

Abstract
The function of G protein‐coupled receptors (GPCRs) can be modulated by compounds that bind to other sites than the endogenous orthosteric binding site, so‐called allosteric sites. Structure elucidation of a number of GPCRs has revealed the presence of a sodium ion bound in a conserved allosteric site. The small molecule amiloride and analogs thereof have been proposed to bind in this same sodium ion site. Hence, this review seeks to summarize and reflect on the current knowledge of allosteric effects by amiloride and its analogs on GPCRs. Amiloride is known to modulate adenosine, adrenergic, dopamine, chemokine, muscarinic, serotonin, gonadotropin‐releasing hormone, GABAB, and taste receptors. Amiloride analogs with lipophilic substituents tend to be more potent modulators than amiloride itself. Adenosine, α‐adrenergic and dopamine receptors are most strongly modulated by amiloride analogs. In addition, for a few GPCRs, more than one binding site for amiloride has been postulated. Interestingly, the nature of the allosteric effect of amiloride and derivatives varies considerably between GPCRs, with both negative and positive allosteric modulation occurring. Since the sodium ion binding site is strongly conserved among class A GPCRs it is to be expected that amiloride also binds to class A GPCRs not evaluated yet. Investigating this typical amiloride‐GPCR interaction further may yield general insight in the allosteric mechanisms of GPCR ligand binding and function, and possibly provide new opportunities for drug discovery.
Keywords: allosteric modulation; amiloride; drug discovery; G protein‐coupled receptors; 5‐(N,N‐hexamethylene)amiloride
Abbreviations
- 5‐HT
5‐hydroxy‐tryptamine
- 8‐OH‐DPAT
8‐hydroxy‐2‐(di‐n‐propylamino)tetralin
- Bmax
maximum number of binding sites
- koff
dissociation rate constant
- A‐EIA‐AS
(N‐2‐aminoethyl‐N‐isopropyl)amiloride‐N‐(4‐azidosalicylamide)
- AB‐MECA
N6‐(4‐aminobenzyl)‐N‐methylcarboxamidoadenosine
- BLT1
leukotriene B4 receptor
- CBDMB
5‐(N‐4‐chlorobenzyl)‐2′,4′‐dimethylbenzamil
- CCL2
C–C motif chemokine ligand 2
- CCR2
C–C chemokine receptor type 2
- Cryo‐EM
cryogenic electron microscopy
- DCB
3′,4′‐dichlorobenzamil
- DMA
5‐(N,N‐dimethyl)amiloride
- DPCPX
dipropylcyclopentylxanthine
- EC50
half‐maximal effective concentration
- EIA
5‐(N‐ethyl‐N‐isopropyl)amiloride
- Emax
maximum efficacy
- EMPA
N‐ethyl‐2‐[(6‐methoxy‐pyridin‐3‐yl)‐(toluene‐2‐sulfonyl)‐amino]‐N‐pyridin‐3‐yl‐methyl‐acetamide
- FD‐1
furan derivative‐1
- GABAB
γ‐aminobutyric acid‐B
- GnRH
gonadotropin‐releasing hormone
- GPCRs
G protein‐coupled receptors
- GTPγS
guanosine 5′‐O‐(γ‐thio)triphosphate
- hA2AAR
human adenosine A2A receptor
- HMA
5‐(N,N‐hexamethylene)amiloride
- IC50
half‐maximal inhibitory concentration
- Ki
equilibrium inhibition constant
- LTB4
leukotriene B4
- MBA
5‐(N‐methyl‐N‐butyl)amiloride
- MGCMA
5‐(N‐methyl‐N‐guanidinocarbonyl‐methyl)amiloride
- MIBA
5‐(N‐methyl‐N‐isobutyl)amiloride
- NECA
5′‐(N‐ethylcarboxamido)adenosine
- NMR
nuclear magnetic resonance
- OX2R
orexin‐2 receptor
- PIA
(‐)‐N6‐(R‐phenylisopropyl)‐adenosine
- SEM
standard error of the mean
- T1R2
taste receptor type 2
- T1R2‐HD
taste receptor type 2‐heptahelical domain
- T1R3
taste receptor type 3
- WT
wild‐type
1. INTRODUCTION
G protein‐coupled receptors (GPCRs) form a family of receptors with approximately 800 members that are responsible for many different physiological functions such as regulation of sleep, vision, blood pressure, central nervous system activity, taste, and olfaction.1 This is reflected by the fact that they are directly or indirectly targeted by 30% to 40% of therapeutic drugs currently in the market.2, 3 GPCRs are grouped according to their structural and genomic characteristics in five main groups: rhodopsin‐like (class A), secretin‐like (class B), glutamate‐like (class C), adhesion, and frizzled/taste2, with class A being the largest group.4, 5
The precise mechanisms of action of these receptors have been studied for a long time, but due to the complexity of their structures, they are not yet fully understood. Novel pharmacological concepts have been introduced that reflect this complexity. For the purpose of this review, the concept of allosteric modulation is particularly relevant, which has been excellently reviewed elsewhere.6, 7, 8 The recent increase in high‐resolution GPCR crystal and cryo‐EM structures also allows a better understanding of how GPCRs function.9, 10, 11 Cocrystallization with orthosteric ligands such as agonists and antagonists allows the study of the orthosteric binding sites, that is the sites for endogenous hormones and neurotransmitters. However, to study allosteric binding sites cocrystallization with allosteric modulators is desired, which is a challenge due to their often low affinities. Adding high concentrations of sodium ions is a common procedure in the crystallization of GPCRs to stabilize the protein, which makes it possible for these ions to bind to low‐affinity sites. However, sodium ions are relatively small and need a high resolution (<2 Å) to be visualized. In recent crystal structures of several GPCRs the resolution was sufficiently high to locate a sodium ion bound in a site which is highly conserved amongst class A GPCRs.12 Currently solved crystal structures with a sodium ion bound in this allosteric site are of the human adenosine A2A receptor,13 the β1‐adrenergic receptor,14, 15 the human δ‐opioid receptor,16 and the human protease‐activated receptor 1.17 The common residues that interact with the sodium ion in these crystal structures, either directly or through water‐mediated hydrogen bond interactions, are Asp2.50, Ser3.39 Trp6.48, Asn7.45, and Asn7.49 (numbering according to Ballesteros‐Weinstein18). The negatively charged amino acid Asp2.50 makes a strong salt bridge with the positively charged sodium ion and is essential for its binding in this site, which confirmed previous “pre‐crystal structure” research.19 It is also the most conserved residue of the sodium ion site amongst GPCRs. The high conservation of the sodium ion pocket amongst class A GPCRs makes it probable that more structures with sodium ions bound in this site will emerge. There is little if any conservation present in the other GPCR classes, which makes it improbable that such a sodium ion binding site exists in these GPCRs.
Amiloride is primarily known as a potassium‐sparing diuretic drug, acting through the blockade of renal epithelial sodium channels.20 Amiloride and its analogs have also been found to bind to the sodium ion site of several GPCRs, modulating orthosteric ligand binding.21 The negatively charged carboxylate of sodium ion site residue Asp2.50 supposedly interacts with the positively charged guanidinium group present in all amilorides. The binding of amilorides into the sodium ion site of class A GPCRs renders these compounds potential pharmacological tools to probe molecular mechanisms of GPCR allosteric modulation. The chemical structures of amiloride and its analogs discussed in this review are depicted in Figures 1, 2. Effects of the amilorides are represented in Table 1 categorized per GPCR and orthosteric ligands used. Most of the receptors in Table 1 are discussed in the main text.
Figure 1.
Chemical structures of amiloride and its 5′‐amino substituted analogs DMA, EIA, MIBA, MBA, HMA, and MGCMA. DMA, 5‐(N,N‐dimethyl)amiloride; EIA, 5‐(N‐ethyl‐N‐isopropyl)amiloride; HMA, 5‐(N,N‐hexamethylene)amiloride; MBA, 5‐(N‐methyl‐N‐butyl)amiloride; MGCMA, 5‐(N‐methyl‐N‐guanidinocarbonyl‐methyl)amiloride; MIBA, 5‐(N‐methyl‐N‐isobutyl)amiloride
Figure 2.
Chemical structures of 2‐guanidino substituted amiloride analogs phenamil, benzamil, DCB, CBDMB, and A‐EIA‐AS. A‐EIA‐AS, (N‐2‐aminoethyl‐N‐isopropyl)amiloride‐N‐(4‐azidosalicylamide); CBDMB, 5‐(N‐4‐chlorobenzyl)‐2',4'‐dimethylbenzamil; DCB, 3',4'‐dichlorobenzamil
Table 1.
Modulation of G protein‐coupled receptors by amiloride and amiloride analogs. The given values reflect a) inhibitory potency or affinity for ligand displacement in radioligand binding assays or inhibition of ligand‐induced receptor activation in functional assays, b) modulatory potency of their effect on radioligand dissociation, and c) fold change of dissociation rates of orthosteric ligands. References are in numbered superscript
| Receptor | Orthosteric ligand antagonist; agonist | Amiloride (analog) | Inhibitory potency or affinity a ‐ Displacement of orthosteric ligand by amiloride (analog) in IC50 or Ki ± SEM (µM) | Modulatory potency ‐ Concentration‐effect on dissociation of orthosteric ligand by amiloride (analog) in EC50 ± SEM (µM) | Effect on dissociation of orthosteric ligand by 100 µM b amiloride (analog) in koff/koff(control) > 1, Increase < 1, Decrease |
|---|---|---|---|---|---|
| Adenosine A1 | [3H]DPCPX | Amiloride | 2.0 ± 0.2B24 | … | 1.5Rd26 |
| 197 ± 23R26 | |||||
| Benzamil | 0.65 ± 0.04B24 | … | … | ||
| CBDMB | 1.2 ± 0.1B24 | … | … | ||
| DCB | 1.6 ± 0.1B24 | … | … | ||
| DMA | 8 ± 2R26 | … | 1.9R26 | ||
| HMA | 0.41 ± 0.03B24 | … | 1.7R26 | ||
| 22 ± 4R26 | |||||
| MBA | 0.070 ± 0.004B24 | … | … | ||
| MGCMA | 22 ± 1B24 | … | … | ||
| MIBA | 0.16 ± 0.01B24 | … | … | ||
| 13 ± 1R26 | |||||
| Phenamil | 1.5 ± 0.1B24 | … | … | ||
| [ 3 H]PIA | Amiloride | 2.4 ± 0.1B24 | … | No effectRd26 | |
| Benzamil | 0.85 ± 0.03B24 | … | … | ||
| CBDMB | 4.0 ± 0.4B24 | … | … | ||
| DCB | 2.7 ± 0.2B24 | … | … | ||
| DMA | … | … | No effectR26 | ||
| HMA | 0.50 ± 0.03B24 | … | No effectR26 | ||
| MBA | 0.09 ± 0.01B24 | … | … | ||
| MGCMA | 16 ± 1B24 | … | |||
| MIBA | 0.20 ± 0.01B24 | … | … | ||
| Phenamil | 2.3 ± 0.1B24 | … | … | ||
| Adenosine A2A | [3H]ZM‐241,385 | Amiloride | 9.7 ± 1.1R25 | … | 1.2Rd25 |
| Benzamil | 2.2 ± 0.3R25 | … | 2.4Rd25 | ||
| HMA | 3.3 ± 0.5R25 | … | 12Rd25 | ||
| MGCMA | 89 ± 13R25 | … | 1.2Rd25 | ||
| MIBA | 3.0 ± 0.2R25 | … | 5.7Rd25 | ||
| Phenamil | 2.6 ± 0.4R25 | … | 1.9Rd25 | ||
| [ 3 H]CGS‐21,680 | Amiloride | … | … | No effectRd26 | |
| DMA | … | … | No effectR26 | ||
| HMA | … | … | No effectR26 | ||
| Adenosine A3 | [3H]PSB‐11 | Amiloride | 82 ± 7H26 | … | No effectHd26 |
| DMA | 13 ± 2H26 | … | 1.3H26 | ||
| HMA | 6 ± 1H26 | … | 2.3H26 | ||
| MIBA | 8 ± 1H26 | … | 1.6H26 | ||
| [ 125 I]I‐AB‐MECA | Amiloride | >100R26 | … | No effectHd26 | |
| DMA | 20 ± 3R26 | … | 0.80 H26 | ||
| HMA | 7 ± 1R26 | … | 0.53 H26 | ||
| MIBA | 7 ± 2R26 | … | 0.59 H26 | ||
| α1A‐Adrenergic | [3H]Prazosin | Amiloride | 11 ± 2H37 | … | 1.2H37 |
| Benzamil | 0.8 ± 0.1H37 | … | 1.7H37 | ||
| DMA | 0.82 ± 0.03 H37 | … | 1.5H37 | ||
| EIA | 2.7 ± 0.3 H37 | … | 2.2H37 | ||
| HMA | 1.1 ± 0.2H37 | … | 5.5H37 | ||
| MIBA | 0.49 ± 0.07H37 | … | 2.4H37 | ||
| α2A‐Adrenergic | [3H]Yohimbine | Amiloride | 30 ± 2H42 | … | 2.0He42 |
| A‐EIA‐AS | … | 40P41 | >1P41 | ||
| Benzamil | 3.5 ± 0.7H42 | … | No effectHe42 | ||
| DMA | 3.6 ± 0.1H42 | … | 5.3He42 | ||
| [3H]Rauwolscine | … | … | 6.3He42 | ||
| [3H]RX‐821,002 | … | … | 7.1He42 | ||
| [3H]Yohimbine | EIA | 1.7 ± 0.2H42 | 50P41 | >1P41 | |
| 155He42 | |||||
| HMA | 0.21 ± 0.00H42 | … | 138He42 | ||
| [3H]Rauwolscine | … | … | 57He42 | ||
| [3H]Yohimbine | MIBA | 0.56 ± 0.01H42 | … | 101He42 | |
| [ 3 H]UK‐14,304 | Amiloride | 25 ± 0.2H43 | … | 0.67 He43 | |
| DMA | 3.2 ± 0.2H43 | … | 0.77 He43 | ||
| HMA | 0.18 ± 0.02H43 | … | 0.37 He43 | ||
| α2B‐Adrenergic | [3H]Rauwolscine | CBDMB | … | … | <1 R44 |
| EIA | … | … | >R44 | ||
| MIBA | … | … | >1R44 | ||
| β1‐Adrenergic | [125I]Iodocyano‐pindolol | Amiloride | 83 ± 14R36 | … | … |
| β2‐Adrenergic | [125I]Iodocyano‐pindolol | Amiloride | 60R36 | … | … |
| CCR2 | [3H]INCB3344 | Amiloride | No effectHf45 | … | … |
| Benzamil | No effectHf45 | … | … | ||
| HMA | 79H45 | … | 1.25H45 | ||
| MCGMA | No effectHf45 | … | … | ||
| MIBA | 158H45 | … | … | ||
| Phenamil | No effectHf45 | … | … | ||
| [3H]CCR2‐RA‐[R] c | Amiloride | No effectHf45 | … | … | |
| Benzamil | No effectHf45 | … | … | ||
| HMA | 79H45 | … | 1.36H45 | ||
| MCGMA | No effectHf45 | … | … | ||
| MIBA | 126H45 | … | … | ||
| Phenamil | No effectHf45 | … | … | ||
| [ 125 I]CCL2 | HMA | … | … | 9.7H45 | |
| Dopamine D1 | [3H]SCH‐23,390 | Amiloride | 49 ± 1H50 | >1000H50 | … |
| Benzamil | 1.6 ± 0.5H50 | 74 ± 8H50 | … | ||
| MIBA | 4.4 ± 0.2H50 | 13 ± 1H50 | 26He50 | ||
| Dopamine D2 | [125I]Epidepride | Amiloride | … | … | 2.5Ri51 |
| [3H]Spiperone | 390 ± 4H50 | 215 ± 35R53 | 1.5Ri51 | ||
| … | 100 ± 10H50 | 2.7Rj53 | |||
| Benzamil | 25 ± 2H50 | 46 ± 4R53 | 4.8Rk53 | ||
| 29 ± 7H50 | |||||
| DMA | … | 76 ± 8R53 | 8.4Rk53 | ||
| EIA | … | 20 ± 5R53 | 18Rl53 | ||
| HMA | … | 10 ± 2R53 | 16Rl53 | ||
| MIBA | 6.6 ± 0.4H50 | 14 ± 1R53 | 14Rl53 | ||
| 2.1 ± 0.2H50 | 88He50 | ||||
| Dopamine | Amiloride | 29Rm54 | … | … | |
| DMA | 1.4Rm54 | … | … | ||
| MIBA | 0.9Rm54 | … | |||
| 0.6 ± 0.2Hn50 | |||||
| Dopamine D3 | [3H]Spiperone | Amiloride | 120 ± 7H50 | 43 ± 3 H50 | … |
| Benzamil | 16 ± 1H50 | 15 ± 2H50 | … | ||
| MIBA | 1.7 ± 0.1H50 | 0.29 ± 0.14H50 | 18He50 | ||
| Dopamine | MIBA | 1.8Ro54 | … | … | |
| Dopamine D4 | [3H]Spiperone | Amiloride | 280 ± 30H50 | 420 ± 4H50 | … |
| Benzamil | 6.1 ± 0.4H50 | 28 ± 2H50 | … | ||
| MIBA | 1.3 ± 0.2H50 | 22 ± 5H50 | >1He50 | ||
| GnRH | [ 125 I]Triptorelin | Amiloride | >100H63 | … | … |
| Benzamil | >100H63 | … | … | ||
| DCB | 30 ± 3 H63 | … | 1.7H63 | ||
| MIBA | 39 ± 7H63 | … | 2.1H63 | ||
| HMA | 29 ± 3 H63 | 49 ± 7H63 | 2.5H63 | ||
| MCGMA | >100H63 | … | … | ||
| Phenamil | >100H63 | … | … | ||
| Histamine H1 | [3H]Mepyramine | Amiloride | >10R21 | … | … |
| Benzamil | 3.2 ± 0.2R21 | … | … | ||
| HMA | 5.6 ± 1.2R21 | … | … | ||
| Muscarinic M1 | [3H]Pirenzepine | Amiloride | >10R21 | … | … |
| Benzamil | 2.9 ± 0.7R21 | … | … | ||
| HMA | 3.6 ± 1.0R21 | … | … | ||
| Muscarinic M2 | [3H]N‐methyl‐scopolamine | Amiloride | >10R21 | … | … |
| Benzamil | 5.8 ± 1.1R21 | … | … | ||
| HMA | 2.9 ± 0.5R21 | … | … | ||
| Muscarinic M3 | [3H]N‐methyl‐scopolamine | Amiloride | 50R67 | … | … |
| Benzamil | 2.8 ± 0.5R21 | … | … | ||
| HMA | 4.7 ± 0.8R21 | … | … | ||
| Acetylcholine | Amiloride | 478Rq65 | … | … | |
| δ‐Opioid | [ 3 H]DADLE | Amiloride | >10R21 | … | … |
| Benzamil | >10R21 | … | … | ||
| HMA | 1.0 ± 0.2R21 | … | … | ||
| κ‐Opioid | [ 3 H]Ethyl‐ketazocine | Amiloride | >10R21 | … | … |
| Benzamil | >10R21 | … | … | ||
| HMA | 3.9 ± 0.6R21 | … | … | ||
| µ‐Opioid | [3H]Naloxone | Amiloride | >10R21 | … | … |
| Benzamil | 1.1 ± 0.4R21 | … | … | ||
| HMA | 0.06 ± 0.02R21 | … | … | ||
| Serotonin 5‐HT1A | [ 3 H]8‐OH‐DPAT | Amiloride | >10R21 | … | … |
| Benzamil | 1.9 ± 0.3R21 | … | … | ||
| HMA | >10R21 | … | … | ||
| Serotonin 5‐HT1B | [ 3 H]5‐Carboxa‐midotryptamine | Amiloride | 20H73 | … | … |
| Sumatriptan | 35Hs73 | … | … | ||
| [ 3 H]Serotonin | Benzamil | >10R21 | … | … | |
| [ 3 H]5‐Carboxa‐midotryptamine | EIA | 13H73 | … | … | |
| [ 3 H]Serotonin | HMA | >10R21 | … | … | |
| Serotonin 5‐HT1C | [ 3 H]Serotonin | Amiloride | >10R21 | … | … |
| Benzamil | >10R21 | … | … | ||
| HMA | 6.7 ± 1.2R21 | … | … | ||
| Serotonin 5‐HT1D | [ 3 H]Serotonin | Amiloride | >10R21 | … | … |
| Benzamil | >10R21 | … | … | ||
| HMA | >10R21 | … | … | ||
| Serotonin 5‐HT2 | [ 3 H]Serotonin | Amiloride | >10R21 | … | … |
| Benzamil | 1.4 ± 0.1R21 | … | … | ||
| HMA | 0.40 ± 0.06R21 | … | … |
Abbreviations: B, bovine receptor; CBDMB, 5‐(N‐4‐chlorobenzyl)‐2',4'‐dimethylbenzamil; c, in presence of 100 µM amiloride (analogue) except when stated otherwise; d, in presence of 1 mM amiloride (analog); DCB, 3',4'‐dichlorobenzamil; DMA, 5‐(N,N‐dimethyl)amiloride; e, calculated for the amiloride (analog) occupied receptor; EIA, 5‐(N‐ethyl‐N‐isopropyl)amiloride; f, no displacement of orthosteric ligand by 100 µM amiloride (analog); g, [3H]CCR2‐RA‐[R] is an ‘intracellular antagonist’ as it binds intracellularly to the chemokine CCR2 receptor; HMA, 5‐(N,N‐hexamethylene)amiloride; H, human receptor; MGCMA, 5‐(N‐methyl‐N‐guanidinocarbonyl‐methyl)amiloride; i, in presence of 500 µM amiloride (analog); j, in presence of 3.16 mM amiloride (analog); k, in presence of 1 mM amiloride (analog); MIBA, 5‐(N‐methyl‐N‐isobutyl)amiloride; q, modulation by amiloride of acetylcholine‐induced contractions of rat tracheal smooth muscle, which expresses the muscarinic M3 receptor; R, rat receptor; m, inhibition by amiloride (analog) of dopamine‐stimulated increase in extracellular acidification rate in cells expressing the dopamine D2 receptor; n, inhibition by MIBA of dopamine‐stimulated [35S]GTPγS binding to dopamine D2 receptors; s, inhibition by amiloride of the sumatriptan‐induced reduction of cAMP formation stimulated by forskolin in cells expressing the Serotonin 5‐HT1B receptor; SEM, standard error of mean.
IC50 values determined with concentrations of orthosteric radioligands around their KD.
In presence of 100 µM amiloride (analog) except when stated otherwise.
[3H]CCR2‐RA‐[R] is an ‘intracellular antagonist’ as it binds intracellularly to the chemokine CCR2 receptor.
2. ADENOSINE RECEPTORS
Adenosine receptors have been studied extensively, and as a result, many orthosteric22 and allosteric23 ligands have been discovered. Amiloride interactions with adenosine receptors were discovered in the early days of adenosine receptor research.24 Since the effects of amiloride binding to adenosine receptors appeared to be closely tied to sodium ion interactions, it was necessary to investigate and exclude the involvement of Na+/H+ exchange proteins (one of the main targets of amiloride) in these interactions.21 In this study, Garritsen et al21 found inhibition of antagonist [3H]DPCPX and agonist [3H]PIA at the calf adenosine A1 receptor by amiloride, its 5′‐amino‐substituted analogs 5‐(N,N‐hexamethylene)amiloride (HMA), 5‐(N‐methyl‐N‐butyl)amiloride (MBA), 5‐(N‐methyl‐N‐guanidinocarbonyl‐methyl) amiloride (MCGMA), and 5‐(N‐methyl‐N‐isobutyl)amiloride (MIBA), and its 2‐guanidino substituted analogs benzamil, 5‐(N‐4‐chlorobenzyl)‐2′,4′‐dimethylbenzamil (CBDMB), 3′,4′‐dichlorobenzamil (DCB), and phenamil.
Gao and IJzerman25 found that amiloride analogs benzamil, HMA, MCGMA, MIBA, and phenamil increased the dissociation rate of the antagonist [3H]ZM‐241,385 at the rat A2A receptor, and that they were more potent than amiloride itself (Figure 3). However, the affinity (defined by radioligand displacement in equilibrium) and the allosteric potency (defined by the concentration‐dependent effect on the radioligand dissociation rate) did not correlate. This indicated a mixed competitive (ie, mutually exclusive displacement) and noncompetitive behavior of amilorides, in which amilorides and orthosteric ligands bind to the receptor at the same time, whereas amiloride influences the orthosteric ligand's dissociation rate. The amiloride analogs HMA and MIBA, with a lipophilic moiety on the 5′‐position, proved to be the most potent compounds in increasing the dissociation rate of the orthosteric ligand, whereas they had equal affinities to benzamil and phenamil in displacing it. In contrast to the effect of amilorides, sodium ions decreased the dissociation rate of [3H]ZM‐241,385. Still, sodium ions and HMA appeared to compete for the same allosteric site.
Figure 3.
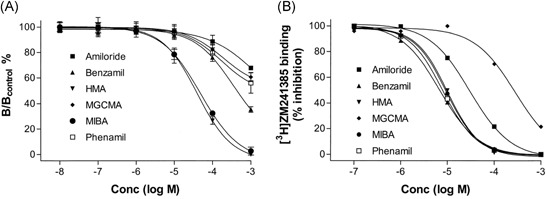
Concentration dependence of amiloride and its analogs for A, increase of [3H]ZM‐241,385 dissociation and B displacement of [3H]ZM‐241,385 after reaching binding equilibrium at adenosine A2A receptors. In A, [3H]ZM‐241,385 binding was allowed to first reach equilibrium at the receptor before its dissociation was induced by addition of an excess of antagonist, in the absence and presence of increasing concentrations of amiloride (analog). The results are expressed as a ratio between the binding of [3H]ZM‐241,385 after 120 minutes in the presence (“B”) and in the absence (Bcontrol) of amiloride (analog). Reproduced with permission from Gao and IJzerman.25 HMA, 5‐(N,N‐hexamethylene)amiloride; MBA, 5‐(N‐methyl‐N‐butyl)amiloride; MGCMA, 5‐(N‐methyl‐N‐guanidinocarbonyl‐methyl)amiloride
In a study by Gao et al26 it appeared that adenosine receptor agonizts and antagonists are differently affected by amilorides. Amilorides increased the dissociation rates of antagonists [3H]DPCPX at the rat adenosine A1 and [3H]PSB‐11 at the human A3 receptors, just as with [3H]ZM‐241,385 at the rat A2A receptor. However, they did not affect the dissociation rates of agonizts [3H]R‐PIA from the rat A1 and [3H]CGS‐21,680 from the rat A2A receptors. Amilorides decreased the dissociation rate of agonist [125I]‐AB‐MECA at the rat adenosine A3 receptor, revealing that amilorides can also act as positive allosteric modulators depending on the radiolabeled probe used.26 Furthermore the amilorides exhibited selectivity for the different adenosine receptor subtypes. Amiloride and 5‐(N,N‐dimethyl)amiloride (DMA) were more potent at the A1 receptor in accelerating antagonist dissociation, whereas HMA was the most potent at the A2A receptor and to a lesser extent at the A3 receptor.
Solving the crystal structure of the adenosine A2A receptor at a resolution of 1.8 Å provided a sufficiently high resolution to detect a sodium ion bound in its allosteric binding site for the first time (Figure 4A).13 The amino acids interacting with the sodium ion in this site are highly conserved amongst other GPCRs which confirmed previous studies in which modulation by sodium ions was tied to the same amino acids for different GPCRs.12 The most conserved amino acid is a negatively charged aspartic acid (Asp522.50) which interacts directly with the positively charged sodium ion by means of a salt bridge. In molecular dynamics simulations, Gutiérrez‐de‐Terán et al27 observed that the interaction of the sodium ion with Asp522.50 is highly stable in the receptor's inactive conformation. The presence of the ion also avoids rotamer changes in two other highly conserved residues, Trp2466.48 and Asn2807.45. Interestingly, an active receptor conformation caused the site to contract to expel the sodium ion from this allosteric binding site. These calculations agree very well with radioligand binding studies on A2AAR (Figure 5A).13, 27, 28 Sodium ions induced an increase in [3H]ZM‐241,385 antagonist binding, but inhibited [3H]NECA agonist binding in a concentration dependent‐manner (Figure 5A),27 suggesting among others that the binding of agonist and sodium ions can be considered as “mutually exclusive”.29 Interestingly, the IC50 value of NaCl to inhibit agonist binding was approximately 50 mM, suggesting that under physiological conditions ([NaCl] = 140 mM) the receptor is predominantly in an inactive state.
Figure 4.
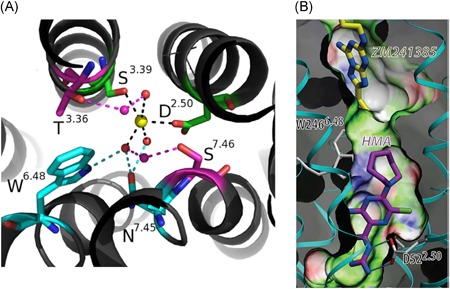
A, The Na+‐distorted octahedral coordination in the A2AAR crystal structure (PDB: 4EIY): the first shell is occupied by two conserved polar residues (green) and three water molecules (small spheres), which contact with the second shell of residues (cyan), or with a second layer of water molecules connecting with a third shell of residues (magenta). B, Docking of HMA in the sodium ion binding site. The guanidinium group of HMA has a salt bridge interaction with Asp522.50 whereas the 5′‐azepane moiety of HMA clashes with Trp2466.48. ZM‐241,385 is the orthosteric antagonist. Reproduced with permission from Gutiérrez‐de Terán et al27 [Color figure can beviewed at wileyonlinelibrary.com]
Figure 5.
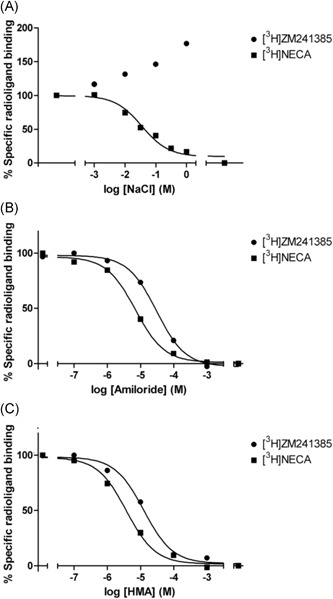
Equilibrium displacement of [3H]ZM‐241,385 (antagonist) and [3H]NECA (agonist) binding to A2AAR by allosteric modulators. A, NaCl, B, amiloride, and C, HMA. Reproduced with permission from Gutiérrez‐de Terán et al.27 HMA, 5‐(N,N‐hexamethylene)amiloride; NECA, 5'‐(N‐ethylcarboxamido)adenosine
The positively charged guanidinium moiety of amiloride and its analog HMA may also interact with Asp522.50 in a manner similar to sodium ions, as inferred from docking studies (Figure 4B). Radioligand binding studies with antagonist [3H]ZM‐241 385 and agonist [3H]NECA demonstrated amiloride and more strongly so HMA to reduce radioligand binding, with greater potency on agonist binding for both (Figure 5B and 5C).27
In a subsequent study, Massink et al30 introduced amino acid mutations in the sodium ion binding site to assess the key residues in the interaction between amiloride/HMA and A2AAR.30 Mutation of the polar residues in the pocket was shown to either abrogate (D52A2.50 and N284A7.49) or reduce (S91A3.39, W246A,6.48 and N280A7.45) the negative allosteric effect of sodium ions on agonist binding. The D52A2.50 mutation also decreased the potency of amilorides with respect to ligand displacement, for example, an 18‐fold reduction in HMA's IC50 value for [3H]ZM‐241,385 binding. Conversely, a big potency gain was observed on the W246A6.48 mutant. HMA's IC50 value increased 25‐fold from 8.9 to 0.36 µM; a similar gain was observed for amiloride, from 63 to 2.6 µM. Apparently, this tryptophan residue, part of a so‐called activation micro‐switch,31 hinders amilorides to bind in hA2AR (and possibly other GPCRs). Indeed, at the adenosine A3 receptor, the mutation of Trp2436.48 into Ala increased the affinity of HMA as well.32
These findings fueled the ambition to design and synthesize novel amiloride/HMA derivatives. The 5′‐substitution of amiloride with phenylethyl (compound 12 in Massink et al33) yielded the largest decrease in antagonist [3H]ZM‐241,385 binding to both the wild‐type and W246A6.48 mutant receptors compared to other substituents and carbon chain elongations. Further derivatization of the phenylethyl moiety yielded 4‐ethoxyphenylethyl derivative 12l (Figure 6), the most potent amiloride derivative of the series. This compound displaced [3H]ZM‐241,385 binding from the wild‐type A2AAR with an IC50 value of 3.4 µM, which was lower than HMA (5.1 µM). Derivative 12l also showed an increased potency compared to that of HMA for the W246A6.48 mutant receptor, 19‐fold compared to WT for HMA in this study and 76‐fold for 12l.33
Figure 6.
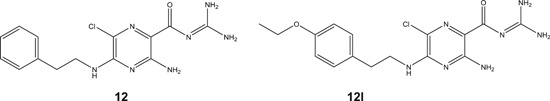
Amiloride derivatives 12 and 12l 33
The conformational flexibility of the adenosine A2A receptor was examined further in a 19F NMR study, providing evidence for the occurrence of four different states of activation. Interestingly, both HMA and a partial agonist favored the population of an active state (S3), still different from the S3′ active state induced by full agonists.34 In a later study by the same team, the effects of NaCl were analyzed, leading to the conclusion that sodium ions reinforce an inactive ensemble of states (S1‐2), as well as the partial‐agonist, stabilized state (S3). HMA competed with the sodium ions, reflected in its effects on both line broadening and chemical shift perturbations in the 23Na NMR binding isotherm.35
In conclusion, the effects of amiloride and derivatives have been most extensively studied on adenosine A2A receptors, through a number of orthogonal approaches. They all hint in the same direction, that is the amilorides compete with sodium ions at the allosteric sodium ion binding site in which Asp2.50 is the central amino acid. The evidence for other GPCRs is less exhaustive but suggests similar conclusions, which will be discussed below.
3. ADRENERGIC RECEPTORS
One of the first indications that amiloride inhibited the binding of orthosteric ligands at α‐ and β‐adrenergic receptors were found in 1987 by Howard et al,36 which was followed by many studies with amiloride and its analogs at a number of adrenergic receptor subtypes. At the human α1A‐adrenergic receptor amiloride and its analogs benzamil, DMA, 5‐(N‐ethyl‐N‐isopropyl)‐amiloride (EIA), MIBA, and HMA increased the dissociation rate of antagonist [3H]prazosin, and the analogs with bulky lipophilic 5′‐moieties were more potent in doing so.37, 38 Amiloride itself was characterized as an allosteric modulator acting at one allosteric site, but all the amiloride analogs appeared to bind to two different allosteric sites. The authors speculated that these allosteric sites could be present on one receptor or on a receptor dimer, but could not further confirm this.37 The allosteric interaction by amilorides was seemingly in contradiction with previous results at rat and mouse α1‐adrenergic receptors in which amiloride only showed a competitive interaction with antagonist [3H]prazosin binding but did not influence its dissociation rate.36
α2‐Adrenergic receptors are allosterically modulated by amilorides as well. At rat, human, bovine, and porcine α2A‐adrenergic receptors amiloride increased the dissociation rate of the antagonists [3H]rauwolscine36, 39 and [3H]yohimbine.40 Amiloride analogs also increased antagonist dissociation from the α2A‐adrenergic receptor, which was found for (N‐2‐aminoethyl‐N‐isopropyl)amiloride‐N‐(4‐azidosalicylamide; A‐EIA‐AS) at the porcine receptor,41 and DMA, EIA, MIBA, and HMA at the human receptor, in relation to [3H]yohimbine, [3H]rauwolscine, and [3H]RX‐821,002 dissociation.42 It is noteworthy that A‐EIA‐AS has no affinity for the Na+/H+ exchange protein, making it a GPCR selective amiloride. EIA, HMA, and MIBA were exceptionally strong negative allosteric modulators of antagonist binding, being 50‐ to 80‐fold more potent than amiloride in increasing the dissociation rate of [3H]yohimbine, showing that bulky lipophilic moieties at the 5′‐position of amiloride increase the allosteric potency at the α2A‐adrenergic receptor considerably. The apparent affinities of these amilorides were not correlating at all with their derived allosteric potencies in this study, cautioning to not confuse these two different pharmacological properties with each other.
In contrast to their effect on antagonists, amiloride, DMA, and HMA decreased the dissociation rate of agonist [3H]UK‐14 304 at the human α2A‐adrenergic receptor, with HMA having the largest effect.43 The dissociation‐slowing effect on agonist binding (2.7‐fold slower dissociation by HMA vs control) was considerably smaller though than the dissociation‐accelerating effect on antagonist binding (140‐fold faster dissociation by HMA). Although they slowed agonist dissociation, amilorides acted as negative allosteric modulators of α2A receptor agonist activation, because amiloride, DMA, and HMA decreased the potency of norepinephrine and UK‐41,304 in [35S]GTPγS binding experiments. This paradoxical behavior was in line with previous findings that amilorides displace the orthosteric ligand competitively from the α2A receptor in addition to their allosteric effects.42 Moreover, the addition of sodium ions increased the affinity of amiloride in doing so.36 This led to the conclusion that at α2A‐adrenergic receptors amilorides bind to two different sites, namely the orthosteric site and an allosteric sodium ion site. Howard et al36 hypothesized that amiloride binding in the orthosteric site was enhanced by binding of a sodium ion in the allosteric site, whereas amiloride binding in the allosteric site increased the dissociation rate of other orthosteric ligands. In a later study by Leppik et al,42 observed variations in the affinity of several amiloride analogs for the antagonist‐occupied and unoccupied receptor led to two different hypotheses. Either the amilorides bind to both the allosteric and orthosteric sites, or binding of an antagonist to the orthosteric site modified the conformation of the allosteric binding site in such a way that amiloride's affinity decreased.42
At the α2B subtype, however, amilorides both increased and decreased the dissociation rate of antagonists. The 5′‐substituted amilorides EIA and MIBA increased the dissociation rate of [3H]rauwolscine binding, whereas the guanidino‐substituted amiloride CBDMB decreased it.44
The interaction of amiloride with β‐adrenergic receptors has only been studied by Howard et al in 1987. At both the β1‐ and β2‐adrenergic receptors amiloride displaced the antagonist [125I]iodocyanopindolol competitively, because their binding was mutually exclusive.36 Addition of sodium ions did not compete with amiloride binding, and it was concluded that amiloride did bind to the orthosteric site rather than to an allosteric sodium ion site. Despite the lack of modulation of β‐adrenergic receptors by sodium ions and amiloride, a sodium ion site was found in the crystal structure of the β1‐adrenergic receptor.14 The amino acids forming the sodium ion sites of the β1‐adrenergic and the adenosine A2A receptor are the most similar of the solved GPCR crystal structures with such a site.12 That makes the difference in modulation by sodium ions and amilorides between these receptors remarkable and it is probably due to differences in the overall architecture of the two receptors.
4. CHEMOKINE RECEPTORS
Amiloride interactions with the chemokine receptor family have only been studied by Zweemer et al45 on the chemokine CCR2 receptor. The sodium ion site was the third binding site found on this receptor, next to the more extracellularly located orthosteric and an intracellular allosteric site.46, 47, 48, 49 Amiloride analogs MIBA and HMA inhibited binding of the antagonist [3H]INCB3344 binding to the orthosteric site and antagonist [3H]CCR2‐RA‐[R] binding to the intracellular site.45 Moreover, HMA inhibited binding of the orthosteric agonist [125I]CCL2. Amiloride, benzamil, MCGMA, and phenamil did however not displace any of these radioligands.
The increased dissociation rates of the orthosteric antagonist [3H]INCB3344, the intracellular antagonist [3H]CCR2‐RA‐[R], and the orthosteric agonist [125I]CCL2 induced by HMA indicate a noncompetitive allosteric interaction. Remarkably, the dissociation rate of the agonist [125I]CCL2 increased more (9.7‐fold) than of the antagonists (1.25‐ and 1.36‐fold) in the presence of HMA. Saturation binding assays revealed that HMA had a mixed competitive/noncompetitive interaction with the orthosteric antagonist [3H]INCB3344, because the radioligand's Bmax value decreased and KD value increased. HMA had a purely noncompetitive interaction with the intracellular antagonist [3H]CCR2‐RA‐[R], causing a decrease in this radioligand's Bmax value only.
The allosteric effect of HMA was diminished by mutation of sodium ion site residues Asp882.50 and His2977.45 into Ala. Mutation of Trp2566.48 even completely abolished HMA's allosteric effect, which is in contrast to the observed increase of HMA's affinity by the same mutation in adenosine receptors as discussed above.32 Amino acid His2977.45 is different from most class A GPCRs which usually harbor an Asn at the same position, but is conserved amongst chemokine receptors. The binding of HMA in CCR2s sodium ion binding site indicates that amiloride binding allows for a certain variation in the amino acids that constitute this binding cavity.
5. DOPAMINE RECEPTORS
The general trend amongst the dopamine receptor subtypes is an increase of the dissociation rate of orthosteric ligands by amiloride and its analogs, as found in a comprehensive study of the effect of amiloride, benzamil, and MIBA.50 MIBA had the largest effect on the dissociation rates of the antagonists [3H]SCH‐23,390 at the human D1 dopamine receptor and [3H]spiperone at the human D2(short), D2(long), D3, and D4 dopamine receptors. As with other GPCRs, the analogs with lipophilic moieties at the 5′‐position were more potent than amiloride itself. At the D1, D2(short), D2(long), and D3 dopamine receptors the amilorides displaced the orthosteric antagonist [3H]spiperone in both a noncompetitive and competitive manner. This may indicate binding of the amilorides to both the orthosteric and allosteric sites. The authors suggested a positive homotropic cooperativity due to a high Hill coefficient of the effect curves, i.e. amilorides binding at the allosteric site enhance the binding of amilorides to the orthosteric site.
The results at the D2 receptor complemented results from other studies, in which similar dissociation rate‐increasing effects and mixed competitive/noncompetitive behavior were found. Amiloride competed with and increased the dissociation rate of antagonists [3H]spiperone and [125I]epidepride binding.51 Amiloride, DMA, benzamil, EIA, MIBA, and HMA did so as well to the antagonist [3H]spiperone at both the rat52 and human53 D2 dopamine receptors, and of these amilorides HMA was the most potent amiloride (Figure 7). Agonists were modulated similarly as antagonists by amilorides at the rat D2 and D3 dopamine receptors, because amiloride, DMA, and MIBA decreased the potency of the agonist dopamine in inducing receptor activation in functional assays.50, 54 At the D4 receptor the allosteric effect of amiloride and its analogs was too small to be measured accurately, but an increase in antagonist [3H]spiperone dissociation rate was still detected. As amilorides still inhibited binding of the orthosteric ligand the displacement was more competitive in nature.50
Figure 7.
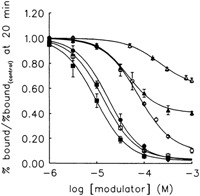
Concentration dependent dissociation modulation by amiloride and its analogs of [3H]spiperone binding at the dopamine D2 receptor after 20 minutes. (Δ‐amiloride, ▲‐benzamil, ○‐DMA, ●‐EIA, □‐MIBA, ■‐HMA). Amiloride modulates dissociation the least, whereas HMA and MIBA are the most effective modulators of dissociation. Reproduced with permission from Hoare and Strange.53 DMA, 5‐(N,N‐dimethyl)amiloride; EIA, 5‐(N‐ethyl‐N‐isopropyl)amiloride; HMA, 5‐(N,N‐hexamethylene)amiloride; MIBA, 5‐(N‐methyl‐N‐isobutyl)amiloride
The amino acids forming the sodium ion site in the dopamine receptors are conserved as well. Computational and mutagenesis studies at the D2 receptor have confirmed the importance of Asp802.50, Ser1213.39, Asn4197.45, and Asn4237.49 for the allosteric effects by sodium ions.55, 56, 57 At the D4 dopamine receptor mutation of Asp802.50 into Asn decreased MIBA affinity,58 indicating that amilorides bind in the sodium ion binding site as well. It may be assumed that amilorides also bind in the sodium ion binding site of the other dopamine receptors, but this has not been confirmed yet.
6. GONADOTROPIN‐RELEASING HORMONE RECEPTOR
The gonadotropin‐releasing hormone (GnRH) receptor, also known as luteinizing hormone‐releasing hormone receptor, is targeted by various drugs in the market for the treatment of sex‐hormone‐dependent diseases such as breast or prostate cancer.59, 60 These drugs are mostly peptidic agonists and antagonists that need to be administered by subcutaneous or intramuscular injections. The development of small‐molecule ligands that may replace these peptidic ligands is therefore desirable.61 Earlier results had indicated allosteric modulation of GnRH‐stimulated luteinizing hormone release by sodium ions and amilorides.62 In that light, the allosteric effects of amilorides on the GnRH receptor were investigated by Heitman et al63 Amiloride, benzamil, MCGMA, and phenamil had a negligible effect on the displacement of the peptide agonist [125I]triptorelin from the GnRH receptor. However, DCB, MIBA, and HMA increased the dissociation rate of [125I]triptorelin, with HMA having the strongest effect. In a luciferase assay, HMA acted as a purely insurmountable noncompetitive allosteric modulator as it only decreased the efficacy (Emax) of GnRH receptor activation by triptorelin and the endogenous ligand GnRH. Furthermore, it was demonstrated that the GnRH receptor harbors a second allosteric site other than the amiloride binding site, because HMA did not compete with FD‐1, another allosteric modulator of the GnRH receptor with a distinct chemical structure.
7. MUSCARINIC RECEPTORS
Amiloride effects have been observed on muscarinic receptors in rat tissue preparations. Benzamil and HMA inhibited [3H]pirenzepine binding at the muscarinic M1 and [3H]N‐methylscopolamine binding at the muscarinic M2 and M3 receptors.21 In rat trachea amiloride inhibited muscarinic M3 receptor‐mediated smooth muscle contraction64 by the endogenous agonist acetylcholine, by an insurmountable noncompetitive interaction as its efficacy (Emax) was reduced.65 In rat parotic acini, which express the muscarinic M3 receptor,66 amiloride inhibited binding of the muscarinic receptor antagonist [3H]N‐methylscopolamine in a competitive manner.67 In the recent, relatively low‐resolution crystal structures of the muscarinic M2 and M3 receptors sodium ion binding was not detected,68, 69, 70 but the amino acids making up the sodium ion site are perfectly conserved when compared to adenosine and adrenergic receptors,12 making amiloride binding to this site likely. In a recent molecular dynamics study sodium ion binding to (deprotonated), Asp2.50 in the muscarinic M3 receptor was suggested, keeping the receptor in an inactive state.71 Along a similar vein, the egress pathway of a sodium ion from Asp2.50 in the muscarinic M2 receptor into the cytosol was also simulated in molecular dynamics calculations.72
8. SEROTONIN RECEPTORS
Amiloride and analogs have been found to inhibit orthosteric ligand binding to serotonin receptors. Benzamil inhibited agonist [3H]8‐OH‐DPAT binding at the rat 5‐HT1A receptor.21 Amiloride and EIA inhibited agonist [3H]5‐carboxamidotryptamine binding at the human 5‐HT1B receptor.73 In functional assays at the same receptor, amiloride inhibited receptor activation by agonist sumatriptan in a competitive manner, whereas EIA displayed partial agonistic activity as it inhibited forskolin‐stimulated cAMP formation, albeit with a 15‐fold higher EC50 value (200 µM) compared to its Ki in inhibiting [3H]5‐carboxamidotryptamine binding (13 µM).73 Endogenous agonist [3H]serotonin binding was inhibited by HMA at the rat 5‐HT1C receptor and by benzamil and HMA at the rat 5‐HT2 receptor.21 Crystal structures of the agonist bound 5‐HT1B receptor74 and the 5‐HT2B receptor,75 again at relatively low resolution, did not reveal a bound sodium ion, but the well‐conserved amino acids of the sodium ion site compared to the other class A GPCRs12 makes the binding of amiloride in the same location likely.
9. OREXIN RECEPTORS
Suno et al76 determined the crystal structure of the human orexin 2 (OX2) receptor in complex with the subtype‐selective antagonist N‐ethyl‐2‐[(6‐methoxy‐pyridin‐3‐yl)‐(toluene‐2‐sulfonyl)‐amino]‐N‐pyridin‐3‐yl‐methyl‐acetamide (EMPA) at 1.96 Å resolution.76 This high‐resolution structure enabled the authors to inspect the putative sodium ion binding site around Asp1002.50, better than in an earlier crystal structure of this receptor.77 Interestingly, and somewhat at odds with this review, the authors identified two water molecules rather than a sodium ion in the vicinity of this aspartic acid residue. Triggered by this absence they performed additional radioligand binding studies in which no effects were observed from the addition of sodium ions or amiloride derivatives, whereas such effects were found in a control experiment the authors performed on the hA2AR.
The receptors discussed above all belong to the class A family of GPCRs. Finally, we should like to discuss the evidence, admittedly limited and inconclusive, of amiloride interaction with two class C receptors.
10. GABAB RECEPTORS
The GABAB receptor is activated by γ‐aminobutyric acid (GABA) and it's derivative, baclofen (β‐4‐chlorophenyl‐GABA). This receptor is coupled to potassium and calcium channels through Gi/Go proteins.78 Ong and Kerr explored the interaction of amiloride and its analogs with baclofen‐induced depression of spontaneous discharges in rat isolated neocortical slices in Mg2+‐free medium. The effect of baclofen (10 µM) was blocked by amiloride (200 µM), which increased the frequency of discharges and slightly reduced their amplitude when applied alone. These effects persisted upon wash‐out and baclofen remained ineffective on the discharges until 30 to 60 minutes after a switch to amiloride‐free medium. Analogs of amiloride, DMA and MIBA, showed a similar mode of action, whereas they were at least twice as potent than amiloride in preventing the effect of baclofen on neocortical spontaneous discharges. DMA alone increased the discharge frequency and slightly reduced the amplitude in a concentration of 100 µM. Analogs lacking the guanidine moiety were ineffective. The authors explicitly stated, however, that an indirect effect of the amilorides via functional antagonism of coactivated adenosine A1 receptors cannot be ruled out.79
11. T1R2/T1R3 RECEPTORS
The heterodimeric T1R2 and T1R3 taste receptor acts as a sweet taste sensor with multiple binding sites for sweeteners.80 Amiloride (3 mM) were found to significantly reduce the responses to sweeteners such as sugar, artificial sweeteners, and sweet protein. Moreover, response inhibition of 1 mM aspartame by amiloride was observed in a concentration‐dependent manner with an IC50 value of 0.87 ± 0.20 mM. A study of the specificity towards the response mediated by the human sweet taste receptors showed that the suppression of receptor activity by amiloride is specific for hT1R2/hT1R3. Inhibitory effects of lactisole, a known hT1R2/hT1R3 inhibitor, and amiloride on the cellular response to aspartame were examined in cells expressing hT1R3 mutants (hT1R2/hT1R3‐A733V and hT1R2/hT1R3‐F778A). Lactisole was less active on the mutants, whereas amiloride did not show such a differential effect. These results suggest that the binding site of amiloride is distinct from that of lactisole.81 Amiloride inhibited the response of perillartine as a sweet activator on hT1R2/T1R3, T1R2, and T1R2‐heptahelical domain (HD). Molecular modeling suggested that perillartine and amiloride occupy the same binding pocket on the extracellular side of the hT1R2‐HD.82
12. FUTURE DIRECTIONS FOR DRUG DISCOVERY
It is increasingly realized that GPCRs have multiple binding sites that may influence each other in allosteric ways. The surge in crystal structures over the last decade has taught that ligands, including marketed drugs and clinical candidates, may have very different binding sites indeed. From this review, it has become obvious that the sodium ion binding site is yet another receptor domain to tune the ligand response, and that amiloride and its derivatives are prototypic small molecules that intervene with that site.
Does this offer options for future drug discovery? One might argue that the generic nature of the site and the evolutionary conservation of the amino acids aligning it are a drawback rather than an opportunity. In that view amilorides are another class of chemical probes that serve to unveil the complexities of GPCR functioning. A recent development, however, may prove this hypothesis wrong.
The crystal structure of the leukotriene B4 (LTB4) receptor BLT1 in complex with antagonist/inverse agonist BIIL260 has recently been reported.83 Chemically, BIIL260 has four phenyl rings, three of which are bound in the orthosteric binding site near the extracellular domain. The fourth (a protonated benzamidine moiety) is penetrating deeper into the transmembrane domain and interacts with Asp662.50, with which it forms a salt bridge. Hydrogen bonds are present with the hydroxyl groups of Ser1063.39 and Ser2767.45 (Figure 8A). Mutation of Asp2.50 or Ser7.45 to alanine markedly reduced the affinity of BIIL260 for the receptor providing also pharmacological evidence for the BIIL260's binding to the sodium ion binding site. Furthermore, benzamidine itself, as well as NaCl, served as negative allosteric modulators of radiolabeled agonist ([3H]LTB4) binding (Figure 8B), suggesting their capability of forcing the receptor in an inactive state.83 The chemical resemblance of amiloride's guanidine moiety and benzamidine might be a good starting point to further study the effects of amiloride and its analogs on the BLT1 receptor.
Figure 8.
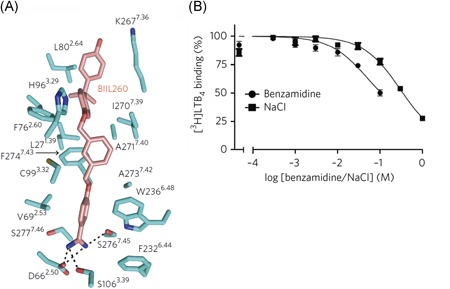
A, Structure of BIIL260 binding site in BLT1 receptor (PDB: 5X33); B, competition binding assay of benzamidine and NaCl to 0.5 nM [3H]LTB4. Reproduced with permission from Hori et al (2018).83 [Color figure can beviewed at wileyonlinelibrary.com]
13. CONCLUDING REMARKS
This review summarizes the current knowledge of the allosteric effects of amiloride and its analogs on GPCRs. Allosteric effects of amilorides have been found on class A GPCRs (adenosine receptors, α‐adrenergic receptors, the CCR2 chemokine receptor, dopaminergic receptors, the gonadotropin‐releasing hormone receptor, the histamine H1 receptor, muscarinic receptors, opioid receptors, and serotonin receptors), and, less convincingly, on class C receptors (GABAB and T1R2/3 receptors).
Amiloride and its analogs seem to follow a few general “rules” in their activity on these receptors. The propensity of amilorides to bind to the well‐conserved sodium ion site amongst GPCRs may explain these common behaviors. For most receptors, amiloride analogs with bulky lipophilic moieties on the 5′‐position have greater affinity and potency than the unsubstituted parent compound. This has not been explained fully, but it is clear that in most GPCRs there is a hydrophobic pocket above the sodium ion site that can accommodate these lipophilic moieties. Most receptors allow substitution on the guanidinium group as well, with a good affinity in displacing orthosteric ligands, but with less or no (allosteric) effect on the dissociation of orthosteric ligands.
Another general “rule” is the importance of Asp2.50 for amiloride binding, just as for sodium ions. In the docking studies performed, the binding mode of amiloride and HMA was predicted in the sodium ion site of the adenosine A2A receptor crystal structure and a CCR2 chemokine receptor homology model. The positively charged guanidinium group has a strong salt bridge interaction with Asp2.50, underlining the great importance of this residue for amiloride binding as found before in mutagenesis studies. Trp6.48 interacts with amilorides as well, in some cases hampering and in other cases accommodating amiloride binding. These interactions of amilorides with the amino acids of the sodium ion site are of interest because these have been shown to be important in receptor functionality, with Asp2.50 and Trp6.48 as most noticeable examples. Mutation of Asp2.50 silences receptor activation in many GPCRs.84 Trp6.48 is noteworthy as part of an “activation micro‐switch” between the active and inactive states of GPCRs,31, 85 and in docking studies of the adenosine A2A receptor amiloride and HMA seem to toggle this amino acid from one rotamer to another. Although not very likely, amilorides may also influence the oligomerization of class A receptors. The interface for receptor dimerization often involves transmembrane domains 4 and 5 that are not part of the sodium ion binding site. In some cases, however, other domains such as TM6, which also flanks the sodium ion binding site, play a role.86
In contrast with these general “rules,” differences in the affinities, potencies, and modulatory behaviors of amilorides can be quite outspoken, even between receptors where the sodium ion site harbors the same amino acids (i.e. adenosine, adrenergic, dopamine, and muscarinic receptors). To appreciate these differences it is important to discern between the different properties by which the allosteric effect of amilorides on orthosteric ligand binding may be described. In Table 1 we collected values for the different amilorides, of their affinity in displacing orthosteric ligands (IC50 or Ki), their (allosteric) effect on the dissociation of orthosteric ligand (koff/koff(control)), and their potency for these dissociation effects (EC50). This information also helps to understand whether the interaction of a particular amiloride with an orthosteric ligand is competitive or noncompetitive. If amiloride inhibits orthosteric ligand binding but does not affect its dissociation rate, the binding is mutually exclusive and the interaction is defined as competitive. If the dissociation rate is changed though, both the orthosteric ligand and amiloride can bind to the receptor at the same time and the interaction is deemed noncompetitive. Another way to confirm a noncompetitive interaction is by showing insurmountability of the inhibiting effect in radioligand saturation (Bmax decrease) or functional assays (Emax decrease), as discussed for the chemokine CCR2, muscarinic M3, and gonadotropin‐releasing hormone receptor. However, these assays have been conducted far less than dissociation assays in amiloride research so we did not include these in Table 1.
In some cases, amilorides behave only as purely competitive inhibitors, whereas in other cases they behave as noncompetitive negative modulators, and a mixed behavior has also been observed. For some receptors the cause for mixed competitive/noncompetitive behavior was explained by a tendency of amilorides to bind both orthosteric and allosteric sites, but also in these cases the observed effect may be caused by binding in the sodium ion site only, where the competitive “fraction” of the allosteric effect is caused by either an overlap of binding with the orthosteric site or a conformational change of the receptor by amiloride binding. The latter option is quite likely from the structural evidence provided by the recently elucidated crystal structures.
At some of the discussed receptors, the modulatory effect by amilorides is probe‐dependent, which has been described in other cases of allosteric modulation as well.87, 88 Amilorides act as positive allosteric modulators for agonist binding and as negative modulators for antagonists at the α2A‐adrenergic and adenosine A3 receptors. Thus, in some cases, amilorides may also influence receptor signaling after agonist activation with consequences for effector bias or functional selectivity, for instance between G protein and β‐arrestin signaling.89, 90 This has, however, not been demonstrated yet. At the α2B‐adrenergic receptor different amilorides even exhibit both positive and negative modulatory effects on the same orthosteric probe. Some of the differences in affinity and modulatory effect may be caused by differences in the sodium ion site itself, but the substantial conservation of the sodium ion site residues amongst GPCRs makes it more likely that these differences are caused by variations in receptor conformations.
Clinical application of amilorides targeting GPCRs is not self‐evident due to their micromolar affinities and lack of selectivity. However, it may be feasible to synthesize amiloride analogs with variations on the 5′‐position to improve their affinity and selectivity for GPCRs. In that sense, the recent structure elucidation of the BLT1/leukotriene B4 receptor in complex with BILL260 (Figure 8) is noteworthy. BIIL260 is a selective, high‐affinity antagonist for this receptor, occupying both the sodium ion and the orthosteric binding site. With the ongoing expansion of the crystal structure pool of GPCRs, further study and knowledge of the mechanisms of amiloride modulation will help in understanding and appreciating the allosteric mechanism in GPCR functioning and may pave the way for the design of antagonists forcing the receptor in a deeply inactive state.
ACKNOWLEDGMENTS
The authors acknowledge the financial support by the Netherlands Organization for Scientific Research—Chemical Sciences (NWO‐TOP #714.011.001; A. M., A. P. IJ.) and the student fellowship provided by the Indonesia Endowment Fund for Education (LPDP), Ministry of Finance, Indonesia (T. A.).
Massink A, Amelia T, Karamychev A, IJzerman AP. Allosteric modulation of G protein‐coupled receptors by amiloride and its derivatives. Perspectives for drug discovery? Med Res Rev. 2020;40:683–708. 10.1002/med.21633
Arnault Massink and Tasia Amelia share first‐authorship.
Present address Arnault Massink, Quantib, Westblaak 106, 3012 KM Rotterdam, The Netherlands. Tasia Amelia, Bandung Institute of Technology, Jalan Ganesha No.10, Bandung 40132, Indonesia.Alex Karamychev, AbbVie BV, Wegalaan 9, 2132 JD Hoofddorp, The Netherlands.
References
REFERENCES
- 1. Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G‐protein‐coupled receptors. Nature. 2009;459(7245):356‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rask‐Andersen M, Almén MS, Schiöth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discovery. 2011;10(8):579‐590. [DOI] [PubMed] [Google Scholar]
- 3. Chan HCS, Li Y, Dahoun T, Vogel H, Yuan S. New binding sites, new opportunities for GPCR drug discovery. Trends Biochem Sci. 2019;44(4):312‐330. [DOI] [PubMed] [Google Scholar]
- 4. Foord SM, Bonner TI, Neubig RR, et al. International Union of Pharmacology. XLVI. G protein‐coupled receptor list. Pharmacol Rev. 2005;57(2):279‐288. [DOI] [PubMed] [Google Scholar]
- 5. Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G‐protein‐coupled receptors in the human genome form five main families. Mol Pharmacol. 2003;63(6):1256‐1272. [DOI] [PubMed] [Google Scholar]
- 6. Christopoulos A. Advances in G protein‐coupled receptor allostery: from function to structure. Mol Pharmacol. 2014;86(5):463‐478. [DOI] [PubMed] [Google Scholar]
- 7. Christopoulos A, Changeux JP, Catterall WA, et al. International Union of Basic and Clinical Pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev. 2014;66(4):918‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thal DM, Glukhova A, Sexton PM, Christopoulos A. Structural insights into G‐protein‐coupled receptor allostery. Nature. 2018;559(7712):45‐53. [DOI] [PubMed] [Google Scholar]
- 9. Shonberg J, Kling RC, Gmeiner P, Löber S. GPCR crystal structures: medicinal chemistry in the pocket. Bioorg Med Chem. 2015;23(14):3880‐3906. [DOI] [PubMed] [Google Scholar]
- 10. Katritch V, Cherezov V, Stevens RC. Structure‐function of the G protein‐coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Munk C, Mutt E, Isberg V, et al. An online resource for GPCR structure determination and analysis. Nat Methods. 2019;16(2):151‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci. 2014;39(5):233‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu W, Chun E, Thompson AA, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337(6091):232‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller‐Gallacher JL, Nehmé R, Warne T, et al. The 2.1 Å resolution structure of cyanopindolol‐bound β1‐adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand‐free receptor. PLOS One. 2014;9(3):e92727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Christopher JA, Brown J, Doré AS, et al. Biophysical fragment screening of the β1‐adrenergic receptor: identification of high affinity arylpiperazine leads using structure‐based drug design. J Med Chem. 2013;56(9):3446‐3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fenalti G, Giguere PM, Katritch V, et al. Molecular control of δ‐opioid receptor signalling. Nature. 2014;506(7487):191‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang C, Srinivasan Y, Arlow DH, et al. High‐resolution crystal structure of human protease‐activated receptor 1. Nature. 2012;492(7429):387‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ballesteros JA, Weinstein H. Integrated methods for the construction of three‐dimensional models and computational probing of structure‐function relations in G protein‐coupled receptors Methods in Neurosciences. 25 San Diego, CA: Academic Press; 1995:366‐428. [Google Scholar]
- 19. Horstman DA, Brandon S, Wilson AL, Guyer CA, Cragoe EJ, Limbird LE. An aspartate conserved among G‐protein receptors confers allosteric regulation of α2‐adrenergic receptors by sodium. J Biol Chem. 1990;265(35):21590‐21595. [PubMed] [Google Scholar]
- 20. Warnock DG, Kusche‐Vihrog K, Tarjus A, et al. Blood pressure and amiloride‐sensitive sodium channels in vascular and renal cells. Nat Rev Nephrol. 2014;10(3):146‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garritsen A, IJzerman AP, Tulp MTM, Cragoe EJ Jr., Soudijn W. Receptor binding profiles of amiloride analogues provide no evidence for a link between receptors and the Na+/H+ exchanger, but indicate a common structure on receptor proteins. J Recept Res. 1991;11(6):891‐907. [DOI] [PubMed] [Google Scholar]
- 22. Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors — an update. Pharmacol Rev. 2011;63(1):1‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Göblyös A, IJzerman AP.Allosteric modulation of adenosine receptors. Biochim Biophys Acta. 2011;1808(5):1309‐1318. [DOI] [PubMed] [Google Scholar]
- 24. Garritsen A, IJzerman AP, Beukers MW, Cragoe EJ Jr., Soudijn W. Interaction of amiloride and its analogues with adenosine A1 receptors in calf brain. Biochem Pharmacol. 1990;40(4):827‐834. [DOI] [PubMed] [Google Scholar]
- 25. Gao ZG, IJzerman AP. Allosteric modulation of A2A adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol. 2000;60(5):669‐676. [DOI] [PubMed] [Google Scholar]
- 26. Gao ZG, Melman N, Erdmann A, et al. Differential allosteric modulation by amiloride analogues of agonist and antagonist binding at A1 and A3 adenosine receptors. Biochem Pharmacol. 2003;65(4):525‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gutierrez‐de‐Teran H, Massink A, Rodriguez D, et al. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein‐coupled receptor. Structure. 2013;21(12):2175‐2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao ZG, I jzerman AP. Allosteric modulation of A(2A) adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol. 2000;60(5):669‐676. [DOI] [PubMed] [Google Scholar]
- 29. Neubig RR, Spedding M, Kenakin T, Christopoulos A. International Union of Pharmacology Committee on Receptor N, Drug C. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev. 2003;55(4):597‐606. [DOI] [PubMed] [Google Scholar]
- 30. Massink A, Gutiérrez‐de‐Terán H, Lenselink EB, et al. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol Pharmacol. 2015;87(2):305‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro‐switches in 7TM receptor structures. Trends Pharmacol Sci. 2009;30(5):249‐259. [DOI] [PubMed] [Google Scholar]
- 32. Gao ZG, Kim SK, Gross AS, Chen A, Blaustein JB, Jacobson KA. Identification of essential residues involved in the allosteric modulation of the human A3 adenosine receptor. Mol Pharmacol. 2003;63(5):1021‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Massink A, Louvel J, Adlere I, et al. 5′‐Substituted amiloride derivatives as allosteric modulators binding in the sodium ion pocket of the adenosine A2A receptor. J Med Chem. 2016;59(10):4769‐4777. [DOI] [PubMed] [Google Scholar]
- 34. Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS. Activation of the A2A adenosine G‐protein‐coupled receptor by conformational selection. Nature. 2016;533(7602):265‐268. [DOI] [PubMed] [Google Scholar]
- 35. Ye L, Neale C, Sljoka A, et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein‐coupled receptor by physiological cations. Nat Commun. 2018;9(1):1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Howard MJ, Hughes RJ, Motulsky HJ, Mullen MD, Insel PA. Interactions of amiloride with α‐ and β‐adrenergic receptors: amiloride reveals an allosteric site on α2‐adrenergic receptors. Mol Pharmacol. 1987;32(1):53‐58. [PubMed] [Google Scholar]
- 37. Leppik RA, Mynett A, Lazareno S, Birdsall NJM. Allosteric interactions between the antagonist prazosin and amiloride analogs at the human α1A‐adrenergic receptor. Mol Pharmacol. 2000;57(3):436‐445. [DOI] [PubMed] [Google Scholar]
- 38. Ciolek J, Maïga A, Marcon E, Servent D, Gilles N. Pharmacological characterization of zinc and copper interaction with the human alpha1A‐adrenoceptor. Eur J Pharmacol. 2011;655(1‐3):1‐8. [DOI] [PubMed] [Google Scholar]
- 39. Jagadeesh G, Cragoe EJ Jr., Deth RC. Modulation of bovine aortic alpha‐2 receptors by Na+, 5′‐guanylylimidodiphosphate, amiloride and ethylisopropylamiloride: evidence for receptor G‐protein precoupling. J Pharmacol Exp Ther. 1990;252(3):1184‐1196. [PubMed] [Google Scholar]
- 40. Nunnari JM, Repaske MG, Brandon S, Cragoe EJ Jr., Limbird LE. Regulation of porcine brain α2‐adrenergic receptors by Na+,H+ and inhibitors of Na+/H+ exchange. J Biol Chem. 1987;262(25):12387‐12392. [PubMed] [Google Scholar]
- 41. Wilson AL, Womble SW, Prakash C, Cragoe EJ Jr., Blair IA, Limbird LE. Novel amiloride analog allosterically modulates the α2‐adrenergic receptor but does not inhibit Na+/H+ exchange. Mol Pharmacol. 1992;42(2):175‐179. [PubMed] [Google Scholar]
- 42. Leppik RA, Lazareno S, Mynett A, Birdsall NJM. Characterization of the allosteric interactions between antagonists and amiloride analogues at the human α2A‐adrenergic receptor. Mol Pharmacol. 1998;53(5):916‐925. [PubMed] [Google Scholar]
- 43. Leppik RA, Birdsall NJM. Agonist binding and function at the human α2A‐adrenoceptor: allosteric modulation by amilorides. Mol Pharmacol. 2000;58(5):1091‐1099. [DOI] [PubMed] [Google Scholar]
- 44. Wilson AL, Seibert K, Brandon S, Cragoe EJ Jr., Limbird LE. Monovalent cation and amiloride analog modulation of adrenergic ligand binding to the unglycosylated α2B‐adrenergic receptor subtype. Mol Pharmacol. 1991;39(4):481‐486. [PubMed] [Google Scholar]
- 45. Zweemer AJM, Hammerl DM, Massink A, et al. Allosteric modulation of the chemokine receptor CCR2 by amiloride analogues and sodium ions. The ins and outs of ligand binding to CCR2. 29763 Leiden University Repository; 2014:98‐119. https://openaccess.leidenuniv.nl/bitstream/handle/1887/29763/05.pdf?sequence=10 [Google Scholar]
- 46. Zweemer AJM, Nederpelt I, Vrieling H, et al. Multiple binding sites for small‐molecule antagonists at the CC chemokine receptor 2. Mol Pharmacol. 2013;84(4):551‐561. [DOI] [PubMed] [Google Scholar]
- 47. Zweemer AJM, Bunnik J, Veenhuizen M, et al. Discovery and mapping of an intracellular antagonist binding site at the chemokine receptor CCR2. Mol Pharmacol. 2014;86(4):358‐368. [DOI] [PubMed] [Google Scholar]
- 48. Ortiz Zacarias NV, Lenselink EB, IJzerman AP, Handel TM, Heitman LH. Intracellular receptor modulation: novel approach to target GPCRs. Trends Pharmacol Sci. 2018;39(6):547‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng Y, Qin L, Zacarias NV, et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature. 2016;540(7633):458‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hoare SRJ, Coldwell MC, Armstrong D, Strange PG. Regulation of human D1, D2(long), D2(short), D3 and D4 dopamine receptors by amiloride and amiloride analogues. Br J Pharmacol. 2000;130(5):1045‐1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Neve KA. Regulation of dopamine D2 receptors by sodium and pH. Mol Pharmacol. 1991;39(4):570‐578. [PubMed] [Google Scholar]
- 52. Hoare SRJ, Strange PG. Allosteric regulation of the rat D2 dopamine receptor. Biochem Soc Trans. 1995;23(1):92S. [DOI] [PubMed] [Google Scholar]
- 53. Hoare SRJ, Strange PG. Regulation of D2 dopamine receptors by amiloride and amiloride analogs. Mol Pharmacol. 1996;50(5):1295‐1308. [PubMed] [Google Scholar]
- 54. Chio CL, Lajiness ME, Huff RM. Activation of heterologously expressed D3 dopamine receptors: comparison with D2 dopamine receptors. Mol Pharmacol. 1994;45(1):51‐60. [PubMed] [Google Scholar]
- 55. Neve KI, Cumbay MG, Thompson KR, et al. Modeling and mutational analysis of a putative sodium‐binding pocket on the dopamine D2 Receptor. Mol Pharmacol. 2001;60(2):373‐381. [DOI] [PubMed] [Google Scholar]
- 56. Michino M, Free RB, Doyle TB, Sibley DR, Shi L. Structural basis for Na+‐sensitivity in dopamine D2 and D3 receptors. Chem Commun (Camb). 2015;51(41):8618‐8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Selent J, Sanz F, Pastor M, De Fabritiis G. Induced effects of sodium ions on dopaminergic G‐protein coupled receptors. PLOS Comput Biol. 2010;6(8):e1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schetz JA, Sibley DR. The binding‐site crevice of the D4 dopamine receptor is coupled to three distinct sites of allosteric modulation. J Pharmacol Exp Ther. 2001;296(2):359‐363. [PubMed] [Google Scholar]
- 59. Conn PM, Crowley WF Jr. Gonadotropin‐releasing hormone and its analogs. Annu Rev Med. 1994;45:391‐405. [DOI] [PubMed] [Google Scholar]
- 60. Kiesel LA, Rody A, Greb RR, Szilágyi A. Clinical use of GnRH analogues. Clin Endocrinol (Oxf). 2002;56(6):677‐687. [DOI] [PubMed] [Google Scholar]
- 61. Armer RE, Smelt KH. Non‐peptidic GnRH receptor antagonists. Curr Med Chem. 2004;11(22):3017‐3028. [DOI] [PubMed] [Google Scholar]
- 62. McArdle CA, Cragoe EJ Jr., Poch A. Na+ dependence of gonadotropin‐releasing hormone action: characterization of the Na+/H+ antiport in pituitary gonadotropes. Endocrinology. 1991;128(2):771‐778. [DOI] [PubMed] [Google Scholar]
- 63. Heitman LH, Ye K, Oosterom J, IJzerman AP. Amiloride derivatives and a nonpeptidic antagonist bind at two distinct allosteric sites in the human gonadotropin‐releasing hormone receptor. Mol Pharmacol. 2008;73(6):1808‐1815. [DOI] [PubMed] [Google Scholar]
- 64. Eglen R. M. Handbook of experimental pharmacology Overview of muscarinic receptor subtypes. 238 Berlin, Heidelberg, Germany: Springer; 2012:3‐28. [DOI] [PubMed] [Google Scholar]
- 65. Santacana GE, Silva WI. Differential antagonism by amiloride and pirenzepine of the muscarinic receptors of rat tracheal smooth muscle. Bol Asoc Med P R. 1990;82(9):403‐406. [PubMed] [Google Scholar]
- 66. Proctor GB. Muscarinic receptors and salivary secretion. J Appl Physiol. 2006;100(4):1103‐1104. [DOI] [PubMed] [Google Scholar]
- 67. Dehaye JP, Verhasselt V. Interaction of amiloride with rat parotid muscarinic and alpha‐adrenergic receptors. Gen Pharmacol. 1995;26(1):155‐159. [DOI] [PubMed] [Google Scholar]
- 68. Haga K, Kruse AC, Asada H, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482(7386):547‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kruse AC, Ring AM, Manglik A, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504(7478):101‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kruse AC, Hu J, Pan AC, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482(7386):552‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Miao Y, Caliman AD, McCammon JA. Allosteric effects of sodium ion binding on activation of the m3 muscarinic g‐protein‐coupled receptor. Biophys J. 2015;108(7):1796‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vickery ON, Carvalheda CA, Zaidi SA, Pisliakov AV, Katritch V, Zachariae U. Intracellular transfer of Na(+) in an active‐state G‐protein‐coupled receptor. Structure. 2018;26(1):171‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pauwels PJ. Competitive and silent antagonism of recombinant 5‐HT1B receptors by amiloride. Gen Pharmacol. 1997;29(5):749‐751. [DOI] [PubMed] [Google Scholar]
- 74. Wang C, Jiang Y, Ma J, et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340(6132):610‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wacker D, Wang C, Katritch V, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340(6132):615‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Suno R, Kimura KT, Nakane T, et al. Crystal structures of human orexin 2 receptor bound to the subtype‐selective antagonist EMPA. Structure. 2018;26(1):7‐19. [DOI] [PubMed] [Google Scholar]
- 77. Yin J, Mobarec JC, Kolb P, Rosenbaum DM. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature. 2015;519(7542):247‐250. [DOI] [PubMed] [Google Scholar]
- 78. Bowery NG. GABAB receptor pharmacology. Annu Rev Pharmacol Toxicol. 1993;33:109‐147. [DOI] [PubMed] [Google Scholar]
- 79. Ong J, Kerr DI. Suppression of GABAB receptor function in rat neocortical slices by amiloride. Eur J Pharmacol. 1994;260(1):73‐77. [DOI] [PubMed] [Google Scholar]
- 80. Cui M, Jiang P, Maillet E, Max M, Margolskee RF, Osman R. The heterodimeric sweet taste receptor has multiple potential ligand binding sites. Curr Pharm Des. 2006;12(35):4591‐4600. [DOI] [PubMed] [Google Scholar]
- 81. Imada T, Misaka T, Fujiwara S, Okada S, Fukuda Y, Abe K. Amiloride reduces the sweet taste intensity by inhibiting the human sweet taste receptor. Biochem Biophys Res Commun. 2010;397(2):220‐225. [DOI] [PubMed] [Google Scholar]
- 82. Zhao M, Xu XQ, Meng XY, Liu B. The heptahelical domain of the sweet taste receptor T1R2 is a new allosteric binding site for the sweet taste modulator amiloride that modulates sweet taste in a species‐dependent manner. J Mol Neurosci. 2018;66(2):207‐213. [DOI] [PubMed] [Google Scholar]
- 83. Hori T, Okuno T, Hirata K, et al. Na(+)‐mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat Chem Biol. 2018;14(3):262‐269. [DOI] [PubMed] [Google Scholar]
- 84. Parker MS, Wong YY, Parker SL. An ion‐responsive motif in the second transmembrane segment of rhodopsin‐like receptors. Amino Acids. 2008;35(1):1‐15. [DOI] [PubMed] [Google Scholar]
- 85. Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation‐a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481‐519. [DOI] [PubMed] [Google Scholar]
- 86. Guidolin D, Marcoli M, Tortorella C, Maura G, Agnati LF. Receptor‐receptor interactions as a widespread phenomenon: novel targets for drug development? Front Endocrinol. 2019;10:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discovery. 2005;4(11):919‐927. [DOI] [PubMed] [Google Scholar]
- 88. Keov P, Sexton PM, Christopoulos A. Allosteric modulation of G protein‐coupled receptors: a pharmacological perspective. Neuropharmacology. 2011;60(1):24‐35. [DOI] [PubMed] [Google Scholar]
- 89. Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G‐protein‐coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35(7):308‐316. [DOI] [PubMed] [Google Scholar]
- 90. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17(4):243‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]