simulations for ER regimens, respectively.
At the time of conducting this work, in order to estimate the absorption parameters of the ER formulation to enable simulations for ER regimens using the exposure–response models, the final pharmacokinetic model for the IR formulation was fitted to data that became available from a study in healthy subjects who received the ER formulation23 to estimate the absorption and bioavailability parameters (relative to the IR formulation) for upadacitinib ER formulation. Summary of the model parameter estimates for the ER formulation are presented in Table S4 . The absorption of upadacitinib ER formulation was adequately described by mixed first‐order and zero‐order processes with lag time with upadacitinib bioavailability of 74% from the ER relative to the IR formulation (Figure S1 ). These results were in agreement with a later analysis conducted using a larger dataset for the ER formulation from upadacitinib RA phase III trials.24 Summary of upadacitinib model‐predicted plasma exposures for both formulations in patients with CD based on simulations are provided in Table 2.
Exposure–response relationships for upadacitinib efficacy in CELEST study
The clinical and endoscopic efficacy end points included in exposure–response analyses were clinical response (≥ 30% reduction from baseline in very soft/liquid stool frequency (SF) and/or abdominal pain (AP) score, neither worse than baseline), clinical remission 2.8 of 1.0 (very soft/liquid SF ≤ 2.8 and AP score ≤ 1.0, neither worse than baseline, among patients with baseline very soft/liquid SF ≥ 4.0 or AP score ≥ 2.0), CDAI < 150, endoscopic response 25% (≥ 25% decrease in SES‐CD from baseline), endoscopic response 50% (> 50% decrease in SES‐CD from baseline or endoscopic remission, defined as SES‐CD ≤ 4 point and ≥ 2 point reduction from baseline with no subscore > 1), and endoscopic remission.
The percentage of subjects achieving clinical response, clinical remission 2.8/1.0, and CDAI < 150 at week 16 increased with increasing upadacitinib plasma concentrations (Figure 1). Similarly, the percentage of patients achieving endoscopic end points at week 12 of 16 also increased with increasing upadacitinib plasma concentrations (Figure 2). Efficacy for clinical and endoscopic end points was trending toward the plateau, but the plateau of response was not clearly established within the range of upadacitinib plasma exposures evaluated in the CELEST study, particularly for the endoscopic end points. Summary of the parameter estimates from the Markov models for clinical end points and the regression models for the endoscopic end points are provided in Tables S5 and S6 , respectively.
Figure 1.
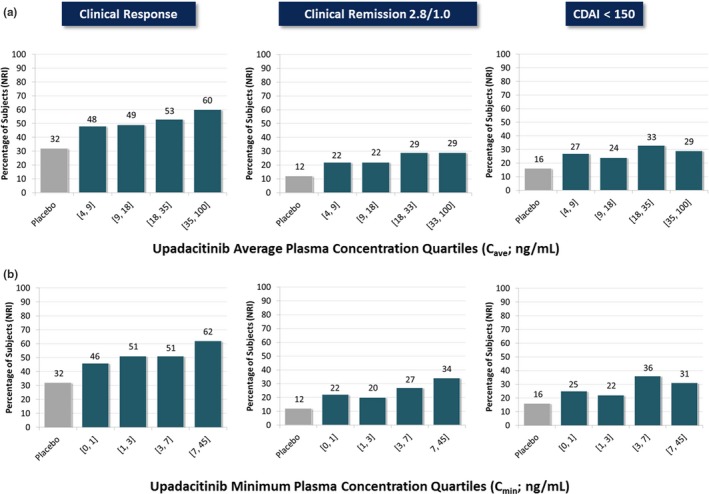
Observed relationships between upadacitinib (a) average plasma concentration during a dosing interval (Cave) or (b) minimum plasma concentration (Cmin) in the phase II CELEST study and the percentages of subjects achieving clinical end points at week 16. CDAI, Clinical Disease Activity Index; NRI, nonresponder imputation. [Colour figure can be viewed at https://www.wileyonlinelibrary.com]
Figure 2.

Observed (symbols) and model‐predicted percentage (solid line and shaded areas) of subjects who achieved endoscopic end points at week 12 or 16 vs. upadacitinib average plasma concentration during a dosing interval (Cave). Symbols and error bars represent the observed NRI response by quartile of upadacitinib Cave; solid line and shaded areas represent the model‐predictions; horizontal lines represent the spread of exposure for different doses. NRI, nonresponder imputation. [Colour figure can be viewed at https://www.wileyonlinelibrary.com]
The relationships between upadacitinib plasma concentration and clinical efficacy end points were characterized using a continuous‐time Markov modeling approach,25 similar to the approach used previously to analyze the efficacy for upadacitinib in patients with RA.26 The relationships between upadacitinib plasma exposures and endoscopic end points (endoscopic response 25%, endoscopic response 50%, and endoscopic remission at week 12 of 16) were characterized using regression analyses.
Upadacitinib exposure–response relationships were described by maximum effect (Emax) models with clinical response and endoscopic end points using time‐varying upadacitinib plasma concentrations and average upadacitinib plasma concentrations over the dosing interval, respectively, as the exposure metrics that correlated with response, whereas the CDAI < 150 and clinical remission using upadacitinib minimum plasma concentration as the exposure metric correlated with response. For endoscopic end points, upadacitinib half‐maximal effective concentration (EC50) values were 40 ng/mL for endoscopic response 25%, 26 ng/mL for endoscopic response 50%, and 27 ng/mL for endoscopic remission. It is noteworthy that the EC50 values for the clinical end points are not directly interpretable because they are for transition rates between states in the Markov models. The model‐estimated vs. observed responses are presented in Figure 2 for the endoscopic efficacy end point, in Figure 3 for clinical response, and in Figure S2 for CDAI < 150 and clinical remission 2.8/1.0.
Figure 3.
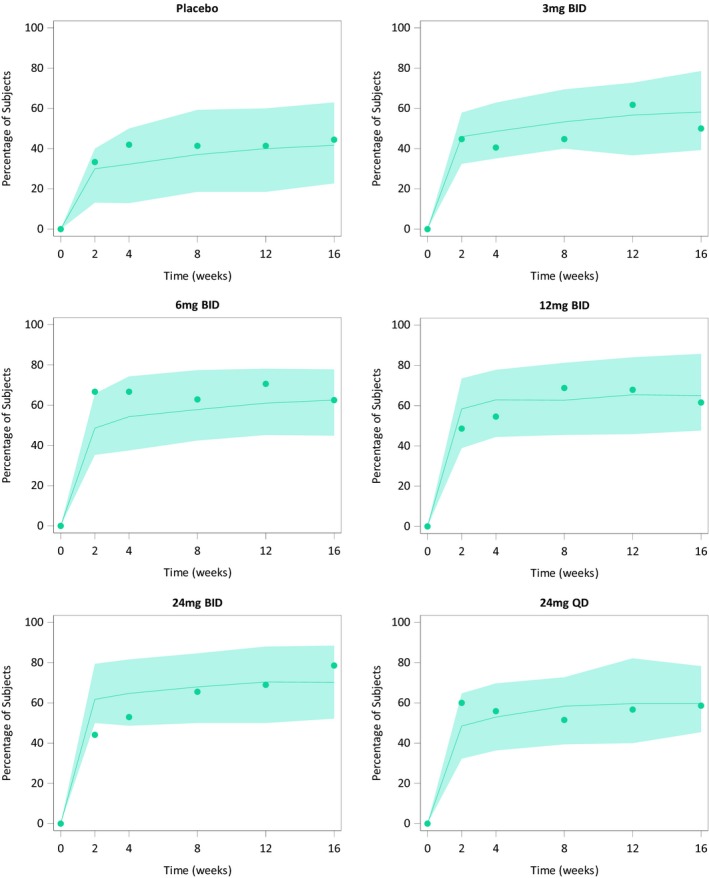
Observed and model‐predicted clinical response vs. time stratified by upadacitinib immediate‐release dose evaluated in the CELEST study. Symbols represent the observed time course of the percentage of patients achieving each of the clinical end points. Solid lines and shaded areas represent the exposure–response model‐predicted median and 90% prediction intervals, respectively. [Colour figure can be viewed at https://www.wileyonlinelibrary.com]
Efficacy simulations for upadacitinib IR and ER formulation regimens
The estimated clinical and endoscopic efficacy for upadacitinib dosing regimens using the IR and ER formulations based on simulations from the exposure–response models are shown in Figure 4. The simulated efficacy responses for IR dosing regimens compared with the responses observed in the CELEST study are show in Supplemental Figure S4 . Upadacitinib doses of 7.5, 15, 30, 45, and 60 mg q.d. regimens using the ER formulation are predicted to provide similar efficacy to 3, 6, 12, 18, and 24 mg b.i.d. regimens, respectively, using the IR formulation. Overall, upadacitinib doses of 45–60 mg q.d. were estimated to provide greater efficacy for the clinical end points at week 12 and endoscopic end points at weeks 12 and 16 compared with lower doses.
Figure 4.

Predicted percentage of subjects achieving the different clinical and endoscopic response/remission end points during the induction period for different upadacitinib immediate‐release b.i.d. and extended‐release q.d. regimens based on exposure–response analyses of the phase II CELEST study. Data are presented as medians and 5th and 95th percentiles of predictions from 100 replicates. Simulations represent 220 subjects for each dose group. [Colour figure can be viewed at https://www.wileyonlinelibrary.com]
Exposure–response relationships for safety parameters with upadacitinib in the CELEST study
The relationships between upadacitinib average plasma concentrations over a dosing interval and selected safety parameters, which are considered of clinical relevance for JAK inhibitors were evaluated (Figure 5). There was no observed trend for a relationship between upadacitinib plasma exposures and decreases in hemoglobin (≥ 2 g/dL) at week 16 from baseline, lymphopenia (grade 1, 2, or 3) at week 16, or with herpes zoster infection, pneumonia, or serious infections during 16 weeks of treatment in this phase II trial in CD.
Figure 5.
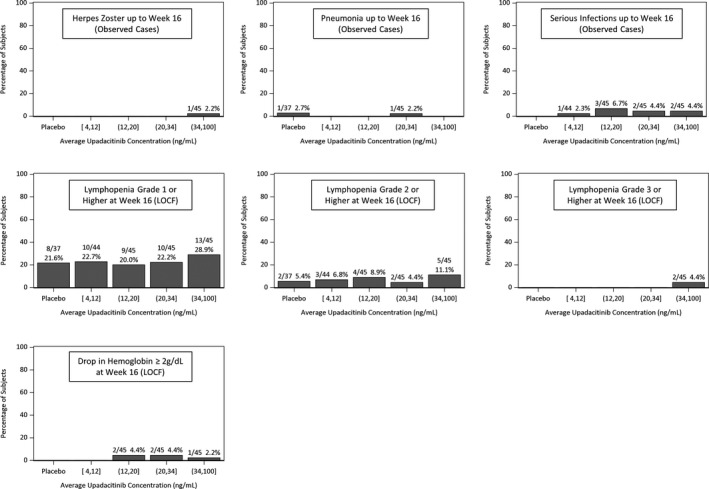
Observed relationships between upadacitinib average plasma concentration during a dosing interval (Cave) and the percentages of subjects experiencing clinically relevant safety events or changes in laboratory parameters at week 16 in the phase II CELEST study.
Discussion
This work characterized the relationships between the upadacitinib plasma exposures in patients with moderate‐to‐severe active CD who were evaluated in the CELEST study induction period and its efficacy as assessed by clinical and endoscopic end points.19 Additionally, the relationships among upadacitinib plasma exposures and clinically relevant safety events and changes in laboratory parameters were evaluated. The characterized efficacy relationships were used to select the optimal exposures for evaluation in phase III trials and to predict the efficacy of q.d. regimens using the planned phase III ER formulation. Results from these exposure–response analyses demonstrated that the percentage of subjects achieving the different clinical and endoscopic end points increased with increasing upadacitinib plasma exposures. Upadacitinib exposures associated with 18 mg to 24 mg b.i.d. using the IR formulation (or 45–60 mg q.d. doses using the ER formulation), as induction treatments were estimated to have greater efficacy in patients with CD compared with lower doses. No trend for exposure–response relationships was observed for the evaluated clinically relevant safety events in this study. Therefore, the optimal upadacitinib exposures in patients with CD were selected to maximize efficacy within the range of exposures evaluated in a phase II study. These analyses, together with the favorable efficacy and acceptable safety profile observed in CELEST19 guided the dose selection and enabled advancement of upadacitinib ER formulation into phase III and obviated the need for separate phase II assessment for upadacitinib ER formulation q.d. regimens in CD.
Population pharmacokinetic analyses showed that upadacitinib exposure was approximately dose‐proportional over the dose range of 3–24 mg b.i.d., which is in agreement with prior analyses in healthy subjects and in subjects with RA.21, 22 Upadacitinib oral clearance in subjects with CD was not statistically significantly different from that observed in healthy subjects.21, 22 It has been previously reported that oral drug absorption can be different in patients with inflammatory bowel diseases compared with other patient populations.27, 28 The consistency of upadacitinib pharmacokinetics between subjects with CD and healthy subjects was reassuring and indicated that CD had no significant impact on upadacitinib absorption.
As pointed out earlier, the analysis did not detect a statistically significant difference in upadacitinib clearance between patients with CD and healthy subjects, whereas subjects with RA had 22% lower clearance compared with non‐RA subjects. Elevated levels of IL‐6, a pro‐inflammatory cytokine that has been reported to suppress CYP3A metabolic activity, is one of the routes contributing to upadacitinib clearance.30, 31 IL‐6 is known to also be elevated in inflammatory diseases, such as RA and CD, the comparative magnitude of this elevation is uncertain.32 Upadacitinib pharmacokinetics in patients with RA were shown to be time‐independent,33 indicating that normalizing levels of IL‐6 with treatment does not impact upadacitinib clearance. The exact mechanism for the ~ 20% lower clearance in patients with RA compared with non‐RA patients is not clear, but it is noteworthy that a similar or larger difference in clearance between patients with RA and healthy subjects has also been observed for other JAK inhibitors (e.g., tofacitinib, baricitinib, and filgotinib), despite having different elimination pathways from upadacitinib.34, 35 As we described previously,22, 33 it is plausible that the lower clearance observed in patients with RA is due to a combination of factors (e.g., older age and lower metabolic capacity) presented collectively in the RA population. As for subjects with CD, additional analyses with a larger number of subjects from phase III trials and analyses from other autoimmune disorders can provide additional insights. Overall, the differences in clearance discussed above are relatively small and are not deemed to be clinically relevant.
The ER formulation of upadacitinib was developed to enable q.d. dosing of upadacitinib while having comparable Cave, maximum plasma concentration (Cmax), and Cmin to the selected b.i.d. IR regimens. At the time of conducting the analyses, the only data that became available for the pharmacokinetics of the ER formulation in humans were from a phase I study in healthy subjects. This phase I study demonstrated that upadacitinib ER regimens of 15 mg and 30 mg q.d. provided equivalent Cave and similar Cmax and Cmin to 6 mg b.i.d. and 12 mg b.i.d., respectively, using the IR formulation.23 Data from this study were used to estimate the absorption parameters of the ER formulation to enable simulation of upadacitinib pharmacokinetic profiles for ER q.d. regimens in patients with CD. The present analysis estimated the bioavailability of upadacitinib ER formulation to be 74% relative to the IR formulation. This estimate is in close agreement with the relative bioavailability estimate (76%) based on a more extensive dataset for the ER formulation in subjects with RA, which became available after this analysis was conducted.33 The lower bioavailability of upadacitinib from the ER compared to the IR formulations is potentially due to incomplete release from the ER formulation in vivo. It was noted that administration of upadacitinib after a high‐fat meal, which increases the transit time within the gastrointestinal tract, increases its bioavailability from the ER formulation by ~ 20%.23 Upadacitinib plasma exposures were dose‐proportional over the range of 3–48 mg using the IR formulation or 7.5–45 mg using the ER formulation21,23 (data on file at AbbVie).
The Markov modeling approach of categorical end points captures the full‐time course of interrelated events providing a robust characterization of the exposure–response relationship.37 Such integration of the full time‐course is especially important with categorical data and the relatively small sample sizes typical of phase II trials. Additionally, dropouts can be simply incorporated in the model as one of the Markov states, consequently enabling use of the model to simulate nonresponder imputation responses. For the endoscopic end points, regression, rather than Markov, analyses were conducted given that endoscopic end points were only assessed at one time point after baseline for each subject (at week 12 or 16). Other analysis methodologies (such as repeated‐measures logistic regression models for the clinical end points and evaluation of joint modeling of the three endoscopic end points) can be alternatives for evaluation in similar analyses.38, 39 No covariates were evaluated for their effect on upadacitinib exposure–response relationships in this study; this assessment is warranted using data from phase III trials, which will include a larger sample size across multiple studies. Graphical evaluation of the observed and model‐predicted end point outcomes demonstrate that the models adequately describe the observed clinical and endoscopic responses in the CELEST study and the variability over time for the clinical end points (Figure 2 and 3 and Figure S1 ). It is worth noting that, given that the efficacy plateau for clinical and endoscopic end points was not clearly established within the range of upadacitinib plasma concentrations evaluated in the study, the interpretability of the model‐estimated EC50 values is limited and the developed models are not to be used for extrapolation of responses at exposures that exceed the exposure range (for 24 mg IR b.i.d. or 60 mg ER q.d.) used to develop these models.
In the CELEST study,19 24 mg q.d. using the IR formulation resulted in lower numerical efficacy for clinical remission 2.8/1.0 and CDAI < 150 compared with 12 mg b.i.d. (no formal statistical comparison between the two doses), despite delivering the same Cave. Upadacitinib 24 mg q.d. is predicted to result in clinical remission 2.8/1.0 and CDAI < 150 responses that are more similar to ~ 6 mg b.i.d. than the 12 mg b.i.d. dose using the IR formulation. This can be attributed to the lower Cmin with 24 mg q.d. regimen (2.0 ng/mL) compared with 12 mg b.i.d. regimen (4.8 ng/mL) using the IR formulation. In the exposure–response models for clinical remission 2.8/1.0 and for CDAI < 150, upadacitinib Cmin was a better plasma exposure measure to correlate with efficacy compared with upadacitinib estimated plasma concentration vs. time (Cp). This suggests that achieving clinical remission may require maintaining adequate plasma levels of upadacitinib throughout the day. The ER formulation enables q.d. dosing of upadacitinib while maintaining adequate plasma levels throughout the day.24, 40
In subjects with RA, upadacitinib doses of 15–30 mg q.d. achieved the plateau for efficacy with very limited benefit for the 30 mg over 15 mg q.d. regimen.15, 16, 18, 26 However, in subjects with CD, upadacitinib doses higher than 30 mg q.d. using the ER formulation (or 12 mg b.i.d. using the IR formulation) are predicted to provide incremental efficacy benefit during the induction period, particularly for endoscopic end points. Based on the exposure–response analyses, upadacitinib doses of 45–60 mg q.d. using the ER formulation are predicted to result in 5–11% higher percentage of subjects achieving endoscopic response 25% and endoscopic response 50% compared with 30 mg q.d. The clinical benefit of doses higher than the optimal RA doses (15–30 mg q.d.) was also demonstrated in subjects with moderate‐to‐severe ulcerative colitis.17 This difference between the optimal dose needed for the different immune diseases can possibly be driven by the inflammatory burden and different primary sites of inflammation among diseases as well as the cytokines involved; IL‐17, IL‐23, and IL‐32 are upregulated in CD, whereas in RA, TNF‐α, IL‐1, IL‐6, IL‐12, and IL‐17 seem to be key cytokines involved in the inflammatory process.41, 42 There were no meaningful differences in the predicted clinical outcomes between weeks 12 and 16, indicating that a treatment duration of 12 weeks with upadacitinib would be sufficient to assess induction of remission in CD. Of note, another JAK inhibitor (tofacitinib, targeting JAK3 > JAK1 > JAK2) did not demonstrate efficacy in two phase II studies in patients with CD, although it is efficacious in the treatment of RA; however, relatively high placebo responses in these studies may have contributed to the lack of difference between treatment arms and placebo.43, 44 Filgotinib, another JAK inhibitor more selective than tofacitinib for JAK1 has shown efficacy in CD.45 Therefore, it is possible that given the differential involvement of various cytokines in the pathophysiology of different autoimmune disease, some JAK inhibitors with different selectivity profiles may be efficacious in one disease but not in the other.41, 42, 46 Moreover, efficacious exposures of the same drug can vary among autoimmune diseases, as observed with upadacitinib in the CELEST study as well as with biologic therapies.47
In conclusion, the percentage of subjects achieving different clinical and endoscopic end points in patients with CD evaluated during the CELEST phase II trial induction period increased with increasing upadacitinib plasma exposures. Based on the exposure–response analyses, plasma exposures associated with doses of 45–60 mg q.d. of the ER formulation are estimated to have greater efficacy of upadacitinib in patients with moderate‐to‐severe active CD compared with lower doses. An induction trial duration of 12 weeks was estimated to be adequate for assessing the clinical and endoscopic efficacy of upadacitinib in the short‐term. This work informed the phase III dose selection for upadacitinib in CD and represents a case study for the use of model‐informed drug development in this disease.
Methods
Study design and participants
The study was conducted in accordance with Good Clinical Practice Guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocol was approved by the institutional review board or ethics committee for each site and each patient provided written informed consent before any study‐related procedures were performed.
The CELEST study was a phase II, dose‐ranging, multicenter, randomized, double‐blind, placebo‐controlled study in patients with moderate‐to‐severe active CD.19 Patients eligible for the study included those with a CDAI score ≥ 220 and ≤ 450, an average daily very soft/liquid SF ≥ 2.5 or average daily AP score of ≥ 2.0, a SES‐CD ≥ 6 (or ≥ 4 for subjects with disease limited to the ileum), and a history of inadequate response or intolerance to an immunomodulator or anti‐TNF therapy. Subjects with prior exposure to JAK inhibitors were excluded. Patients were randomized in equal proportions to double‐blind induction oral treatment with IR upadacitinib 3, 6, 12, or 24 mg b.i.d. or 24 mg q.d., or placebo for 16 weeks, followed by re‐randomization into a double‐blind extension phase for 36 weeks. Data from the induction period were included in the exposure–response analyses. Starting at week 2, patients receiving oral corticosteroids initiated a mandatory corticosteroid taper following a protocol‐specified schedule until discontinuation.
Pharmacokinetic, clinical, and endoscopic assessments
Sparse blood samples for determination of upadacitinib plasma concentrations were collected during the study visits at weeks 2, 4, 8, 12, and 16. The date and time of blood collection relative to the last dose of upadacitinib that was taken before sampling were recorded and used in the analyses. Plasma concentrations of upadacitinib were measured using a validated liquid chromatography method with mass spectrometric detection, as previously described.21
Patients’ symptoms were assessed for clinical efficacy assessments at baseline and weeks 2, 4, 8, 12, and 16. Inflammation of the intestinal mucosa was assessed by ileocolonoscopies performed during screening and randomly assigned at either week 12 or 16 and the SES‐CD determined based on central reading. The SES‐CD values at weeks 12 and 16 were combined for the endoscopic end points.
Pharmacokinetic and exposure–response analyses
Population pharmacokinetic analyses
A population pharmacokinetic model for upadacitinib was previously developed for healthy subjects and patients with RA22 and was used as a starting model in this analysis. Details of the population pharmacokinetic analyses are provided in the Supplemental Methods.
Exposure–response analyses of efficacy
The relationships between upadacitinib plasma concentration and clinical end points were characterized using a continuous‐time Markov modeling approach.25 A continuous time Markov model approach was used to describe the relationship between upadacitinib plasma exposures on achievement of different responses in the CELEST study. The schematic of the model is shown in Figure S3 . The transition states of the Markov chain were defined as: no response = state 0, response = state 1 (whether clinical response, CDAI < 150, or clinical remission 2.8/1.0), and dropout = state D. Separate models were developed for exposure–response analyses of clinical response, CDAI < 150, and clinical remission 2.8/1.0. Further details of the Markov analyses methodology are provided in Supplemental Methods.
The relationships between upadacitinib plasma exposures and endoscopic end points (endoscopic response 25%, endoscopic response 50%, and endoscopic remission at week 12/16) were characterized using regression analyses with SAS version 9.4 (SAS Institute, Cary, NC).
The exposure–response models were used to simulate the efficacy of b.i.d. regimens using the IR formulation and q.d. regimens using the upadacitinib ER formulation in a sample size of 220 subjects with CD (using subject baseline characteristics of CELEST study subjects) for each regimen, which is representative of a phase III‐like sample size. To enable simulation of efficacy for ER formulation regimens, the parameters characterizing the absorption of upadacitinib from the ER formulation were estimated using data from a phase I study of the ER formulation in healthy volunteers40 and incorporated into the exposure–response models to predict efficacy of upadacitinib ER formulation regimens.
Exposure–response analyses of safety
Exposure–response relationships were evaluated for the following clinically relevant safety end points: decreases in hemoglobin by ≥ 2 g/dL at week 16 (last observation carried forward) from baseline, lymphopenia grade 1 (less than the lower limit of normal to 0.8 × 109/L) at week 16, lymphopenia grade 2 (< 0.8 to 0.5 × 109/L) at week 16, lymphopenia grade 3 (< 0.5 to 0.2 × 109/L) at week 16, herpes zoster infections during the during 16‐week induction period, pneumonia during the 16‐week induction period, or serious infections during the 16‐week induction period. The relationship between upadacitinib average plasma exposure during a dosing interval and the percentage of subjects who experienced each of these end points were assessed through plotting the events observed within each quartile of plasma exposures. If a trend was observed for an exposure–response relationship with one of the safety events, further assessment was to be conducted through regression analyses to further characterize the relationship and use in simulations.
Funding
This study was funded by AbbVie. AbbVie contributed to the study design, research, and interpretation of the data and the writing, review, and approval of the manuscript.
Conflict of Interest
Mohamed‐Eslam F. Mohamed, Ben Klünder, Ana P. Lacerda, and Ahmed A. Othman are employees and shareholders of AbbVie.
Author Contributions
M.‐E.F.M., B.K., A.P.L., and A.A.O. wrote the manuscript. M.‐E.F.M. and A.A.O. designed the research. M.‐E.F.M., B.K., A.P.L., and A.A.O. performed the research. M.‐E.F.M., B.K., and A.A.O. analyzed the data.
Data Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan and execution of a Data Sharing Agreement. Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Supporting information
Figure S1. Visual predictive check showing simulated and observed upadacitinib exposures vs. time since last dose from the study in healthy subjects receiving the ER formulation stratified by formulation.
Figure S2. Observed and model‐predicted a) CDAI < 150 and b) clinical remission 2.8/1.0 vs. time stratified by upadacitinib immediate‐release dose evaluated in CELEST.
Figure S3. Illustration of the continuous‐time Markov model used for exposure–response analyses of clinical efficacy in the CELEST study.
Figure S4. Simulated percentage of subjects achieving the different clinical end points at week 16 and endoscopic end points at week 12/16 during the induction period for different upadacitinib immediate‐release regimens overlaid by the observed responses in CELEST study.
Table S1. Summary of studies used for in the population pharmacokinetics analyses.
Table S2. Demographics and baseline characteristics of subjects included in population pharmacokinetic analyses.
Table S3. Population pharmacokinetics parameter estimates and variability for upadacitinib IR formulation from healthy subjects, rheumatoid arthritis patients, and Crohn's disease patients*.
Table S4. Parameter estimates for upadacitinib pharmacokinetic model applied to data from a study in healthy subjects using the extended‐release formulation.
Table S5. Summary of the Markov model parameter estimates for clinical response, clinical remission 2.8/1.0, and CDAI < 150 in subjects with Crohn's disease.
Table S6. Parameter estimates (95% CI) of nonlinear regression of models for the relationships between upadacitinib average exposure and endoscopic efficacy endpoints at weeks 12/16.
Table S7. Final NONMEM model control streams.
Acknowledgments
The authors thank AbbVie employees, Allison Kitten and Wesley Wayman, for medical writing support.
References
- 1. Eustace, G.J. & Melmed, G.Y. Therapy for Crohn's disease: a review of recent developments. Curr. Gastroenterol. Rep. 20, 19 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Cholapranee, A. , Hazlewood, G.S. , Kaplan, G.G. , Peyrin‐Biroulet, L. & Ananthakrishnan, A.N. Systematic review with meta‐analysis: comparative efficacy of biologics for induction and maintenance of mucosal healing in Crohn's disease and ulcerative colitis controlled trials. Aliment. Pharmacol. Ther. 45, 1291–1302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma, C. et al Heterogeneity in definitions of efficacy and safety endpoints for clinical trials of Crohn's disease: a systematic review for development of a core outcome set. Clin. Gastroenterol. Hepatol. 16, 1407–1419 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Danese, S. , Fiorino, G. & Peyrin‐Biroulet, L. Early intervention in Crohn's disease: towards disease modification trials. Gut 66, 2179–2187 (2017). [DOI] [PubMed] [Google Scholar]
- 5. Bouguen, G. & Peyrin‐Biroulet, L. Surgery for adult Crohn's disease: what is the actual risk? Gut 60, 1178–1181 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Frolkis, A.D. et al Risk of surgery for inflammatory bowel diseases has decreased over time: a systematic review and meta‐analysis of population‐based studies. Gastroenterology 145, 996–1006 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Sanchez‐Munoz, F. , Dominguez‐Lopez, A. & Yamamoto‐Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 14, 4280–4288 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghoreschi, K. , Laurence, A. & O'Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 228, 273–287 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Vries, L.C.S. , Wildenberg, M.E. , De Jonge, W.J. & D'Haens, G.R. The future of Janus kinase inhibitors in inflammatory bowel disease. J. Crohns Colitis 11, 885–893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olivera, P. , Danese, S. & Peyrin‐Biroulet, L. JAK inhibition in inflammatory bowel disease. Expert Rev. Clin. Immunol. 13, 693–703 (2017). [DOI] [PubMed] [Google Scholar]
- 11. AbbVie . A study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to biologic therapy <https://clinicaltrials.gov/ct2/show/NCT03345836?term=NCT03345836&draw=2&rank=1>. Accessed April 20, 2018.
- 12. AbbVie . A study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to conventional therapies but have not failed biologic therapy <https://ClinicalTrials.gov identifier NCT03345849>. Accessed April 20, 2018.
- 13. AbbVie . A maintenance and long‐term extension study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with Crohn's disease who completed the studies M14–431 or M14–433 <https://clinicaltrials.gov/ct2/show/NCT03345823?term=NCT03345823&draw=2&rank=1>. Accessed March 26, 2019.
- 14. Guttman‐Yassky, E. et al Primary results from a phase 2b, randomized, placebo‐controlled trial of upadacitinib for patients with atopic dermatitis [Abstract #6533]. American Academy of Dermatology Annual Meeting, San Diego, CA, USA. February 16–20, 2018. [Google Scholar]
- 15. Burmester, G.R. et al Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease‐modifying anti‐rheumatic drugs (SELECT‐NEXT): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 391, 2503–2512 (2018). [DOI] [PubMed] [Google Scholar]
- 16. Genovese, M.C. et al Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease‐modifying anti‐rheumatic drugs (SELECT‐BEYOND): a double‐blind, randomised controlled phase 3 trial. Lancet 391, 2513–2524 (2018). [DOI] [PubMed] [Google Scholar]
- 17. Sandborn, W. et al Efficacy and safety of upadacitinib as an induction therapy for patients with moderately‐to‐severely active ulcerative colitis: data from the phase 2b study u‐achieve" to be presented as an oral presentation (OP 195) at UEGW 2018.
- 18. Smolen, J. et al Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT‐MONOTHERAPY): a randomised, placebo‐controlled, double‐blind phase 3 study. Lancet 393, 2303–2311 (2019). [DOI] [PubMed] [Google Scholar]
- 19. Sandborn, W.J. Safety and efficacy of ABT‐494 (Upadacitinib), an oral Jak1 inhibitor, as induction therapy in patients with Crohn's disease: results from CELEST. Gastroenterology 152, S1308–S1309 (2017). [Google Scholar]
- 20. Panes, J. et al Efficacy and safety of upadacitinib maintenance treatment for moderate to severe Crohn's disease: results from the CELEST study. Gastroenterology 154, S178–S179 (2018). [Google Scholar]
- 21. Mohamed, M.F. , Camp, H.S. , Jiang, P. , Padley, R.J. , Asatryan, A. & Othman, A.A. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin. Pharmacokinet. 55, 1547–1558 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Klunder, B. , Mohamed, M.F. & Othman, A.A. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I and II clinical trials. Clin. Pharmacokinet. 57, 977–988 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mohamed, M.F. , Zeng, J. , Marroum, P.J. , Song, I.H. & Othman, A.A. Pharmacokinetics of upadacitinib with the clinical regimens of the extended‐release formulation utilized in rheumatoid arthritis phase 3 trials. Clin. Pharmacol. Drug Dev. 8, 208–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klünder, B. , Mittapalli, R.K. , Mohamed, M.F. , Friedel, A. , Noertersheuser, P. & Othman, A.A. Population pharmacokinetics of upadacitinib using the immediate‐release and extended‐release formulations in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I‐III clinical trials. Clin. Pharmacokinet. 58, 1045–1058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lacroix, B.D. , Karlsson, M.O. & Friberg, L.E. Simultaneous exposure‐response modeling of ACR20, ACR50, and ACR70 improvement scores in rheumatoid arthritis patients treated with certolizumab pegol. CPT Pharmacometrics Syst. Pharmacol. 3, e143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mohamed, M.F. , Klunder, B. , Camp, H. & Othman, A.A. Exposure‐response analyses of upadacitinib efficacy in phase 2 trials in rheumatoid arthritis and basis for phase 3 dose selection. Clin. Pharmacol. Ther. 10.1002/cpt.1671. [e-pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fallingborg, J. , Pedersen, P. & Jacobsen, B.A. Small intestinal transit time and intraluminal pH in ileocecal resected patients with Crohn's disease. Dig. Dis. Sci. 43, 702–705 (1998). [DOI] [PubMed] [Google Scholar]
- 28. Fischer, M. , Siva, S. , Wo, J.M. & Fadda, H.M. Assessment of small intestinal transit times in ulcerative colitis and Crohn's disease patients with different disease activity using video capsule endoscopy. AAPS PharmSciTech 18, 404–409 (2017). [DOI] [PubMed] [Google Scholar]
- 29. Castiglione, F. et al Orocecal transit time and bacterial overgrowth in patients with Crohn's disease. J. Clin. Gastroenterol. 31, 63–66 (2000). [DOI] [PubMed] [Google Scholar]
- 30. Dickmann, L.J. , Patel, S.K. , Rock, D.A. , Wienkers, L.C. & Slatter, J.G. Effects of interleukin‐6 (IL‐6) and an anti‐IL‐6 monoclonal antibody on drug‐metabolizing enzymes in human hepatocyte culture. Drug Metab. Dispos. 39, 1415–1422 (2011). [DOI] [PubMed] [Google Scholar]
- 31. Xu, Y. , Hijazi, Y. , Wolf, A. , Wu, B. , Sun, Y.N. & Zhu, M. Physiologically based pharmacokinetic model to assess the influence of blinatumomab‐mediated cytokine elevations on cytochrome P450 enzyme activity. CPT Pharmacometrics Syst. Pharmacol. 4, 507–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nancey, S. , Hamzaoui, N. , Moussata, D. , Graber, I. , Bienvenu, J. & Flourie, B. Serum interleukin‐6, soluble interleukin‐6 receptor and Crohn's disease activity. Dig. Dis. Sci. 53, 242–247 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Klünder, B. , Mittapalli, R.K. , Mohamed, M.F. , Friedel, A. , Noertersheuser, P. & Othman, A.A. Population pharmacokinetics of upadacitinib using the immediate‐release and extended‐release formulations in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I‐III clinical trials. Clin. Pharmacokinet. 58, 1045–1058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. US Food and Drug Administration (FDA) .Center for Drug Evaluation and Research . Clinical pharmacology and biopharmaceutics review(s): tofacitinib. Application number 203214Orig1s000. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203214Orig1s000ClinPharmR.pdf> (2011).
- 35. US Food and Drug Administration (FDA) . Clinical pharmacology and biopharmaceutics review(s)—baricitinib. (Center for Drug Evaluation and Research, Silver Spring, MD, 2018). [Google Scholar]
- 36. Namour, F. et al Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of Filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin. Pharmacokinet. 54, 859–874 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diggle, P. et al. Analysis of Longitudinal Data (OUP, Oxford, 2002). [Google Scholar]
- 38. Agresti, A. Modelling ordered categorical data: recent advances and future challenges. Stat. Med. 18, 2191–2207 (1999). [DOI] [PubMed] [Google Scholar]
- 39. Hu, F.B. , Goldberg, J. , Hedeker, D. , Flay, B.R. & Pentz, M.A. Comparison of population‐averaged and subject‐specific approaches for analyzing repeated binary outcomes. Am. J. Epidemiol. 147, 694–703 (1998). [DOI] [PubMed] [Google Scholar]
- 40. Mohamed, M.‐F. , Zeng, T. , Marroum, P.J. , Song, I. & Othman, A.A. Pharmacokinetics of upadacitinib with the clinical regimens of the extended‐release formulation utilized in rheumatoid arthritis phase 3 trials. Clin. Pharmacol. Drug Develop. 8, 208–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nemeth, Z.H. , Bogdanovski, D.A. , Barratt‐Stopper, P. , Paglinco, S.R. , Antonioli, L. & Rolandelli, R.H. Crohn's disease and ulcerative colitis show unique cytokine profiles. Cureus 9, e1177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alunno, A. , Carubbi, F. , Giacomelli, R. & Gerli, R. Cytokines in the pathogenesis of rheumatoid arthritis: new players and therapeutic targets. BMC Rheumatol. 1, 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sandborn, W.J. et al A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn's disease. Clin. Gastroenterol. Hepatol. 12, 1485–1493.e2 (2014). [DOI] [PubMed] [Google Scholar]
- 44. Panes, J. et al Tofacitinib for induction and maintenance therapy of Crohn's disease: results of two phase IIb randomised placebo‐controlled trials. Gut 66, 1049–1059 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vermeire, S. et al Clinical remission in patients with moderate‐to‐severe Crohn's disease treated with filgotinib (the FITZROY study): results from a phase 2, double‐blind, randomised, placebo‐controlled trial. Lancet 389, 266–275 (2017). [DOI] [PubMed] [Google Scholar]
- 46. Murray, P.J. The JAK‐STAT signaling pathway: input and output integration. J. Immunol. 178, 2623–2629 (2007). [DOI] [PubMed] [Google Scholar]
- 47. Reimold, A.M. The role of adalimumab in rheumatic and autoimmune disorders: comparison with other biologic agents. Open Access Rheumatol. 4, 33–47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Visual predictive check showing simulated and observed upadacitinib exposures vs. time since last dose from the study in healthy subjects receiving the ER formulation stratified by formulation.
Figure S2. Observed and model‐predicted a) CDAI < 150 and b) clinical remission 2.8/1.0 vs. time stratified by upadacitinib immediate‐release dose evaluated in CELEST.
Figure S3. Illustration of the continuous‐time Markov model used for exposure–response analyses of clinical efficacy in the CELEST study.
Figure S4. Simulated percentage of subjects achieving the different clinical end points at week 16 and endoscopic end points at week 12/16 during the induction period for different upadacitinib immediate‐release regimens overlaid by the observed responses in CELEST study.
Table S1. Summary of studies used for in the population pharmacokinetics analyses.
Table S2. Demographics and baseline characteristics of subjects included in population pharmacokinetic analyses.
Table S3. Population pharmacokinetics parameter estimates and variability for upadacitinib IR formulation from healthy subjects, rheumatoid arthritis patients, and Crohn's disease patients*.
Table S4. Parameter estimates for upadacitinib pharmacokinetic model applied to data from a study in healthy subjects using the extended‐release formulation.
Table S5. Summary of the Markov model parameter estimates for clinical response, clinical remission 2.8/1.0, and CDAI < 150 in subjects with Crohn's disease.
Table S6. Parameter estimates (95% CI) of nonlinear regression of models for the relationships between upadacitinib average exposure and endoscopic efficacy endpoints at weeks 12/16.
Table S7. Final NONMEM model control streams.