

Abstract
This review aims to assemble many years of research and clinical experience in the fields of neurodevelopment and neuroscience to present an up-to-date understanding of the clinical presentation, molecular and brain pathology associated with Fragile X syndrome, a neurodevelopmental condition that develops with the full mutation of the FMR1 gene, located in the q27.3 loci of the X chromosome, and Fragile X-associated tremor/ataxia syndrome a neurodegenerative disease experienced by aging premutation carriers of the FMR1 gene. It is important to understand that these two syndromes have a very distinct clinical and pathological presentation while sharing the same origin: the mutation of the FMR1 gene; revealing the complexity of expansion genetics.
Keywords: Fragile X syndrome, Fragile X-associated tremor/ataxia syndrome, FXTAS, FMR1 gene, neuropathology
Introduction
The expansion of the trinucleotide CGG above normal range (greater than 54 repeats) in the non-coding region of the Fragile X Mental Retardation 1 (FMR1) gene (Fig. 1) is responsible for the development of the fragile X- associated disorders in those carrying the premutation (55–200 CGG repeats), including fragile-X associated tremor/ataxia syndrome (FXTAS)(1, 2), fragile X-associated primary ovarian insufficiency (FXPOI) (3) and fragile X-associated neuropsychiatric disorders (FXAND)(4); and the presence of fragile X syndrome (FXS) in those carrying the full mutation (greater than 200 CGG repeats). This review details the clinical presentation and neuropathology of the two entities affecting normal brain function: FXTAS, a neurodegenerative disease that commonly develops during the seventh decade of life in 40% of premutation male carriers and 16% of female carriers (5); and FXS, a neurodevelopmental disorder found in 1:7000 males and 1:11000 females (6) causing intellectual disability and Autism Spectrum Disorder (ASD) in more than half of those affected (7).
Figure 1. FMR1 GGG repeat length and gene expression.
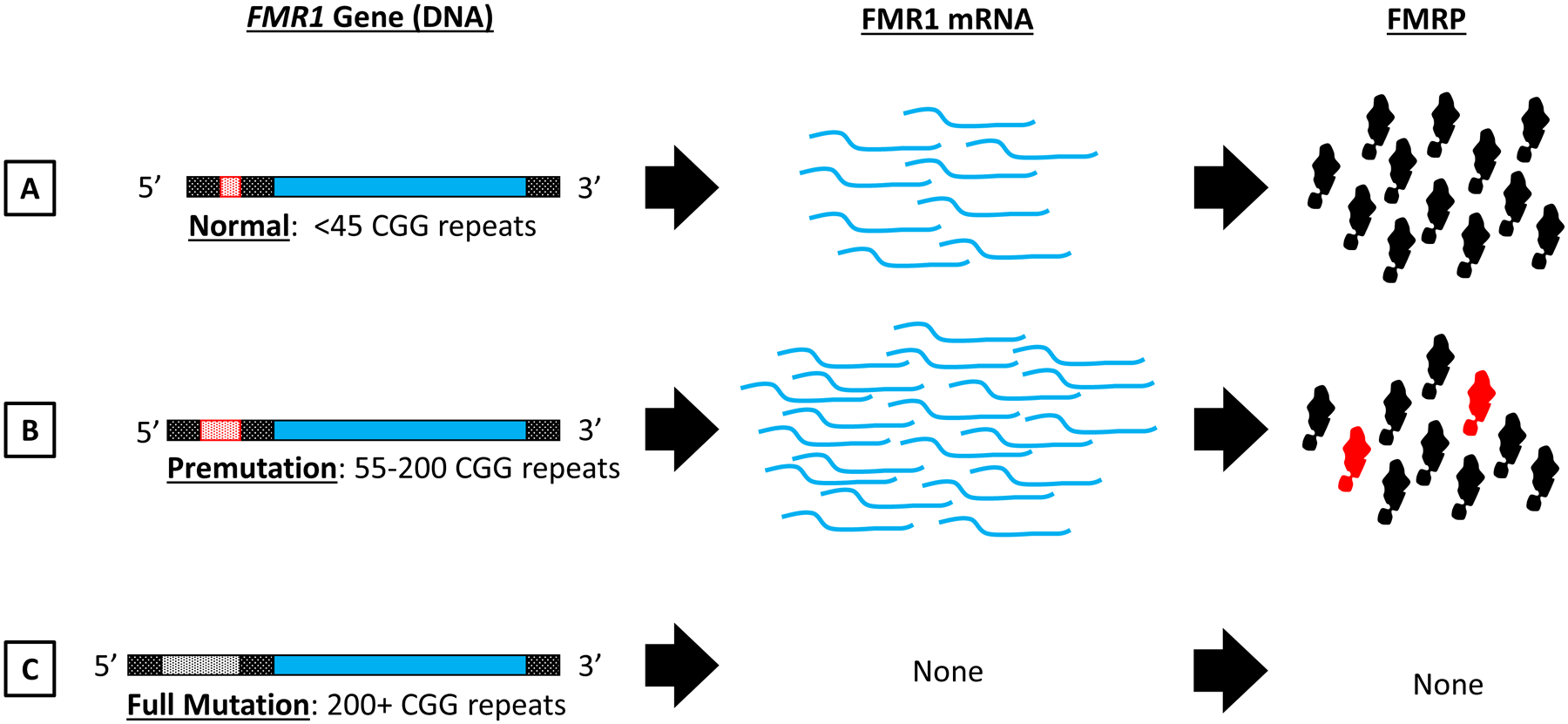
Overview of the relationship between CGG repeat length (A,B,C) within the FMR1 gene (left column) and its effects on FMR1 mRNA (middle) and FMRP protein synthesis (right). A) FMR1 alleles bearing less than 45 CGG repeats are considered in the normal range. B) CGG repeat expansion into the premutation range (containing 55–200 CGG repeats) causes an upregulation in FMR1 mRNA transcripts. For most premutation cases FMRP levels (black shapes) are not altered, although some individuals may show a modest reduction. Additionally, RAN translation of FMR1 mRNA produces toxic FMRpolyG protein species (red shapes). C) CGG repeat expansion into the full mutation range (200+ repeats) causes hypermethylation of the FMR1 gene, resulting in full transcriptional and translational silencing. Figure Key: FMR1 Gene: Open reading frame indicated with solid blue, non-coding 5’ and 3’ regions indicated with shaded black pattern. CGG repeat is located in the 5’ untranslated region (red shaded in A and B, white shaded in C to represent hypermethylation of the gene). FMR1 mRNA transcripts indicated with curved blue lines. FMRP protein represented as black shapes and FMRpolyG is represented as red shapes.
Fragile X Syndrome
Clinical Aspects
FXS is caused by the lack or deficiency of the FMR1 protein FMRP in both males and females with a full mutation. With CGG repeats of >200 there is typically silencing of FMR1 through methylation. The subsequent lack of FMRP, a regulator of translation, leads to dysregulation of hundreds of proteins that affect synaptic plasticity and connectivity in the developing brain leading to intellectual disability (ID) and other clinical features of the syndrome (8–12). Along with the variable presentation of ID, 60% of boys and 20% of girls with FXS are also diagnosed with ASD (13). The complexity of the clinical presentation is accentuated with a well reported psychiatric profile including general anxiety, social avoidance and hyperactive behaviors. These characteristics are commonly seen in those with and without the comorbid presentation of FXS and ASD (13–15). Other comorbid conditions frequently diagnosed during childhood in FXS are seizures (16), recurrent otitis media, strabismus and obesity (17). A Prader-Willi like phenotype, with obsessive/compulsive behaviors, delayed puberty, small genitalia, hyperphagia and lack of satiation after meals leading to severe obesity, has also been described in less than 10% of boys with FXS (18–20).
The physical characteristics of FXS include an elongated face, broad forehead, high palate, prominent ears, hyperextensible finger joints, flat feet and macroorchidism (during and after puberty) (17). However, classic facial characteristics have differences inherent to age and ethnicity (21). In addition to commonly recognized characteristics, patients can present with a variable presentation of connective tissue alterations. Their presence is attributed to FMRP dysregulation of essential components of the extracellular matrix including elastin. Phenotypic findings related to connective problems include soft velvet-like skin, joint hyperextensibility, particularly in the fingers, double jointed thumbs, flat feet with pronation, mitral valve prolapse, dilated aortic root, and occasional scoliosis (22).
After puberty, there is a tendency for improvement of the most problematic behaviors during childhood, including aggression, hyperactivity and irritability; however, behavior and comorbidities can also worsen in those exposed to exogenous neurotoxins (23). During adulthood, patients with FXS seem to have an increased risk of hypertension, obesity, gastrointestinal disorders, parkinsonism, mood disorders, anxiety and in some cases dementia (24, 25). However, patients with FXS have a normal life span. The female phenotype differs from the males since they have the benefit of an unaffected X chromosome. Their cognition includes 30% with an IQ less than 70 (intellectual disability), 30% with an IQ in the borderline range (70–79) and 30% with an IQ in the normal range (above 80), but anxiety and attentional problems can occur in all groups. (7).
Diagnostic Criteria
The diagnosis of FXS can only be confirmed using genetic testing. Southern blot analysis reports an expansion of the CGG trinucleotide number greater than 200 repeats in the 5’ untranslated region in the FMR1 gene located on the X chromosome. The result is a full methylation of the gene and its subsequent silencing (17). However, in some cases individuals can present with mosaicism, showing variability in CGG allele size and in methylation patterns within and between different cell lines. This particular genotype benefits the clinical phenotype by improving both the cognitive and behavioral profiles of males and female patients with FXS (26).
Cell and Molecular Pathology
The origin of all changes that lead to the molecular, pathological and clinical symptoms shown by individuals with FXS is the loss of functional FMRP (Fig. 1C). While CGG expansion leading to hypermethylation and functional silencing of FMR1 is by far the most common genetic cause of FXS, loss of a functional FMRP due deletions or point mutation can also occur (27).
While FMRP expression is ubiquitous, it is expressed at highest levels in the brain and testes (28, 29). FMRP expression has been detected in neurons, astrocytes, microglia, and oligodendrocyte precursor cells (11, 30, 31), and it is largely localized in the cytosol of neurons, in close association with ribosomes of the endoplasmic reticulum, and at high levels in dendritic spines (32). FMRP can also appear in cytoplasmic granules that are transported to dendrites, axons, and pre-synaptic terminals in some neurons (33–38), enabling localized translation (39–41). FMRP granules are also present in axon growth cones during development, likely playing a role in axon guidance, circuit formation, and synaptogenesis (36, 38, 39).
FMRP is an RNA binding protein that regulates translation of numerous associated mRNAs. FMRP is largely considered a translational repressor that suppresses translation initiation and elongation of nascent proteins (reviewed extensively in 42, 43). FMRP also binds and regulates miRNA and miRNA machinery (43, 44), thus exerting translational control through a separate but complimentary molecular mechanism. Accordingly, due to a loss of FMRP mediated translational repression, there is a modest (10–20%) but functionally significant elevation in FMRP-regulated proteins in FXS patients and in FMR1 KO mice (43).
Some key FMRP regulated mRNAs/proteins include second messenger proteins involved in mGluR1 and mGluR5 signal transduction (EIF4E and S6K), (34, 35, 45), GABAA and GABAB receptor subunits (46–48), numerous voltage gated ion channels (49–51), Bone morphogenic protein receptor 2 (BMPR2) (52), matrix metalloproteinase 9 (MMP9) (53), and amyloid precursor protein (APP) (54, 55). Many of these affected mRNA/protein species play a direct role in synaptic transmission. FXS dendrites in the cortex and hippocampus show increased spine density and size, and reduced spine maturity (36, 56, 57). FMRP mediated suppression of the FMPR2-Cofilin pathway is necessary for normal dendrite formation and maturation – and accordingly disruption in this pathway through a loss of FMRP contributes to dendritic abnormalities in FXS. Suppression of MMP9, which is upregulated in the FMR1 KO mouse, normalizes dendritic spine morphology and synapse formation (58). Additionally, loss of FMRP leads to excess soluble APP levels, which also contributes to a lack of dendrite maturation (55). Normalizing APP levels in FMR1 KO mice rescues alterations in synaptic spines, LTP deficits, and reduces audiogenic seizures (54).
The excitation/inhibition imbalance hypothesis has been proposed to explain how cellular and circuit-level alterations in excitatory/inhibitory signaling may lead to clinical symptomology in idiopathic ASD (59). Given the tremendous overlap in FXS and ASD symptomology, as well as the high rates of co-diagnosis in patients with FXS, this hypothesis is believed to largely apply to FXS as well. One way in which excitation and inhibition balance may be disrupted in FXS is through dysregulation of glutamatergic and GABAergic signal transduction. FMRP binds and regulates second messenger proteins that mediate metabotropic glutamate receptor I family (mGluR1 and mGluR5) signal transduction. When FMRP is absent, there is increased phosphorylation of two such downstream effectors - eukaryotic translation initiation factor 4E (EIF4E) and ribosomal protein S6 kinase (S6K), which leads to excess translation of mRNAs that are typically bound and regulated by FMRP (45, 60, 61). There is an increase in synaptic long-term depression (LTD) in FMR1 KO mice, which is believed to be related to dysregulation in mGluR1 signaling. Another consequence of mGluR1 signaling dysfunction in FXS is a reduction in inhibitory retrograde endocannabinoid signaling by mGluR1+ dendrites (7, 62), which likely leads to increased glutamatergic signaling from upstream pre-synaptic glutamatergic neurons and increased excitatory tone. Deficits in GABA signaling have also been characterized in the FMR1 KO mouse, suggesting that a lack of inhibitory GABAergic tone could also lead to hyperexcitability in the FXS CNS.
Neuropathology
Structural MRI studies have identified a pattern of regional volume alterations in patients with FXS, characterized by an enlargement in the caudate nucleus (63–67) and lateral ventricles (63, 64, 66), and a reduction in cerebellar vermis (63, 66, 68). Alterations in caudate and cerebellar vermis appear as early as one year of age (69), and persist into adulthood (63, 66). There also appears to be a moderate and region-specific alteration in cortical lobe grey matter volume, with modest reductions in temporal (63, 67) and frontal lobes (63, 65), and a modest increase in the parietal (63, 65) and occipital lobes (63, 65). Although less consistent, volumetric reductions of amygdala (63, 70) and enlargement of hippocampus (71) have sometimes been observed. White matter volumetric alterations have also been detected, including increased white matter volume in the septal fornix (67), increased brainstem-hippocampus tract and cingulate-corpus callosum tract volume (65), and decreases in frontal lobe (65) and cerebellar white matter (67). Most human FXS structural abnormalities are not recapitulated in the FMR1 KO mouse - striatal volume is unaltered (72) or reduced (73, 74), and there is no change in cerebellar vermis volume (72–74) or cortical lobe volume (72, 73).
The neuropathological correlates of these structural abnormalities in the human FXS brain are poorly characterized – there only exist a handful of such studies and all typically have very small sample sized (n ≤ 3 for all). The earliest and most well characterized finding demonstrated that there are alterations in dendrites and synaptic spines in the postmortem FXS brain. More specifically, FXS cortical tissue in the occipital and temporal cortices have more dendritic spines (56), and these spines are longer and immature (57, 75). Ultrastructural analysis also shows a reduction in synaptic size at dendritic contacts (57). Cerebellar Purkinje cells are reduced in number (76, 77) and in dendritic arbor complexity (76), and the hippocampal structure presents with restricted hyperplasia in the CA1 region (76). Structural and functional MRI studies have both been able to correlate abnormal activation patterns with specific symptom domains in FXS patients. For example, intellectual functioning, as indicated by IQ, is inversely correlated with caudate volume (65) and positively correlated with cerebellar vermis volume (63).
Fragile X-associated Tremor/Ataxia Syndrome
Clinical Aspects
First described in a publication in 2001, fragile X-associated tremor/ ataxia syndrome (FXTAS) is a neurodegenerative disease that primarily affects premutation carriers (55 to 200 CGG repeats) and clinically presents with the core features of intention tremor and/or cerebellar gait ataxia (1). While different in etiology, FXTAS like FXS is more common in males due to the X-linked etiology of the FMR1 gene. It is estimated from one study in United States that 1 in every 403 men are FMR1 premutation carriers and of those, 40% will be diagnosed with FXTAS by their seventh decade. In comparison, women premutation carriers have a prevalence of 1 in every 209 and only an estimated 16% chance of developing FXTAS due to their having one regular functioning X chromosome (78, 79). It is worth noting that while premutation carrier women develop FXTAS at lower rates, they are at risk for fragile X- associated primary ovarian insufficiency (FXPOI), the most heritable form of premature menopause or early ovarian failure. In addition, female premutation carriers report higher rates of psychiatric symptoms including anxiety, ADHD, depression, insomnia, chronic fatigue, and chronic pain. Which fall under the umbrella term of FXAND (4). While not all premutation carriers go on to develop FXTAS the prevalence for FXTAS does increase in age with one study of premutation men showing 17% being affected at 50 years, 38% at 60 years, 47% at 70 years, and 75% at 80 years of age (80)
The hallmark radiological sign of FXTAS is an increased signal on a T2 flair MRI sequence in cerebral white matter especially on the middle cerebellar peduncles (MCP) (81). This characteristic sign is not often seen in women; however, MRI scans of female FXTAS brains reveal increased signal in the splenium of the corpus callosum and in the pons (82). Moderate to severe cortical and general atrophy and increased ventricular volumes is seen in both genders. One study that analyzed 322 magnetic resonance imaging scans confirmed that overall brain and cerebellar volumes were statistically smaller in premutation carrier males with FXTAS as compared to premutation carrier males without FXTAS and controls (83). Another study in young asymptomatic premutation carriers found no differences in measures of executive function with aged matched controls however, the premutation carriers showed a significantly longer manual movement and reaction times. Suggesting that these cerebellar changes might underlie motor deceleration that occurs before symptoms are detected (84).
Diagnostic Criteria
After the FMR1 mutation is confirmed as a premutation carrier status, diagnosis is often made after patients approach their physician with complaints of an action tremor and/or an increase in falls and unsteadiness or ataxia. A definite diagnosis of FXTAS is given if the FMR1 premutation carrier presents with at least one major radiological sign (refer to Table 1) along with at least one major clinical symptom (85, 86). Other frequently seen but more minor symptoms are parkinsonism (bradykinesia, muscle rigidity, masked facies and slowed speech) and cognitive decline in executive function and moderate to severe short-term deficits. Other comorbidities seen in premutation carriers with FXTAS include autonomic dysfunction, thyroid disease, peripheral neuropathy including symptoms of numbness, tingling and pain, fibromyalgia, migraines, hypertension, bradycardia, sleep apnea, and irritability or depression. (5).
Table 1.
Major and minor radiological, clinical, and neuropathological signs of FXTAS. The molecular criteria include an FMR1 premutation. Adapted from Berry-Kravis et al 2007 and Hall et al 2014 (86, 127)
| Major (Signs/Symptoms) | Minor (Signs/Symptoms) | |
|---|---|---|
| Radiological | Radiological sign: T2 FLAIR MRI Middle Cerebellar Peduncle (MCP) | Radiological sign: T2 FLAIR MRI white matter lesions in cerebral white matter Radiological sign: T2 FLAIR MRI Moderate-to-severe generalized brain atrophy Radiological sign: T2 FLAIR MRI white matter hyperintensity in Splenium of Corpus Callosum (CCS) |
| Clinical Signs | Intention tremor Cerebellar ataxia |
Parkinsonism (bradykinesia, shuffling gait, masked facies) Neuropathy Executive function and memory deficits |
| Neuropathological signs | Intranuclear inclusions in CNS and PNS |
Currently there is no cure for FXTAS and treatment is based on alleviation of symptoms, such as the use of primidone or beta blockers for the tremor and SSRIs for irritability or depression.
Molecular Pathology
In contrast to FMR1 full mutation, which leads to transcriptional silencing of FMR1 mRNA and a concomitant loss of FMRP, in FXTAS there is not a substantial alteration in FMRP levels – only a modest reduction in the high premutation repeat range (Fig 1B). However, in premutation cases there is a dramatic increase in FMR1 mRNA (Fig. 1B). There are three primary molecular mechanisms by which FMR1 premutation excess mRNA is believed to lead to FXTAS neuropathology (5, 87), including: 1) Sequestration of proteins and RNAs into inclusion bodies (Fig. 2D/E) that leads to impaired cell function due to loss of these RNA and protein species; 2) R-loop formation leading to DNA damage; and 3) RAN translation leading to the production of toxic FMRpolyG protein (Fig. 1B). In addition, elevated mRNA levels lead to elevated levels of Ca+2 in the neuron and subsequent mitochondrial dysfunction which worsens as FXTAS develops (88). These proposed mechanisms are not mutually exclusive, and there may remain other yet unidentified molecular mechanisms by which FMR1 premutation leads to pathogenesis.
Figure 2. FXTAS Neuropathology.
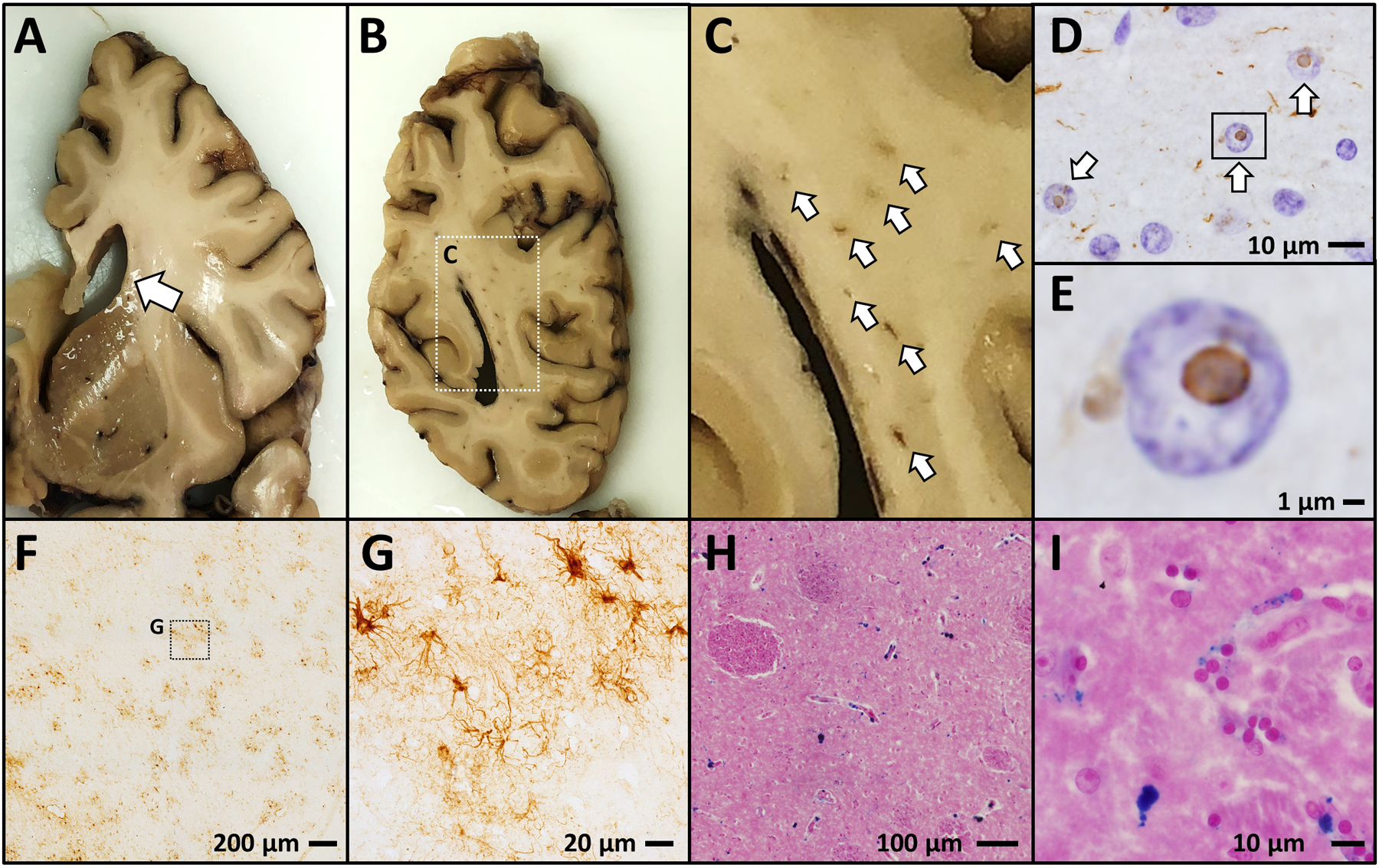
Postmortem neuropathological analysis from human FXTAS cases show a characteristic neurodegenerative phenotype which includes ventricular enlargement (A); focal white matter lesions (B,C); ubiquitinated intranuclear inclusion bodies (D,E – ubiquitin IHC with hematoxylin nuclear counterstain); patches of astrogliosis (F,G - GFAP IHC); and excessive iron accumulation (H,I – ferric iron stained using Perl’s method, Eosin counterstain).
The first mechanism proposed for how FMR1 premutation leads to FXTAS is that FMR1 premutation mRNA sequesters other RNA and protein species into intranuclear inclusion bodies, which in turns disrupts essential cellular processes dependent on these molecules. Inclusion bodies are the hallmark neuropathological indicator of FXTAS; they contain FMR1 mRNA but lack FMRP (87, 89), and are exclusively located within the nucleus (90–93). Inclusions are almost always present as a single body (90, 91, 93, 94), with the exception of Purkinje cells that sometimes form twin inclusions (95). FACS sorting in combination with inclusion autofluorescence has enabled improved isolation and purification of inclusion bodies, revealing some of their basic biochemistry (87). Inclusions are predominantly an aggregate of protein, composed of a heterogenous assortment of many proteins which are particularly enriched in RNA-binding, DNA-binding, and protein turnover regulating proteins (87). The inclusions also contain mRNA species, but to a much lower degree than protein, and inclusions do not contain DNA (87). Five proteins are particularly enriched: SUMO2, p62/SQSTM1, Myeloid Leukemia Factor 2 (MLF2), Ubiquitin, and Myelin Basic Protein (MBP) (87). The authors emphasize that the protein species found in the inclusion are indicative of inclusions forming in response to oxidative stress, and suggest that this may lead to an impairment in DNA damage response and an impairment in protein autophagy, both of which may lead to toxicity and neurodegeneration (87). However, while the specific mechanisms by which inclusion mediated protein sequestration leads to disease are an area of active investigation, these mechanisms remain poorly understood.
The second proposed molecular mechanism involves the formation of R-loops during FMR1 premutation transcription leading to DNA damage. CGG expansion at the FMR1 locus results in an increase in the number and size of R-loops formed (96), during which time the non-template DNA strand is vulnerable to DNA damage (97, 98). DNA damage should be corrected by the DNA damage response (DDR) molecular signaling pathway (99, 100) that appears to be impaired in FXTAS (101), possibly through sequestration of DDR molecules by FXTAS inclusions (87, 89). Additionally, high levels of oxidative stress and mitochondrial dysfunction occurs in FXTAS (101, 102), which could independently lead to further DNA damage, or possibly exacerbate DNA damage caused by R-loop formation.
The third proposed molecular mechanism in FXTAS is RAN (non-AUG) translation (reviewed extensively in 103, 104). RAN translation is a common feature in triplet repeat expansion disorders, whereby regions of an mRNA containing the triplet repeat become prone to errors in translation and protein synthesis is initiated outside of the traditional AUG start codon (105–109) (107, 109–111). In FXTAS, the non-coding region of FMR1 premutation mRNA is translated into multiple RAN translation proteins, including the FMRpolyG protein being the most highly expressed and thus likely the largest contributor to pathology (105, 106). Sellier et al. (105) reported that FMRpolyG protein is present in inclusion bodies that form in the cytosol, and that these FMRpolyG positive inclusions are subsequently transported into the nucleus where they disrupt the nuclear lamina protein LAP2β, leading to toxicity. They also reported that this toxicity only occurs when FMRpolyG proteins are produced from FMR1 premutation mRNA in transgenic rodent models, and that increasing expression of LAP2β ameliorates FMRpolyG toxicity (105). Numerous studies have identified the presence of intranuclear FMRpolyG+ and FMRpolyA+ inclusions in FXTAS postmortem human brain (105, 112–114) and peripheral tissue (114, 115), as well as in FMR1 premutation rodent models (105, 116, 117). Multiple studies in FXTAS rodent, fly, and cell culture models have also demonstrated that expression of FMR1 RAN translation products are necessary for inclusion formation and toxicity (105, 106, 118, 119). However while it is clear that RAN translation does occur in humans with FXTAS, whether it is a central driver of pathogenesis remains unclear. In a recent study utilizing mass spectroscopy to analyze human FXTAS inclusion composition, Ma et al (87) indeed detected RAN translation products in human FXTAS brain tissue, but also found using parallel reaction monitoring that they occurred in very low abundance and were not localized within FXTAS inclusion bodies. Future studies are needed to expand upon and further clarify the relative contribution of these three proposed mechanisms of FMR1 premutation toxicity, in addition to identifying other possible molecular mechanisms that may also co-occur.
Neuropathology
The hallmark pathological change in FXTAS is the presence of intranuclear inclusion bodies (Fig. 2D/E) that are ubiquitin and FMR1 mRNA positive (90, 91, 120). FXTAS inclusion burden is positively correlated with CGG repeat length within the premutation range (90). In the CNS, FXTAS inclusions occur in astrocytes and neurons, although they tend to be larger and occur at a higher frequency in astrocytes in male cases (90, 91, 120). There is a regional variability in the proportion of neurons and astrocytes bearing inclusions. At one extreme, there tend to be no inclusion+ neurons in the pons, although inclusion+ astroctyes are abundant occurring in 9–30% of cells (90). Inclusion+ neurons and astrocytes are particularly prevalent in the hippocampus (up to 40% of both cell types) and to a lesser degree in the frontal and temporal cortex (≤10% of neurons and ≤20% of astrocytes) (90, 91, 120). Neuronal and astrocytic inclusions also occur in the putamen, globus pallidus, substantia nigra, and amygdala (91, 121). Inclusion bodies are also present in the periphery, including in the peripheral nervous system, enteric nervous system, endocrine glands, heart, and kidney (94, 122).
Gross pathological assessment of the FXTAS brain indicates severe white matter disease, atrophy of both grey and white matter, and ventricular enlargement (Fig. 2A) (90, 91, 120). Spongiosis and discoloration of cerebellar white matter is present in the vast majority of FXTAS cases (Fig. 2B/C) (90, 91, 120), including the MCP (90). Cerebral white matter disease is also common, but less consistently than in the cerebellum (90). Microscopic evaluation of white matter demonstrates a loss of myelin, as well as axonal degeneration (90). Grey matter atrophy of frontal cortex, cerebellum, and the pons have been qualitatively documented and consistently occur in FXTAS cases (90, 91). While grey matter neuronal and possibly astrocytic cells loss is presumed, the relative contributions of cellular atrophy and cell loss to regional volumetric reductions have not been directly assessed. Grey matter neuronal loss has so far only been demonstrated to occur in Purkinje cells (90, 91), and to date has not been systematically assessed in other brain regions or cell types. Female FXTAS cases that present clinically with dementia symptomology also tend to show high rates of comorbid Alzheimer’s neuropathology, including significant amyloid plaques and neurofibrillary tangles (120). Finally, postmortem FXTAS brains show high levels of iron accumulation in brain capillaries and parenchyma (Fig. 2H/I), as well as choroid plexus, which can occur as extracellular or intracellular deposits (123–125). Striatal iron accumulation is prevalent and severe (123), while cerebellar iron accumulation occurs in a smaller subset of patients (<25%) and at lower levels (125). The iron binding protein ceruloplasmin is also dramatically reduced in neurons and astrocytes, and to a lesser degree in oligodendrocytes (123), suggesting that FXTAS associated dysregulation of iron metabolic pathways may underlie iron accumulation. In contrast, ceruloplasmin and transferrin expression is increased in microglia, as well as intracellular iron deposits, suggesting that microglia may be actively attempting to counteract iron accumulation (123). Astrocytes show profound reactive gliosis and profound microglial activation occurs in a majority of FXTAS cases (Fig. 2F/G) (126). Microglial senescence also occurs in FXTAS suggesting that disease-associated microglial impairment may further exacerbate FXTAS neuropathology (126).
Conclusion
Many physicians confuse FXS and FXTAS so it is essential to remember that these are 2 very different disorders, one causing ID and autism and the other leading to neurodegeneration in otherwise normally developed individuals who are aging. Each disorder has differing levels of CGG repeats with deficient levels of FMRP in FXS and elevated levels of FMR1 mRNA in FXTAS. However, these disorders are usually found in the same families and often multiple individuals with each of these disorders can be found. Therefore, when one individual is identified with a fragile X mutation the whole family through multiple generations are at risk for one or more of these mutations. Cascade testing for these mutations are necessary throughout the family tree either by the physician who identified the initial mutation or by referral to genetics so that the whole family can understand and be tested for these disorders.
Acknowledgements:
This research was supported by NICHD R01 HD036071, the MIND Institute Intellectual and Developmental Disabilities Research Center funded by NICHD U54 HD079125. This publication was also made possible by National Center for Advancing Translational Sciences, the National Institutes of Health (grant UL1 TR001860), the National Institute of Neurological Disorders and Stroke (NINDS) grant R01 1NS107131 and Shriners Hospitals for Children - Northern California
References
- 1.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57(1):127–30. [DOI] [PubMed] [Google Scholar]
- 2.Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72(4):869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97(3):189–94. [DOI] [PubMed] [Google Scholar]
- 4.Hagerman RJ, Protic D, Rajaratnam A, Salcedo-Arellano MJ, Aydin EY, Schneider A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front Psychiatry. 2018;9:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagerman RJ, Hagerman P. Fragile X-associated tremor/ataxia syndrome - features, mechanisms and management. Nat Rev Neurol. 2016;12(7):403–12. [DOI] [PubMed] [Google Scholar]
- 6.Hunter J, Rivero-Arias O, Angelov A, Kim E, Fotheringham I, Leal J. Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am J Med Genet A. 2014;164A(7):1648–58. [DOI] [PubMed] [Google Scholar]
- 7.Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr., Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Primers. 2017;3:17065. [DOI] [PubMed] [Google Scholar]
- 8.Danesi C, Achuta VS, Corcoran P, Peteri U-K, Turconi G, Matsui N, et al. Increased Calcium Influx through L-type Calcium Channels in Human and Mouse Neural Progenitors Lacking Fragile X Mental Retardation Protein. Stem cell reports. 2018;11(6):1449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gatto CL, Pereira D, Broadie K. GABAergic circuit dysfunction in the Drosophila Fragile X syndrome model. Neurobiol Dis. 2014;65:142–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higashimori H, Morel L, Huth J, Lindemann L, Dulla C, Taylor A, et al. Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum Mol Genet. 2013;22(10):2041–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Ku L, Osterhout DJ, Li W, Ahmadian A, Liang Z, et al. Developmentally-programmed FMRP expression in oligodendrocytes: a potential role of FMRP in regulating translation in oligodendroglia progenitors. Hum Mol Genet. 2004;13(1):79–89. [DOI] [PubMed] [Google Scholar]
- 12.Pilaz LJ, Lennox AL, Rouanet JP, Silver DL. Dynamic mRNA Transport and Local Translation in Radial Glial Progenitors of the Developing Brain. Curr Biol. 2016;26(24):3383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailey DB Jr., Raspa M, Olmsted M, Holiday DB. Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A. 2008;146A(16):2060–9. [DOI] [PubMed] [Google Scholar]
- 14.Thurman AJ, McDuffie A, Hagerman R, Abbeduto L. Psychiatric symptoms in boys with fragile X syndrome: a comparison with nonsyndromic autism spectrum disorder. Research in developmental disabilities. 2014;35(5):1072–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDuffie A, Thurman AJ, Hagerman RJ, Abbeduto L. Symptoms of Autism in Males with Fragile X Syndrome: A Comparison to Nonsyndromic ASD Using Current ADI-R Scores. Journal of autism and developmental disorders. 2015;45(7):1925–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, Bailey DB. Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil. 2010;115(6):461–72. [DOI] [PubMed] [Google Scholar]
- 17.Hagerman R, Hagerman P. Fragile X Syndrome. Baltimore: The John Hopkins University Press; 2002. [Google Scholar]
- 18.McLennan Y, Polussa J, Tassone F, Hagerman R. Fragile x syndrome. Curr Genomics. 2011;12(3):216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nowicki ST, Tassone F, Ono MY, Ferranti J, Croquette MF, Goodlin-Jones B, et al. The Prader-Willi Phenotype of Fragile X Syndrome. 2007;28(2):133–8. [DOI] [PubMed] [Google Scholar]
- 20.Fryns JP, Haspeslagh M, Dereymaeker AM, Volcke P, van den Berghe H. A peculiar subphenotype in the fra(X) syndrome: extreme obesity-short stature-stubby hands and feet-diffuse hyperpigmentation. Further evidence of disturbed hypothalamic function in the fra(X) syndrome? 1987;32(6):388–92. [DOI] [PubMed] [Google Scholar]
- 21.Lubala TK, Lumaka A, Kanteng G, Mutesa L, Mukuku O, Wembonyama S, et al. Fragile X checklists: A meta-analysis and development of a simplified universal clinical checklist. Molecular genetics & genomic medicine. 2018;6(4):526–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramirez-Cheyne JA, Duque GA, Ayala-Zapata S, Saldarriaga-Gil W, Hagerman P, Hagerman R, et al. Fragile X syndrome and connective tissue dysregulation. Clin Genet. 2018. [DOI] [PubMed] [Google Scholar]
- 23.Salcedo-Arellano MJ, Lozano R, Tassone F, Hagerman RJ, Saldarriaga W. Alcohol use dependence in fragile X syndrome. Intractable & rare diseases research. 2016;5(3):207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauna-Aho O, Bjelogrlic-Laakso N, Siren A, Arvio M. Signs indicating dementia in Down, Williams and Fragile X syndromes. Mol Genet Genomic Med. 2018;6(5):855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Utari A, Adams E, Berry-Kravis E, Chavez A, Scaggs F, Ngotran L, et al. Aging in fragile X syndrome. Journal of neurodevelopmental disorders. 2010;2(2):70–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunter J, Berry-Kravis E, Hipp H, Todd P. FMR1 Disorders In: Adam MPAH, Pagon RA, et al. , ed. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019. [Google Scholar]
- 27.Quartier A, Poquet H, Gilbert-Dussardier B, Rossi M, Casteleyn AS, Portes VD, et al. Intragenic FMR1 disease-causing variants: a significant mutational mechanism leading to Fragile-X syndrome. Eur J Hum Genet. 2017;25(4):423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bakker CE, de Diego Otero Y, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, et al. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res. 2000;258(1):162–70. [DOI] [PubMed] [Google Scholar]
- 29.Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4(4):335–40. [DOI] [PubMed] [Google Scholar]
- 30.Pacey LK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia. 2007;55(15):1601–9. [DOI] [PubMed] [Google Scholar]
- 31.Gholizadeh S, Halder SK, Hampson DR. Expression of fragile X mental retardation protein in neurons and glia of the developing and adult mouse brain. Brain Res. 2015;1596:22–30. [DOI] [PubMed] [Google Scholar]
- 32.Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17(5):1539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akins MR, Leblanc HF, Stackpole EE, Chyung E, Fallon JR. Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J Comp Neurol. 2012;520(16):3687–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antar LN, Dictenberg JB, Plociniak M, Afroz R, Bassell GJ. Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4(6):350–9. [DOI] [PubMed] [Google Scholar]
- 36.Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32(1–2):37–48. [DOI] [PubMed] [Google Scholar]
- 37.Price TJ, Flores CM, Cervero F, Hargreaves KM. The RNA binding and transport proteins staufen and fragile X mental retardation protein are expressed by rat primary afferent neurons and localize to peripheral and central axons. Neuroscience. 2006;141(4):2107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christie SB, Akins MR, Schwob JE, Fallon JR. The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci. 2009;29(5):1514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, Bassell GJ, Sasaki Y. Fragile X Mental Retardation Protein is Involved in Protein Synthesis-Dependent Collapse of Growth Cones Induced by Semaphorin-3A. Front Neural Circuits. 2009;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee A, Ifrim MF, Valdez AN, Raj N, Bassell GJ. Aberrant RNA translation in fragile X syndrome: From FMRP mechanisms to emerging therapeutic strategies. Brain Res. 2018;1693(Pt A):24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29(11):2276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci. 2013;16(11):1530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16(10):595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Lin L, Jin P. The microRNA pathway and fragile X mental retardation protein. Biochim Biophys Acta. 2008;1779(11):702–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gkogkas CG, Khoutorsky A, Cao R, Jafarnejad SM, Prager-Khoutorsky M, Giannakas N, et al. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 2014;9(5):1742–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braat S, Kooy RF. Insights into GABAAergic system deficits in fragile X syndrome lead to clinical trials. Neuropharmacology. 2015;88:48–54. [DOI] [PubMed] [Google Scholar]
- 47.D’Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA, et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121(1):238–45. [DOI] [PubMed] [Google Scholar]
- 48.Wahlstrom-Helgren S, Klyachko VA. GABAB receptor-mediated feed-forward circuit dysfunction in the mouse model of fragile X syndrome. J Physiol. 2015;593(22):5009–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci. 2010;13(7):819–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferron L Fragile X mental retardation protein controls ion channel expression and activity. J Physiol. 2016;594(20):5861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferron L, Nieto-Rostro M, Cassidy JS, Dolphin AC. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat Commun. 2014;5:3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kashima R, Roy S, Ascano M, Martinez-Cerdeno V, Ariza-Torres J, Kim S, et al. Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci Signal. 2016;9(431):ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sidhu H, Dansie LE, Hickmott PW, Ethell DW, Ethell IM. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J Neurosci. 2014;34(30):9867–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Westmark CJ, Westmark PR, O’Riordan KJ, Ray BC, Hervey CM, Salamat MS, et al. Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS One. 2011;6(10):e26549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pasciuto E, Ahmed T, Wahle T, Gardoni F, D’Andrea L, Pacini L, et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron. 2015;87(2):382–98. [DOI] [PubMed] [Google Scholar]
- 56.Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98(2):161–7. [DOI] [PubMed] [Google Scholar]
- 57.Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, et al. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 1985;67(3–4):289–95. [DOI] [PubMed] [Google Scholar]
- 58.Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46(2):94–102. [DOI] [PubMed] [Google Scholar]
- 59.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2(5):255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sawicka K, Pyronneau A, Chao M, Bennett MV, Zukin RS. Elevated ERK/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in fragile X mice. Proc Natl Acad Sci U S A. 2016;113(41):E6290–E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11(3):332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Busquets-Garcia A, Gomis-Gonzalez M, Guegan T, Agustin-Pavon C, Pastor A, Mato S, et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med. 2013;19(5):603–7. [DOI] [PubMed] [Google Scholar]
- 63.Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O’Hara R, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP). Ann Neurol. 2008;63(1):40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB. Neurodevelopmental effects of the FMR-1 full mutation in humans. Nat Med. 1995;1(2):159–67. [DOI] [PubMed] [Google Scholar]
- 65.Hallahan BP, Craig MC, Toal F, Daly EM, Moore CJ, Ambikapathy A, et al. In vivo brain anatomy of adult males with Fragile X syndrome: an MRI study. Neuroimage. 2011;54(1):16–24. [DOI] [PubMed] [Google Scholar]
- 66.Lee AD, Leow AD, Lu A, Reiss AL, Hall S, Chiang MC, et al. 3D pattern of brain abnormalities in Fragile X syndrome visualized using tensor-based morphometry. Neuroimage. 2007;34(3):924–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sandoval GM, Shim S, Hong DS, Garrett AS, Quintin EM, Marzelli MJ, et al. Neuroanatomical abnormalities in fragile X syndrome during the adolescent and young adult years. J Psychiatr Res. 2018;107:138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mostofsky SH, Mazzocco MM, Aakalu G, Warsofsky IS, Denckla MB, Reiss AL. Decreased cerebellar posterior vermis size in fragile X syndrome: correlation with neurocognitive performance. Neurology. 1998;50(1):121–30. [DOI] [PubMed] [Google Scholar]
- 69.Hoeft F, Carter JC, Lightbody AA, Cody Hazlett H, Piven J, Reiss AL. Region-specific alterations in brain development in one- to three-year-old boys with fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107(20):9335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kates WR, Abrams MT, Kaufmann WE, Breiter SN, Reiss AL. Reliability and validity of MRI measurement of the amygdala and hippocampus in children with fragile X syndrome. Psychiatry Res. 1997;75(1):31–48. [DOI] [PubMed] [Google Scholar]
- 71.Reiss AL, Lee J, Freund L. Neuroanatomy of fragile X syndrome: the temporal lobe. Neurology. 1994;44(7):1317–24. [DOI] [PubMed] [Google Scholar]
- 72.Kooy RF, Reyniers E, Verhoye M, Sijbers J, Bakker CE, Oostra BA, et al. Neuroanatomy of the fragile X knockout mouse brain studied using in vivo high resolution magnetic resonance imaging. Eur J Hum Genet. 1999;7(5):526–32. [DOI] [PubMed] [Google Scholar]
- 73.Ellegood J, Pacey LK, Hampson DR, Lerch JP, Henkelman RM. Anatomical phenotyping in a mouse model of fragile X syndrome with magnetic resonance imaging. Neuroimage. 2010;53(3):1023–9. [DOI] [PubMed] [Google Scholar]
- 74.Lai JK, Lerch JP, Doering LC, Foster JA, Ellegood J. Regional brain volumes changes in adult male FMR1-KO mouse on the FVB strain. Neuroscience. 2016;318:12–21. [DOI] [PubMed] [Google Scholar]
- 75.Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41(3):289–94. [DOI] [PubMed] [Google Scholar]
- 76.Greco CM, Navarro CS, Hunsaker MR, Maezawa I, Shuler JF, Tassone F, et al. Neuropathologic features in the hippocampus and cerebellum of three older men with fragile X syndrome. Mol Autism. 2011;2(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sabaratnam M. Pathological and neuropathological findings in two males with fragile-X syndrome. J Intellect Disabil Res. 2000;44 (Pt 1):81–5. [DOI] [PubMed] [Google Scholar]
- 78.Tassone F, Iong KP, Tong TH, Lo J, Gane LW, Berry-Kravis E, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012;4(12):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xuncla M, Badenas C, Kulisevsky J, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009;17(10):1359–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, Brunberg JA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291(4):460–9. [DOI] [PubMed] [Google Scholar]
- 81.Brunberg JA, Jacquemont S, Hagerman RJ, Berry-Kravis EM, Grigsby J, Leehey MA, et al. Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am J Neuroradiol. 2002;23(10):1757–66. [PMC free article] [PubMed] [Google Scholar]
- 82.Hall DA, Birch RC, Anheim M, Jonch AE, Pintado E, O’Keefe JA, et al. Erratum: Emerging topics in FXTAS. J Neurodev Disord. 2015;7(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang JY, Hessl D, Hagerman RJ, Simon TJ, Tassone F, Ferrer E, et al. Abnormal trajectories in cerebellum and brainstem volumes in carriers of the fragile X premutation. Neurobiol Aging. 2017;55:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shickman R, Famula J, Tassone F, Leehey M, Ferrer E, Rivera SM, et al. Age- and CGG repeat-related slowing of manual movement in fragile X carriers: A prodrome of fragile X-associated tremor ataxia syndrome? Mov Disord. 2018;33(4):628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, et al. Fragile X-associated tremor/ataxia syndrome: Clinical features, genetics, and testing guidelines. 2007;22(14):2018–30. [DOI] [PubMed] [Google Scholar]
- 86.Hall DA, Birch RC, Anheim M, Jønch AE, Pintado E, O’Keefe J, et al. Emerging topics in FXTAS. Journal of neurodevelopmental disorders. 2014;6(1):31-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ma L, Herren AW, Espinal G, Randol J, McLaughlin B, Martinez-Cerdeno V, et al. Composition of the Intranuclear Inclusions of Fragile X-associated Tremor/Ataxia Syndrome. Acta Neuropathol Commun. 2019;7(1):143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giulivi C, Napoli E, Tassone F, Halmai J, Hagerman R. Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem J. 2016;473(21):3871–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129(Pt 1):256–71. [DOI] [PubMed] [Google Scholar]
- 90.Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain. 2006;129(Pt 1):243–55. [DOI] [PubMed] [Google Scholar]
- 91.Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125(Pt 8):1760–71. [DOI] [PubMed] [Google Scholar]
- 92.Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet. 2004;41(4):e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wenzel HJ, Hunsaker MR, Greco CM, Willemsen R, Berman RF. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010;1318:155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hunsaker MR, Greco CM, Spath MA, Smits AP, Navarro CS, Tassone F, et al. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011;122(4):467–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ariza J, Rogers H, Monterrubio A, Reyes-Miranda A, Hagerman PJ, Martinez-Cerdeno V. A Majority of FXTAS Cases Present with Intranuclear Inclusions Within Purkinje Cells. Cerebellum. 2016;15(5):546–51. [DOI] [PubMed] [Google Scholar]
- 96.Loomis EW, Sanz LA, Chedin F, Hagerman PJ. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014;10(4):e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roy D, Lieber MR. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol Cell Biol. 2009;29(11):3124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ginno PA, Lim YW, Lott PL, Korf I, Chedin F. GC skew at the 5’ and 3’ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013;23(10):1590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Silva AR, Santos AC, Farfel JM, Grinberg LT, Ferretti RE, Campos AH, et al. Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS One. 2014;9(6):e99897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barzilai A DNA damage, neuronal and glial cell death and neurodegeneration. Apoptosis. 2010;15(11):1371–81. [DOI] [PubMed] [Google Scholar]
- 101.Kaplan ES, Cao Z, Hulsizer S, Tassone F, Berman RF, Hagerman PJ, et al. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J Neurochem. 2012;123(4):613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alvarez-Mora MI, Rodriguez-Revenga L, Madrigal I, Guitart-Mampel M, Garrabou G, Mila M. Impaired Mitochondrial Function and Dynamics in the Pathogenesis of FXTAS. Mol Neurobiol. 2017;54(9):6896–902. [DOI] [PubMed] [Google Scholar]
- 103.Glineburg MR, Todd PK, Charlet-Berguerand N, Sellier C. Repeat-associated non-AUG (RAN) translation and other molecular mechanisms in Fragile X Tremor Ataxia Syndrome. Brain Res. 2018;1693(Pt A):43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boivin M, Willemsen R, Hukema RK, Sellier C. Potential pathogenic mechanisms underlying Fragile X Tremor Ataxia Syndrome: RAN translation and/or RNA gain-of-function? Eur J Med Genet. 2018;61(11):674–9. [DOI] [PubMed] [Google Scholar]
- 105.Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P, et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron. 2017;93(2):331–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78(3):440–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108(1):260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cleary JD, Pattamatta A, Ranum LPW. Repeat-associated non-ATG (RAN) translation. J Biol Chem. 2018;293(42):16127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cleary JD, Ranum LP. New developments in RAN translation: insights from multiple diseases. Curr Opin Genet Dev. 2017;44:125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ayhan F, Perez BA, Shorrock HK, Zu T, Banez-Coronel M, Reid T, et al. SCA8 RAN polySer protein preferentially accumulates in white matter regions and is regulated by eIF3F. EMBO J. 2018;37(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Banez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, et al. RAN Translation in Huntington Disease. Neuron. 2015;88(4):667–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Krans A, Skariah G, Zhang Y, Bayly B, Todd PK. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathol Commun. 2019;7(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sacino AN, Prokop S, Walsh MA, Adamson J, Subramony SH, Krans A, et al. Fragile X-associated tremor ataxia syndrome with co-occurrent progressive supranuclear palsy-like neuropathology. Acta Neuropathol Commun. 2019;7(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buijsen RA, Sellier C, Severijnen LA, Oulad-Abdelghani M, Verhagen RF, Berman RF, et al. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol Commun. 2014;2:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Buijsen RA, Visser JA, Kramer P, Severijnen EA, Gearing M, Charlet-Berguerand N, et al. Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum Reprod. 2016;31(1):158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wenzel HJ, Murray KD, Haify SN, Hunsaker MR, Schwartzer JJ, Kim K, et al. Astroglial-targeted expression of the fragile X CGG repeat premutation in mice yields RAN translation, motor deficits and possible evidence for cell-to-cell propagation of FXTAS pathology. Acta Neuropathol Commun. 2019;7(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gohel D, Sripada L, Prajapati P, Singh K, Roy M, Kotadia D, et al. FMRpolyG alters mitochondrial transcripts level and respiratory chain complex assembly in Fragile X associated tremor/ataxia syndrome [FXTAS]. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1379–88. [DOI] [PubMed] [Google Scholar]
- 118.Hoem G, Bowitz Larsen K, Overvatn A, Brech A, Lamark T, Sjottem E, et al. The FMRpolyGlycine Protein Mediates Aggregate Formation and Toxicity Independent of the CGG mRNA Hairpin in a Cellular Model for FXTAS. Front Genet. 2019;10:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Oh SY, He F, Krans A, Frazer M, Taylor JP, Paulson HL, et al. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum Mol Genet. 2015;24(15):4317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tassone F, Greco CM, Hunsaker MR, Seritan AL, Berman RF, Gane LW, et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 2012;11(5):577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Louis E, Moskowitz C, Friez M, Amaya M, Vonsattel JP. Parkinsonism, dysautonomia, and intranuclear inclusions in a fragile X carrier: a clinical-pathological study. Mov Disord. 2006;21(3):420–5. [DOI] [PubMed] [Google Scholar]
- 122.Gokden M, Al-Hinti JT, Harik SI. Peripheral nervous system pathology in fragile X tremor/ataxia syndrome (FXTAS). Neuropathology. 2009;29(3):280–4. [DOI] [PubMed] [Google Scholar]
- 123.Ariza J, Rogers H, Hartvigsen A, Snell M, Dill M, Judd D, et al. Iron accumulation and dysregulation in the putamen in fragile X-associated tremor/ataxia syndrome. Mov Disord. 2017;32(4):585–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ariza J, Steward C, Rueckert F, Widdison M, Coffman R, Afjei A, et al. Dysregulated iron metabolism in the choroid plexus in fragile X-associated tremor/ataxia syndrome. Brain Res. 2015;1598:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rogers H, Ariza J, Monterrubio A, Hagerman P, Martinez-Cerdeno V. Cerebellar Mild Iron Accumulation in a Subset of FMR1 Premutation Carriers with FXTAS. Cerebellum. 2016;15(5):641–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martinez Cerdeno V, Hong T, Amina S, Lechpammer M, Ariza J, Tassone F, et al. Microglial cell activation and senescence are characteristic of the pathology FXTAS. Mov Disord. 2018;33(12):1887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, et al. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord. 2007;22(14):2018–30, quiz 140. [DOI] [PubMed] [Google Scholar]