

Summary
Background
A clinical decision support tool (CDST) has been validated for predicting treatment effectiveness of vedolizumab (VDZ) in Crohn’s disease.
Aim
To assess the utility of this CDST for predicting exposure‐efficacy and disease outcomes.
Methods
Using data from three independent datasets (GEMINI, GETAID and VICTORY), we assessed clinical remission rates and measured VDZ exposure, rapidity of onset of action, response to dose optimisation and progression to surgery by CDST‐defined response groups (low, intermediate and high).
Results
A linear relationship existed between CDST‐defined groups, measured VDZ exposure, rapidity of onset of action and efficacy in GEMINI through week 52 (P < 0.001 at all time points across three CDST‐defined groups). In GETAID, CDST predicted differences in clinical remission at week 14 (AUC = 0.68) and rapidity of onset of action (P = 0.04) between probability groups. The high‐probability patients did not benefit from shortening of infusion intervals, and differences in onset of action between the high‐intermediate and low‐probability groups within GETAID were no longer significant when including low‐probability patients who received a week 10 infusion. CDST predicted a twofold increase in surgery risk over 12 months of VDZ therapy among low‐ to intermediate‐probability vs high‐probability patients (adjusted HR 2.06, 95% CI 1.33‐3.21).
Conclusions
We further extended the clinical utility of a previously validated VDZ CDST, which accurately predicts at baseline exposure‐efficacy relationships and rapidity of onset of action and could be used to help identify patients who would most benefit from interval shortening and those most likely to require surgery while on active therapy.
1. INTRODUCTION
In the GEMINI 2 pivotal phase 3 clinical trial of vedolizumab (VDZ) for Crohn’s disease (CD), approximately one‐third of patients with active CD achieved corticosteroid‐free remission at week 52.1 Similar results were recently reported in a meta‐analysis of observational studies with an estimated 1 year corticosteroid‐free remission rate of 31% (95% confidence interval [CI] 20‐45%).2, 3 These data underscore that while a substantial proportion of treatment‐resistant patients respond to VDZ therapy, the majority do not. Although this circumstance is likely multifactorial, variability in VDZ pharmacokinetics (PK) is a potential explanation in some patients. Specifically, high drug clearance resulting in inadequate drug exposure may be responsible for suboptimal results in some.
Multiple studies in patients with active CD have shown a correlation between VDZ exposure and response, and higher clinical and endoscopic remission rates when stratified by drug exposure.4, 5, 6, 7, 8, 9, 10 These findings hold out the possibility that dose intensification in patients with low VDZ trough concentrations during induction may result in higher remission rates. In support of this notion, observational data suggest that empiric administration of an additional drug dose in patients with suboptimal response to induction may improve outcomes.11 It is also relevant to note that a perception exists that VDZ induction therapy has a slower onset of action and is generally less effective in CD than in ulcerative colitis (UC). Notwithstanding that prior exposure to a TNF antagonist has been consistently associated with low response and remission rates,12 no single clinical factor accurately predicts which patients will respond quickly to VDZ therapy or will benefit from therapeutic drug monitoring and/or dose intensification. Accurate identification of these patients could allow a personalised medicine approach to induction therapy and greater treatment efficiency.
The VICTORY consortium investigators previously developed and validated a clinical decision support tool (CDST) that classifies CD patients according to low, intermediate and high probability of response to VDZ.13 In the current analysis, we used the GEMINI 2 clinical trial data (NCT00783692) to assess whether these differences were related to differences in measured VDZ concentrations (exposure‐efficacy) and whether the CDST predicts differences in rapidity of onset of action. We subsequently performed a second external validation of the CDST based on data from a prospective cohort study (GETAID) and then, using data from both the GETAID and VICTORY cohorts, assessed whether the CDST accurately identified patients who might benefit from dose intensification. Finally, we evaluated whether the CDST estimated the likelihood of surgery for CD while on VDZ, which is of importance when determining the incremental value of aggressive treatment‐monitoring approaches.
2. METHODS
2.1. Data Sources and Participants
Methodology for the development and validation of our CDST has been published previously.13 In the current study, individual participant data from the phase 3 VDZ in CD trial (GEMINI 2) were used in combination with observational cohort data from the VICTORY consortium and GETAID collaboration.14, 15 A treat‐straight‐through cohort was created from the GEMINI 2 clinical trial programs to mimic an observational cohort design. Patients from VICTORY and GETAID were included if they had started VDZ therapy for clinically or endoscopically active CD, had a follow‐up clinical or endoscopic assessment of disease activity after VDZ initiation, and had baseline data available to calculate the CDST.
2.2. Clinical decision support tool
The CDST is calculated using the following five variables:
No prior bowel surgery (+2 points).
No prior TNF‐antagonist therapy (+3 points).
No prior fistulising disease (+2 points).
Baseline albumin (+0.4 points per g/L).
Baseline C‐reactive protein (−0.5 points if 3.0‐10.0 mg/L; −3.0 points if >10 mg/L).
Patients with a score of 13 points or less are classified as low probability, >13 to 19 points as intermediate probability and >19 points as high probability.13
2.3. Outcomes
Our main objectives were to determine whether the previously created and validated CDST predicted measured VDZ concentrations (trough and peak) in the 52‐week GEMINI 2 clinical trial and whether differences in measured drug exposure corresponded to differences in drug efficacy and rapidity of onset of action as assessed by reductions in Harvey‐Bradshaw Index (HBI) over time (exposure‐efficacy). The HBI was chosen given its widespread use internationally in routine practice, its availability in the GETAID cohort dataset, and its good correlation with the Crohn’s Disease Activity Index.16 Secondary objectives were to (a) externally validate the CDST in an independent multicentre cohort (GETAID collaboration) and (b) determine whether the CDST identified patients most likely to benefit from VDZ dose intensification for apparent lack of response. Finally, we assessed whether differences in predicted exposure‐efficacy correlated with achievement of endoscopic remission and the likelihood of undergoing surgery for CD.
2.4. VDZ Pharmacokinetics
VDZ concentrations were assessed in the GEMINI 2 trials using serum samples with a direct VDZ capture PK assay. A sandwich ELISA assay was used for quantifying VDZ in human serum. Serum concentrations of VDZ were determined in accordance with good laboratory practice. The lower limit of detection was 0.125 µg/mL. Time points for trough concentration assessments taken 30 minutes before VDZ infusions were weeks 0, 2, 6, 22 and 46. Additional concentration assessments were taken at weeks 4, 14, 38 and 52. Time points for peak concentration assessments taken 2 hours post‐infusion were weeks 0, 2, 6, 22 and 46.
2.5. Statistical analysis
First, we evaluated the relationship between CDST‐defined probability groups, changes in HBI scores and measured VDZ concentrations using the entire 52‐week GEMINI 2 study dataset (exposure‐efficacy relationship). Differences in median concentrations at each time point among the three probability groups were first assessed using nonparametric testing (Kruskal‐Wallis); pairwise comparisons were subsequently performed for each group at each time point. A closed testing procedure was used to control the overall type I error such that each of the pairwise comparisons was conducted at the 0.05 level with no P value adjustments if the initial omnibus hypotheses that all of the probability groups showed equal (a) mean HBI scores and (b) median measured VDZ concentrations were first rejected at the 0.05 level. If the omnibus comparison was not significant at the 5% level, subsequent pairwise comparisons were not performed.
Second, we re‐validated the CDST in the GETAID cohort for predicting differences in week 14 remission rates between patients classified as low probability and intermediate‐high probability. Intermediate‐ and high‐probability patients were pooled in the GETAID cohort for comparison because of the low number of patients being classified as high probability (<10%) in this cohort. Week 14 was chosen for analysis because it is specified in US Food and Drug Administration (FDA) labelling as the most appropriate time for evaluation of the success of induction therapy. Furthermore, over 90% of the GETAID cohort had prior TNF‐antagonist exposure, and prior subgroup analyses of GEMINI have observed that these patients require at least 10 weeks of exposure to observe meaningful differences in remission rates compared to placebo.17 Secondary analyses were performed comparing changes in HBI over time and rates of clinical remission and corticosteroid‐free remission at weeks 6, 14, 22 and 30. Sensitivity analyses were done limiting the analyses to patients receiving Q8 week VDZ maintenance, as European labelling allows for an additional dose to be given at week 10 in patients with a suboptimal induction response. Categorical data were compared using chi‐square or Fisher’s exact test.
We then assessed response to VDZ dose intensification in the GETAID cohort and VICTORY consortium according to the CDST‐defined baseline probability of response (low vs intermediate‐high) to confirm whether the exposure‐efficacy relationship observed could be modified by higher predicted drug exposure. The decision to dose escalate was made clinically by treating providers without consideration for CDST‐defined probability of response as the providers were unaware of how the different variables were used to generate a score and how that CDST score might classify a patient’s probability of response. Our a priori hypothesis was that the low‐probability and possibly the intermediate‐probability groups would most likely benefit from an extra infusion at week 10 or interval shortening to Q4 or Q6 weeks given that these patients would have lower drug exposure than the high‐probability group. In the GETAID cohort, response to interval shortening was assessed using pre‐ and post‐interval shortening HBI scores. In the VICTORY consortium, response was assessed using the physician global assessment, with a clinically meaningful response defined as a >50% reduction in symptom activity post‐interval shortening. Within‐patient and within‐group changes in HBI were assessed using repeated‐measure analysis of variance with the group‐time interaction function.
Finally, in our prior publication, we observed differences in week 26 endoscopic remission rates according to CDST strata. Using data from the most recent VICTORY consortium cohort database, we assessed differences in 52‐week cumulative rates of endoscopic remission (absence of ulcers) across probability groups among patients undergoing endoscopic follow‐up, and whether these differences in endoscopic remission corresponded to differences in rates of surgery between the high‐probability group and the intermediate‐ or low‐probability groups (exposure‐efficacy‐complication relationship). This relationship was initially assessed by groupwise and pairwise log‐rank analyses and univariable Cox proportional hazard analyses. Adjustment for hazard ratio (HR) estimates was then performed for the covariates known to influence risk of surgery that were not already included in the baseline prediction model, including disease duration >2 years, ileal disease location, age >60 years, prior CD‐related hospitalisation and smoking status.
2.6. Ethics compliance statement
VICTORY consortium and GETAID collaboration datasets were collected after ethics/IRB approval at all participating sites. GEMINI data were collected as part of the phase 3 clinical trial (NCT00783692) with corresponding ethics/IRB approval. All authors had access to the study data results and have reviewed and approved the final manuscript.
3. RESULTS
3.1. Patient characteristics
The VICTORY consortium and GETAID populations had higher proportions of TNF‐antagonist–exposed and female patients, and the participants were slightly older with longer disease duration at the time of VDZ treatment than subjects in the GEMINI 2 clinical trial (Table 1). Importantly, of the 173 CD patients in the GETAID cohort, only 55 had all the necessary baseline variables for calculation of the CDST. However, the patients with complete data had characteristics similar to the excluded patients (P > 0.20 for all comparisons; Table 1).
Table 1.
Demographics of cohorts used for current analyses
| GEMINI 2 (n = 814) | VICTORY Consortium | GETAID cohort | |||
|---|---|---|---|---|---|
| Entire cohort (n = 659) | Included cohort (n = 501) | Entire cohort (n = 173) | Included cohort (n = 55) | ||
| Female sex, n (%) | 435 (53) | 381 (58) | 280 (56) | 109 (63) | 34 (62) |
| Mean age, y (SD) | 35.5 (11.9) | 40 (15.4) | 39 (15.7) | 37.3 (11.8) | 36.4 (10.8) |
| Mean BMI, kg/m2 (SD) | 24.0 (6.0) | 25.5 (6.9) | 25.3 (6.9) | 20.9 (3.9) | 20.2 (3.5) |
| Mean disease duration, y (SD) | 9.1 (7.5) | 14.2 (11.2) | 13.7 (11.1) | 12.2 (7.6) | 13.2 (8.8) |
| Prior TNFα‐antagonist exposure, n (%) | 535 (66) | 598 (91) | 452 (90) | 172 (99) | 55 (100) |
| Prior TNFα‐antagonist failure, n (%) | 497 (61) | 497 (75) | 379 (76) | 134 (78) | 41 (75) |
| Median CRP, mg/L (IQR) | 10.6 (4.5‐31.6) | 4.7 (1‐16.9) | 4.4 (1‐16.9) | 18.4 (8.0‐45.0) | 33 (8.0‐50.0) |
| Mean albumin, g/L (SD) | 34.9 (5.7) | 38.7 (5.4) | 38.4 (5.6) | 31.4 (7.6) | 30.2 (6.8) |
| Disease location, n (%) | |||||
| Ileum only | 141 (17) | 104 (16) | 74 (15) | 31 (18) | 10 (18) |
| Colon only | 230 (28) | 138 (21) | 105 (21) | 37 (21) | 16 (29) |
| Ileocolonic | 443 (54) | 413 (63) | 318 (63) | 94 (54) | 29 (53) |
| Prior surgery for CD, n (%) | 355 (44) | 400 (61) | 293 (59) | 84 (49) | 27 (49) |
| Prior fistulising disease, n (%) | 297 (36) | 240 (36) | 178 (36) | 57 (33) | 20 (36) |
Patients were excluded from the VICTORY and GETAID cohorts for not having baseline laboratory test values (CRP or albumin) to calculate the CDST. Patients in the VICTORY consortium were classified as follows: high‐probability group (n = 131), intermediate‐probability group (n = 281), low‐probability group (n = 89). Patients in the GETAID cohort were classified as follows: high‐probability group (n = 3); intermediate‐probability group (n = 24); low‐probability group (n = 28). Because of the small sample size of the high‐probability group in GETAID (a highly refractory population early in the period during which vedolizumab became available), it was combined with the intermediate‐probability group for analyses.
3.2. VDZ Exposure and Onset of Action
In the GEMINI 2 cohort, a significant linear trend was observed for measured VDZ concentrations when stratified by the CDST (Figure 1, Table S1). This observation was significant through week 52 of the study and was associated with significant differences in rapidity of onset of action and reduction in HBI (Figure 2, Table S2). Rates of anti‐drug antibody formation were comparable between the low (n = 9/226, 3.98%), intermediate (n = 20/414, 4.83%) and high (n = 4/174, 2.30%) probability of response groups.
Figure 1.
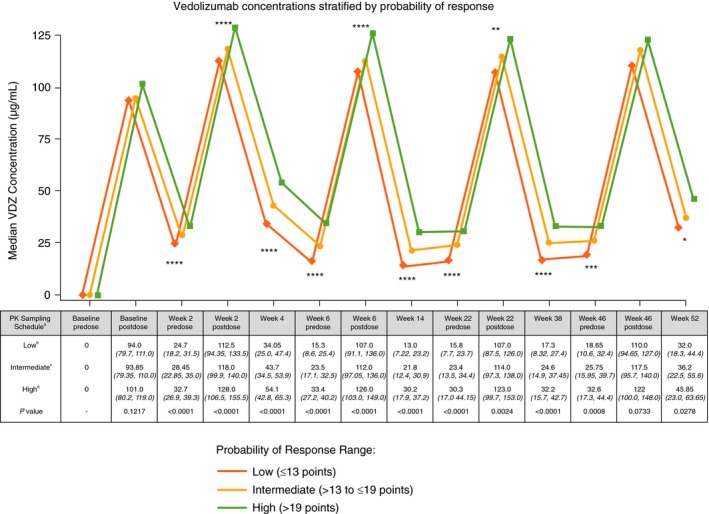
GEMINI 2 clinical trial 52‐week vedolizumab serum drug concentrations stratified by CDST. aAll values in table are median VDZ concentration (µg/mL) (IQR); post‐dose concentration was measured 2 h after dosing. bLow probability; ≤13 points in CDST model at baseline. cIntermediate probability; >13 to ≤19 points in CDST model at baseline. dHigh probability; >19 points in CDST model at baseline. ****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05. Bolded P values are statistically significant. CDST, clinical decision support tool; IQR, interquartile range; PK, pharmacokinetics; VDZ, vedolizumab
Figure 2.
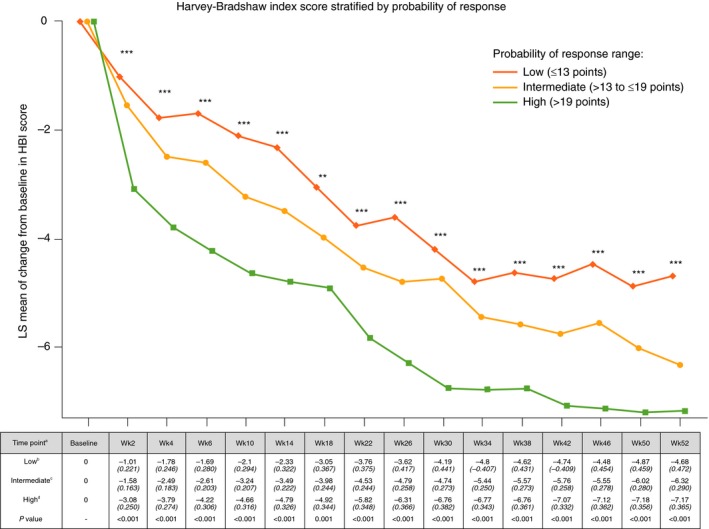
GEMINI 2 clinical trial 52‐week reduction in Harvey‐Bradshaw Index stratified by CDST aAll values in table are mean HBI (SE). bLow probability; ≤13 points in CDST model at baseline. cIntermediate probability; >13 to ≤19 points in CDST model at baseline. dHigh probability; >19 points in CDST model at baseline. ****P < 0.0001, ***P < 0.001, and **P < 0.01. Bolded P values are statistically significant. CDST, clinical decision support tool; HBI, Harvey‐Bradshaw Index; LS, least‐squares; SE, standard error; Wk, week
There was no catch‐up in HBI reductions in the low‐probability group compared with the intermediate, or the intermediate compared with the high‐probability group, and significant differences in HBI reductions from baseline remained at week 52. No significant differences between the probability groups were observed for concomitant use of steroids or immunomodulators.
3.3. GETAID Cohort
Rates of clinical remission at week 14 in the GETAID cohort were significantly higher among the intermediate or high‐probability patients compared with the low‐probability patients (P = 0.04). Similar trends were also observed at weeks 22 and 30 for both clinical remission and corticosteroid‐free clinical remission (Figure 3A,B). Analysis of changes in mean HBI scores over time showed a significant reduction in HBI in the low‐probability and intermediate‐ or high‐probability groups (P < 0.01). Among patients receiving standard VDZ induction and every 8‐week maintenance dosing (with no week 10 infusion), a significant group‐time interaction for week 14 HBI reductions was observed, and patients in the intermediate‐ or high‐probability groups had a more significant reduction in HBI during the first 14 weeks of therapy than those in the low‐probability group (P = 0.045) (Figure 3C).
Figure 3.
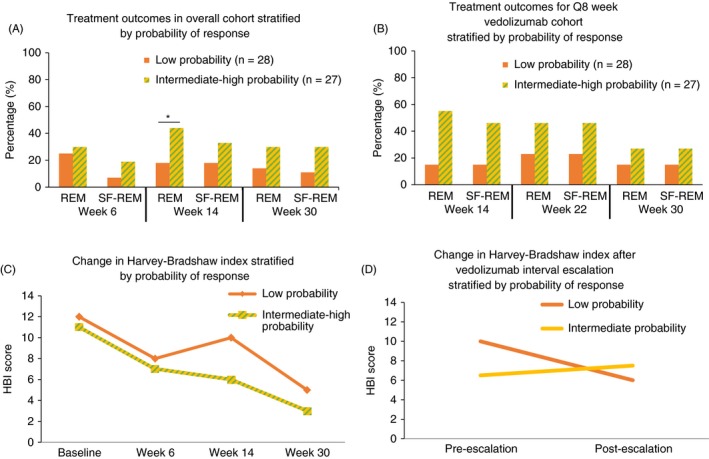
GETAID vedolizumab cohort treatment outcomes stratified by CDST. (A) Treatment outcomes stratified by CDST in overall GETAID cohort. (B) Treatment outcomes stratified by CDST in GETAID cohort on Q8 week vedolizumab maintenance. (C) Reduction in HBI stratified by CDST. (D) Reduction in HBI after vedolizumab interval shortening (escalation) stratified by CDST. Low probability; ≤13 points in CDST model at baseline. Intermediate probability; >13 to ≤19 in CDST model at baseline. High probability; >19 points in CDST model at baseline. *P < 0.05. Abbreviations: CDST, clinical decision support tool; HBI, Harvey‐Bradshaw index; Q8, every 8 weeks; REM, remission; SF‐REM, steroid‐free remission. High‐probability group (n = 3); intermediate‐probability group (n = 24); low‐probability group (n = 28). Because of the small sample size of the high‐probability group (a highly refractory population early in the period during which vedolizumab became available), it was combined with the intermediate‐probability group for analyses
3.4. Response to dose optimisation or interval shortening for lack of response
The group‐time interaction for reduction in HBI between the intermediate‐ or high‐probability groups and the low‐probability group was no longer significant after including low‐probability patients who were dose optimised early with a week 10 VDZ infusion (exposure‐efficacy‐optimisation). There was also a numeric reduction in HBI among low‐probability patients requiring interval shortening, with no change in HBI among intermediate‐probability patients requiring interval shortening; however, this did not reach statistical significance (P > 0.20) (Figure 3D). None of the high‐probability patients in GETAID required interval shortening for apparent lack of response.
In the VICTORY consortium, 38 patients underwent interval shortening for apparent lack of response (n = 11 low probability, n = 18 intermediate probability and n = 9 high probability). A clinical response was seen in 46% (n = 5/11) of the low‐probability group, 39% (n = 7/18) of the intermediate‐probability group and 0% (n = 0/9) of the high‐probability group (P = 0.038 for comparison of high‐ or intermediate‐ vs low‐probability groups).
3.5. Endoscopic remission and progression to surgery
In the VICTORY consortium, cumulative 12‐month rates for endoscopic remission were numerically lower in the low‐probability group (35%) compared with the intermediate‐ (42%) or high‐probability groups (53%). Likewise, cumulative 12‐month rates for progression to surgery were numerically higher in the low‐probability group (21%) compared with the intermediate‐ (17%) or high‐probability groups (12%) (Figure 4).
Figure 4.
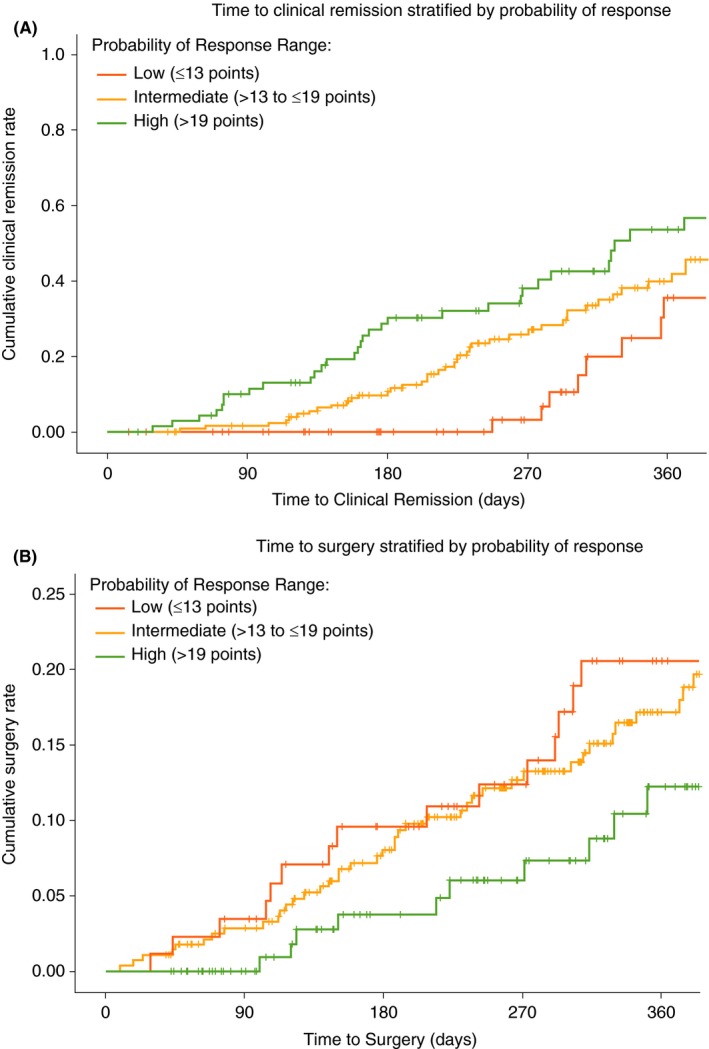
VICTORY consortium vedolizumab‐treated rates of endoscopic remission and progression to surgery stratified by CDST. A, Cumulative rates of endoscopic remission. B, Cumulative rates of progression to surgery. Low probability; ≤13 points in CDST model at baseline. Intermediate probability; >13 to ≤19 in CDST model at baseline. High probability; >19 points in CDST model at baseline. High‐probability group (n = 131), intermediate‐probability group (n = 281), low‐probability group (n = 89). Abbreviation: CDST, clinical decision support tool. Analysis of endoscopic remission limited to those patients with follow‐up endoscopic assessments (n = 326; high probability n = 84; intermediate probability n = 172; low probability n = 70). Endoscopic remission defined as absence of ulcerations. Pairwise log‐rank comparisons across the three probability groups for endoscopic remission: high vs low P < 0.001; high vs intermediate P = 0.076; low vs intermediate P = 0.002. Pairwise log‐rank comparisons across the three probability groups for progression to surgery: high vs low P = 0.024; high vs intermediate P = 0.076; low vs intermediate P = 0.264
After adjusting for disease duration >2 years, ileal disease location history, age >60 years, prior CD‐related hospitalisation, and smoking status, the adjusted HR (aHR) for high‐ vs low‐ or intermediate‐probability groups remained significant for stratification of achieving endoscopic remission (aHR 2.06, 95% CI 1.33‐3.21) and risk of surgery while on VDZ (aHR 0.50, 95% CI 0.26‐0.95). The intermediate‐probability group was significantly more likely to achieve endoscopic remission (aHR 2.47, 95% CI 1.26‐4.87) vs the low‐probability group; however, no significant difference was observed in risk of surgery between these two groups (aHR 0.86, 95% CI 0.51‐1.47).
4. DISCUSSION
Although the introduction of biologic therapy has greatly improved the management of CD, fewer than one‐third of patients treated with a TNF antagonist, VDZ, or ustekinumab achieve corticosteroid‐free clinical remission 1 year following initiation of therapy. The reasons for this unsatisfactory circumstance are complex and include disease heterogeneity, delayed initiation of therapy, drug sensitisation, development of disease‐related complications and adverse effects of medical therapy. However, irrespective of specific causes, low efficacy rates translate into poor incremental cost‐effectiveness estimates and reluctance by payers to fund these therapies. One of the fundamental concepts of personalised medicine is identification of patients who are more likely to respond to a specific therapy. Accordingly, we previously developed and validated a CDST for predicting response to VDZ therapy in CD. In the current study, we extend the clinical utility of this CDST through several additional analyses and an international collaboration between the VICTORY consortium and GETAID investigators, which also benefited from access to the phase 3 GEMINI trial data.
In the GEMINI 2 trial dataset, the CDST‐defined response categories predicted highly significant differences in measured VDZ exposure throughout the 52‐week study, and this was observed irrespective of whether VDZ exposure was measured at trough (pre‐dose), peak (2‐hour post‐dose) or midway between infusions. Most notably, the measured week 6 trough VDZ concentrations in the CDST‐defined low‐ and high‐probability response groups from the GEMINI 2 cohort had non‐overlapping interquartile ranges, with a twofold higher median concentration being observed in the high‐probability group. In the GEMINI 2 trial dataset, we also observed that the CDST‐defined response categories predicted highly significant differences in rapidity of onset of action and absolute reductions in HBI throughout the 52‐week study. Specifically, throughout the first 14 weeks of VDZ treatment, the high‐probability response group had at least a twofold increased reduction in HBI from baseline compared with the low‐probability response group (P < 0.001). Together these observations demonstrate a relationship between CDST‐defined probability groups, VDZ exposure, and rapidity of onset of action and an ability to identify patients undergoing VDZ therapy who will have low drug exposure and slower onset of action, and therefore may benefit from early dose intensification.
In the VICTORY and GETAID cohorts, we observed the CDST to predict differences in VDZ response comprising both rapidity of onset of action and overall effectiveness as defined by both symptomatic and endoscopic remission rates. In the VICTORY cohort, rates of endoscopic remission were significantly different between probability groups as defined by the CDST, and this predicted difference between CDST‐defined response groups remained significant even after accounting for factors known to influence disease outcomes. In the GETAID cohort, rates of clinical remission at week 14 were significantly different between the CDST‐defined low‐ and intermediate‐ or high‐probability groups, and we observed significant differences in rapidity of onset of action between these groups as measured by reductions in HBI through 14 weeks of therapy. Using the GETAID cohort, we expanded on our observations from the GEMINI 2 cohort by observing that these differences in onset of action between the CDST‐defined low and intermediate or high probability of response groups appeared to be overcome by administration of a week 10 infusion. Using the VICTORY cohort, we also observed that only the low and intermediate probability of response groups benefited from VDZ interval shortening for nonresponse. Collectively, these findings provide further evidence of the validity of the CDST across diverse CD patient populations and its potential application to identify CD patients starting VDZ therapy who could potentially benefit from early dose optimisation through a week 10 infusion and/or shortening of VDZ maintenance intervals. However, this strategy requires further validation in prospective studies. It is also important to clarify that CD labelling in the European Union includes administration of an additional week 10 dose of VDZ; however, this is not authorised in labelling in North America, including the United States.
Another important finding of the study was that the CDST seemed to effectively predict “hard endpoints” such as endoscopic remission and CD‐related surgery. Although other tools have been developed to prognosticate overall risk of complications among CD patients to help guide patient discussions for starting biologics,18, 19 it is difficult to know who remains at risk for disease‐related complications after biologic initiation.20 We observed the low and intermediate CDST groups of VDZ therapy patients to be significantly less likely to achieve endoscopic remission and more likely to undergo surgery relative to the high CDST group. Given the observations made for variability in measured VDZ exposure, onset of action, response to interval shortening for the low‐probability patients, and endoscopic remission across CDST groups, it could be hypothesised that VDZ dose optimisation in low and intermediate CDST groups could be effective in off‐setting the increased risk for surgery observed by optimising the achievement of endoscopic remission, an endpoint associated with risk of disease‐related complications. This concept, however, requires formal assessment in a well‐designed phase 3 trial similar to REACT,21 and it is worth noting that despite a significant difference in endoscopic remission rates between the low and intermediate CDST groups, no significant difference was observed for risk of surgery between these groups.
Our study has several strengths, including the multinational validation in mixed practice settings and extension of CDST predictions to VDZ exposure, onset of action, response to interval shortening and risk of surgery. Several limitations, however, require acknowledgment. Post hoc analyses of clinical trial datasets have inherent limitations that prevent definitive conclusions, and real‐world data have inherent limitations in collection methods and consistency of assessments that may have biased the results. No specific or consistent timing was applied for the assessment of response to interval shortening in the VICTORY consortium, and the physician global assessment was used, which carries a risk for misclassification. The GETAID cohort was an early, treatment‐refractory population, and only a subset had all necessary data to calculate the CDST. Therefore, the analyses were limited to a subset of patients, which could still introduce a selection bias, and significance in comparisons was not observed beyond week 14. Further analyses are likely therefore still needed to fully capture the validity of this CDST to assist in treatment optimisation, particularly with regard to the use of a week 10 dose for optimisation. Accordingly, well‐designed phase 3 trials focusing on optimisation of disease outcomes and treatment response for VDZ using the CDST as an enrichment or stratification tool could help overcome the current gap in personalised medicine for inflammatory bowel disease.
In conclusion, the previously built CDST for VDZ in CD appears to be valid across multiple cohorts and has significant prognostic and predictive capacity to guide therapeutic decisions in routine practice. Patients deemed low probability for response to VDZ may potentially benefit from a week 10 dose to optimise drug exposure and rapidity of onset of action. When implementing aggressive treat‐to‐target monitoring strategies, low‐ or intermediate‐probability patients may benefit most from this strategy, and healthcare systems may consider stratified follow‐up intervals based on probability of response. Finally, among high‐probability patients, we observed rapid onset of action and high drug concentrations. If these patients fail to respond to VDZ, it may be related to an immunologic or genetic mechanism, and further studies are needed to help identify additive biomarkers to further optimise the predictive capacity of the CDST for VDZ.
AUTHORSHIP
Guarantor of the article: PSD acts as guarantor.
Author contributions: PSD, AA and KL were responsible for study concept and design. PSD was responsible for statistical analysis. PSD and AA were responsible for drafting of the manuscript. PSD and KL were responsible for study supervision. All authors were responsible for acquisition of data and for critical revision of the manuscript for important intellectual content. All authors had access to the study results and reviewed and approved the final manuscript. PSD accepts responsibility for the integrity of this work as a whole.
Supporting information
ACKNOWLEDGEMENT
Declaration of personal interests: PSD has received research support, honorarium and travel support from Takeda and research support from Pfizer; and has served on an advisory board for Janssen. AA has received consulting fees from AbbVie, Hospira, Takeda, Gilead and Biocodex, as well as lecture fees and travel accommodations from AbbVie, Janssen, Biocodex, Hospira, Ferring, Takeda and MSD. AA has also received advisory board fees from Gilead, Takeda and AbbVie. LPB has received consulting fees from Merck, AbbVie, Janssen, Genentech, Ferring, Tillots, Vifor, Pharmacosmos, Celltrion, Takeda, Biogaran, Boehringer Ingelheim, Lilly, Pfizer, Index Pharmaceuticals, Amgen, Sandoz, Celgene, Biogen, Samsung Bioepis, Alma, Sterna, Nestlé and Enterome. VJ has received consulting fees from AbbVie, Janssen, Takeda, Sandoz, Ferring, Pfizer, GSK, Robarts Clinical Trials, Eli Lilly and Arena, and speaker fees from Takeda, Ferring, Janssen and Shire. MS has received speaking fees from AbbVie and Ferring. JF has received lecture fees from AbbVie, Ferring, Janssen, MSD, Pfizer and Takeda, and consulting fees from AbbVie, Janssen, MSD, Takeda and Vifor, and has served on advisory boards for Biogen, Janssen and Takeda. SS has received research support from Pfizer and support from the American College of Gastroenterology and the Crohn’s and Colitis Foundation. BP has received consulting fees from AbbVie, MSD and Pfizer, and lecture fees from AbbVie, MSD, Ferring and Takeda. EVL has served as a consultant for Janssen, Takeda, AbbVie, UCB, Amgen, Pfizer, Salix, Mesoblast, Eli Lilly, Celgene and CVS Caremark, and has received research support from Janssen, Takeda, AbbVie, UCB, Amgen, Pfizer, Genentech, Gilead, Receptos, Celgene, MedImmune, Seres Therapeutics and Robarts Clinical Trials. XR has reported a relationship with AbbVie, MSD, Janssen Cilag and Takeda. SK has served as a consultant to AbbVie, Janssen, Merck, Spherix Health, Pfizer and UCB and as a board member of ABIM, and has received research support from UCB. A Buisson has received research funding from Pfizer and lecture fees from AbbVie, Ferring, Hospira, MSD, Janssen, Sanofi‐Aventis, Takeda and Vifor Pharma, and has served as a consultant for AbbVie, Biogen, Janssen, Pfizer and Takeda. CAS has served as a consultant for AbbVie, Amgen, Celgene, Janssen, Lilly, Pfizer, Prometheus, Sandoz and Takeda, and as a speaker for CME activities for AbbVie, Janssen, Pfizer and Takeda; and has received grant support from AbbVie, Janssen, Pfizer and Takeda. YB has received lecture and consulting fees from AbbVie, Biogaran, Boehringer Ingelheim, CTMA, Ferring, Gilead, Hospira, ICON, Inception IBD, Janssen, Lilly, Mayoly Spindler, Merck, MSD, Norgine, Pfizer, Robarts Clinical Trials, Roche, Sanofi, Shire, Takeda, UCB and Vifor Pharma, and owns stock in Inception IBD, San Diego, CA, USA. WJS has received personal fees from Actavis, ActoGeniX NV, Adheron Therapeutics, Akros Pharma, AM Pharma BV, Ardelyx Inc., Arena Pharmaceuticals, Ambrx Inc., Avaxia Biologics, Baxter Healthcare, Biogen, Catabasis Pharmaceuticals, Celgene, Cellular Therapeutics, Chiasma, Cosmo Pharmaceuticals, Dr. August Wolff, Eisai, Eli Lilly, Ferring Pharmaceuticals, Ferring Research Institute, Forward Pharma, Galapagos, Immune Pharmaceuticals, InDex Pharmaceuticals, Ironwood Pharmaceuticals, Kyowa Hakko Kirin, Lexicon Pharmaceuticals, Lipid Therapeutics GmbH, Luitpold Pharmaceuticals, MedImmune (AstraZeneca), Mesoblast, Millennium Pharmaceuticals, Nestle, Novo Nordisk, Orexigen, Palatin, Qu Biologics, Regeneron, Ritter Pharmaceuticals, Salix Pharmaceuticals, Santarus, Seattle Genetics, Seres Health, Shire, Sigmoid Biotechnologies, Teva Pharmaceuticals, Theradiag, Theravance, TiGenix, Tillotts Pharma, Toray Industries Inc., UCB Pharma, University of Western Ontario (owner of Robarts Clinical Trials), Vascular Biogenics, Vertex Pharmaceuticals, Warner Chilcott, and Zyngenia; grants and personal fees from AbbVie, Amgen, Atlantic Pharmaceuticals, Boehringer Ingelheim, Bristol‐Myers Squibb, Genentech, Gilead Sciences, GlaxoSmithKline, Nutrition Science Partners, Prometheus Laboratories, Takeda, Pfizer and Receptos; grants, personal fees and nonfinancial support from Janssen; and grants from American College of Gastroenterology, Broad Foundation, Exact Sciences. KL and MR are employees of Takeda Pharmaceuticals U.S.A., Inc. BGF has received grant support from AbbVie, Amgen, AstraZeneca, Bristol‐Myers Squibb, Roche, Genentech, J&J, Janssen, Millennium, Pfizer, Receptos, Tillotts and UCB; and has served as a consultant or advisory board member for AbbVie, ActoGeniX, Akros, Albireo, Amgen, AstraZeneca, Avaxia Biologics, Avir Pharma, Baxter Healthcare Corp, Biogen Idec, Boehringer Ingelheim, Bristol‐Myers Squibb, Calypso Biotech, Celgene, Elan/Biogen, enGene, Ferring Pharmaceuticals, Galapagos, Genentech/Roche, GiCare Pharma, Gilead, Given Imaging, GSK, Inception IBD Inc, Ironwood Pharmaceuticals, J&J, Janssen, Japan Tobacco, Kyowa Hakko Kirin Co Ltd, Lexicon, Lilly, Lycera Biotech, Merck, Mesoblast Ltd, Millennium, Nektar, Nestlé, Novartis, Novo Nordisk, Pfizer, Prometheus Therapeutics & Diagnostics, Protagonist, Receptos, Salix, Shire, Sigmoid Pharma, Synergy Pharmaceuticals Inc, Takeda, Teva Pharmaceutical Industries Ltd, TiGenix, Tillotts, UCB, Vertex Pharmaceuticals, VHsquared Ltd, Warner Chilcott, Wyeth, Zealand Pharma and Zyngenia. DB is an employee of Takeda Pharmaceuticals International AG. CTP has received lecture fees from MSD, Takeda, Janssen, AbbVie, Vifor Pharma and Norgine, as well as travel accommodations from Biogaran, Takeda, Hospira, MSD and Mayoly Spindler. BS has served as a consultant for AbbVie, Janssen, Robarts Clinical Trials, Salix, Takeda and Theravance. RA has received board or lectures fees from AbbVie, Janssen, Pfizer and Takeda. BES has served as a consultant for and received research support from Amgen, Celgene, Janssen, Pfizer, Prometheus Laboratories and Takeda; and has served as a consultant for AbbVie, Akros Pharma, Arena Pharmaceuticals, AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Cowen Services Company, Forest Research Institute, Forward Pharma, Immune Pharmaceuticals, Lilly, Receptos, Salix Pharmaceuticals, Shire, Synergy Pharmaceuticals, Theravance Biopharma R&D, TiGenix, TopiVert Pharma, UCB, Vivelix Pharmaceuticals, Target PharmaSolutions and Allergan. JFC has served as a consultant or advisory board member for AbbVie, Amgen, Boehringer Ingelheim, Celgene Corporation, Celltrion, Enterome, Ferring, Genentech, Janssen & Janssen, MedImmune, Merck & Co., Pfizer, Protagonist, Second Genome, Seres, Takeda and Theradiag and as a speaker for AbbVie, Ferring, Takeda and Shire; has received research support from AbbVie, Janssen & Janssen, Genentech, Takeda; and has stock options in Intestinal Biotech Development and Genfit. FC has served as a speaker for AbbVie, BMS, Enterome, Janssen, Mayoly‐Spindler, MSD, Pfizer and Takeda.
APPENDIX 1. Collaborators: GETAID OBSERV‐IBD and VICTORY cohorts collaboration
Cleveland Clinic Foundation: Gursimran Kochhar. Division of Gastroenterology, University of California, San Diego, La Jolla, CA, USA: Joseph Meserve, Maria Barsky, Brigid S. Boland. Department of Gastroenterology, Henri Mondor University Hospital, Creteil, France: Charlotte Gagniere. INSERM U954 and Department of Gastroenterology, University of Lorraine, Nancy, France: Marc‐Andre Bigard, Camille Zallot. Aix‐Marseille University, Marseille, France: Jean‐Charles Grimaud. Department of Gastroenterology and Clinical Nutrition, Nice University Hospital, Nice, France: Xavier Hebuterne. Department of Gastroenterology, Claude Huriez Hospital, Lille, France: Maria Nachury, Pierre Desreumaux. Department of Gastroenterology, University Hospital of Saint‐Etienne, Saint‐Etienne, France: Emilie Del Tedesco. Department of Hepato‐Gastroenterology, Clermont‐Ferrand University Hospital, Clermont‐Auvergne University, INSERM U1071 M2iSH, INRA2018, 3iHP, Clermont‐Ferrand, France: Gilles Bommelaer. Dartmouth‐Hitchcock Medical Center, Lebanon, NH, USA: Jenna L. Koliani‐Pace. Department of Gastroenterology, IBD and Nutrition Support, Beaujon Hospital, Clichy, France: Carmen Stefanescu. Department of Gastroenterology, University Hospital of Nantes, Nantes, France: Arnaud Boureille. Icahn School of Medicine at Mount Sinai, New York, NY, USA: Robert Hirten, Ryan Ungaro. Department of Gastroenterology, Bicetre University Hospital, Paris, France: Thibaud Vaysse. Indiana University, Indianapolis, IN, USA: Matthew Bohm, Sashidhar Varma, Monika Fischer. New York University, New York, NY, USA: David Hudesman, Shannon Chang. Department of Gastroenterology, Saint‐Antoine University Hospital, Paris, France: Anne Bourrier, Philippe Seksik, Laurent Beaugerie, Jacques Cosnes, Harry Sokol, Cecilia Landman. Montefiore Medical Center, New York, NY, USA: Dana Lukin, Aaron Weiss. Medicosurgical Department of Digestive Diseases, Lariboisière Hospital, Paris, France: Philippe Marteau, Xavier Dray. Department of Gastroenterology, Claude Bernard University Lyon 1, Lyon, France: Stephane Nancey, Gilles Boschetti. Department of Hepato‐Gastroenterology, University Hospital of Bordeaux, Bordeaux, France: David Laharie, Florian Poullenot. Department of Gastroenterology, Saint Louis Hospital, Paris, France: Matthieu Allez, Jean‐Marc Gornet, Clautilde Baudry. Department of Gastroenterology, University Hospital of Rouen, Rouen, France: Guillaume Savoye. Department of Gastroenterology, Rangueil Hospital, Toulouse, France: Jacques Moreau. Department of Gastroenterology, University Hospital of Besançon, Besançon, France: Lucine Vuitton, Stephane Koch. Department of Gastroenterology, University Hospital of Caen, Caen, France: Stephanie Viennot. Department of Gastroenterology, Trousseau Hospital, Saint‐Avertin, France: Alexandre Aubourg, Laurence Picon. Department of Hepato‐Gastroenterology, Bichat‐Claude Bernard Hospital, Paris, France: Anne‐Laure Pelletier, Gaelle Sickersen. Department of Gastroenterology, Rennes University Hospital, Rennes, France: Guillaume Bouguen. Department of Gastroenterology, Cochin Hospital, Paris, France: Vered Abitbol, Stanislas Chaussade. Department of Hepato‐Gastroenterology, Le Raincy Montfermeil Intercommunal Hospital Group, Montfermeil, France: Stephane Nahon. Department of Hepato‐Gastroenterology, Amiens‐Picardie University Hospital, Amiens, France: Mathurin Fumery. Department of Hepato‐Gastroenterology, Hospital of Belfort‐Montbéliard, Belfort, France: Betsy Winkfield. Department of Hepato‐Gastroenterology, Reims University Hospital, Reims, France: Hedia Brixi‐Benmansour. Department of Hepato‐Gastroenterology, Edouard Herriot Hospital, Lyon, France: Rodica Gincul. Department of Hepato‐Gastroenterology, Bagatelle Hospital, Talence, France: Jean‐Christophe Barberis. Department of Hepato‐Gastroenterology, University Hospital of Grenoble, Grenoble, France: Bruno Bonaz. Department of Hepato‐Gastroenterology, Dijon Bourgogne University Hospital, Dijon, France: Christophe Michiels. Department of Hepato‐Gastroenterology, Saint‐Andre Hospital, Bordeaux, France: Frank Zerbib. Department of Hepato‐Gastroenterology, Gabriel Martin Hospital, Réunion, France: Marie Bourrier de Beauregard. Department of Hepato‐Gastroenterology, Est Francilien Hospital, Meaux, France: Christophe Locher. Department of Hepato‐Gastroenterology, Le Corbusier Hospital, Firminy, France: Sophie Davin‐Couve. Department of Hepato‐Gastroenterology, Hospital of Hyères, Hyères, France: Armelle Poirette. Department of Hepato‐Gastroenterology, Elbeuf‐Louviers‐Val du Reuil Intercommunal Hospital, Elbeuf, France: Laurence Guillem. Department of Hepato‐Gastroenterology, Eure‐Seine Intercommunal Hospital, Évreux, France: Monica Stetiu‐Mocanu. Department of Hepato‐Gastroenterology, University Hospital of Poitiers, Poitiers, France: Beau Philippe. Department of Hepato‐Gastroenterology, Clinique de la Sauvegarde, Lyon, France: Sylvain Beorchia. Department of Hepato‐Gastroenterology, Louis Pasteur Hospital, Chartres, France: Jawad Al Qaddi. Lenox Hill Hospital, New York, NY, USA: Arun Swaminath.
APPENDIX 2. The complete list of authors’ affiliations
Parambir S. Dulai, Siddharth Singh, and William J. Sandborn, Division of Gastroenterology, University of California, San Diego, La Jolla, CA, USA. Aurelien Amiot, Henri Mondor University Hospital, Creteil, France. Laurent Peyrin‐Biroulet, INSERM U954 and Department of Gastroenterology, University of Lorraine, Nancy, France. Vipul Jairath and Brian G. Feagan, University of Western Ontario, London, ON, Canada. Melanie Serrero, Aix‐Marseille University, Marseille, France. Jerome Filippi, Nice University Hospital, Nice, France. Benjamin Pariente, Claude Huriez Hospital, Lille, France. Edward V. Loftus Jr. and Sunanda Kane Mayo Clinic, Rochester, MN, USA. Xavier Roblin, University Hospital of Saint‐Etienne, Saint‐Etienne, France. Anthony Buisson, Clermont‐Ferrand University Hospital, Clermont‐Auvergne University, INSERM U1071 M2iSH, Clermont‐Ferrand, France. Corey A. Siegel, Dartmouth‐Hitchcock Medical Center, Lebanon, NH, USA. Yoram Bouhnik, Beaujon Hospital, Clichy, France. Karen Lasch and Maria Rosario, Takeda Pharmaceuticals U.S.A., Inc., Deerfield, IL, USA. Daniela Bojic, Takeda Pharmaceuticals International AG, Zurich, Switzerland. Caroline Trang‐Poisson, University Hospital of Nantes, Nantes, France. Bo Shen, Cleveland Clinic Foundation, Cleveland, OH, USA. Romain Altwegg, University Hospital of Montpellier Saint‐Eloi, Montpellier, France. Bruce E. Sands and Jean‐Frederic Colombel, Icahn School of Medicine at Mount Sinai, New York, NY, USA. Franck Carbonnel, Bicetre University Hospital, Paris, France.
Dulai PS, Amiot A, Peyrin‐Biroulet L, et al; on Behalf of the GETAID OBSERV‐IBD, VICTORY Cohorts Collaboration* . A clinical decision support tool may help to optimise vedolizumab therapy in Crohn’s disease. Aliment Pharmacol Ther. 2020;51:553–564. 10.1111/apt.15609
Contributor Information
Parambir S. Dulai, Email: pdulai@ucsd.edu.
the GETAID OBSERV‐IBD, VICTORY Cohorts Collaboration*:
Gursimran Kochhar, Joseph Meserve, Maria Barsky, Brigid S Boland, Charlotte Gagniere, Marc‐Andre Bigard, Camille Zallot, Jean‐Charles Grimaud, Xavier Hebuterne, Maria Nachury, Pierre Desreumaux, Emilie Del Tedesco, Gilles Bommelaer, Jenna L Koliani‐Pace, Carmen Stefanescu, Arnaud Boureille, Robert Hirten, Ryan Ungaro, Thibaud Vaysse, Matthew Bohm, Sashidhar Varma, Monika Fischer, David Hudesman, Shannon Chang, Anne Bourrier, Philippe Seksik, Laurent Beaugerie, Jacques Cosnes, Harry Sokol, Cecilia Landman, Dana Lukin, Aaron Weiss, Philippe Marteau, Xavier Dray, Stephane Nancey, Gilles Boschetti, David Laharie, Florian Poullenot, Matthieu Allez, Jean‐Marc Gornet, Clautilde Baudry, Guillaume Savoye, Jacques Moreau, Lucine Vuitton, Stephane Koch, Stephanie Viennot, Alexandre Aubourg, Laurence Picon, Anne‐Laure Pelletier, Gaelle Sickersen, Guillaume Bouguen, Vered Abitbol, Stanislas Chaussade, Stephane Nahon, Mathurin Fumery, Betsy Winkfield, Hedia Brixi‐Benmansour, Rodica Gincul, Jean‐Christophe Barberis, Bruno Bonaz, Christophe Michiels, Frank Zerbib, Marie Bourrier de Beauregard, Christophe Locher, Sophie Davin‐Couve, Armelle Poirette, Laurence Guillem, Monica Stetiu‐Mocanu, Beau Philippe, Sylvain Beorchia, Jawad Al Qaddi, and Arun Swaminath
REFERENCES
- 1. Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. 2013;369:711–721. [DOI] [PubMed] [Google Scholar]
- 2. Engel T, Ungar B, Yung DE, Ben‐Horin S, Eliakim R, Kopylov U. Vedolizumab in IBD‐lessons from real‐world experience: a systematic review and pooled analysis. J Crohns Colitis. 2018;12:245–257. [DOI] [PubMed] [Google Scholar]
- 3. Schreiber S, Dignass A, Peyrin‐Biroulet L, et al. Systematic review with meta‐analysis: real‐world effectiveness and safety of vedolizumab in patients with inflammatory bowel disease. J Gastroenterol. 2018;53:1048–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dreesen E, Verstockt B, Bian S, et al. Evidence to support monitoring of vedolizumab trough concentrations in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2018;16:1937–1946. [DOI] [PubMed] [Google Scholar]
- 5. Ungar B, Kopylov U, Yavzori M, et al. Association of vedolizumab level, anti‐drug antibodies, and alpha4beta7 occupancy with response in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2018;16:697–705.e7. [DOI] [PubMed] [Google Scholar]
- 6. Rosario M, French JL, Dirks NL, et al. Exposure‐efficacy relationships for vedolizumab induction therapy in patients with ulcerative colitis or Crohn's disease. J Crohns Colitis. 2017;11:921–929. [DOI] [PubMed] [Google Scholar]
- 7. Guidi L, Pugliese D, Tonucci TP, et al. Early vedolizumab trough levels predict treatment persistence over the first year in inflammatory bowel disease. United Eur Gastroenterol J. 2019;7:1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh S, Dulai PS, Vande Casteele N, et al. Systematic review with meta‐analysis: association between vedolizumab trough concentration and clinical outcomes in patients with inflammatory bowel diseases. Aliment Pharmacol Ther. 2019;50:848–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Verstockt B, Mertens E, Dreesen E, et al. Influence of drug exposure on vedolizumab‐induced endoscopic remission in anti‐TNF naive and anti‐TNF exposed IBD patients. J Crohns Colitis. 2019. 10.1093/ecco-jcc/jjz151. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 10. Danese S, Sandborn WJ, Colombel J‐F, et al. Endoscopic, radiologic, and histologic healing with vedolizumab in patients with active Crohn's disease. Gastroenterology. 2019;157:1007–1018.e7. [DOI] [PubMed] [Google Scholar]
- 11. Williet N, Boschetti G, Fovet M, et al. Association between low trough levels of vedolizumab during induction therapy for inflammatory bowel diseases and need for additional doses within 6 months. Clin Gastroenterol Hepatol. 2017;15:1750–1757.e3. [DOI] [PubMed] [Google Scholar]
- 12. Feagan BG, Lasch K, Lissoos T, et al. Rapid response to vedolizumab therapy in biologic‐naive patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2018;17:130–138.e7. [DOI] [PubMed] [Google Scholar]
- 13. Dulai PS, Boland BS, Singh S, et al. Development and validation of a scoring system to predict outcomes of vedolizumab treatment in patients with Crohn's disease. Gastroenterology. 2018;155:687–695.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amiot A, Grimaud JC, Peyrin‐Biroulet L, et al. Effectiveness and safety of vedolizumab induction therapy for patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2016;14:1593–1601.e2. [DOI] [PubMed] [Google Scholar]
- 15. Amiot A, Serrero M, Peyrin‐Biroulet L, et al. One‐year effectiveness and safety of vedolizumab therapy for inflammatory bowel disease: a prospective multicentre cohort study. Aliment Pharmacol Ther. 2017;46:310–321. [DOI] [PubMed] [Google Scholar]
- 16. Best WR. Predicting the Crohn's disease activity index from the Harvey‐Bradshaw Index. Inflamm Bowel Dis. 2006;12:304–310. [DOI] [PubMed] [Google Scholar]
- 17. Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn's disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147:618–627.e3. [DOI] [PubMed] [Google Scholar]
- 18. Siegel CA, Horton H, Siegel LS, et al. A validated web‐based tool to display individualised Crohn's disease predicted outcomes based on clinical, serologic and genetic variables. Aliment Pharmacol Ther. 2016;43:262–271. [DOI] [PubMed] [Google Scholar]
- 19. Guizzetti L, Zou G, Khanna R, et al. Development of clinical prediction models for surgery and complications in Crohn's disease. J Crohns Colitis. 2018;12:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dulai PS, Singh S, Ohno‐Machado L, Sandborn WJ. Population health management for inflammatory bowel disease. Gastroenterology. 2018;154:37–45. [DOI] [PubMed] [Google Scholar]
- 21. Khanna R, Bressler B, Levesque BG, et al. Early combined immunosuppression for the management of Crohn's disease (REACT): a cluster randomised controlled trial. Lancet. 2015;386:1825–1834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials