

Abstract
Background:
Fibronectin splicing variant containing extra domain A (Fn-EDA), which is an endogenous ligand for toll-like-receptor 4 (TLR4), is present in negligible amounts in the plasma of healthy humans, but markedly elevated in patients with comorbid conditions including diabetes and hyperlipidemia, which are risk factors for myocardial infarction (MI). Very little is known about the role of Fn-EDA in the pathophysiology of acute MI under these comorbid conditions.
Methods:
We determined the role of Fn-EDA in myocardial ischemia/reperfusion (I/R) injury in the hyperlipidemic apolipoprotein E-deficient (Apoe−/−) mice. Infarct size, plasma cardiac troponin I (cTnI) levels, intravascular thrombosis (CD41-positive), neutrophil infiltration (Ly6 B.2-positive), neutrophil extracellular traps (citrullinated H3-positive) and myocyte apoptosis (TUNEL-positive) were assessed in myocardial I/R injury model (1-hour ischemia/ 23 hours of reperfusion).
Results:
Irrespective of gender, Fn-EDA−/−Apoe−/− mice exhibited smaller infarct size and decreased cTnI levels concomitant with reduced postischemic intravascular thrombi, neutrophils influx, neutrophil extracellular traps and myocyte apoptosis (P<0.05 vs. Apoe−/− mice). Genetic deletion of TLR4 attenuated myocardial I/R injury in Apoe−/− mice (P<0.05 vs. Apoe−/− mice), but did not further reduce in Fn-EDA−/− Apoe−/− mice suggesting that Fn-EDA requires TLR4 to mediate myocardial I/R injury. Bone marrow transplantation experiments revealed that Fn-EDA exacerbates myocardial I/R injury through TLR4 expressed on the hematopoietic cells. Infusion of a specific inhibitor of Fn-EDA, 15 minutes post-reperfusion, into Apoe−/− mice attenuated myocardial I/R injury.
Conclusions:
Fn-EDA exacerbates TLR4-dependent myocardial I/R injury by promoting postischemic thrombo-inflammatory response. Targeting Fn-EDA may reduce cardiac damage following coronary artery recanalization after acute MI.
Keywords: Fibronectins, thrombosis, inflammation, myocardial ischemia, hyperlipidemia
Introduction
Acute myocardial infarction (MI) occurs most often because of coronary artery thrombosis caused by an unstable atherosclerotic plaque rupture. Despite advances in therapy, MI continues to be the common threat to life and health worldwide.(1) Reperfusion is the standard therapy following acute MI. Although reperfusion of blocked coronary artery either by percutaneous coronary intervention or fibrinolysis has shown promising clinical outcomes, evidence from several patients and animal models suggest that I/R injury by itself can further aggravate myocardial death in the peri-infarct region.(2) Potential mediators of I/R injury include endothelial dysfunction, oxidative stress, increased cell calcium, microvascular thrombosis, inflammation and other factors.(3)
Following MI, extracellular matrix (ECM) remodeling occurs that is known to play a critical role in the regulation of cellular responses, which mediate cardiac repair.(4) Accumulating evidence suggests that ECM protein fibronectins (Fns) promote thrombosis and inflammation,(5–8) two potential mediators of acute MI.(3) Fns play a pivotal role in several cellular processes through integrin-mediated actions.(9) Fn has multiple isoforms that are generated by alternative splicing of a single primary transcript at three sites: Extra Domain A (EDA), Extra Domain B (EDB), and the Type III Homologies Connecting Segment (IIICS). Two forms of Fns exist; plasma Fn (pFN) synthesized by hepatocytes and circulates as a soluble dimer in plasma, and cellular Fn (cFn) synthesized by several cell types including fibroblast, endothelial cells, and vascular smooth muscle cells and deposited as insoluble multimeric fibrils in the tissue ECM. cFN contains EDA (Fn-EDA) or EDB (Fn-EDB) or both in different proportions. Fn-EDA, but not Fn-EDB, is an endogenous ligand for innate immune receptor toll-like-receptor 4 (TLR4).(10) Notably, Fn-EDA is present in negligible amounts in the plasma of healthy humans but markedly elevated in patients with comorbid conditions including diabetes and hyperlipidemia.(11, 12)
Studies done in animal models have shown that Fn-EDA accumulates more rapidly, within 24 hours, in the infarcted area of reperfused hearts when compared to hearts of animal with permanent ligation.(13, 14) A previous study showed that deletion of the Fn-EDA in healthy wild-type (WT) mice prevent post-infarct remodeling and heart function at day 7 and 28 in permanent ischemia model; (15) however, molecular mechanisms remain to be elucidated yet. Although experiments done in WT mice are very useful in dissecting mechanisms of myocardial I/R injury, the inbred strains of WT mice do not model the effects of comorbid conditions such as hyperlipidemia. Notably, human MI occurs in the context of comorbid conditions such as hypertension, diabetes, and hyperlipidemia. Recent evidence from our and others lab suggests that Fn-EDA is pro-thrombotic, pro-inflammatory and contributes to atherosclerosis and ischemic stroke.(5, 8, 16–21) Based on recent pieces of evidence, we hypothesized that Fn-EDA contributes to acute myocardial I/R injury in the comorbid condition of hyperlipidemia. We found that Fn-EDA exacerbates myocardial I/R injury in the comorbid condition of hyperlipidemia by promoting postischemic thrombo-inflammation through TLR4 expressed on hematopoietic cells.
Materials and Methods
Animals
Fn-EDA−/−, Fn-EDA−/−Apoe−/−, TLR4−/−Apoe−/−, Fn-EDA−/−TLR4−/−Apoe−/− mice have been described previously.(16, 17, 21) Whenever possible, littermate control mice were studied. Mice were genotyped by PCR according to protocols from the Jackson Laboratory, and as described.(22) All the mice used in the present study were on the C57BL/6J background. Both male and female mice (littermates of age approximately 12-14 weeks) weighing 25-30 grams were utilized. The University of Iowa Animal Care and Use Committee approved all procedures.
Myocardial ischemia and reperfusion injury
We used left anterior descending coronary artery ligation model as described.(23) The mouse was anesthetized by injecting subcutaneously 0.15 ml/20 g body weight of a cocktail consisting of Ketamine (100 mg/kg body weight) and Xylazine (10 mg/kg body weight). The anesthetized mouse was placed in a supine position with its paws taped on a Plexiglas board before endotracheal intubation. Body temperature was maintained at 37°C ± 1 using a heating pad and monitoring with a rectal temperature probe. A middle cervical incision was made, and an endotracheal tube was passed through the exposed trachea. Mechanical ventilation (mini-vent, Harvard Apparatus, Boston, MA) was set to a stroke volume of 250 μL and 125 strokes per minute. The left pectoralis major muscles were retracted towards the right shoulder, and the left rectus thoracic and serratus anterior muscles were retracted towards the left with microretractors. Thoracotomy was performed in the intercostal space between 4th and 5th ribs, and self-retaining microretractors were used to expose the operating region while preserving rib integrity. A sterile polyamide 8-0 silk suture was passed under the left anterior descending (LAD) coronary artery, 2 mm distal to the left atrial appendage immediately inferior to the bifurcation of the major left coronary artery. For induction of ischemia, a short segment of PE-10 tubing was placed between the LAD and suture to protect the artery against traumatic injury, and the LAD was ligated. Mice were subjected to 60 minutes of ischemia, which was confirmed by blanching of the left ventricle. At the end of the ischemic period, the LAD ligation was removed, and reperfusion was confirmed by visual inspection of the left ventricle. The chest was closed with running sutures (one muscle suture layer and one skin suture layer) with sterile silk (6-0) suture. The mouse was gently removed from the ventilator and placed on a heating pad and monitored until fully recovered from anesthesia. Saline (300 μl) was injected intraperitoneally (IP) at the end of the surgical procedure. Sham-operated animals underwent identical surgical procedure except that the suture around the LAD coronary artery was not fastened. After 23 hours of reperfusion period, heparinized blood samples were collected for measuring plasma cardiac troponin I (cTnI) levels.
Assessment of Area at Risk and Infarct area
After reperfusion, myocardial infarct size was determined using a double-staining technique and a digital imaging system (infarct area/area at risk), as described previously with slight modification.(24) After 23 hours of reperfusion, coronary blood flow was again blocked with a sterile polyamide 8-0 silk suture, and Evans blue (2%) prepared in phosphate buffer saline (PBS) was injected into the right ventricle. For morphometric measurement, 1-mm serial sections were cut using a mouse Heart Matrix (Roboz surgical instrument). Sections were stained with 4% triphenyl-2, 3, 5-tetrazolium-chloride (TTC) for 20 min at 37° C. Sections were scanned on both sides, digitalized and infarct areas on each side was measured using the NIH Image J software. Evans blue stained area (blue staining, non-ischemic area), TTC stained area (red staining, ischemic area) and non-TTC stained areas (white, infarct area) were analyzed with Image J software. Myocardial infarct size (infarct area/area at risk %) was calculated as a percentage of the left ventricle area.
Troponin I levels
Plasma cardiac troponin I (cTnI) levels were measured with commercially available quantitative sandwich enzyme-linked immunoassay assay (ELISA) kit (Life Diagnostic) according to the manufacturer’s instructions.
Immunohistochemistry
All sections were deparaffinized, rehydrated, and subjected to heat-induced antigen retrieval as described.(16, 25) Briefly, sections were blocked with 5% serum at room temperature (RT), from the species in which the secondary antibody was raised. Endogenous peroxidase activity was quenched with 0.1% hydrogen peroxide in methanol for 15 min. Sections were stained with primary antibodies for platelet (anti-CD41 antibody, rat monoclonal, 1:100, Abcam), neutrophil (rat anti-mouse Ly6B.2; 1:100; Bio-rad) in the presence of 5% rabbit serum. After overnight incubation at 4°C, slides were washed PBS for 5 minutes and incubated with biotinylated secondary antibody for 1 hour at RT. Slides were then incubated with streptavidin-HRP for 40 minutes at RT, washed and incubated with DAB substrate until color develops. Slides were then washed and counter-stained with hematoxylin, mounted using aqueous mounting medium and examined under a light microscope (Olympus). Incubation without primary antibodies and with isotype-matched immunoglobulins was used as a negative control for immunostaining.
Quantification:
In four different regions of the infarct and surrounding area extravascular neutrophils (400X magnification) were quantified by counting the immunoreactive cells (brown color staining). Platelet (CD41 positive) thrombi were visualized at 200X magnification. CD41 positive thrombi area is reported as thrombi area index (%) using the following formula: (CD41 positive area/ total area of the section) X 100. NIH Image J software (with the plugin for individual cell analysis) was used for neutrophil and thrombi area quantification. Each mouse represents a mean of 16 fields from 4 serial sections (separated by 30 μm).
Neutrophil extracellular trap staining
All sections were subjected to heat-induced antigen retrieval. Sections were blocked with 5% normal goat serum in Tris-buffered saline at room temperature (RT). Sections were stained with the neutrophil antibody (1:50, rat Ly6 B.2, Biorad). After overnight incubation at 4°C, slides were washed thrice with PBS for 5 minutes and again incubated with the Citrullinated H3 antibody (1:200, citrulline R2 + R8 + R17; Abcam) for 6 hours RT. After washing, goat anti-rat IgG 488 (4 μg/ml, Invitrogen), goat anti-rabbit IgG 546 (4 μg/ml, Invitrogen) was added together. Nuclei were stained using an Antifade-mounting medium with Hoechst. Isotype-matched immunoglobulins were used as a negative control. Images were taken using Olympus BX51 fluorescent microscope. ImageJ software (NIH ImageJ, USA) was used for all the quantifications.
In situ apoptosis assay
Terminal deoxynucleotidyl-transferase mediated dUTP nick-end labeling (TUNEL) staining was performed at 23 hours after reperfusion injury.(24) Briefly, hearts were fixed for 24 hours in 4 % paraformaldehyde and paraffin embedded. Five μm thick sections were cut and treated with an in situ cell death detection kit (Roche Diagnostics) according to manufacturer’s instruction. The TUNEL signal was detected by an anti-fluorescein antibody conjugated with alkaline phosphatase, which generates a red-colored product, using Vector Red substrate kit (Vector Laboratories). Slides were counterstained with hematoxylin, mounted with an aqueous-based mounting medium, and examined under a light microscope (Olympus). Apoptotic cells were quantified by Image J (plugin individual cell analysis) software in four random fields of the infarcted and surrounding region at 400X magnification. The apoptotic index (%) is defined as: (number of TUNEL positive cell nuclei/ number of total cell nuclei) X 100. Each mouse represents a mean of 16 fields from four serial sections (separated by 30 μm).
Quantification of cellular Fn-EDA in plasma samples
Cellular Fn-EDA levels in the plasma were measured by sandwich enzyme-linked immunosorbent assay (ELISA) as described.(16) Briefly, microtiter plates were coated overnight at 4°C with primary antibody for cellular Fn-EDA (IST-9, 10 μg/mL, Abcam) diluted in 50 mM sodium carbonate buffer. 50 μl of plasma samples (diluted 1:2 in PBS) were incubated for 2 h in the coated wells at RT. After five washes biotinylated secondary antibody to FN (2 μg/ml diluted in blocking buffer) was added to wells and incubated for 1 h at RT. Following five washes avidin HRP solution (1:1000) in blocking buffer was added to wells and incubated for 30 minutes. Microtiter plates were washed five times, before adding 3, 3′, 5, 5′-tetramethylbenzidine substrate solution (Sigma) to the wells and the colorimetric reaction was stopped with H2SO4 (2M) after 10 min. Results were read in an ELISA microplate reader at A450 nm. Human cellular Fn (Sigma) was used for standards.
Bone marrow transplantation
The donor and recipient mice utilized for bone marrow BMT experiments were males (7-8 weeks in age) on the C57BL/6J background as described.(16) Recipient mice were irradiated with two doses of 6.5-Gy at an interval of 4 hours between the first and second irradiations. BM cells were aseptically extracted from excised femurs and tibias of euthanized donor mice. Bone marrow cells (1 X 107) under sterile environment were suspended in sterile PBS and injected into the retro-orbital venous plexus of lethally irradiated recipient mice. Post-transplantation, mice were maintained in sterile cages and fed autoclaved food and water ad libitum. Following different sets of BMT experiments were performed (Figure SI): 1) irradiated Apoe−/− mice reconstituted with BM from either Apoe−/− or TLR4−/−Apoe−/− donors, 2) irradiated Fn-EDA−/−Apoe−/− mice reconstituted with BM from either Fn-EDA−/−Apoe−/− or Fn-EDA−/−TLR4−/−Apoe−/− donors. Successful BMT was confirmed after four weeks by PCR for the presence of the genomic DNA of the respective donor mice in peripheral blood cells (not shown). Total blood cell counts were obtained using automated veterinary hematology analyzer (ADVIA) to ascertain that BMT did not affect the number of BM-derived blood cells (Table SI).
Treatment with an anti-fibronectin cellular antibody
Mice were infused with either anti-cellular fibronectin antibody (100 μg/mouse, monoclonal Fn-3E2; #F6140, Sigma, USA) or with control Ig isotype (100 μg/mouse, Rockland antibodies, and assays, USA) intravenously, 15 minutes after reperfusion.
Statistics
Results are reported as mean ± SEM. The number of experimental animals in each group was based on power calculations for the primary parameter (infarct area) with mean differences and standard deviations taken from pilot data at power 80% with the alpha of 0.05. For statistical analysis, Graph Pad Prism (version 7.03) was used. Shapiro-Wilk test was used to check normality and Bartlett’s test was used to check equal variance. The statistical significance was assessed using either unpaired Student’s t-test (normally distributed) or analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison tests (more than two groups). P<0.05 was considered to be statistically significant.
Results
Genetic deletion of Fn-EDA in Apoe−/− mice, but not in WT mice, reduces acute myocardial I/R injury
Just as in healthy humans, Fn-EDA is absent in the circulation and healthy arteries of wild-type (WT) mice.(20, 22) However, Fn-EDA reappears in the plasma and arteries of hyperlipidemic Apoe−/− mice, a response that is similar to that seen in human patients with atherosclerosis (Figure SII).(20, 26) First, we determined the effect of Fn-EDA deletion in wild-type (WT, C57Bl6/J) mice in myocardial I/R injury model (60 minutes of myocardial ischemia/ 23 hours of reperfusion). Infarct size and cTnI levels (an index of myocyte injury) in the plasma were comparable between Fn-EDA−/− and WT male mice (Figure SIII). To determine whether the elevated plasma levels of Fn-EDA present in the comorbid condition of hyperlipidemia contribute to acute myocardial I/R injury, we examined the effect of Fn-EDA deletion in Apoe−/− mice. To minimize the potential confounding effect of atherosclerotic lesions in Apoe−/− mice, which can impair blood flow and indirectly exacerbate the effect of myocardial I/R injury, Fn-EDA−/−Apoe−/− and Apoe−/− mice were fed a normal chow diet until 12-14 weeks of age, an age at which minimal vascular lesions are found (not shown). Earlier we have reported that total plasma cholesterol, triglycerides, and blood pressure are comparable between these groups.(16) In agreement with previous observations,(16, 20) we observed significantly elevated Fn-EDA levels in the plasma of Apoe−/− mice. Fn-EDA was not detected in the plasma of Fn-EDA−/−Apoe−/− mice (Figure SII). Infarct size (%) was markedly decreased (~ 47%) in Fn-EDA−/−Apoe−/− mice when compared with Apoe−/− mice (59.8 ± 2.0 % vs. 31.9 ± 1.3 %, P<0.05; Figure 1A). Reduced infarct size in Fn-EDA−/−Apoe−/− mice was concomitant with decreased cTnI levels (17.8 ± 1.8 vs. 9.8 ± 1.5, P<0.05; Figure 1B). Myocardial infarcts were not observed in sham animals (not shown). Similar to male mice, female Fn-EDA−/−Apoe−/− mice exhibited significantly smaller infarct size and decreased cTnI levels when compared with Apoe−/− mice (P<0.05, Figure 1CD) suggesting that, irrespective of gender, Fn-EDA exacerbates acute myocardial I/R injury.
Figure 1. Irrespective of gender, deletion of Fn-EDA in Apoe−/− mice improves acute myocardial I/R injury.
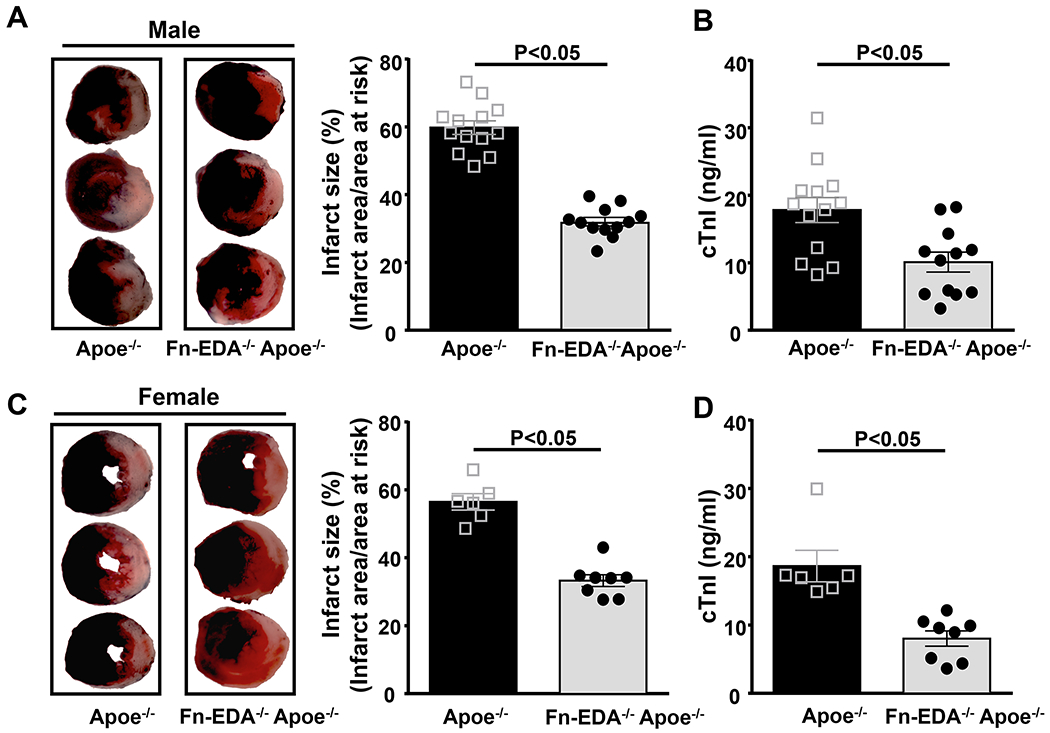
A&C. Left panels show representative 2, 3, 5-triphenyl-tetrazolium chloride stained serial heart sections from one mouse of each genotype after 60 minutes of ischemia and 23 hours of reperfusion. The infarcted area is white. Right panels show quantification of infarct size (%) in male (N=12-13 mice/group) and female mice (N=6-8 mice/group). B&D. Plasma cardiac troponin I (cTnI) levels in male and female mice as assessed before sacrifice at 24 hours. Each dot represents a single mouse. Data are mean ± SEM. Statistical analysis: unpaired Student’s t-test.
Fn-EDA−/−Apoe−/− mice exhibited reduced postischemic thrombo-inflammation and apoptosis
We and others have previously shown that Fn-EDA promotes thrombosis and inflammation in experimental models.(17, 19, 20) To determine whether attenuated myocardial I/R injury in the Fn-EDA−/−Apoe−/− mice is concomitant with less susceptibility to postischemic thrombosis and inflammation, we quantified intravascular thrombi and neutrophil influx in the infarcted and surrounding regions after 60 minutes of ischemia and 23 hours of reperfusion. Immunostaining revealed a significant decrease in intravascular platelet thrombi (CD41-positive thrombi) and neutrophils influx (Ly-6B.2-positive cells) (Figure 2AB, P<0.05 vs. Apoe−/− mice). Recent evidence suggests that neutrophil extracellular traps (NETs) may exacerbate acute myocardial I/R injury by promoting thrombo-inflammation.(27, 28) Therefore, we quantitated NETs. Immunostaining revealed decreased NETs (citrullinated H3-positive cells) in the infarcted region of Fn-EDA−/−Apoe−/− mice (Figure 2C, P<0.05 versus Apoe−/− mice). Double immunofluorescence analysis confirmed that citrullinated H3-positive cells were neutrophils (Figure S4). Next, we assessed whether attenuated myocardial I/R injury in the Fn-EDA−/−Apoe−/− mice is associated with decreased apoptosis. We quantified apoptotic index (apoptotic cells/total cells) by TUNEL staining. We found a significantly lower apoptotic index in the Fn-EDA−/−Apoe−/− mice when compared to Apoe−/− mice (29.7 ± 1.5 vs. 55.3 ± 1.8, P<0.05, Figure 2D). Double immunostaining (TUNEL/α-actinin-positive cells) revealed that ~85 % of the apoptotic cells were myocytes (Figure S5). Together, these results suggest that Fn-EDA promotes postischemic thrombosis, neutrophil influx, NETs release, and myocyte apoptosis and thereby exacerbates acute myocardial I/R injury in the comorbid condition of hyperlipidemia.
Figure 2. Fn-EDA−/−Apoe−/− mice exhibited reduced postischemic thrombosis, neutrophil influx, neutrophil extracellular traps (NETs), and apoptosis.
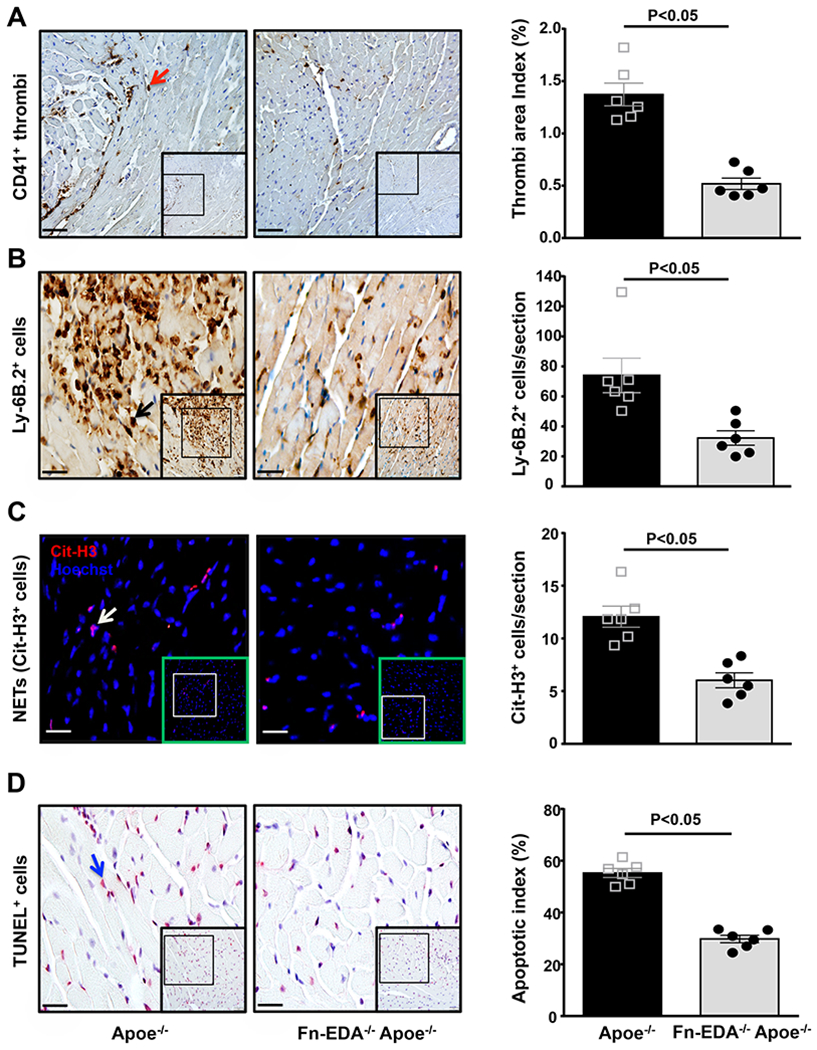
Left panels show representative low-magnification (boxed region) and higher-magnification (insert in boxed region) for A, Platelet thrombi (brown CD41-positive platelets as indicated by red arrow); B, Neutrophils (brown Ly6B.2-positive cells as indicated by black arrow); C, NETs (citrullinated H3-positive cells as indicated by white arrow); D, Apoptotic cells (red TUNEL-positive cells indicated by blue arrow) and counterstained with hematoxylin from one mouse of each genotype. Right panels show quantification of immunoreactive cells (A, B & C) and TUNEL positive cells (D) in the infarcted and surrounding region. Each dot is a mean of 16 fields from four serial sections (separated by 30 μm) per mouse. Data are mean ± SEM. Statistical analysis: unpaired Student’s t-test. Scale bar =A, 50 μm : B, C& D, 20 μm.
TLR4 contributes to the Fn-EDA-mediated acute myocardial I/R injury
To determine the possible mechanism by which Fn-EDA exacerbates acute myocardial I/R injury in the comorbid condition of hyperlipidemia, we focused on TLR4 because it is an endogenous receptor for Fn-EDA,(10) and contributes to myocardial I/R injury.(29) Male Apoe−/− and Fn-EDA−/−Apoe−/− mice on TLR4−/− background were subjected to 60 minutes of myocardial ischemia followed by 23 hours of reperfusion. Control mice were Apoe−/− and Fn-EDA−/−Apoe−/− littermates. Compared with Apoe−/− mice, infarct size (%) was significantly decreased in TLR4−/−Apoe−/− mice (52.3 ± 2.8 versus 34.7 ± 1.1 %, P<0.05; Figure 3A). Reduced infarct size in TLR4−/−Apoe−/− mice was concomitant with reduced cTnI levels (P<0.05; Figure 3B). To rule out the possibility that global deletion of TLR4 may reduce infarct size in Apoe−/− mice independently of Fn-EDA, we compared infract size in Fn-EDA−/−Apoe−/− and Fn-EDA−/−TLR4−/−Apoe−/− mice. Infarct size (%) and cTnI levels were comparable between Fn-EDA−/−Apoe−/− and Fn-EDA−/−TLR4−/−Apoe−/− mice (Figure 3 AB), suggesting that Fn-EDA requires TLR4 for myocardial I/R injury exacerbation. Immunostained sections of the infarcted and peri-infarcted regions revealed a significant decrease in postischemic intravascular thrombosis (CD41-positive), neutrophil influx (Ly-6B.2-positive cells), NETs (citrullinated H3-positive) and apoptotic index (TUNEL staining) in TLR4−/−Apoe−/− mice (P<0.05 vs. Apoe−/− mice, Figure 4 A–D). No significant differences in the postischemic thrombosis, neutrophil influx, NETs, and apoptosis were observed between Fn-EDA−/−TLR4−/−Apoe−/− mice and Fn-EDA−/−Apoe−/− mice (Figure 4 A–D). Together these results suggest that Fn-EDA exacerbates myocardial I/R injury most likely through TLR4.
Figure 3. Genetic ablation of TLR4 improves acute myocardial I/R injury in Apoe−/− mice, but not in Fn-EDA−/−Apoe−/− mice.

A. Left panels show representative 2, 3, 5-triphenyl-tetrazolium chloride stained serial heart sections from one mouse of each genotype after 60 minutes of ischemia and 23 hours of reperfusion. The right panel shows infarct size (%) in male mice of each genotype (N=7-11 mice/group). B. Plasma cTnI levels in male mice as assessed before sacrifice at 24 hours. Each dot represents a single mouse. Data are mean ± SEM. Statistical analysis: ANOVA followed by Bonferroni’s multiple comparison tests. NS= nonsignificant.
Figure 4. Fn-EDA/TLR4 axis exacerbates acute myocardial I/R injury by promoting thrombosis, neutrophil influx, neutrophil extracellular traps (NETs), and apoptosis.
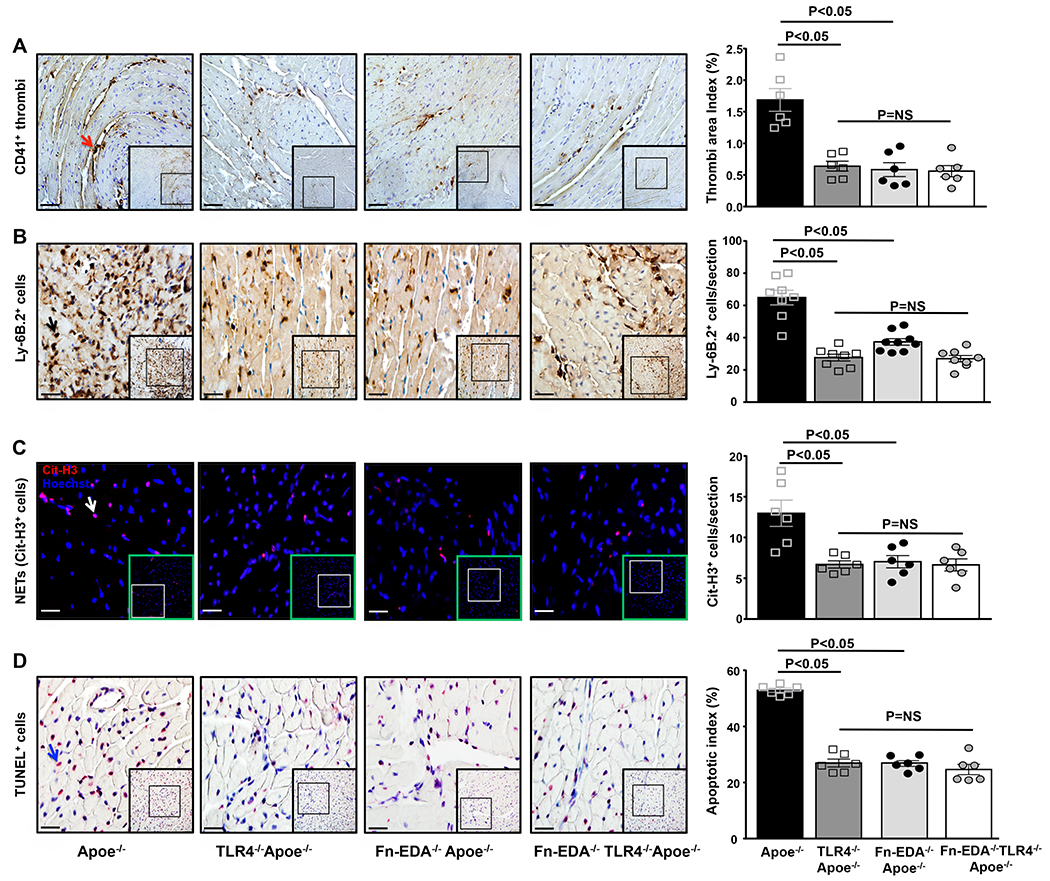
Left panels show representative low-magnification (boxed region) and higher-magnification (insert in boxed region) stained for A, Platelet thrombi (brown CD41-positive platelets as indicated by red arrow); B, Neutrophils (brown Ly6B.2-positive cells as indicated by black arrow); C, NETs (citrullinated H3-positive cells as indicated by white arrow); D, Apoptotic cells (red TUNEL-positive cells indicated by blue arrow) and counterstained with hematoxylin from one mouse of each genotype. Right panels show quantification of immunoreactive cells (A, B & C) and TUNEL-positive cells (D) in the infarcted and surrounding region. Each dot is a mean of 16 fields from four serial sections (separated by 30 μm) per mouse. Data are mean ± SEM. Statistical analysis: ANOVA followed by Bonferroni’s multiple comparison tests. Scale bar =A, 50 μm, B, C&D, 20 μm. NS= nonsignificant.
Fn-EDA exacerbates acute myocardial I/R injury through TLR4 expressed on cells of hematopoietic origin
TLR4 is expressed on hematopoietic cells and non-hematopoietic cells including endothelial cells. To investigate the cellular source of TLR4 that contributes to Fn-EDA-mediated myocardial I/R injury exacerbation, we transplanted irradiated Apoe−/− mice with BM from either Apoe−/− or TLR4−/− Apoe−/− mice. This BMT protocol resulted in chimeric mice that express TLR4 on non-hematopoietic cells, but lack TLR4 in cells of hematopoietic origin (Figure SI). Compared with Apoe−/− BM→ Apoe−/− mice, TLR4−/− Apoe−/−-BM→ Apoe−/− mice showed a significant decrease in infarct size and cTnI levels (Figure 5AB). Reduced infarct size in TLR4−/− Apo−/−-BM→ Apoe−/− mice was concomitant with a decrease in postischemic intravascular thrombosis, neutrophil influx and apoptosis (P<0.05 vs. Apoe−/− BM→ Apoe−/−, Figure 5C–E). To exclude the possibility that these in vivo effects are not simply mediated by a TLR4 deletion in hematopoietic cells, we compared infarct size in Fn-EDA−/−Apoe−/−-BM→Fn-EDA−/−Apoe−/− mice and Fn-EDA−/−TLR4−/−Apoe−/−-BM →Fn-EDA−/−Apoe−/− mice. Infarct size, plasma cTnI levels, intravascular thrombosis, neutrophil influx and apoptosis were comparable between these groups (Figure 5A–E) suggesting that Fn-EDA promotes acute myocardial I/R injury via TLR4 expressed on cells of hematopoietic origin. Complete blood counts were comparable suggesting that BMT did not affect the number of BM-derived blood cells (Table SI).
Figure 5. Non-hematopoietic cells-derived Fn-EDA exacerbates myocardial I/R injury through TLR4 expressed on cells of hematopoietic origin.
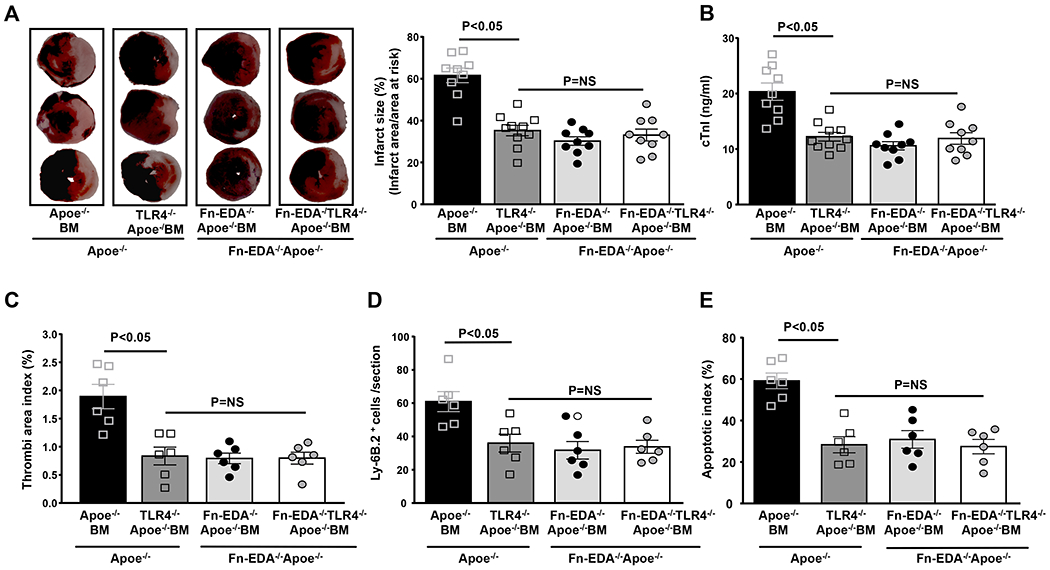
A. Left panels show representative 2, 3, 5-triphenyl-tetrazolium chloride stained serial heart sections from one mouse of each genotype after 60 minutes of ischemia and 23 hours of reperfusion. The right panel shows infarct size (%) in male mice of each genotype (N=9-10 mice/group). B. Plasma cTnI levels in male mice as assessed before sacrifice at 24 hours. C, D & E. Quantification of microthrombi (CD41-positive), neutrophils (Ly6B.2-positive) and apoptotic cells (TUNEL-positive). Each dot is a mean of 16 fields from four serial sections (separated by 30 μm) per mouse. Data are mean ± SEM. Statistical analysis: ANOVA followed by Bonferroni’s multiple comparison tests. NS= nonsignificant.
Infusion of anti-Fn-EDA Ig attenuates myocardial I/R injury in the Apoe−/− mice
We next evaluated the therapeutic potential of targeting plasma Fn-EDA using specific antibodies to the EDA of Fn. Male Apoe−/− mice were subjected to 60 minutes of ischemia followed by 23 hours of reperfusion. To mimic clinical conditions of acute therapy, we infused anti-Fn-EDA Ig intravenously into Apoe−/− mice 15 minutes after reperfusion. Apoe−/− mice treated with control Ig served as controls. Both groups of mice appeared normal during and after treatment. Infusion of anti-Fn-EDA Ig or control Ig did not significantly affect total Fn levels in the plasma (data not shown) and as reported.(16) Body weight was comparable among treated and control mice (not shown). Infarct size (37.82 ± 2.15 versus 58.79 ± 3.49 %, P<0.05) and cTnI levels were significantly reduced in anti-Fn-EDA Ig-treated Apoe−/− mice compared with control Ig-treated Apoe−/− mice (Figure 6).
Figure 6. Targeting plasma Fn-EDA with anti-Fn-EDA Ig after reperfusion significantly reduces infarct size.
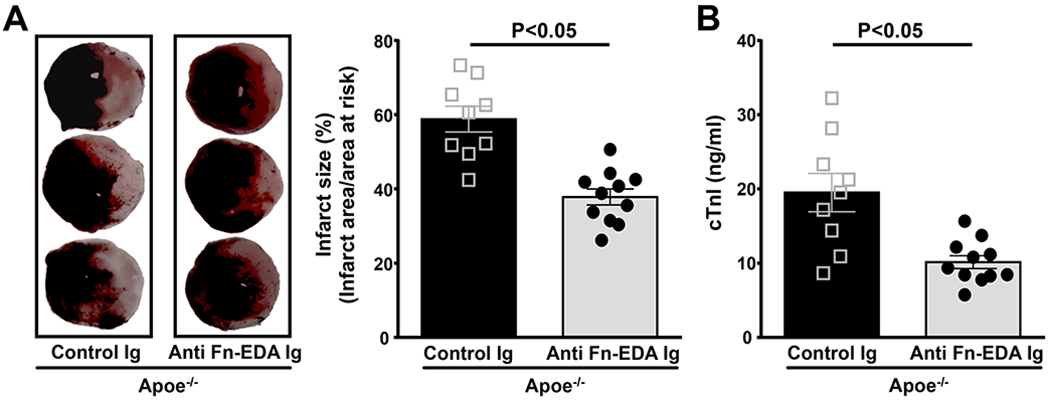
A. The left panels show representative 2, 3, 5-triphenyl-tetrazolium chloride stained serial heart sections from control and anti-Fn-EDA Ig treated Apoe−/− male mice. The right panel shows quantification (N=9-11 mice/group). B. cTnI levels as assessed before sacrifice at 24 hours. Data are mean ± SEM. Statistical analysis: unpaired Student’s t-test.
Discussion
We believe that the findings of this study may have clinical significance for the following reasons: First, human MI occurs in the context of comorbid conditions including hyperlipidemia. Fn-EDA, which is negligible present in the plasma of healthy humans, is elevated in patients with hyperlipidemia and atherosclerotic disease. We provide genetic evidence that irrespective of gender Fn-EDA promotes TLR4-dependent acute myocardial I/R injury in the comorbid condition of hyperlipidemia most likely by promoting thrombo-inflammation. Second, mechanistically, we show that Fn-EDA promotes acute myocardial I/R injury through TLR4 expressed on the hematopoietic cells. Third, as a potential therapeutic strategy, we show that infusion of an antibody specific to Fn-EDA 15 minutes after reperfusion attenuated myocardial I/R injury in the comorbid condition of hyperlipidemia.
Previous studies suggested that Fn-EDA rapidly accumulates in the infarcted area of reperfused hearts within 24 hours in animal models of myocardial I/R injury when compared to permanent ligation model.(13, 14) Therefore, we determined the role of Fn-EDA in the pathophysiology of MI in I/R injury model. We found that Fn-EDA−/− mice on hyperlipidemic Apoe−/− background, but not on WT, exhibited significantly reduced infarct size at 24 hours. Our results in WT mice are in agreement with the previous study that showed comparable infarct size in WT and Fn-EDA−/− mice in the permanent coronary artery ligation model.(15) However, in this study authors reported that lack of Fn-EDA in WT mice promotes survival, prevents adverse remodeling and heart function deterioration at later stages (day 7 and 28) concomitant with reduced fibrosis and inflammation. The explanation for the apparent discrepancies in the infarct size at 24 hours in the WT and Apoe−/− mice could be related to the presence of elevated levels of Fn-EDA in the plasma of Apoe−/−, but not in WT mice. Increased vascular permeability in the infarcted region would have resulted in extravasation of plasma Fn-EDA that will form the provisional matrix, which will serve as a scaffold for infiltrating inflammatory cells thus exacerbating infarct size.
Following reperfusion of the acute myocardial ischemia, several factors including endothelial dysfunction and intravascular thrombosis are known to influence infarct size.(3) We found that Fn-EDA−/−Apoe−/− mice exhibited reduced intravascular thrombosis in the reperfused heart at 24 hours. We speculate that Fn-EDA may promote myocardial infarct exacerbation by promoting intravascular thrombosis because of following evidence. First, several studies have shown a prothrombotic role for Fn-EDA in experimental models.(5, 7, 16) Second, a recent study found that Fn-EDA was associated with increased clot density in type 2 diabetic patients, thus favoring a prothrombotic state.(12) Furthermore, we showed that Fn-EDA promotes intravascular thrombosis in the reperfused heart through TLR4. Previously, we have demonstrated Fn-EDA by interacting with platelet-specific TLR4 promotes experimental thrombosis.(7) Platelets have all the molecular machinery necessary for signal transduction downstream of TLR4, including components of the canonical NF-κB pathway, which has been shown to play a role in platelet function.(30) The possibility that TLR4-independent mechanisms, such as an increased tendency of Fn-EDA to deposit as fibrils,(31) or an alteration in the arginylglycylaspartic acid (RGD) motif present in the Fn type III-10 domain that results in enhanced binding to platelet integrin’s αIIbβ3, ανβ3 and α5β1,(9, 31) may also contribute to Fn-EDA-mediated intravascular thrombosis cannot be ruled out.
I/R injury elicits a strong post-MI inflammatory response that further promotes myocyte damage in peri-infarcted regions.(3) TLR4 is known to exacerbate myocardial I/R by promoting neutrophil influx, lipid peroxides, and complement deposition.(29) We found that Fn-EDA promotes TLR4-dependent neutrophil influx in the infarcted and peri-infarcted regions. These results are in line with other studies that have shown a role of Fn-EDA/TLR4 axis in promoting inflammation.(16, 17) Several studies suggest that neutrophils recruited to the site of infarction within hours of ischemia exacerbate myocardial I/R injury.(32–34) The possible mechanisms by which neutrophils could promote myocardial I/R injury includes capillary occlusion, the release of vasoconstrictive mediators, production of reactive oxygen species, release of proteolytic enzymes such as elastase and MMP9, the release of other inflammatory mediators such as platelet-activating factor and arachidonic acid metabolites.(35) Additionally, we found that Fn-EDA mediates TLR4-dependent NETs release in the infarcted and peri-infarcted regions. Platelet TLR4 is known to induce NETs release and recent evidence from murine studies suggests that NETs exacerbate acute myocardial I/R injury most likely by promoting thrombo-inflammation.(28, 36) Future studies utilizing bone marrow transplantation from platelet-specific TLR deficient mice are required to elucidate whether Fn-EDA exacerbates myocardial I/R injury through platelet TLR4. Although our studies indicate that Fn-EDA/TLR4 axis contributes to the neutrophil influx, the possibility that Fn-EDA by directly interacting with leukocyte integrin’s including α4β1 and α9β1 could promote neutrophil infiltration could not be ruled out.
Recently, we have reported that Fn-EDA/TLR4 axis promoted apoptosis in advanced atherosclerotic plaques.(21) Myocardial I/R injury is known to promote early myocyte apoptosis that was associated with increased neutrophil accumulation in the infarcted region.(37) We speculated that Fn-EDA/TLR4 axis might exacerbate myocardial I/R injury by promoting apoptosis. Indeed, we found Fn-EDA modulates TLR4-dependent myocyte apoptosis in the reperfused hearts. Apoptosis is regulated by multiple factors, such as the production of reactive oxygen species by myeloperoxidase and NADPH oxidase, the release of inflammatory cytokines TNF-α and IL-6, and neutrophils.(38, 39) Hematopoietic and non-hematopoietic cells express TLR4. Using BMT studies, we found that Fn-EDA contributes to myocardial I/R injury via TLR4 expressed on hematopoietic cells. Finally, we evaluated whether specific inhibition of Fn-EDA during reperfusion will attenuate myocardial I/R injury. We found that infusion of anti-Fn-EDA Ig 15 minutes after reperfusion significantly reduced myocardial infarct size. Future studies will be needed to determine whether the combination of anti-Fn-EDA Ig with thrombolytic therapy will reduce infarct size at early and later stages following MI.
Our studies have some limitations. First, the present study did not define the specific cell type that contributes to Fn-EDA-mediated MI exacerbation. More complex studies using endothelial cell-specific Fn-EDA−/− or smooth muscle cell-specific Fn-EDA−/− mice are required to precisely dissect the specific role of each cell type in Fn-EDA-mediated MI exacerbation. Second, we have not studied heart function and other parameters of remodeling at later time points (day 7 and 28). Nevertheless, our findings demonstrate that Fn-EDA plays a causative role in myocardial I/R injury exacerbation in the comorbid condition of hyperlipidemia most likely by promoting thrombo-inflammation. The mechanistic insights provided by the current study suggest that Fn-EDA should not only be considered a marker of vascular injury but an important factor that may exacerbate MI following coronary artery recanalization in hyperlipidemic patients.
Supplementary Material
What is known in this topic
Fn-EDA, an endogenous ligand for TLR4, is markedly elevated in patients with comorbid conditions including diabetes and hyperlipidemia, which are risk factors for myocardial infarction.
Deletion of the Fn-EDA in WT mice prevents post-infarct remodeling and heart function at day 7 and 28 in permanent ischemia model; however, molecular mechanisms remain to be elucidated yet.
What this paper adds
We provide genetic evidence that Fn-EDA exacerbates TLR4-dependent myocardial I/R injury in the comorbid condition of hyperlipidemia by promoting postischemic thrombo-inflammatory response.
Targeting Fn-EDA may reduce cardiac damage following reperfusion therapy after acute myocardial infarction.
Acknowledgment
A.K.C lab is supported by grants from the National Heart, Lung and Blood Institute of the National Institutes of Health grants (R35HL139926) and by Established Investigator Award 18EIA33900009 from American Heart Association.
Footnotes
Conflicts of Interest
None.
References
- 1.Writing Group M, Mozaffarian D, Benjamin EJ, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016; 133(4): e38–60. [DOI] [PubMed] [Google Scholar]
- 2.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007; 357(11): 1121–35. [DOI] [PubMed] [Google Scholar]
- 3.Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol 2010; 106(3): 360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest 2017; 127(5): 1600–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chauhan AK, Kisucka J, Cozzi MR, et al. Prothrombotic effects of fibronectin isoforms containing the EDA domain. Arterioscler Thromb Vasc Biol 2008; 28(2): 296–301. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Reheman A, Spring CM, et al. Plasma fibronectin supports hemostasis and regulates thrombosis. J Clin Invest 2014; 124(10): 4281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prakash P, Kulkarni PP, Lentz SR, et al. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll-like receptor 4. Blood 2015; 125(20): 3164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Gallant RC, Ni H. Extracellular matrix proteins in the regulation of thrombus formation. Curr Opin Hematol 2016; 23(3): 280–7. [DOI] [PubMed] [Google Scholar]
- 9.White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. J Pathol 2008; 216(1): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 2001; 276(13): 10229–33. [DOI] [PubMed] [Google Scholar]
- 11.Kanters SD, Banga JD, Algra A, et al. Plasma levels of cellular fibronectin in diabetes. Diabetes Care 2001; 24(2): 323–7. [DOI] [PubMed] [Google Scholar]
- 12.Konieczynska M, Bryk AH, Malinowski KP, et al. Interplay between elevated cellular fibronectin and plasma fibrin clot properties in type 2 diabetes. Thromb Haemost 2017; 117(9): 1671–8. [DOI] [PubMed] [Google Scholar]
- 13.Carlyle WC, Jacobson AW, Judd DL, et al. Delayed reperfusion alters matrix metalloproteinase activity and fibronectin mRNA expression in the infarct zone of the ligated rat heart. J Mol Cell Cardiol 1997; 29(9): 2451–63. [DOI] [PubMed] [Google Scholar]
- 14.Knowlton AA, Connelly CM, Romo GM, et al. Rapid expression of fibronectin in the rabbit heart after myocardial infarction with and without reperfusion. J Clin Invest 1992; 89(4): 1060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arslan F, Smeets MB, Riem Vis PW, et al. Lack of Fibronectin-EDA Promotes Survival and Prevents Adverse Remodeling and Heart Function Deterioration After Myocardial Infarction. Circ Res 2011; 108(5): 582–92. [DOI] [PubMed] [Google Scholar]
- 16.Dhanesha N, Ahmad A, Prakash P, et al. Genetic Ablation of Extra Domain A of Fibronectin in Hypercholesterolemic Mice Improves Stroke Outcome by Reducing Thrombo-Inflammation. Circulation 2015; 132(23): 2237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doddapattar P, Gandhi C, Prakash P, et al. Fibronectin Splicing Variants Containing Extra Domain A Promote Atherosclerosis in Mice Through Toll-Like Receptor 4. Arterioscler Thromb Vasc Biol 2015; 35(11): 2391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gondokaryono SP, Ushio H, Niyonsaba F, et al. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J Leukoc Biol 2007; 82(3): 657–65. [DOI] [PubMed] [Google Scholar]
- 19.Khan MM, Gandhi C, Chauhan N, et al. Alternatively-spliced extra domain a of fibronectin promotes acute inflammation and brain injury after cerebral ischemia in mice. Stroke 2012; 43(5): 1376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan MH, Sun Z, Opitz SL, et al. Deletion of the alternatively spliced fibronectin EIIIA domain in mice reduces atherosclerosis. Blood 2004; 104(1): 11–8. [DOI] [PubMed] [Google Scholar]
- 21.Doddapattar P, Jain M, Dhanesha N, et al. Fibronectin Containing Extra Domain A Induces Plaque Destabilization in the Innominate Artery of Aged Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muro AF, Chauhan AK, Gajovic S, et al. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. The Journal of cell biology 2003; 162(1): 149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michael LH, Entman ML, Hartley CJ, et al. Myocardial ischemia and reperfusion: a murine model. The American journal of physiology 1995; 269(6 Pt 2): H2147–54. [DOI] [PubMed] [Google Scholar]
- 24.Gandhi C, Motto DG, Jensen M, et al. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood 2012; 120(26): 5224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhanesha N, Doddapattar P, Chorawala MR, et al. ADAMTS13 Retards Progression of Diabetic Nephropathy by Inhibiting Intrarenal Thrombosis in Mice. Arterioscler Thromb Vasc Biol 2017; 37(7): 1332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Keulen JK, de Kleijn DP, Nijhuis MM, et al. Levels of extra domain A containing fibronectin in human atherosclerotic plaques are associated with a stable plaque phenotype. Atherosclerosis 2007; 195(1): e83–91. [DOI] [PubMed] [Google Scholar]
- 27.de Boer OJ, Li X, Teeling P, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost 2013; 109(2): 290–7. [DOI] [PubMed] [Google Scholar]
- 28.Savchenko AS, Borissoff JI, Martinod K, et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 2014; 123(1): 141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oyama J, Blais C Jr., Liu X, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 2004; 109(6): 784–9. [DOI] [PubMed] [Google Scholar]
- 30.Spinelli SL, Casey AE, Pollock SJ, et al. Platelets and megakaryocytes contain functional nuclear factor-kappaB. Arterioscler Thromb Vasc Biol 2010; 30(3): 591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guan JL, Trevithick JE, Hynes RO. Retroviral expression of alternatively spliced forms of rat fibronectin. The Journal of cell biology 1990; 110(3): 833–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta J, Dinerman J, Mehta P, et al. Neutrophil function in ischemic heart disease. Circulation 1989; 79(3): 549–56. [DOI] [PubMed] [Google Scholar]
- 33.Litt MR, Jeremy RW, Weisman HF, et al. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation 1989; 80(6): 1816–27. [DOI] [PubMed] [Google Scholar]
- 34.Dreyer WJ, Michael LH, West MS, et al. Neutrophil accumulation in ischemic canine myocardium. Insights into time course, distribution, and mechanism of localization during early reperfusion. Circulation 1991; 84(1): 400–11. [DOI] [PubMed] [Google Scholar]
- 35.Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res 1999; 43(4): 860–78. [DOI] [PubMed] [Google Scholar]
- 36.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007; 13(4): 463–9. [DOI] [PubMed] [Google Scholar]
- 37.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res 1996; 79(5): 949–56. [DOI] [PubMed] [Google Scholar]
- 38.Nian M, Lee P, Khaper N, et al. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res 2004; 94(12): 1543–53. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura M, Wang NP, Zhao ZQ, et al. Preconditioning decreases Bax expression, PMN accumulation and apoptosis in reperfused rat heart. Cardiovasc Res 2000; 45(3): 661–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.