

Abstract
Background
Focal cerebral ischaemia causes release of excitatory amino acid (EAA) neurotransmitters, principally glutamate, with resultant over‐stimulation of EAA receptors and downstream pathways. Excess glutamate release is a pivotal event in the evolution of irreversible ischaemic damage in animal models of ischaemia, and drugs that modulate glutamate action either by inhibiting its release, or blocking post‐synaptic receptors, are potent neuroprotective agents. Many clinical trials with EAA modulating drugs have been conducted, none individually demonstrating efficacy.
Objectives
To synthesise all the available data on all different classes of EAA modulators and to evaluate evidence of effects on outcome systematically.
Search methods
We searched the Cochrane Stroke Group Trials Register (last searched May 2001). In addition, we searched MEDLINE and EMBASE, handsearched conference proceedings from European, International, American Heart Association and Princeton conferences on Stroke; American Neurological Association and American Academy of Neurology meetings from 1992 to 2001; and had direct contact with individual investigators and pharmaceutical companies.
Selection criteria
Randomised, controlled studies giving agents with pharmacological properties that included modification of release of EAAs, or blockade of EAA receptors, in stroke within 24 hours of onset. Efficacy analysis was restricted to trials with a parallel group design: dose escalation studies were excluded. Intention‐to‐treat analyses were performed on all data. Outcome had to be reported in terms of death or dependence one to 12 months after the acute event.
Data collection and analysis
Data were available for 36 of 41 relevant trials identified, involving 11,209 participants. Data were unavailable for 632 participants (517 in trials fulfilling criteria for efficacy analysis). Seven trials did not report disability data, which were available for 29 trials involving 10,802 participants. Twenty‐one of these trials, involving 10,342 participants, were parallel group studies included in the primary efficacy analysis. Efficacy analysis included data derived from nine trials not primarily designed to assess efficacy (1022 participants). The primary (efficacy) end‐point was the proportion of patients dead or disabled at final follow up (defined by Barthel Index < 60 at three months by preference). Mortality was a secondary end‐point. Drugs were considered as individual agents, and also grouped principally into categories of ion channel modulators (glutamate release inhibition) and NMDA antagonists.
Main results
There was no significant heterogeneity of outcome amongst individual drugs, or of drug classes either for the primary efficacy analysis (death or dependence) or for mortality at final follow up. For the primary efficacy analysis, odds of death or dependence were 1.03 (95% confidence interval (CI) 0.96 to 1.12), and for mortality 1.02 (0.92 to 1.12). Neither ion channel modulators (death or dependence 1.02 (0.90 to 1.16)) nor NMDA antagonists (death or dependence 1.05 (0.95 to 1.16)) differed from the principal analysis including all compounds. Trends for increased mortality with three NMDA antagonists were seen ‐ selfotel (OR 1.19 (0.81 to 1.74)), aptiganel (OR 1.32 (0.91 to 1.93)) and gavestinel (OR 1.12 (0.95 to 1.32)) ‐ but this did not achieve significance for the NMDA antagonists considered as a class (1.09 (0.96 to 1.23)). Aptiganel was also associated with a trend towards worse functional outcome (OR 1.20 (0.88 to 1.65)) although this was not the case for either of the other two compounds. No statistically significant detriment of psychotomimetic NMDA antagonists was found, although a trend towards higher mortality in this sub‐group was seen (OR 1.25 (0.96 to 1.64)).
Authors' conclusions
There was no evidence of significant benefit or harm from drugs modulating excitatory amino acid action. Reduction of death or dependence by 8% or more has been excluded for gavestinel and lubeluzole, which contribute most of the data for this review. However, mechanistic understanding of neuroprotection is too poor to extrapolate from these two failed development plans to all glutamate modulators. Further clinical trials of neuroprotective agents remain justified, since confidence limits around estimates of effect remain wide for most agents, and cannot reliably exclude benefit. Although numbers of patients are too small to confirm or refute a trend towards increased mortality with some NMDA antagonists, further commercial development of these agents is exceedingly unlikely.
Plain language summary
Excitatory amino acid antagonists for acute stroke
There is no evidence of benefit from excitatory amino acid antagonists for acute stroke. Most strokes are caused by blockage of an artery with consequent loss of blood supply. Some brain tissue supplied by the artery suffers severe reduction in blood flow, and brain cells quickly die, but some tissue maintains enough blood flow to keep it alive for a period of time ‐ probably hours in most individuals. However, the loss of blood flow sets in motion a series of events that will eventually kill the tissue. The processes that cause damage to progress involve chemical changes, one of which is the release of large amounts of glutamate (an excitatory amino acid), a substance normally used for signalling between brain cells. In large amounts, glutamate is toxic. A number of drugs have been developed to block either the release of glutamate, or the sites to which it binds on brain cells. These drugs were highly effective in animal studies. Individual clinical trials in stroke patients did not confirm benefit for any of the drugs, however. This review confirms that there are no overall benefits for these drugs in stroke, although only two of them have been tested in a large enough population to be reasonably confident that they have no major effects. Some drugs may be harmful. Over 11,000 patients have participated in trials of 13 different drugs that inhibit glutamate release or binding, but two‐thirds of all data are from trials of just two drugs. For most drugs in this class, trials have been too small to provide conclusive evidence of harm or benefit. Major differences among individual drugs mean that it is impossible to conclude that all drugs with this mode of action are ineffective. Further trials remain justified, and several are ongoing.
Background
Experimental focal cerebral ischaemia causes excess release of excitatory amino acid (EAA) neurotransmitters, particularly glutamate, from presynaptic vesicles. Ischaemia also prevents normal reuptake of glutamate, with consequently very high synaptic concentrations. Glutamate acts at post‐synaptic receptors, notably the N‐methyl D‐aspartate (NMDA) receptor complex to promote entry of calcium and sodium to neurones, and the alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid (AMPA) receptor to promote principally sodium entry. Resultant cellular depolarisation and calcium overload activates intracellular second messenger systems with consequent cell death. Glutamate is toxic in neuronal cell culture and in vivo. In preclinical models of stroke, antagonists of glutamate release or of post‐synaptic glutamate receptors significantly reduce the volume of histological neuronal infarction, and improve functional recovery, even when administered up to several hours after the onset of ischaemia (McCulloch 1992). Drugs that modulate EAA toxicity are consistently protective across species, laboratories, investigators, and models.
EAA antagonists encompass a diversity of pharmacological agents and a number of potential mechanisms of action, including principally inhibition of glutamate release, NMDA receptor antagonism, and AMPA receptor antagonism. The NMDA receptor itself has multiple modulatory sites that are amenable to pharmacological modification. Furthermore, many of these drugs also have ancillary properties which may influence both their efficacy and safety (Muir 1995b).
Phase II and phase III clinical trials of several agents have been conducted. Individually, no trial to date has shown evidence of efficacy, and some trials have suggested the possibility of harm from some compounds.
Objectives
The objective of this review is to assess the efficacy and safety of EAA modulatory drugs for acute stroke. The primary outcome chosen for review is the proportion of patients dead or dependent; secondary outcomes are (1) mortality and (2) complete recovery.
Methods
Criteria for considering studies for this review
Types of studies
All controlled clinical trials of drugs modulating EAA release or receptors were considered. Trials were included in the review if adequate randomisation methods were specified, with appropriate allocation concealment from investigators. Efficacy analyses were restricted to trials fulfilling the following criteria:
treatment initiation within 24 hours of stroke onset;
outcome determined between one and 12 months after stroke;
randomisation into parallel groups (two or more) (ie not blocks of ascending dose).
Nevertheless, it is recognised that some trials fulfilling these criteria were not designed to test for efficacy, and interpretation of their results must be cautious. Studies with dose escalation design intended to evaluate tolerability and pharmacokinetics (predominantly phase IIa trials) were excluded from efficacy analyses since investigators were not considered adequately blinded to treatment allocation, with potential bias towards reporting of adverse effects or final outcome, and many participants are likely to have received doses of an agent unlikely to have pharmacological effect or which produced toxicity (Schulz 1995).
Types of participants
Patients randomised with a clinical diagnosis of acute stroke were included. Most trials randomised patients without prior imaging to differentiate ischaemic stroke from intracranial haemorrhage since EAA antagonists in general do not have antihaemostatic activity. Where possible, data were sought for both ischaemic stroke and intracerebral haemorrhage.
Types of interventions
Since rapid attainment of effective drug concentrations in brain is believed essential, only intravenously administered drugs were considered. Drug classes included in literature review and search strategy:
Ionotropic glutamate receptor antagonists:
NMDA receptor antagonists
glutamate recognition site
glycine site
polyamine site
ion channel/magnesium site
AMPA receptor antagonists
Metabotropic glutamate receptor ligands Glutamate release inhibitors / ion channel modulators
Sodium channel blockers
N type calcium antagonists
Adenosine agonists
kappa opioid agonists
Types of outcome measures
The primary outcome measure was death or dependence. The endpoint must have been recorded between four weeks and one year from the date of the stroke, and should have been assessed using a recognised functional outcome score (Barthel index or Rankin scale) (Van Swieten 1988, Rankin 1957, Mahoney 1965), or alternatively the trial should have reported a qualitative outcome such as institutionalisation. Dependence was taken as Barthel score of < 60 by preference, but modified Rankin score of > 3 or the nearest equivalent dichotomous outcomes for trials that did not use these cut offs. A measurement at 90 days was preferred. As a secondary endpoint, a cut off for complete or near‐complete recovery was considered for Barthel > 90 (or Rankin < 2 if Barthel data unavailable). Mortality was recorded.
Search methods for identification of studies
See: 'Specialized register' section in Cochrane Stroke Group
Relevant trials were identified in the Cochrane Stroke Group Trials Register which was last searched by the Review Group Co‐ordinator in May 2001. Standard bibliographic databases (MEDLINE, EMBASE, Ovid from 1966 to May 2001) were searched for all trials or drug studies pertaining to stroke or cerebrovascular disease, using the search terms [(neuroprotect* OR glutamate) AND (stroke OR cerebrovasc* OR trial)], and all individual drug names listed below. Any references in published trials were pursued. Conference Proceedings of the following meetings were searched by hand: European Stroke Conference, International Stroke Conference, American Heart Association, American Academy of Neurology, American Neurological Association (all 1991 to 2001), symposia on Pharmacology of Cerebral Ischemia (1994, 1996, 1998), and Neuroprotective Agents (1995, 1997). The Washington University Stroke Center online Stroke trials Database (www.strokecenter.org/trials/, last accessed January 2002) was searched. Additional data on published studies and information on unpublished studies was sought by direct queries to investigators and pharmaceutical companies, including Janssen‐Cilag, GlaxoWellcome, Cambridge NeuroScience Inc, CeNeS, Novartis, Sanofi‐Synthelabo, Boehringer‐Ingelheim, Pfizer / Parke‐Davis, and Astra‐Zeneca. Drug names included in the search strategy were as follows:
sipatrigine, 619C89, BW619C89
1003C87
lamotrigine
lubeluzole, prosynap, R‐087926
lifarizine, RS87476
fosphenytoin
aptiganel, CNS 1102, cerestat
dizocilpine, MK 801
selfotel, CGS 19755
eliprodil
ifenprodil
gavestinel, GV150526A
remacemide
AR‐R15896AR, FPL15896
licostinel, ACEA 1021
YM872
ZK200775
NPS1506
Dextrorphan
Magnesium, Mg, Mg2+, Mg++
Data collection and analysis
One reviewer prepared a list of potential trials, and the studies were then agreed by both independently. Trials were assessed independently by each reviewer for methodological quality. Data were extracted by one reviewer (KM in most instances) and checked by the other. Data were sought on the number of patients with each outcome measure (where available) by allocated treatment group. Intention‐to‐treat analyses were conducted, and figures were not adjusted to consider subsequent exclusion from the trial for any reason. Where numbers were not presented, the original figures were calculated from percentages. Since treatment group denominator populations vary in some trials according to exclusions and protocol violations, figures calculated for some trials may contain minor inaccuracies. In some instances, numbers with favourable outcomes were subtracted from the number randomised to define the number dead or dependent.
Since EAA modulators cover a diversity of pharmacological mechanisms that may lead to differences in efficacy and safety, individual drugs were considered separately and then considered as groups according to pharmacological action.
Analysis plan
Treatment differences for the specified analyses are expressed as odds ratios (OR) with 95% confidence intervals (CI) calculated by the Peto method using a fixed‐effect model. Heterogeneity was considered to be present at p < 0.05 for the Chi‐squared test, and a possibility of heterogeneity being present if 0.05<p<0.10.
Results
Description of studies
Ion Channel Modulators of EAA Release
Sipatrigine (619C89)
Sipatrigine is a pyrimidine blocker of voltage‐gated sodium and calcium channels, with additional affinity for sigma, musarinic and 5‐HT3 receptors (Hainsworth 2000). Sipatrigine and its related compounds 1003C87 and lamotrigine inhibit sodium‐dependent glutamate release. Sipatrigine is neuroprotective in permanent and transient middle cerebral artery occlusion (MCAO) models in rodents (Smith 1997). Volunteer studies describe pharmacokinetics and tolerability of single intravenous (IV) infusions up to 1mg/kg.
Four phase II clinical trials sponsored by Wellcome were undertaken (Sipatrigine 137‐101; Sipatrigine 137‐121137‐102; Sipatrigine 137‐104). A further development programme undertaken by CeNeS Ltd after taking over the product licence was terminated during recruitment to a further phase II trial which included MRI entry criteria. Results of two trials have been published, both of dose escalation design, and some results for two trials of parallel group design (137‐102, 137‐104) have been made available. No data from the CeNeS trial (CEE 05) are available. From a total of 220 patients exposed to sipatrigine, dose‐related neuropsychiatric effects were seen (principally visual perceptual disturbances (36%), and confusion (16%)). Sipatrigine has a long half‐life and active metabolites, which probably contributed to the high incidence of CNS side effects. Doses in the four completed trials were large (up to 2 g total dose) whereas the recent CeNeS trial programme planned maximum doses of 500 mg delivered as a short infusion rather than over a 72‐hour period.
Lubeluzole
Lubeluzole is a benzothiazole, similar in structure to riluzole (licensed for use in amyotrophic lateral sclerosis), whose mechanism of action includes inhibition of ischaemia‐induced glutamate release due to affinity for voltage‐gated Na+ channels (Scheller 1997). The compound also has affinity for serotonin 5HT1A receptors, sigma receptors and possibly Q‐type voltage‐gated Ca2+ channels, all mechanisms that have been found to be neuroprotective in other agents. Since lubeluzole inhibits neuronal cyclic guanosine monophosphate (cGMP) accumulation, inhibition of neuronal nitric oxide pathways has been proposed as a further mechanism of neuroprotection (Maiese 1997). Since neuroprotection in the photothrombotic rat model used for preclinical work is stereoselective, and only inhibition of glutamate release and inhibition of neuronal cGMP increase share this property, these (particularly the latter), have been promoted as the basis for anti‐ischaemic effects.
Lubeluzole has shown reductions of infarct volume and improved neurological function in a photothrombotic occlusion model in rats (De Ryck 2000), and subsequently in a conventional permanent MCAO model also in rats (Aronowski 1996), although neuroprotective effects in the MCAO model were lost if administration was delayed by more than 30 minutes after onset of ischaemia.
Two small single‐blind trials of lubeluzole in stroke patients (Lub‐Int 2 & Lub‐Bel 4) were followed by four randomised, double‐blind, placebo‐controlled clinical trials in acute stroke patients (Lub‐Int 4; Lub‐Int 5; Lub‐Int 9; Lub‐Int 13). All trials used a one hour loading infusion of 75% of the daily maintenance dose, followed by maintenance continuous iv infusion for a period of five days. One phase II study investigated two doses of lubeluzole (10 mg and 20 mg daily dose): excess deaths in the 20 mg daily group, and potential cardiotoxicity (prolongation of QTc interval and fatalities due to ventricular dysrhythmias) prompted a small cardiac safety study (Lub‐Int 7: 5 mg and 10 mg daily tested) and selection of the 10 mg total daily dose for phase III trials. Further trials, including a trial of lubeluzole in acute intracerebral haemorrhage (Lub‐Int 15, 110 patients enrolled, no data presented) and concurrent use of lubeluzole and rtPA (Lub‐USA 6), were terminated after failure to find efficacy in the Lub‐Int 13 trial.
Lub‐Int 13 recruited up to eight hours after stroke onset, but powered the study on the basis of recruiting a core population of patients within six hours of onset who were younger than 75 years and had mild or moderate strokes. Eighty patients with intracerebral haemorrhage were randomised, but no separate data are available on this group, and no data on overall outcomes in ischaemic stroke patients are available. Trial reports detail the sub‐group core population only. QTc prolongation was seen, but there was no excess of ventricular dysrhythmia or cardiac adverse effects.
The Cochrane systematic review of lubeluzole trials (Gandolfo 2002), involving 3510 patients in five trials (excluding Lub‐USA 6) confirmed lack of benefit and also significant excess risk of cardiac conduction abnormalities at all doses, including the 10 mg dose employed in most trials. Figures in this review do not necessarily correspond to those in that of Gandolfo and colleagues since patients lost to follow up were counted differently: we considered patients lost to follow up as having the least favourable outcome (severe disability/dependence).
Lifarizine
Lifarizine blocks both voltage‐gated sodium and calcium channels. It also has affinity for dopamine D2 receptors and may cause hyperprolactinaemia in animals. A single phase II clinical trial in stroke patients has been conducted (Lifarizine). Blood pressure fell, particularly in older women (by up to 15% compared to baseline), and post‐hoc analysis suggested an association with reduced likelihood of good outcome (p = 0.08 quoted). Non‐significant prolongation of the QTc interval on ECG was also seen, and a higher incidence of agitation compared to placebo. Primary intracerebral haemorrhage was reported in 17 patients in the trial: separate outcome data were not reported. Further clinical development has not been undertaken to date.
Fosphenytoin
Fosphenytoin is a prodrug of phenytoin and blocks glutamate release through inhibition of sodium channels. Sodium channel blockers are likely to have neuroprotective effects independent of glutamate‐mediated toxicity. A phase II study of fosphenytoin in 22 stroke patients (Fosphenytoin IIa) was followed by a phase III efficacy trial in 462 ischaemic stroke patients (Fosphenytoin phase 3). This trial was terminated after interim analysis found no difference in outcome. Results have not been published and further development for stroke was not undertaken. Data for the phase III trial were sought from the sponsors but not made available at the time of writing.
NMDA Antagonists
Selfotel (CGS 19755)
Selfotel (CGS 19755) is a competitive antagonist of the NMDA receptor, acting at the glutamate recognition site. Competitive NMDA antagonists demonstrated a pattern of neuroprotection matching that of the ion channel blocking agents in preclinical work, but penetrate the brain only slowly and are ineffective when administered only a short time after onset of ischaemia. Significant behavioural side effects were also evident with this drug class, and in humans this has proved to be a dose‐limiting factor in all studies involving conscious patients. A single small study in patients undergoing neurosurgical procedures found intolerable neuropsychiatric effects in all patients at doses above 1.5 mg/kg, and in a significant proportion at this dose. In the same study, cerebrospinal fluid concentration achieved by the 1.5 mg/kg dose was only 10% of that required for optimal neuroprotection in animal models (Yenari 1998, Perez‐Pinzon 1995). Trials have been conducted in stroke and traumatic brain injury. In stroke, two phase II studies were conducted (Selfotel IIa; Selfotel IIb), followed by two phase III efficacy trials with matching protocols, one in Europe, Canada, Argentina and Australia (protocol 10) and the other in the USA and Israel (protocol 7) (ASSIST Protocols 7 & 10). The ASSIST (Acute Stroke Trials Involving Selfotel Treatment) studies were terminated on the advice of the Data and Safety monitoring committee after recruitment of only 31% of the planned sample due to concerns over excess early neurological mortality in the selfotel arms, although no significant difference was evident at day 90. Day 30 mortality was significantly higher in selfotel treated patients (54/280 selfotel versus 37/286 placebo, OR 1.61 (1.02 to 2.54), p = 0.05). Patients given selfotel exhibited a side‐effect profile typical of NMDA antagonists (Muir 1995b), with agitation, confusion, reduced conscious level, hallucinations, and hypertension all significantly greater than placebo. Interpretation is confounded somewhat by the observation that selfotel treated patients were more often administered sedatives (benzodiazepines or major tranquillisers) than were placebo patients (39% versus 17%). Approximately 8% of patients had intracerebral haemorrhages on post‐randomisation CT. Separate data for ischaemic stroke and haemorrhages have not been presented.
Two trials in head injury (Morris 1999) were also terminated after recruitment of 693 patients with severe head trauma when the Safety Committees became concerned about possible excess mortality in the treatment arms. Subsequent results showed no significant detriment of treatment, but futility analysis led to termination of the trial programme.
Aptiganel Hydrochloride (CNS 1102)
Aptiganel HCl is a non‐competitive NMDA receptor antagonist with high affinity for the ion channel site. Significant neuroprotective effects similar to other drugs of this class have been found in rodent MCA occlusion models with treatment initiated up to one hour after stroke onset, using both histological and magnetic resonance imaging (MRI) techniques (Minematsu 1993; Meadows 1994). Studies in volunteers confirmed the side effect profile for this class of agent, with elevation of blood pressure (Grosset 1995), peripheral sensory disturbance, nystagmus, disinhibition and ultimately catatonia at doses of 30 to 100 ug/kg intravenously (Muir 1994; Muir 1997). Similar effects were reported in phase II trials, including blood pressure elevation (CNS1102‐010).
Three phase II studies were undertaken (CNS1102‐003; CNS1102‐008; CNS1102‐010), and an efficacy trial commenced in 1997 in the USA and Europe, but was discontinued later the same year after review of safety data (CNS1102‐011). Functional outcome data were unavailable for some of the studies, and in one (CNS1102‐010) no functional outcome measure fulfilling study eligibility for review was collected (final follow up at seven days +/‐ two days). The CNS1102‐011 trial included low (3 mg + 0.5 mg/h for 12 hours) and high (5 mg + 0.75 mg/h for 12 hours) dose treatment arms. The higher dose of aptiganel was associated with higher mortality than placebo, almost reaching statistical significance (41/214 placebo versus 56/214 high dose aptiganel, OR 1.50 (0.95 to 2.36), p = 0.06), but the lower dose did not differ significantly. Somnolence, reduced level of consciousness, confusion, cerebral oedema, hypertension and ventricular dysrhythmias were all more common in aptiganel‐treated patients. The interim analysis that led to trial termination found seven‐day mortality to be 4/113 placebo versus 22/189 aptiganel (OR 3.59 (1.20 to 10.7), p = 0.02). Fifty‐seven patients had CT evidence of intracerebral haemorrhage as the cause of stroke: separate outcome data are unavailable.
Eliprodil
Eliprodil and the related compound ifenprodil bind to the polyamine modulatory site of the NMDA receptor, and are now known to be specific ligands for the NR2B protein subunit. Both are neuroprotective in preclinical models, with a similar pattern and magnitude of effect in focal ischaemia to dizocilpine and other non‐competitive ion channel antagonists (Lekieffre 1997). Based upon animal data and clinical experience to date, it has been suggested that NR2B specific drugs may not share the neuropsychiatric side effect profile of NMDA ion channel antagonists. Eliprodil additionally binds to L, N and P‐type voltage‐gated calcium channels and is a high affinity sigma ligand.
Eleven separate studies in volunteers exposed 167 patients to eliprodil: no study has been published. Oral doses of 0.2 to 60 mg daily and iv doses of 0.25 to 6 mg daily have been studied. No psychotomimetic effects were reported, with reversible QT interval prolongation proving the dose‐limiting effect.
A multicentre randomised, double‐blind placebo‐controlled phase II trial of two dose levels of eliprodil involved 114 patients (40 placebo, 35 low dose and 39 high dose eliprodil patients) treated within 24 hours of onset. Intravenous medication was given for three days (3 or 6 mg/day) and oral medication for a further 11 days. No cardiovascular or psychotomimetic effects were noted, and QT interval was reportedly unaffected at these doses. Follow up at 76 days was conducted but efficacy data are unavailable (Giroux 1994). Three subsequent multicentre randomised, controlled trials were conducted: CVD 715 (875 patients), LES01 (346 patients) and LES02 (126 patients). Interim analyses of the European CVD 715 trial led to termination of the two US protocols (LES 01 and 02) on the basis of a futility analysis. Since protocols differed slightly among the different trials, they are presented separately in the table of included studies, but only pooled data are available for analyses (Eliprodil). Available data indicate the full patient population to be 1347, but outcome data are not given for 95 patients for reasons that are unspecified. Full data have not yet been presented for any study.
Remacemide hydrochloride
Remacemide blocks voltage‐gated sodium channels, and is metabolised to a desglycinyl derivative that is a non‐competitive NMDA antagonist at the ion channel site. Remacemide was neuroprotective in a cat permanent MCA occlusion model (Bannan 1994). One phase II dose escalation tolerability study was conducted in stroke patients. Intravenous treatment was followed by oral therapy, or repeated intravenous infusions. An increasing incidence of CNS side effects typical of NMDA antagonists was seen with increasing dose (agitation, hallucinations, confusion, depressed level of consciousness), potentially due to accumulation of the active desglycinyl metabolite (Remacemide phase 2). Further development of remacemide for stroke is not presently being pursued. A further study evaluating prophylactic neuroprotection in cardiopulmonary bypass found no effect on early or late neuropsychological function in 87 patients receiving remacemide 600 mg daily for five days before and five days after bypass compared to 84 controls (Arrowsmith 1998).
AR‐R15896AR
This compound is a low‐affinity non‐competitive NMDA antagonist which also has affinity for the sigma‐1 receptor. AR‐R15896AR reduced infarct volume in rodents when administered up to one hour after onset of MCA occlusion, but not later (Bialobok 1999). A single phase II dose‐ranging study in stroke patients has been reported (AR‐R15896AR). There was a low incidence of psychotomimetic effects within the dose range studied. Dizziness, nausea and vomiting were more common in actively treated patients, but only 6% experienced hallucinations or reduced consciousness. A high proportion of patients (26/170, 15%) did not have one month outcome data. The proportion of patients with intracerebral haemorrhage was not reported, but CT could have been deferred for up to 72 hours after onset. Clinical development is not presently being pursued.
Gavestinel (GV150526A)
This compound is a competitive antagonist at the glycine recognition site of the NMDA receptor complex. Gavestinel reduces infarct volume and improves neurological functional recovery in rodent permanent MCA occlusion models, but without the behavioural side effects seen with NMDA antagonists that block the receptor complex at other sites (Reggiani 2001). The pharmacokinetics indicate that gavestinel is > 99.9% protein bound and has a low volume of distribution, raising concerns that brain penetrance in acute stroke may be very limited. The absence of psychotomimetic effects typical of other NMDA antagonists in volunteers or in five subsequent phase II trials add to this concern (GLYA2001, GLYB2001, GLYB2002). A single cerebrospinal fluid sample demonstrating rises in CSF gavestinel concentration during treatment was obtained in the phase II development programme (Wester 1985). After ascending‐dose studies to evaluate pharmacokinetics in Europe and North America, a European dose‐ranging study (GLYB2002) was conducted to finalise doses for two phase III efficacy trials (GAIN‐I, GAIN‐A). GlyB2002 found significantly worse outcome for the gavestinel‐treated patients, but these groups had worse baseline stroke severity (median NIH stroke scale score 11.5 in high‐dose gavestinel versus eight for placebo), and 9/10 intracerebral haemorrhages randomised in the trial.
Stratified randomisation in both phase III trials produced groups well balanced for prognostic factors. No significant toxicity was found. There was no evidence of benefit from gavestinel in either trial. Both GAIN‐I and GAIN‐A quoted results for ischaemic stroke patients, excluding 571 patients (17%) randomised with intracerebral haemorrhage. One‐quarter of all patients randomised in GAIN‐Americas also received thrombolytic treatment with rtPA (none in GAIN‐International). Separate analysis of outcome in primary intracerebral haemorrhage patients in GAIN‐I (333 patients) and GAIN‐A (238 patients) found no evidence of benefit or harm (GAIN ICH). Ten out of 128 patients treated in the GlyB2002 protocol also had intracerebral haemorrhage, but data were not presented separately for this population.
Licostinel (ACEA 1021)
This competitive antagonist of the glycine recognition site on the NMDA receptor underwent a single phase II dose escalation study involving 64 participants within 48 hours of stroke onset (Licostinel). The trial identified dose‐related agitation, dizziness and depression of conscious level, together with some GI upset. Results for mortality were included in sensitivity analysis, but delayed treatment precluded inclusion of efficacy data even if this had been available. Further clinical development has not been undertaken due to concerns regarding renal toxicity in animals.
Dextrorphan
This compound has a number of actions in addition to being a non‐competitive NMDA antagonist at the ion channel site. The most relevant is voltage‐gated calcium channel antagonism. A single phase II dose‐ranging study was conducted in 67 stroke patients, with a protracted time window and delayed recruitment. This trial found evidence of significant blood‐pressure lowering by dextrorphan, and dose‐related psychotomimetic effects typical of NMDA antagonists (Dextrorphan). Further clinical development was not undertaken. Safety data are included in sensitivity analysis.
NPS‐1506
This is a non‐competitive NMDA antagonist with a pharmacological profile suggesting dissociation of neuroprotective dose from psychotomimetic side effects in animals. Initial tolerability studies have been conducted in healthy volunteers (NPS 1506), up to doses of 40 mg as iv infusion, and a dose‐escalation study in stroke patients within 48 hours of onset was undertaken. No details of this are available and further development is not currently planned.
AMPA Antagonists
ZK‐200775
This agent is a competitive AMPA antagonist with efficacy in focal ischaemia models in rodents and a prolonged time window of four hours after MCA occlusion (Turski 1998). A phase IIa tolerability study was completed in Europe and safety results published (ZK‐200775). Sixty‐one patients were randomised, 25 to placebo, and 36 to ZK200775 in doses of 525 mg or 262.5 mg over 48 hours, or to 105 mg over six hours. Elevated concentrations of the glial cell marker S‐100B in serum, deterioration in NIH stroke scale score at 48 hours after stroke, and increased incidence of reduced consciousness were reported with the highest dose group. Functional outcome data, although collected, were not published, nor were figures on mortality. One patient in the highest dose group was reported to have died.
YM‐872 (Zonampanel)
This competitive AMPA antagonist is neuroprotective in rodent permanent and transient MCA occlusion models up to two hours after onset of ischaemia (Takahashi 2002). YM872 has undergone preliminary evaluation in volunteers (YM872) and in a phase IIa stroke study, details of which have not been presented. YM872 is the subject of two ongoing trials, one in combination with rtPA in acute ischaemic stroke within three hours of onset (ARTIST+), and an MRI study (ARTIST MRI) treating up to six hours after onset in patients not given rtPA. Over 400 patients had been recruited to these two protocols by mid‐2002.
Agents of uncertain or mixed modes of action
Magnesium sulphate
Magnesium is neuroprotective in rodent models up to six hours after induction of focal ischaemia. Possible modes of action include non‐competitive NMDA antagonism, inhibition of glutamate release, block of voltage‐gated calcium channels, increased cerebral blood flow, vasoconstrictor antagonism at vascular smooth muscle (including attenuation of endothelin‐1 and angiotensin II ‐mediated vasoconstriction), preservation of tissue ATP, enhanced buffering of intracellular calcium ions, and inhibition of deleterious ion shifts in white matter. Six small trials have explored magnesium sulphate in stroke (Wester 1985; Muir 1995a; Muir 1998; IMAGES). One reported trial (510 participants) was excluded due to methodological uncertainty (Galeas 1999). Unfortunately, no useful data could be extracted from this study, including mortality. The study of Lampl and colleagues (Lampl 2001) reported improved outcome in magnesium‐treated patients but the authors did not report results as dichotomous outcomes, and have to date not made available further data to permit inclusion. Magnesium remains the subject of two ongoing trials, IMAGES (IMAGES), with projected sample size of 2700, and FAST‐MAG, which will test magnesium given within two hours of stroke onset. Recruitment to the main IMAGES trial ended in April 2003. An MRI sub‐study (MR‐IMAGES) is likely to include some patients additional to the main IMAGES trial.
Risk of bias in included studies
The majority of studies of glutamate‐modulating drugs in stroke have been sponsored by the pharmaceutical industry and conducted in the 1990s. Most have followed clinical development programmes in which phase I studies in healthy young and/or elderly volunteers established pharmacokinetics and approximate tolerable dosing regimes before proceeding to conduct phase II studies in stroke patients.
Ethical approval was specified for all trials reviewed
Informed consent from patients or next of kin was specified in all trials
Randomisation was satisfactory in all trials included
Trial procedures, including adverse event reporting, complied with ICH Good Clinical Practice requirements
Allocation concealment was specified in the majority of trials and was satisfactory; most employed central telephone randomisation. Dose escalation studies generally randomised patients in sequential blocks of limited size (typically six to eight patients) and were considered quasi‐randomised since investigators were aware of an expectation of more adverse events or drug effect with increasing trial duration. Dose escalation trials all followed acceptable randomisation procedures.
All phase III, and most large phase IIb trials, recorded numbers of patients randomised, treated, and lost to follow up. Few patients were lost to follow up in all large trials.
Time to treatment was detailed in most large trials. In phase IIb and III trials, average treatment time was under five hours after stroke onset in most.
If final outcome visit data were unavailable, most trials included the last observation carried forward.
Effects of interventions
Three trials were excluded through failure to fulfil methodological quality criteria (Lub‐Int 2 & Lub‐Bel 4; Galeas 1999). Forty relevant trials were identified. Data were unavailable from four trials fulfilling methodological selection criteria (eliprodil phase II (Giroux 1994), fosphenytoin phase III (Fosphenytoin phase 3), magnesium (Lampl 2001), and ZK‐200775 phase IIa (ZK‐200775)), including a total of over 676 participants (5% of total participants), of whom 561 were included in trials fulfilling criteria for efficacy analysis (4.8% of total participants).
Efficacy analysis (Comparison 01)
Thirteen dose escalation trials were excluded from analysis of true randomised comparisons (AR‐R15896AR, CNS1102‐003, CNS1102‐010, Dextrorphan, Fosphenytoin IIa, GLYA2001, GLYB2001, GLYB2003, Licostinel, Remacemide phase 2, Selfotel IIa, Sipatrigine 137‐101, Sipatrigine 137‐121; see Selection Criteria). Two small trials of parallel group design did not report functional outcome data (Sipatrigine 137‐102, Lub‐Int 7), leaving 21 of 23 true randomised trials, involving 10,342 patients, to form the basis for primary efficacy analysis. Efficacy analysis included data derived from nine (generally parallel group phase IIb) trials, involving 1071 patients, not primarily designed to assess efficacy (CNS1102‐008, GLYA2005, GLYB2002, Lifarizine, Lub‐Int 4, Muir 1998, Selfotel IIb, Sipatrigine 137‐104, Wester 1985).
Primary endpoint
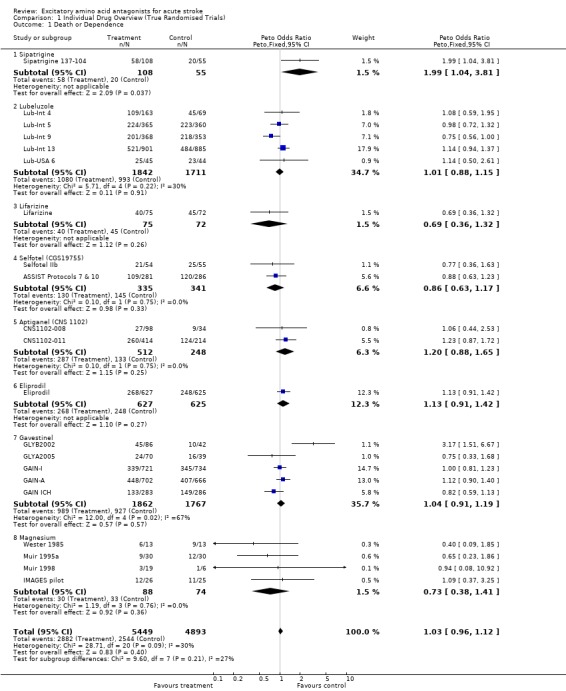
There was no significant effect on death or dependence at final follow up (OR 1.03 (0.96 to 1.12)). There was a non‐significant trend towards heterogeneity of outcome amongst individual drugs (chi‐squared = 28.7, df = 20, p = 0.09).
Secondary endpoints
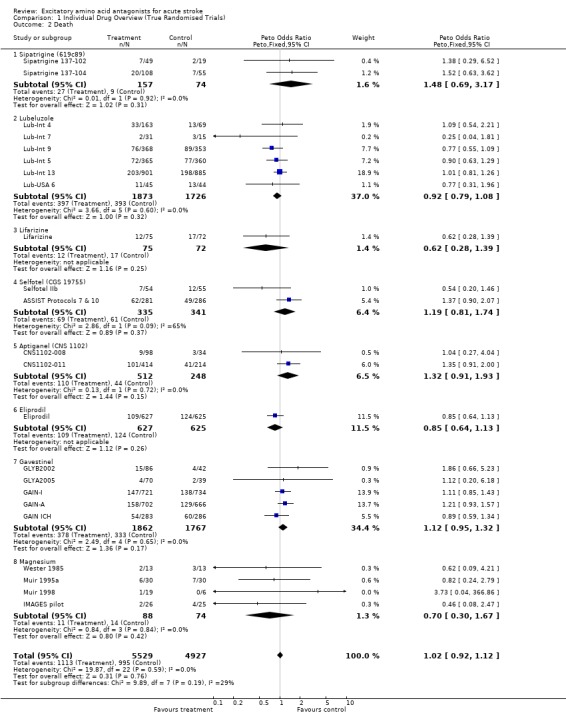
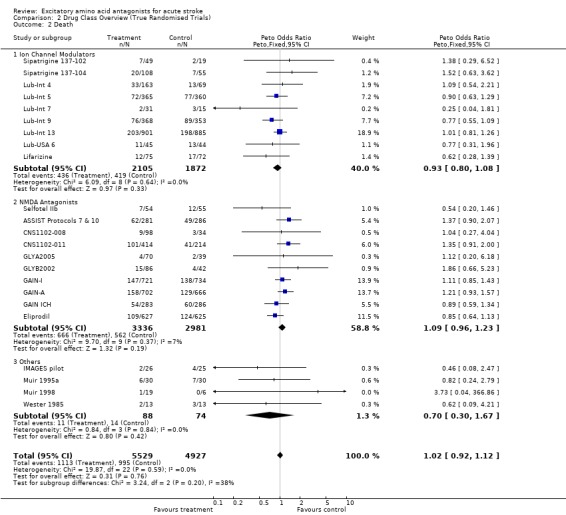
Mortality was unaffected by EAA antagonists (overall OR 1.02 (0.92 to 1.12)). There was no heterogeneity among trials.
Sub‐group analyses
Individual drugs
Trends for increased mortality with three NMDA antagonists were seen ‐ selfotel (OR 1.19 (0.82 to 1.75)), aptiganel (OR 1.29 (0.87 to 1.90)) and gavestinel (OR 1.13 (0.95 to 1.33)) ‐ but this did not achieve significance for the NMDA antagonists considered as a class. Aptiganel was also associated with a trend towards worse functional outcome (OR 1.21 (0.88 to 1.66)) although this was not the case for either of the other two compounds. Exploratory analysis of NMDA antagonists with psychotomimetic side effects at the doses studied versus those without psychotomimetic effects disclosed a trend towards higher mortality with psychotomimetic drugs (aptiganel and selfotel), but not combined death or dependence.
Drug classes
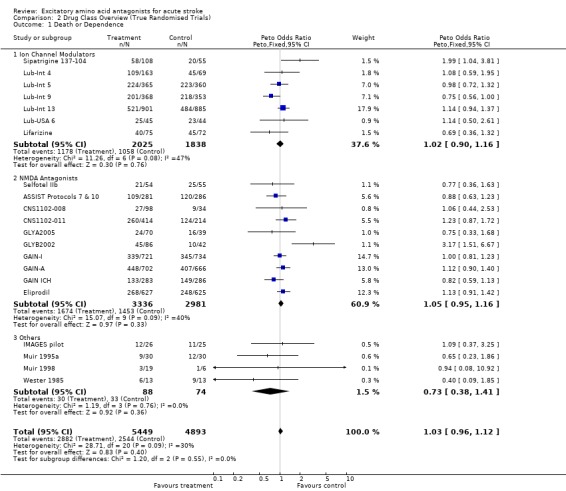
Trends towards heterogeneity for death or dependence (but not mortality) among both ion channel modulators (chi‐squared = 11.6, df = 6, p = 0.08) and NMDA antagonists (chi‐squared = 15.1, df = 9, p = 0.09) suggested that broad classification into these modes of action may be inappropriate. Individual drug analysis may be more informative.
Sensitivity analysis (Comparison 04)
Thirty‐six trials were included, involving 11,209 patients: of these trials, six did not report disability data, which were available for 29 trials involving 10,802 patients.
Efficacy analysis
There was no effect on death or dependence (OR 1.04 (0.96 to 1.12)). Heterogeneity among individual drugs was just significant (chi‐squared 41.4, df = 28, p = 0.05), with a suggestion of worsened outcome with some compounds (eg sipatrigine, AR‐R15896AR) and improved outcome with others (eg magnesium).
Secondary Endpoints
Mortality
Mortality was unaffected (OR 1.01 (0.92 to 1.11)). Eighteen trials involving 5143 patients reported detailed outcome data including full or nearly full recovery (Barthel > 90). There was no effect of treatment on this outcome (OR 1.03 (0.90‐1.17)). Some individual compounds tested only in small trials (eg magnesium, sipatrigine) showed divergent outcomes depending upon whether Barthel <60 or Barthel<95 was chosen.
Ischaemic stroke
Data were limited to 4699 patients in 10 trials (including studies excluded from efficacy analysis) for ischaemic stroke alone: most trials did not distinguish from intracerebral haemorrhage in reporting results. There was no evidence of benefit (odds of death or dependence 0.97 (0.87 to 1.10)) or harm (odds of death 1.03 (0.89 to 1.18)). This analysis may not contribute greatly to the main efficacy analysis, since, although separate outcome data for ischaemic versus haemorrhagic stroke were rarely presented, the proportion of patients with intracerebral haemorrhage randomised in most large trials (the exception being the GAIN trials) was small, and unlikely to have swayed the overall estimates of treatment effect.
Discussion
Neuroprotection, despite consistent effects in preclinical model systems, has yet to show clinical benefit. EAA antagonist drugs join a long list of drug classes abandoned through neutral (eg calcium antagonists (Horn 2001), clomethiazole (Wahlgren 1999)) or detrimental (eg tirilazad (Tirilazad)) effects on outcome after stroke. Many potential reasons for trial failure have been discussed, including inadequate preclinical evaluation (STAIR), inadequate sample size (Dorman 1996), stroke heterogeneity (Muir 2002a), or insensitive outcome data analysis (Muir 1999).
The range of compounds for which antagonism of excitatory amino acid neurotoxicity is a component is large, and encompasses a diversity of pharmacological properties. It is uncertain whether considering all compounds included in this review together is justifiable. Certain drugs have predominant pharmacological actions (eg selfotel, aptiganel, gavestinel) that permit some confidence regarding an overview of their results; others have a diversity of possible actions, for which antagonism of EAA release or action may be a minor, or indeed irrelevant, component of their neuroprotective efficacy in animal models (eg magnesium, lubeluzole, eliprodil, remacemide, lifarizine). The diverse pharmacology also cautions against over‐interpretation of adverse effect profiles, with some clear examples of dose‐limiting effects being unrelated to the EAA antagonist properties (eg QT prolongation with eliprodil and lubeluzole, hypotension with dextrorphan).
Data from 11,207 patients randomised in 36 clinical trials of agents recognised to antagonise EAAs are included in this review, representing 95% of relevant data identified by the search strategy. Data were unavailable for a small number of trials known to have satisfactory randomisation procedures, and one of uncertain methodological quality (510 patients receiving magnesium) (Galeas 1999). The final efficacy analysis was based on data from 10,342 patients, and safety data from 10,456 patients. Both ischaemic stroke and primary intracerebral haemorrhage were included in the efficacy analysis since many trials have not reported results separately, and there was no a priori reason to expect detriment to intracerebral haemorrhage patients.
Trials were well‐conducted and of high methodological quality in almost all cases. Time to treatment, known to be linked closely to treatment benefit in both animals and man (eg with thrombolysis (NINDS 1995)), averaged under five hours in many trials, although only a minority of patients were treated in three hours or less after stroke onset. Exclusion of the nine phase IIb trials, generally with longer time windows, smaller sample sizes, less rigorous selection criteria with respect to stroke severity, and greater likelihood of imbalance of groups for prognostic markers, makes no difference to estimates of treatment effect. The majority of true randomised trials were well balanced at baseline. However, some of the smaller phase IIb trials included in efficacy analyses (eg GLYB2002) had imbalance of stroke severity and pathological type at baseline that account for their status as outliers.
Considering data for individual compounds, there are too few patients included in clinical trials to make any definitive statement regarding potential benefit or harm for the majority of agents reviewed. There are only two exceptions, lubeluzole and gavestinel, for which data on over 3000 patients each are included, and for which there is no evidence of significant benefit (or harm). Results in both cases exclude an effect greater than 8% change in relative risk of death or dependence.
The diverse pharmacology of EAA antagonists is emphasised by borderline significance in tests for heterogeneity in several analyses. There do appear to be differences in individual compounds, for example with a trend towards worse outcome for sipatrigine (OR 1.99 (1.04 to 3.81)) and better outcome for magnesium (OR 0.73 (0.38 to 1.41)), but these possible differences must be interpreted with caution due to small numbers of patients, issues around dosing (especially for sipatrigine), and incomplete data availability. Further, divergent effects on outcome were seen when a different end‐point was analysed (analysis 4.3). The results of this analysis therefore do not permit confident identification of promising neuroprotective compounds, nor do they identify with certainty agents that may be harmful. Interpretation of trial data requires detailed review of the protocol and clinical pharmacology of the drug in question in addition to analysis of figures for outcome, and must be especially cautious for small trials.
Considering agents grouped by their probable main mode of action into ion channel modulators of glutamate release and NMDA receptor antagonists, the results are dominated by the comparatively large numbers of patients included in the lubeluzole and gavestinel trials (lubeluzole represents 92% of data on ion channel modulators and gavestinel 58% of data on NMDA antagonists). A trend towards higher mortality (but not worse functional outcome) for psychotomimetic NMDA antagonists is not statistically significant, but of concern. An increase in early mortality led to the termination of the selfotel trials, and both higher mortality and poorer functional outcome led to termination of the aptiganel trial programme. Although with hindsight, completion of these trial protocols would have increased the chance of more definitive estimates of potential harm being made, trial safety committees are obliged to act on such data for novel compounds, particularly where neurological toxicity is a known concern. Data do not suggest that safety concerns necessarily apply to all agents acting at the NMDA receptor, consistent with the diverse pharmacology of the receptor complex. Compounds acting at some components (eg selective NR2B subunit antagonists) do not possess the same profile of toxicity in preclinical evaluation as non‐selective ion channel blockers or competitive antagonists.
In the absence of consistent sub‐group analyses or individual patient data being available, this review cannot presently address many of the issues around the possible reasons for failure of neuroprotective trials to find benefit. Important sub‐groups that should be addressed are ischaemic stroke versus haemorrhage, cortical versus lacunar infarction, elderly versus young patients, and early treatment versus late. It is hoped that further data may become available to permit such analyses.
Authors' conclusions
Implications for practice.
No neuroprotective drug has yet been found to have significant clinical benefits. Further use of any of these agents should occur only in the context of a clinical trial.
Implications for research.
Further clinical trials of new neuroprotective agents remain justified. This review cannot presently be used to draw conclusions leading directly to changes in trial design or selection of a target population, but emphasises the importance of adequate sample sizes in the planning of future trials. Early termination of trials on the basis of futility analysis is often criticised since it leaves continuing uncertainty over potential harm from compounds such as psychotomimetic NMDA antagonists; nevertheless, it is difficult to see how a trial steering committee could react differently when faced with the possibility (with aptiganel) or likelihood (with selfotel) of significant harm and extremely low probability of eventual benefit. Although the combined trials with these two compounds included approximately 1000 patients, the confidence limits around the observed trend towards increased mortality remain wide. Where larger trials were allowed to run to completion, with gavestinel and lubeluzole, a relative risk reduction of 8% for disability has effectively been excluded: further trials with gavestinel or lubeluzole would now be difficult to pursue. Even so, the high incidence and devastating nature of stroke would justify a search for a neuroprotective effect of under 5% relative risk reduction. Our understanding of the mechanisms by which some drugs effect their neuroprotection in the laboratory is too poor to allow extrapolation from two failed development plans to all members of this disparate class, particularly when pharmacokinetic and trial design issues may also have influenced the outcome. We now have data from acute stroke trials involving several thousands of patients and one of the challenges is to ensure that we make full use of such resources by prospective plans to make datasets available for bona fide research.
What's new
| Date | Event | Description |
|---|---|---|
| 20 August 2008 | Amended | Converted to new review format. |
Acknowledgements
The authors thank the following individuals and companies for providing unpublished data for this review: Elkan Gamzu, Laima Mathews (Cambridge NeuroScience Inc); Christelle Depierre (Sanofi‐Synthelabo); Steven Hobbiger, Simon Coggins (CeNeS), Tom Wessel (Janssen‐Cilag), Hans‐Christoph Diener, James Grotta.
Data and analyses
Comparison 1. Individual Drug Overview (True Randomised Trials).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death or Dependence | 21 | 10342 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.03 [0.96, 1.12] |
| 1.1 Sipatrigine | 1 | 163 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.99 [1.04, 3.81] |
| 1.2 Lubeluzole | 5 | 3553 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.01 [0.88, 1.15] |
| 1.3 Lifarizine | 1 | 147 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.69 [0.36, 1.32] |
| 1.4 Selfotel (CGS19755) | 2 | 676 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.86 [0.63, 1.17] |
| 1.5 Aptiganel (CNS 1102) | 2 | 760 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [0.88, 1.65] |
| 1.6 Eliprodil | 1 | 1252 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.13 [0.91, 1.42] |
| 1.7 Gavestinel | 5 | 3629 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.04 [0.91, 1.19] |
| 1.8 Magnesium | 4 | 162 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.73 [0.38, 1.41] |
| 2 Death | 23 | 10456 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.02 [0.92, 1.12] |
| 2.1 Sipatrigine (619c89) | 2 | 231 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.48 [0.69, 3.17] |
| 2.2 Lubeluzole | 6 | 3599 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.92 [0.79, 1.08] |
| 2.3 Lifarizine | 1 | 147 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.62 [0.28, 1.39] |
| 2.4 Selfotel (CGS 19755) | 2 | 676 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.19 [0.81, 1.74] |
| 2.5 Aptiganel (CNS 1102) | 2 | 760 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.32 [0.91, 1.93] |
| 2.6 Eliprodil | 1 | 1252 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.64, 1.13] |
| 2.7 Gavestinel | 5 | 3629 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.12 [0.95, 1.32] |
| 2.8 Magnesium | 4 | 162 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.70 [0.30, 1.67] |
1.1. Analysis.
Comparison 1 Individual Drug Overview (True Randomised Trials), Outcome 1 Death or Dependence.
1.2. Analysis.
Comparison 1 Individual Drug Overview (True Randomised Trials), Outcome 2 Death.
Comparison 2. Drug Class Overview (True Randomised Trials).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death or Dependence | 21 | 10342 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.03 [0.96, 1.12] |
| 1.1 Ion Channel Modulators | 7 | 3863 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.02 [0.90, 1.16] |
| 1.2 NMDA Antagonists | 10 | 6317 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.05 [0.95, 1.16] |
| 1.3 Others | 4 | 162 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.73 [0.38, 1.41] |
| 2 Death | 23 | 10456 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.02 [0.92, 1.12] |
| 2.1 Ion Channel Modulators | 9 | 3977 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.93 [0.80, 1.08] |
| 2.2 NMDA Antagonists | 10 | 6317 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.09 [0.96, 1.23] |
| 2.3 Others | 4 | 162 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.70 [0.30, 1.67] |
2.1. Analysis.
Comparison 2 Drug Class Overview (True Randomised Trials), Outcome 1 Death or Dependence.
2.2. Analysis.
Comparison 2 Drug Class Overview (True Randomised Trials), Outcome 2 Death.
Comparison 3. NMDA Antagonists.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death or Dependence | 10 | 6317 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.05 [0.95, 1.16] |
| 1.1 Psychotomimetic | 4 | 1436 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.01 [0.81, 1.26] |
| 1.2 Non‐psychotomimetic | 6 | 4881 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.06 [0.95, 1.19] |
| 2 Death | 10 | 6317 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.09 [0.96, 1.23] |
| 2.1 Psychotomimetic | 4 | 1436 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.25 [0.96, 1.64] |
| 2.2 Non‐psychotomimetic | 6 | 4881 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.05 [0.91, 1.21] |
3.1. Analysis.
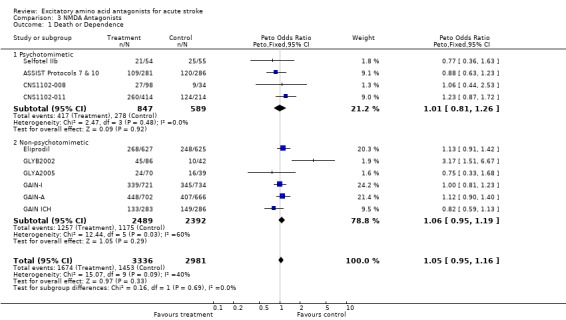
Comparison 3 NMDA Antagonists, Outcome 1 Death or Dependence.
3.2. Analysis.
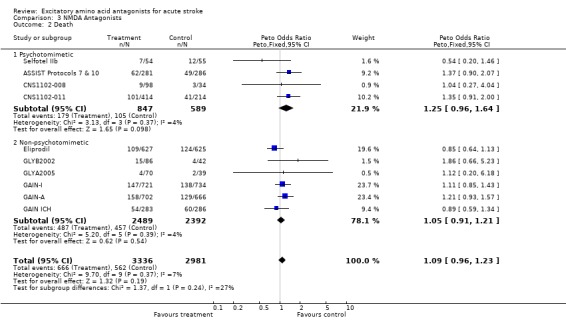
Comparison 3 NMDA Antagonists, Outcome 2 Death.
Comparison 4. Individual Drug Overview (All Trials).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death or Dependence | 29 | 10802 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.04 [0.96, 1.12] |
| 1.1 Sipatrigine (619c89) | 3 | 238 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.73 [0.99, 3.02] |
| 1.2 Lubeluzole | 5 | 3553 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.01 [0.88, 1.15] |
| 1.3 Lifarizine | 1 | 147 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.69 [0.36, 1.32] |
| 1.4 Selfotel (CGS19755) | 3 | 708 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.81 [0.60, 1.09] |
| 1.5 Aptiganel (CNS1102) | 2 | 760 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [0.88, 1.65] |
| 1.6 Eliprodil | 1 | 1252 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.13 [0.91, 1.42] |
| 1.7 Remacemide | 1 | 61 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.18 [0.38, 3.68] |
| 1.8 AR‐R15896AR | 1 | 170 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.34 [1.18, 4.64] |
| 1.9 Gavestinel (GV150526) | 8 | 3751 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.04 [0.91, 1.18] |
| 1.10 Magnesium | 4 | 162 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.73 [0.38, 1.41] |
| 2 Death | 36 | 11209 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.01 [0.92, 1.11] |
| 2.1 Sipatrigine (619c89) | 4 | 306 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [0.61, 2.34] |
| 2.2 Lubeluzole | 6 | 3599 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.92 [0.79, 1.08] |
| 2.3 Lifarizine | 1 | 147 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.62 [0.28, 1.39] |
| 2.4 Fosphenytoin | 1 | 22 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.11 [0.00, 4.20] |
| 2.5 Selfotel (CGS19755) | 3 | 708 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.18 [0.81, 1.73] |
| 2.6 Aptiganel (CNS1102) | 4 | 900 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.32 [0.92, 1.91] |
| 2.7 Eliprodil | 1 | 1252 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.64, 1.13] |
| 2.8 Remacemide | 1 | 61 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.93 [0.24, 3.51] |
| 2.9 AR‐R15896AR | 1 | 170 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.25, 2.52] |
| 2.10 Gavestinel (GV150526) | 8 | 3751 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.12 [0.95, 1.32] |
| 2.11 Licostinel (ACEA 1021) | 1 | 64 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.28 [0.06, 293.84] |
| 2.12 Dextrorphan | 1 | 67 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.72 [0.04, 368.99] |
| 2.13 Magnesium | 4 | 162 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.70 [0.30, 1.67] |
| 3 Not fully recovered (BI < 95) | 18 | 5143 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.03 [0.90, 1.17] |
| 3.1 Sipatrigine (619C89) | 3 | 238 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.73 [0.38, 1.37] |
| 3.2 Lubeluzole | 1 | 89 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.94 [0.39, 2.23] |
| 3.3 Aptiganel (CNS 1102) | 2 | 760 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.03 [0.72, 1.46] |
| 3.4 AR‐R15896AR | 1 | 170 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.81 [0.90, 3.62] |
| 3.5 Gavestinel | 8 | 3751 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.00 [0.87, 1.16] |
| 3.6 Magnesium | 3 | 135 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.61 [0.79, 3.29] |
4.1. Analysis.
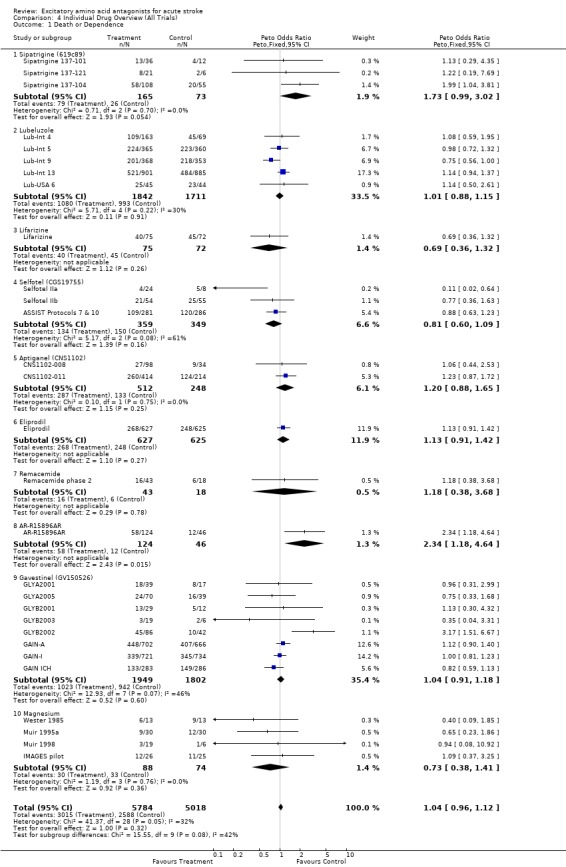
Comparison 4 Individual Drug Overview (All Trials), Outcome 1 Death or Dependence.
4.2. Analysis.
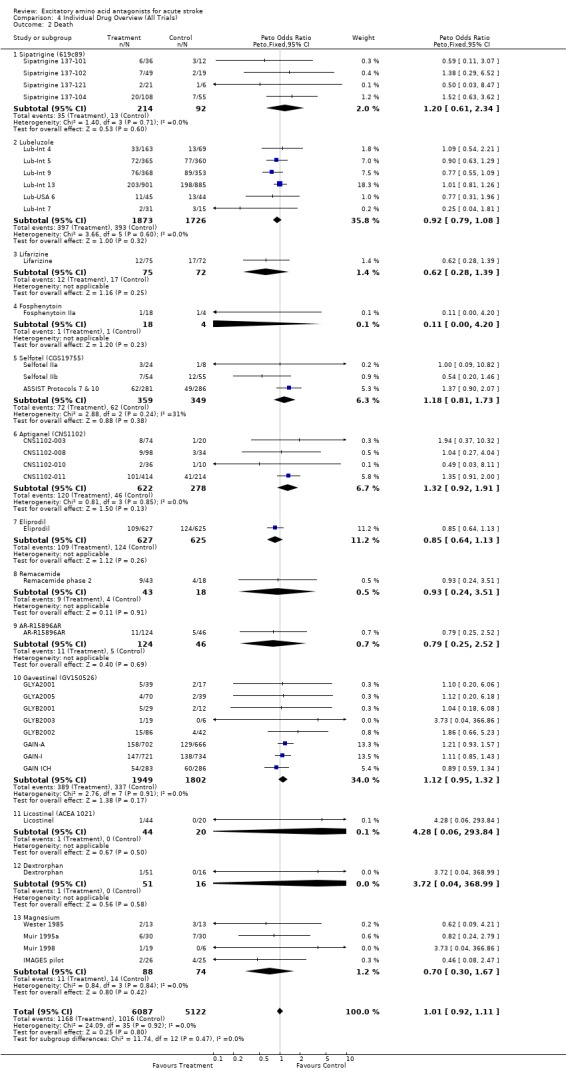
Comparison 4 Individual Drug Overview (All Trials), Outcome 2 Death.
4.3. Analysis.
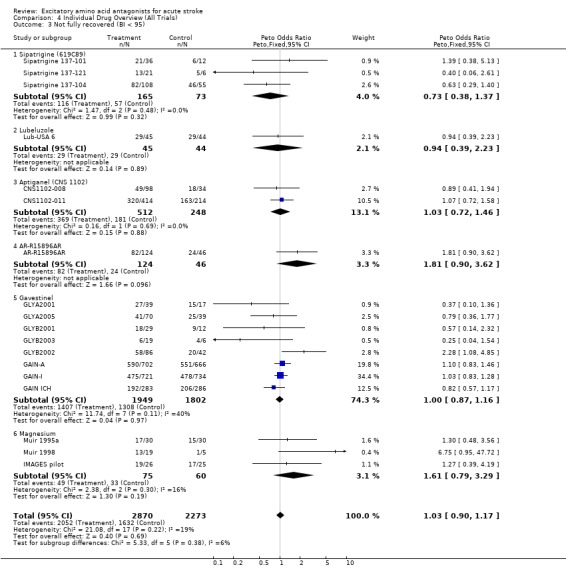
Comparison 4 Individual Drug Overview (All Trials), Outcome 3 Not fully recovered (BI < 95).
Comparison 5. Ischaemic Stroke (All Trials).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death or Dependence | 10 | 4699 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.97 [0.87, 1.10] |
| 2 Death | 10 | 4699 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.03 [0.89, 1.18] |
5.1. Analysis.
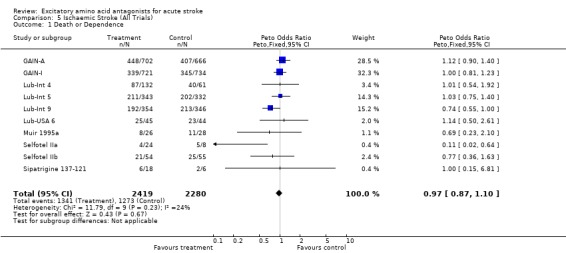
Comparison 5 Ischaemic Stroke (All Trials), Outcome 1 Death or Dependence.
5.2. Analysis.
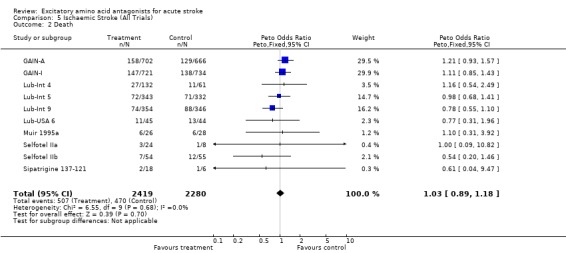
Comparison 5 Ischaemic Stroke (All Trials), Outcome 2 Death.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
AR‐R15896AR.
| Methods | Randomised double‐blind placebo‐controlled ascending dose tolerability and pharmacokinetic evaluation study | |
| Participants | Acute stroke < 24 hours after onset | |
| Interventions | AR‐R15896AR 100 to 300mg loading +/‐ 60 to 120 mg tid iv versus placebo | |
| Outcomes | 4 week Barthel, Rankin and mortality | |
| Notes | Ascending dose study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
ASSIST Protocols 7 & 10.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study Protocols 7 (USA and Canada) and 10 (rest of the world) were combined in the main publication | |
| Participants | Acute clinically diagnosed hemipsheric stroke < 6 hours after onset Aged 40 to 85 years | |
| Interventions | Placebo versus selfotel 1.5 mg iv bolus | |
| Outcomes | 90 day mortality, Barthel, NIHSS and SNSS | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
CNS1102‐003.
| Methods | Randomised, double‐blind placebo‐controlled ascending dose safety and tolerability study | |
| Participants | Ischaemic stroke < 18 hours after onset | |
| Interventions | Placebo versus iv aptiganel 10 to 30 ug/kg bolus then 0 to 20 ug/kg/h iv infusion for 4 or 6 hours | |
| Outcomes | 30 to 60 day Barthel, mortality | |
| Notes | Ascending dose study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
CNS1102‐008.
| Methods | Randomised, double‐blind, placebo‐controlled multiple parallel group study | |
| Participants | Acute stroke patients < 6 hours from onset | |
| Interventions | IV placebo versus aptiganel HCl 30 mcg/kg, 70 mcg/kg or 110 mcg/kg | |
| Outcomes | 90 day Barthel, Rankin, NIHSS, mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
CNS1102‐010.
| Methods | Phase IIb randomised, double blind, placebo controlled, parallel group study | |
| Participants | Acute stroke < 6 hours after onset | |
| Interventions | Aptiganel high dose, aptiganel low dose, placebo | |
| Outcomes | Barthel, Rankin and mortality at 3 months | |
| Notes | Dose escalation study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
CNS1102‐011.
| Methods | Phase III randomised, double‐blind placebo‐controlled parallel group study | |
| Participants | Acute stroke < 6 hours after onset | |
| Interventions | Placebo versus aptiganel 3 mg + 0.5mg/h x 12 hours or 5 mg + 0.75 mg/h x 12 hours | |
| Outcomes | Rankin, Barthel, mortality at 3 months | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
CVD 715.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Acute stroke < 8 hours after onset | |
| Interventions | Placebo versus 3 mg bd iv eliprodil for 3 days, then 10 mg bd orally for 11 days | |
| Outcomes | 3 month Barthel and mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Dextrorphan.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose | |
| Participants | Hemispheric stroke < 48 hours after onset | |
| Interventions | Placebo versus dextrorphan 60 to 260 mg iv over 1 hour then 15 to 135 mg/h iv infusion for 23 hours | |
| Outcomes | 48h NIHSS, 30 day mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
Eliprodil.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group studies | |
| Participants | Acute stroke < 8 hours after onset | |
| Interventions | 3mg bd iv eliprodil for 3 or 5 days, +/‐ oral dosing for 11 days | |
| Outcomes | 3 month Barthel and mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Fosphenytoin IIa.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose safety and tolerability study | |
| Participants | Acute ischaemic stroke < 72 hours after onset | |
| Interventions | Placebo versus fosphenytoin 7.5, 15, 22.5 or 30 mg/kg iv infusion | |
| Outcomes | Mortality | |
| Notes | Ascending dose study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
GAIN ICH.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Acute stroke < 6 hours after onset | |
| Interventions | Gavestinel 800 mg loading then 200 mg 12 hourly x 5 versus placebo | |
| Outcomes | 3 month Barthel | |
| Notes | PICH patients from GAIN‐A and GAIN‐I | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
GAIN‐A.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Acute stroke < 6 hours after onset | |
| Interventions | Gavestinel 800 mg loading then 200 mg 12 hourly x 5 versus placebo | |
| Outcomes | 3 month Barthel | |
| Notes | Only ischaemic stroke results available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
GAIN‐I.
| Methods | Randomised, double blind, placebo‐controlled parallel group study | |
| Participants | Acute stroke < 6 hours after onset | |
| Interventions | Gavestinel 800 mg loading then 200 mg 12 hourly x 5 versus placebo | |
| Outcomes | Barthel at 3 months | |
| Notes | Only ischaemic stroke results available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
GLYA2001.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose safety and pharmacokinetic evaluation study | |
| Participants | Acute stroke < 12 hours after onset | |
| Interventions | Gavestinel 400 or 800 mg loading +/‐ 200 or 400 mg bd for 5 doses versus placebo | |
| Outcomes | NIHSS, Barthel and Rankin at discharge or day 7; 4 week mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
GLYA2005.
| Methods | Randomised, double‐blind, placebo‐controlled tolerability study | |
| Participants | Acute stroke < 12 hours after onsert | |
| Interventions | Gavestinel 800 mg loading then 200 mg bd iv for 5 doses versus placebo | |
| Outcomes | 4 week mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
GLYB2001.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose tolerability and pharmacokinetic study | |
| Participants | Acute stroke < 12 hours after onset | |
| Interventions | Gavestinel iv 50 to 800 mg over 7.5 minutes to 4 hours or placebo | |
| Outcomes | Barthel and NIHSS at 1 month | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
GLYB2002.
| Methods | Randomised, double‐blind, placebo‐controlled dose‐ranging phase IIb study | |
| Participants | Acute stroke < 12 hours after onset | |
| Interventions | Gavestinel iv 800 mg loading then 200 mg or 400 mg bd for 5 days versus placebo | |
| Outcomes | Barthel and mortality at 30 days | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
GLYB2003.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose tolerability and pharmacokinetic study | |
| Participants | Acute stroke < 12 hours after onset | |
| Interventions | Gavestinel 800 mg iv loading then 100 to 400 mg bd for 5 days or placebo | |
| Outcomes | Barthel and NIHSS at 1 month | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
IMAGES pilot.
| Methods | Randomised, double‐blind, placebo controlled parallel group study | |
| Participants | Clinically diagnosed stroke < 12 hours after onset | |
| Interventions | IV magnesium sulphate 16 mmol over 15 minutes then 65 mmol over 24 hours, or placebo | |
| Outcomes | 30 day Barthel and Rankin scores | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
LES 01.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Clinically diagnosed stroke < 8 hours after onset | |
| Interventions | IV eliprodil 3 mg bd for 5 days | |
| Outcomes | 3 month Barthel and mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
LES 02.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Clinically diagnosed stroke < 8 hours after onset | |
| Interventions | IV eliprodil 3 mg bd for 5 days | |
| Outcomes | 3 month Barthel and mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Licostinel.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose safety and tolerability study | |
| Participants | Ischaemic stroke (CT or MRI confirmed) < 48 hours after onset, aged 18 to 80 years | |
| Interventions | Placebo versus iv licostinel 0.03 to 3 mg/kg | |
| Outcomes | Mortality, 1 month NIHSS | |
| Notes | Dose escalation study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
Lifarizine.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | First ever stroke < 12 hours after onset | |
| Interventions | Placebo versus lifarizine 250 ug/kg iv then 60 mg bd orally for 5 days | |
| Outcomes | 3 month Barthel, Rankin, mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Lub‐Int 13.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Acute hemispheric stroke < 8 hours after onset | |
| Interventions | Placebo versus lubeluzole 7.5 mg over 1hour then 10 mg/day | |
| Outcomes | 3 month mortality, Barthel, Rankin | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Lub‐Int 4.
| Methods | Randomised, double‐blind placebo‐controlled multiple parallel group study | |
| Participants | Acute carotid territory stroke < 6 hours after onset More than 50 years of age Upper limb weakness | |
| Interventions | Placebo versus lubeluzole 10 mg/day versus lubeluzole 20 mg/day | |
| Outcomes | 28 day mortality, Barthel score, NIHSS, ESS | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Lub‐Int 5.
| Methods | Randomised, double‐blind placebo‐controlled parallel group study | |
| Participants | Acute hemispheric stroke < 6 hours after onset | |
| Interventions | Placebo versus lubeluzole 7.5 mg over 1 hour then 10 mg/day for 5 days | |
| Outcomes | 3 month mortality, Barthel score, Rankin Score | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Lub‐Int 7.
| Methods | Randomised, double‐blind placebo‐controlled multiple parallel group study for cardiac safety | |
| Participants | Ischaemic stroke < 24 hours after onset without significant cardiac disease | |
| Interventions | Placebo versus lubeluzole 10 mg/day or 5 mg/day for 5 days | |
| Outcomes | 30 day mortality, cardiac safety | |
| Notes | No functional outcome data presented | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Lub‐Int 9.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Acute hemispheric stroke < 6 hours after onset | |
| Interventions | Placebo versus lubeluzole 7.5 mg over 1 hour then 10 mg/day for 5 days | |
| Outcomes | 3 month mortality, Barthel, Rankin | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Lub‐USA 6.
| Methods | Randomised, double‐blind placebo‐controlled parallel group study | |
| Participants | Acute ischaemic stroke treated with iv rtPA < 3 hours after onset | |
| Interventions | Placebo versus lubeluzole 7.5 mg over 1 hour then 10 mg/day for days by continuous ivi, treatment started before end of rtPA infusion | |
| Outcomes | Barthel | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Muir 1995a.
| Methods | Randomised, double‐blind, placebo‐controlled parallel group study | |
| Participants | Clinically diagnosed stroke < 12 hours after onset | |
| Interventions | IV magnesium sulphate 8 mmol over 15 minutes then 65 mmol over 24 hours or placebo | |
| Outcomes | 30 and 90 day Barthel and Rankin scores | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Muir 1998.
| Methods | Randomised, double‐blind, placebo‐controlled multiple parallel group study | |
| Participants | Clinically diagnosed stroke < 24 hours after onset | |
| Interventions | IV magnesium sulphate 8, 12 or 16 mmol over 15 minutes, each followed by 65 mmol over 24 hours, or placebo | |
| Outcomes | 30 and 90 day Barthel and Rankin scores | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Remacemide phase 2.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose study | |
| Participants | Acute stroke < 12 hours after onset | |
| Interventions | Remacemide 100 mg bd x 6 doses. First 2 doses were iv, remainder were oral. Subsequent cohorts received 200 mg bd, 300 mg bd, 400 mg bd, 500 mg bd or 600 mg bd for 3 days; or placebo in 3:1 randomisation schedule. | |
| Outcomes | 1 month Barthel and Canadian Neurological Scale, mortality | |
| Notes | Dose escalation study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
Selfotel IIa.
| Methods | Randomised, double‐blind, placebo‐controlled ascending dose study | |
| Participants | Acute ischaemic stroke < 12 hours after onset CT compatible with ischaemic stroke | |
| Interventions | Placebo versus selfotel 1.0 (two doses), 1.5 (one dose), 1.75 (one dose), or 2.0 (one or two doses) mg/kg iv | |
| Outcomes | 30 and 90 day Barthel and NIHSS | |
| Notes | Ascending dose study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
Selfotel IIb.
| Methods | Randomised, double‐blind placebo‐controlled parallel group study | |
| Participants | Acute ischaemic stroke < 6 hours after onset | |
| Interventions | Placebo versus selfotel 1.5 mg bolus | |
| Outcomes | 90 day NIHSS, Barthel | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Sipatrigine 137‐101.
| Methods | Ascending dose safety and tolerability phase II trial Randomised, double‐blind, placebo controlled | |
| Participants | Clinically diagnosed stroke < 12 hours after onset | |
| Interventions | IV infusion of 619c89 or placebo as loading infusion then repeated maintenance infusions 8 hourly | |
| Outcomes | Barthel, Rankin scales at 90 days | |
| Notes | Ascending dose study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
Sipatrigine 137‐102.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group phase II trial | |
| Participants | Clinically diagnosed stroke < 12 hours | |
| Interventions | IV infusion of sipatrigine 2 mg/kg loading then 1 mg/kg/8 hours x 8 | |
| Outcomes | 3 month Barthel, Rankin, mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Sipatrigine 137‐104.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group trial | |
| Participants | Clinically diagnosed stroke < 12 hours | |
| Interventions | Placebo versus iv sipatrigine 150 or 200 mg loading then 18.75 or 25 mg/h ivi for 72 hours | |
| Outcomes | 12 week NIHSS, Barthel, Rankin, mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Sipatrigine 137‐121.
| Methods | Ascending dose safety and tolerability study Randomised double‐blind, placebo controlled | |
| Participants | Clinically diagnosed stroke < 24 hours after onset | |
| Interventions | IV 619c89 or placebo as loading infusion then continuous ivi | |
| Outcomes | Barthel and Rankin scores at 90 days | |
| Notes | Ascending dose study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | High risk | C ‐ Inadequate |
Wester 1985.
| Methods | Randomised double‐blind placebo‐controlled study | |
| Participants | Acute stroke < 12 hours after onset | |
| Interventions | IV magnesium sulphate 4 mmol loading then 5 mmol/h for 3 days then oral magnesium hydroxide for 5 days | |
| Outcomes | Placement at 6 months | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Fosphenytoin phase 3 | No outcome data available. |
| Galeas 1999 | No information on randomisation method or placebo group. Non‐standard outcome measures. Time of treatment initiation and outcome assessment not known. |
| Giroux 1994 | No outcome data available. |
| Lampl 2001 | No dichotomous outcome data available. |
| Lub‐Int 2 & Lub‐Bel 4 | Single‐blind methodology. |
| Sipatrigine CEE 05 | No outcome data available. Trial terminated for commercial reasons. |
| ZK‐200775 | No outcome data available. |
Characteristics of ongoing studies [ordered by study ID]
ARTIST MRI.
| Trial name or title | ARTIST MRI |
| Methods | |
| Participants | Acute ischaemic stroke patients not treated with rtPA < 6 hours after onset, MRI (DWI) lesion volume 4 to 120cc |
| Interventions | Placebo versus iv YM872 for 24 hours |
| Outcomes | 28 day MRI lesion volume; 90 day clinical outcomes |
| Starting date | 2002 |
| Contact information | kyurkoma@mail.cato.com |
| Notes | Recruitment 108 as of July 2002 |
ARTIST+.
| Trial name or title | ARTIST+ |
| Methods | |
| Participants | Acute ischemic stroke < 3 hours after onset and treated with rtPA |
| Interventions | Placebo versus iv YM 872 for 24 hours |
| Outcomes | 3 month GOS, Barthel, Rankin and NIHSS |
| Starting date | January 2001 |
| Contact information | wlong@mail.cato.com |
| Notes | Recruitment 312 patients as of July 2002 |
IMAGES.
| Trial name or title | IMAGES |
| Methods | |
| Participants | Acute Stroke < 12 hours after onset |
| Interventions | IV magnesium sulphate 8 mmol bolus + 65 mmol over 24 hours versus placebo |
| Outcomes | 3 month Barthel, Rankin and mortality |
| Starting date | 1996 |
| Contact information | IMAGES Co‐ordinating Centre, Western Infirmary , Glasgow |
| Notes |
Contributions of authors
Both reviewers identified the topic for the review, drafted the protocol, identified relevant studies and assessed their quality. KM was primarily responsible for data entry and the text of the review. KL checked data, reviewed and modified the text.
Declarations of interest
Prof Lees was principal investigator in GAIN‐International. Both authors are principal investigators in the Intravenous Magnesium Efficacy in Stroke trial, funded by the UK Medical Research Council. Both authors have received fees or expenses for speaking at symposia and / or consultancy on behalf of pharmaceutical companies in the neuroprotective field, including GlaxoWellcome, AstraZeneca, Boehringer Ingelheim, Pfizer, Janssen‐Cilag, Cambridge NeuroScience, and Roche. Both authors have received sponsorship for attendance at meetings and symposia organised by these companies and received payments for speaking at events related to neuroprotection.
Edited (no change to conclusions)
References
References to studies included in this review
AR‐R15896AR {published and unpublished data}
- Lees KR, Dyker AG, Sharma A, Ford GA, Ardron ME, Grosset DG. Tolerability of the low‐affinity, use‐dependent NMDA antagonist AR‐R15896AR in stroke patients: a dose‐ranging study. Stroke 2001;32:466‐472. [DOI] [PubMed] [Google Scholar]
- Lees KR, Dyker AG, Sharma A, Ford GA, Ardron ME, Grosset DG, Dean A. AR‐R15896AR in patients with acute stroke: a dose‐finding study. Cerebrovascular Diseases 1999;9(Supplement 1):102. [Google Scholar]
ASSIST Protocols 7 & 10 {published and unpublished data}
- Davis SM, Albers GW, Diener HC, Lees KR, Norris J. Termination of Acute Stroke Studies Involving Selfotel Treatment. ASSIST Steering Committee. Lancet 1997;349:32. [DOI] [PubMed] [Google Scholar]
CNS1102‐003 {published data only}
- Block GA. Final results from a dose‐escalating safety and tolerance study of the non‐competitive NMDA antagonist CNS1102 in patients with acute cerebral ischemia. Stroke 1995;26:185. [Google Scholar]
- Fisher M. Cerestat (CNS 1102), a non‐competitive NMDA antagonist, in ischemic stroke patients: dose‐escalating safety study. Cerebrovascular Diseases 1994;4:245. [Google Scholar]
- McBurney RN. Development of the NMDA ion‐channel blocker, aptiganel hydrochloride, as a neuroprotective agent for acute CNS injury. In: Green AR, Cross AJ editor(s). Neuroprotective Agents and Cerebral Ischaemia. Vol. 40, San Diego: Academic Press, 1997:173‐195. [0‐12‐366840‐9] [DOI] [PubMed] [Google Scholar]
CNS1102‐008 {unpublished data only}
- Edwards K. Cerestat (aptiganel hydrochloride) in the treatment of acute ischemic stroke: results of a phase II trial. Neurology 1996;46(Supplement 2):A424. [Google Scholar]
CNS1102‐010 {unpublished data only}
- Dyker AG, Edwards KR, Fayad PB, Hormes JT, Lees KR. Safety and Tolerability Study of Aptiganel Hydrochloride in Patients With an Acute Ischemic Stroke. Stroke 1999;30(10):2038‐2042. [DOI] [PubMed] [Google Scholar]
CNS1102‐011 {published data only}
- CNS 1102‐011 Trial Synopsis. Unpublished 1997.
- Albers GW, Goldstein LB, Hall D, Lesko LM. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA 2001;286(21):2673‐2682. [DOI] [PubMed] [Google Scholar]
CVD 715 {unpublished data only}
- Depierre C. Clinical evaluation of eliprodil in the acute phase of stroke. Sanofi‐Synthelabo Report 2000.
Dextrorphan {published data only}
- Albers GW, Atkinson RP, Kelley RE, Rosenbaum DM. Safety, tolerability, and pharmacokinetics of the N‐methyl‐D‐ aspartate antagonist dextrorphan in patients with acute stroke. Stroke 1995;26:254‐258. [DOI] [PubMed] [Google Scholar]
Eliprodil {unpublished data only}
- Depierre C. Clinical evaluation of eliprodil in the acute phase of stroke. Sanofi‐Synthelabo report 2000.
Fosphenytoin IIa {published data only}
- Tietjen GE, Dombi T, Pulsinelli W, Becske T, Kugler AR, Mann ME. A double‐blind, safety, and tolerance study of single intravenous doses of fosphenytoin in patients with acute ischemic stroke. Neurology 1996;46(Supplement 2):A424‐A425. [Google Scholar]
GAIN ICH {published data only}
- Davis S, Zafar K, Vohora S, Okubo S, Nagashima J, Ito T, Katayama Y. Efficacy and safety of GV150526 in primary intracerebral haemorrhage: data from the GAIN trials (Glycine Antagonist in Neuroprotection). Stroke 2000;31(11):2865. [Google Scholar]
GAIN‐A {published data only}
- Sacco RL. GV150526 in the treatment of acute stroke: results of the GAIN Americas trial. Cerebrovascular Diseases. 2001; Vol. 10, issue Supplement 2:106.
- Sacco RL, DeRosa JT, Haley EC, Jr, Levin B, Ordronneau P, Phillips SJ, et al. Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial. JAMA 2001;285(13):1719‐1728. [DOI] [PubMed] [Google Scholar]
GAIN‐I {published data only}
- Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Orgogozo JM, Whitehead J. Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomised controlled trial. Lancet 2000;355:1949‐1954. [DOI] [PubMed] [Google Scholar]
GLYA2001 {unpublished data only}
- Phase II studies of the glycine antagonist GV150526 in acute stroke : the North American experience. The North American Glycine Antagonist in Neuroprotection (GAIN) Investigators. Stroke 2000;31(2):358‐365. [DOI] [PubMed] [Google Scholar]
GLYA2005 {unpublished data only}
- Phase II studies of the glycine antagonist GV150526 in acute stroke : the North American experience. The North American Glycine Antagonist in Neuroprotection (GAIN) Investigators. Stroke 2000;31(2):358‐365. [DOI] [PubMed] [Google Scholar]
GLYB2001 {unpublished data only}
- Dyker AG, Lees KR. Safety and tolerability of GV150526 (a glycine site antagonist at the N‐ methyl‐D‐aspartate receptor) in patients with acute stroke. Stroke 1999;30(5):986‐992. [DOI] [PubMed] [Google Scholar]
GLYB2002 {unpublished data only}
- GAIN European Study Group. Safety and tolerability of GV150526 in acute stroke (GLYB 2002). Cerebrovascular Diseases 1998;8(Supplement 4):20. [Google Scholar]
- Lees KR, Lavelle JF, Cunha L, Diener HC, Sanders EA, Tack P, et al. Glycine antagonist (GV150526) in acute stroke: a multicentre, double‐blind placebo‐controlled phase II trial. Cerebrovasc Dis 2001;11(1):20‐29. [DOI] [PubMed] [Google Scholar]
GLYB2003 {unpublished data only}
- Dyker AG, Lees KR. Safety and tolerability of GV150526 (a glycine site antagonist at the N‐ methyl‐D‐aspartate receptor) in patients with acute stroke. Stroke 1999;30(5):986‐992. [DOI] [PubMed] [Google Scholar]
IMAGES pilot {published data only}
- IMAGES Study Group, Bradford APJ, Muir KW, Lees KR. IMAGES pilot study of intravenous magnesium in acute stroke. Cerebrovascular Diseases 1998;8(Supplement 4):86. [Google Scholar]
LES 01 {unpublished data only}
- Depierre C. Clinical evaluation of eliprodil in the acute phase of stroke. Sanofi‐Synthelabo report 2000.
LES 02 {unpublished data only}
- Depierre C. Clinical evaluation of eliprodil in the acute phase of stroke. Sanofi‐Synthelabo report 2000.
Licostinel {published data only}
- Albers GW, Clark WM, Atkinson RP, Madden K, Data JL, Whitehouse MJ. Dose escalation study of the NMDA glycine‐site antagonist licostinel in acute ischemic stroke. Stroke 1999;30(3):508‐513. [DOI] [PubMed] [Google Scholar]
Lifarizine {published and unpublished data}
- Squire IB, Lees KR, Pryse‐Phillips W, Kertesz A, Bamford J. The effects of lifarizine in acute cerebral infarction: a pilot safety study. Cerebrovasc Dis 1996;6:156‐160. [Google Scholar]
Lub‐Int 13 {published data only}
- Diener HC, Cortens M, Ford G, Grotta J, Hacke W, Kaste M, et al. Lubeluzole in acute ischemic stroke treatment : A double‐blind study with an 8‐hour inclusion window comparing a 10‐mg daily dose of lubeluzole with placebo. Stroke 2000;31(11):2543‐2551. [DOI] [PubMed] [Google Scholar]
Lub‐Int 4 {published data only}
- Diener HC, Hacke W, Hennerici M, Radberg J, Hantson L, Keyser J. Lubeluzole in acute ischemic stroke. A double‐blind, placebo‐controlled phase II trial. Lubeluzole International Study Group. Stroke 1996;27:76‐81. [DOI] [PubMed] [Google Scholar]
Lub‐Int 5 {published and unpublished data}
- Diener HC. Multinational randomised controlled trial of lubeluzole in acute ischaemic stroke. European and Australian Lubeluzole Ischaemic Stroke Study Group. Cerebrovasc Dis 1998;8:172‐181. [DOI] [PubMed] [Google Scholar]
Lub‐Int 7 {published data only}
- Hacke W, Lees KR, Timmerhuis T, Haan J, Hantson L, Hennerici M, et al. Cardiovascular safety of lubeluzole (prosynap®) in patients with ischemic stroke. Cerebrovasc Dis 1998;8(5):247‐254. [DOI] [PubMed] [Google Scholar]
Lub‐Int 9 {published data only}
- Grotta J. Lubeluzole treatment of acute ischemic stroke. The US and Canadian Lubeluzole Ischemic Stroke Study Group. Stroke 1997;28:2338‐2346. [DOI] [PubMed] [Google Scholar]
Lub‐USA 6 {published data only}
- Grotta J. Combination Therapy Stroke Trial: recombinant tissue‐type plasminogen activator with/without lubeluzole. Cerebrovasc Dis 2001;12(3):258‐263. [DOI] [PubMed] [Google Scholar]
- Grotta JC. Combination therapy stroke trial: rtPA +/‐ lubeluzole. Stroke 2000;31(1):278. [PubMed] [Google Scholar]
Muir 1995a {published data only}
- Muir KW, Lees KR. A randomised, double‐blind, placebo‐controlled pilot trial of intravenous magnesium sulfate in acute stroke. Stroke 1995;26:183‐188. [DOI] [PubMed] [Google Scholar]
Muir 1998 {published data only}
- Muir KW, Lees KR. Dose optimization of intravenous magnesium sulfate after acute stroke. Stroke 1998;29:918‐923. [DOI] [PubMed] [Google Scholar]
Remacemide phase 2 {unpublished data only}
- Dyker AG, Lees KR. Remacemide hydrochloride: a double‐blind, placebo‐controlled, safety and tolerability study in patients with acute ischemic stroke. Stroke 1999;30(9):1796‐1801. [DOI] [PubMed] [Google Scholar]
Selfotel IIa {published data only}
- Grotta J, Clark W, Coull B, et al. Safety and tolerability of the glutamate antagonist CGS 19755 (selfotel) in patients with acute ischemic stroke: Results of a phase IIa randomized trial. Stroke 1995;26:602‐605. [DOI] [PubMed] [Google Scholar]
Selfotel IIb {published data only}
- Markabi S. Selfotel (CGS 19755): the preliminary clinical experience. In: Krieglstein J, Oberpichler‐Schwenk H editor(s). Pharmacology of Cerebral Ischemia. Stuttgart: Medpharm, 1994:635‐642. [Google Scholar]
Sipatrigine 137‐101 {published data only}
- Hussein Z, Fraser IJ, Lees KR, et al. Pharmacokinetics of 619C89, a novel neuronal sodium channel inhibitor, in acute stroke patients after loading and discrete maintenance infusions. Br J Clin Pharmac 1996;41:505‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir KW, Hamilton SJC, Lunnon MW, Hobbiger S, Lees KR. Safety and tolerability of 619C89 after acute stroke. Cerebrovasc Dis 1998;8:31‐37. [DOI] [PubMed] [Google Scholar]
Sipatrigine 137‐102 {unpublished data only}
- Hainsworth AH, Stefani A, Calabresi P, Smith TW, Leach MJ. Sipatrigine (BW619C89) is a neuroprotective agent and a sodium and calcium channel inhibitor. CNS Drug Reviews 2000;6(2):111‐134. [Google Scholar]
Sipatrigine 137‐104 {unpublished data only}
- Hainsworth AH, Stefani A, Calabresi P, Smith TW, Leach MJ. Sipatrigine (BW619C89) is a neuroprotective agent and a sodium and calcium channel inhibitor. CNS Drug Reviews 2000;6(2):111‐134. [Google Scholar]
Sipatrigine 137‐121 {published data only}
- Muir KW, Holzapfel L, Lees KR. Phase II clinical trial of sipatrigine (619C89) by continuous infusion in acute stroke. Cerebrovascular Diseases 2000;10(6):431‐436. [DOI] [PubMed] [Google Scholar]
Wester 1985 {published data only}
- Wester PO, Asplund K, Eriksson S, Hagg E, Lithner F, Strand T. Infusion of magnesium in patients with acute brain infarction. Acta Neurol Scand 1984;70(2):143. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Fosphenytoin phase 3 {published data only (unpublished sought but not used)}
- Multicenter Study to Evaluate the Safety and Efficacy of Intravenous Fosphenytoin in Patients with Acute Ischemic Stroke. Washington Stroke Center (http://www.strokecenter.org/trials/list/trialPage21.htm) 2001.
Galeas 1999 {published data only}
- Galeas T, Contos T, Exarhos P, Galea V, Valotasiou B. The role of magnesium (Mg) a natural calcium (Ca) antagonist in the treatment of acute ischaemic stroke (clinical study). Cerebrovascular Diseases 1999;9(Supplement 1):102. [Google Scholar]
Giroux 1994 {published data only}
- Giroux C, Garreau M, Morselli PL. Glutamate receptor antagonists in stroke patients. Neuropsychopharmacology 1994;10(Supplement 1):213S. [Google Scholar]
Lampl 2001 {published data only}
- Lampl Y, Gilad R, Geva D, Eshel Y, Sadeh M. Intravenous administration of magnesium sulfate in acute stroke: a randomized double‐blind study. Clin Neuropharmacol 2001;24(1):11‐15. [DOI] [PubMed] [Google Scholar]
Lub‐Int 2 & Lub‐Bel 4 {published data only}
- Keyser J, Velde V, Schellens RL, Hantson L, Tritsmans L, Gheuens J, et al. Safety and pharmacokinetics of the neuroprotective drug lubeluzole in patients with ischemic stroke. Clinical Therapeutics 1997;19(6):1340‐1351. [DOI] [PubMed] [Google Scholar]
Sipatrigine CEE 05 {unpublished data only}
- Sipatrigine. Unpublished.
ZK‐200775 {published data only (unpublished sought but not used)}
- Elting JW, Sulter GA, Kaste M, Lees KR, Diener HC, Hommel M, et al. AMPA antagonist ZK200775 in patients with acute ischemic stroke: possible glial cell toxicity detected by monitoring of S‐100B serum levels. Stroke 2002;33(12):2813‐2818. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
ARTIST MRI {published and unpublished data}
- Yurko‐Mauro KA, Prince HC, Williams B, Tempel D, Warach S. ARTIST MRI: AMPA Receptor Antagonist Treatment in Ischemic Stroke Trial‐MRI. Proceedings of the 27th International Stroke Conference. 2002; Vol. Ongoing Clinical Trials:CTP349.
ARTIST+ {unpublished data only}
- Peterson L, Prince HC, Williams B, Tempel D, Grotta JC. ARTIST+: AMPA Receptor Antagonist Treatment in Ischemic Stroke Trial, YM872 + Alteplase. Proceedings of the 27th International Stroke Conference. American Stroke Association, 2002; Vol. Ongoing Clinical Trials:CTP350.
IMAGES {published and unpublished data}
- Bradford APJ, Lees KR. Design of the Intravenous Magnesium Efficacy in Acute Stroke (IMAGES) trial. Current Controlled Trials in Cardiovascular Medicine 2000;1:184‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir KW. Magnesium in stroke treatment. Postgrad Med J 2002;78(925):641‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Aronowski 1996
- Aronowski J, Strong R, Grotta JC. Treatment of experimental focal ischemia in rats with lubeluzole. Neuropharmacology 1996;35(6):689‐693. [DOI] [PubMed] [Google Scholar]
Arrowsmith 1998
- Arrowsmith JE, Harrison MJ, Newman SP, Stygall J, Timberlake N, Pugsley WB. Neuroprotection of the brain during cardiopulmonary bypass: a randomized trial of remacemide during coronary artery bypass in 171 patients. Stroke 1998;29(11):2357‐2362. [DOI] [PubMed] [Google Scholar]
Bannan 1994
- Bannan PE, Graham DI, Lees KR, McCulloch J. Neuroprotective effect of remacemide hydrochloride in focal cerebral ischemia in the cat. Brain Res 1994;664:271‐275. [DOI] [PubMed] [Google Scholar]
Bialobok 1999
- Bialobok P, Cregan EF, Sydserff SG, Eisman MS, Miller JA, Cross AJ, et al. Efficacy of AR‐R15896AR in the rat monofilament model of transient middle cerebral artery occlusion. Journal of Stroke and Cerebrovascular Diseases 1999;8:388‐397. [DOI] [PubMed] [Google Scholar]
De Ryck 2000
- Ryck M, Verhoye M, Linden AM. Diffusion‐weighted MRI of infarct growth in a rat photochemical stroke model: effect of lubeluzole. Neuropharmacology 2000;39(4):691‐702. [DOI] [PubMed] [Google Scholar]
Dorman 1996
- Dorman PJ, Sandercock PA. Considerations in the design of clinical trials of neuroprotective therapy in acute stroke. Stroke 1996;27:1507‐1515. [DOI] [PubMed] [Google Scholar]
Gandolfo 2002
- Gandolfo C, Sandercock P, Conti M. Lubeluzole for acute ischaemic stroke. Cochrane Database of Systematic Reviews 2002, Issue 4. [Art. No.: CD001924. DOI: 10.1002/14651858.CD001924] [DOI] [PubMed] [Google Scholar]
Grosset 1995
- Grosset DG, Muir KW, Lees KR. Systemic and cerebral hemodynamic responses to the non‐competitive NMDA antagonist CNS 1102. J Cardiovasc Pharmacol 1995;25:705‐709. [DOI] [PubMed] [Google Scholar]
Hainsworth 2000
- Hainsworth AH, Stefani A, Calabresi P, Smith TW, Leach MJ. Sipatrigine (BW619C89) is a neuroprotective agent and a sodium and calcium channel inhibitor. CNS Drug Reviews 2000;6(2):111‐134. [Google Scholar]
Horn 2001
- Horn J, Limburg M. Calcium antagonists for ischemic stroke: a systematic review. Stroke 2001;32(2):570‐576. [DOI] [PubMed] [Google Scholar]
Lekieffre 1997
- Lekieffre D, Benavides J, Scatton B, Nowicki JP. Neuroprotection afforded by a combination of eliprodil and a thrombolytic agent, rt‐PA, in a rat thromboembolic stroke model. Brain Res 1997;776(1‐2):88‐95. [DOI] [PubMed] [Google Scholar]
Mahoney 1965
- Mahoney FI, Barhel DW. Functional evaluation: the Barthel Index. Md State Med J 1965;14:61‐65. [PubMed] [Google Scholar]
Maiese 1997
- Maiese K, TenBroeke M, Kue I. Neuroprotection of lubeluzole is mediated through the signal transduction pathways of nitric oxide. J Neurochem 1997;68(2):710‐714. [DOI] [PubMed] [Google Scholar]
McCulloch 1992
- McCulloch J. Excitatory amino acid antagonists and their potential for the treatment of ischaemic brain damage in man. Brit J Clin Pharmacol 1992;34:106‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Meadows 1994
- Meadows M‐E, Fisher M, Minematsu K. Delayed treatment with a noncompetitive NMDA antagonist, CNS 1102, reduces infarct size in rats. Cerebrovasc Dis 1994;4:26‐31. [Google Scholar]
Minematsu 1993
- Minematsu K, Fisher M, Li L, Davis MA, Knapp AG, Cotter RE, et al. Effects of a novel NMDA antagonist on experimental stroke rapidly and quantitatively assessed by diffusion‐weighted MRI. Neurology 1993;43:397‐403. [DOI] [PubMed] [Google Scholar]
Morris 1999
- Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF. Failure of the competitive N‐methyl‐D‐aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg 1999;91:737‐743. [DOI] [PubMed] [Google Scholar]
Muir 1994
- Muir KW, Grosset DG, Gamzu E, Lees KR. Pharmacological effects of the non‐competitive NMDA antagonist CNS 1102 in normal volunteers. Br J Clin Pharmac 1994;38:33‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muir 1995b
- Muir KW, Lees KR. Clinical experience with excitatory amino acid antagonist drugs. Stroke 1995;26(3):503‐513. [DOI] [PubMed] [Google Scholar]
Muir 1997
- Muir KW, Grosset DG, Lees KR. Effects of prolonged infusions of the NMDA antagonist aptiganel hydrochloride (CNS 1102) in normal volunteers. Clin Neuropharmacol 1997;20:311‐321. [DOI] [PubMed] [Google Scholar]
Muir 1999
- Muir KW, Grosset DG. Neuroprotection for acute stroke : making clinical trials work. Stroke 1999;30(1):180‐182. [DOI] [PubMed] [Google Scholar]
Muir 2002a
- Muir KW. Heterogeneity of stroke pathophysiology and neuroprotective clinical trial design. Stroke 2002;33(6):1545‐1550. [DOI] [PubMed] [Google Scholar]
NINDS 1995
- The National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. The New England Journal of Medicine 1995;333(24):1581‐1587. [DOI] [PubMed] [Google Scholar]
NPS 1506
- Brady EM, Wells DS, Kierstead AE, Mueller AL, Newman MK, Sanguinetti EL, Marriott TB. Ascending dose safety and pharmacokinetic trial of NPS 1506, a novel NMDA receptor antagonist, in normal male volunteers. Neurology 1998;50(Supplement 1):A346. [Google Scholar]
Perez‐Pinzon 1995
- Perez‐Pinzon MA, Maier CM, Yoon EJ, Sun GH, Giffard RG, Steinberg GK. Correlation of CGS 19755 neuroprotection against in vitro excitotoxicity and focal cerebral ischemia. J Cereb Blood Flow Metab 1995;15(5):865‐876. [DOI] [PubMed] [Google Scholar]
Rankin 1957
- Rankin J. Cerebral vascular accidents in patients over the age of 60, II: prognosis. Scott Med J 1957;2:200‐215. [DOI] [PubMed] [Google Scholar]
Reggiani 2001
- Reggiani A, Pietra C, Arban R, Marzola P, Guerrini U, Ziviani L, et al. The neuroprotective activity of the glycine receptor antagonist GV150526: an in vivo study by magnetic resonance imaging. Eur J Pharmacol 2001;419(2‐3):147‐153. [DOI] [PubMed] [Google Scholar]
Scheller 1997
- Scheller DK, Ryck M, Kolb J, Szathmary S, Reempts J, Clincke G, et al. Lubeluzole blocks increases in extracellular glutamate and taurine in the peri‐infarct zone in rats. Eur J Pharmacol 1997;338(3):243‐251. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408. [DOI] [PubMed] [Google Scholar]
Smith 1997
- Smith SE, Hodges H, Sowinski P, Man CM, Leach MJ, Sinden JD, et al. Long‐term beneficial effects of BW619C89 on neurological deficit, cognitive deficit and brain damage after middle cerebral artery occlusion in the rat. Neuroscience 1997;77(4):1123‐1135. [DOI] [PubMed] [Google Scholar]
STAIR
- Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999;30(12):2752‐2758. [DOI] [PubMed] [Google Scholar]
Takahashi 2002
- Takahashi M, Kohara A, Shishikura J, Kawasaki‐Yatsugi S, Ni JW, Yatsugi S, et al. YM872: a selective, potent and highly water‐soluble alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid receptor antagonist. CNS Drug Rev 2002;8(4):337‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tirilazad
- Tirilazad International Steering Committee. Tirilazad mesylate in acute ischemic stroke: A systematic review. Stroke 2000;31(9):2257‐2265. [DOI] [PubMed] [Google Scholar]
Turski 1998
- Turski L, Huth A, Sheardown M, McDonald F, Neuhaus R, Schneider HH, et al. ZK200775: a phosphonate quinoxalinedione AMPA antagonist for neuroprotection in stroke and trauma. Proc Natl Acad Sci USA 1998;95(18):10960‐10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Swieten 1988
- Swieten JC, Koudstaal PJ, Visser MC, Schouten HJA, Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 1988;19:604‐607. [DOI] [PubMed] [Google Scholar]
Wahlgren 1999
- Wahlgren NG, Ranasinha KW, Rosolacci T, Franke CL, Erven PM, Ashwood T, et al. Clomethiazole acute stroke study (CLASS) : results of a randomized, controlled trial of clomethiazole versus placebo in 1360 acute stroke patients. Stroke 1999;30(1):21‐28. [DOI] [PubMed] [Google Scholar]
Yenari 1998
- Yenari MA, Bell TE, Kotake AN, Powell M, Steinberg GK. Dose escalation safety and tolerance study of the competitive NMDA antagonist selfotel (CGS 19755) in neurosurgery patients. Clin Neuropharmacol 1998;21(1):28‐34. [PubMed] [Google Scholar]
YM872
- Napoliello MJ, Voss RW. Clinical experience with YM872, a novel AMPA receptor antagonist for acute ischemic stroke. Stroke 1999;30(1):264. [Google Scholar]