

ABSTRACT
Introduction
Turoctocog alfa is a recombinant, B domain‐truncated factor VIII (FVIII) approved for patients with haemophilia A.
Aim
To evaluate the safety and efficacy of turoctocog alfa in previously untreated patients (PUPs) with severe haemophilia A.
Methods
Guardian 4 was a multicentre, multinational, non‐randomized, open‐label phase 3 trial comprising a main and extension phase. The former concluded once ≥ 50 patients had received treatment for ≥ 50 exposure days (EDs) or developed inhibitors. Patients received turoctocog alfa intravenously for prevention and treatment of bleeds. The primary endpoint was the incidence rate of FVIII inhibitors (≥0.6 Bethesda Units) reported during the first 50 EDs.
Results
Of the 58 patients who completed the main phase, 25 (43.1%) patients developed inhibitors (detected within 6‐24 [mean: 14.2] EDs from treatment start). High‐risk mutations were identified in 60% of patients who developed inhibitors in the main phase and were a significant predictor of inhibitor development (P = .003). Of the 21 patients who started immune tolerance induction therapy, 85.7% completed treatment with a negative inhibitor test (note that data on the last 3 patients completing ITI are based on information collated from sites prior to the final database lock). Haemostatic response (including missing values as failure) was rated as ‘excellent’ or ‘good’ for 86.1% of bleeds occurring during prophylaxis. The estimated mean annualized bleeding rate for patients on prophylaxis was 4.26 bleeds/patient/year (95% CI: 3.34 − 5.44).
Conclusions
Turoctocog alfa was effective at preventing and stopping bleeds and was well tolerated. Inhibitor development was within the expected range for this PUP population.
Keywords: annualized bleeding rate, Haemophilia A, immunogenicity, previously untreated patients, recombinant factor VIII, turoctocog alfa
1. INTRODUCTION
Turoctocog alfa is a third‐generation, recombinant, B domain‐truncated human coagulation factor VIII (FVIII): the molecule has been discussed in detail elsewhere.1, 2 Truncation of the B domain relative to endogenous FVIII has not been associated with any impact on the safety or efficacy of turoctocog alfa, which has demonstrated efficacy and safety in Phase 3 trials in previously treated children, adolescents and adults (guardian 1, 2 and 3 clinical trials). Reductions in annualized bleeding rate (ABR) were observed across all age groups with an overall median ABR of 1.37 bleeds/patient/year (3.7 and 3.0 bleeds/patient/year reported for adolescents/adults and children on prophylaxis, respectively).3, 4, 5 Furthermore, no inhibitors were reported in previously treated patients (PTPs) (N = 238) in clinical trials following treatment with turoctocog alfa with a cumulative of 856 patient‐years of exposure.3, 4, 5
Inhibitors occur most frequently in patients with severe haemophilia A,6 and the majority of patients who develop inhibitors are likely to do so within the first 50 exposure days (EDs) of treatment.7 However, inhibitor formation can occur earlier and inhibitors have been detected as early as after 5 EDs.8 In single product and cohort studies of previously untreated patients (PUPs) with haemophilia A, inhibitors have been reported in up to 39% of patients.9, 10, 11 The aim of this trial was to evaluate the safety and efficacy of turoctocog alfa in PUPs with severe haemophilia A.
2. MATERIALS AND METHODS
2.1. Trial design
Guardian 4 was a multicentre, multinational, non‐randomized, open‐label, safety and efficacy trial in a paediatric population of PUPs with haemophilia A (NCT01493778). The trial involved 40 participating sites in Algeria, Austria, China, Denmark, Greece, Hong Kong, Hungary, Japan, Lithuania, Poland, Russian Federation, Serbia, Spain, Turkey and the United States, and began on 17 September 2012. The Last Patient Last Visit was on 27 June 2018.
The trial comprised two phases—a main phase and an extension phase. Once enrolled, five patient visits were scheduled (until the end of the main phase based on the number of EDs reached), including the screening visit (Visit 1) and four subsequent visits (Figure 1). Inhibitor testing was performed at three scheduled visits: Visits 3, 4 and 5 (10‐15, 20‐25 and 50‐55 EDs, respectively) and could be done at any unscheduled visit at the investigators’ discretion. The main phase of the trial concluded once ≥ 50 patients had received treatment for ≥ 50 EDs or developed FVIII inhibitors. Patients who developed inhibitors (confirmed by two positive consecutive tests, preferably within two weeks) during the main or extension phases of the trial could continue treatment with turoctocog alfa, including immune tolerance induction (ITI). The trial was approved by all relevant independent ethics committees and institutional review boards. Written informed consent was obtained from all participants’ legally authorized representatives before any study‐related activities commenced. The trial was conducted in accordance with the declaration of Helsinki12 and Good Clinical Practice.13
Figure 1.

Trial design. *Inhibitor testing was performed at visits 3, 4 and 5 (10‐15, 20‐25 and 50‐55 EDs, respectively) and could be done at any unscheduled visit at the investigators’ discretion. Patients receiving on‐demand treatment were allowed to postpone initiation of their prophylaxis regimen until they had experienced either a minimum of two treatment‐requiring bleeds or once they had reached 2 y of age (whichever came first). On‐demand treatment was initiated as soon as a bleed was identified. EDs, exposure days; EOT, end of end of trial; V, visit
2.2. Participants
Patients were eligible for inclusion if they were as follows: male, age < 6 years, diagnosed with congenital severe haemophilia A (FVIII level ≤ 1% at the time of diagnosis), had no previous use of purified clotting FVIII products, including commercially available NovoEight® (Novo Nordisk); previous exposure of ≤ 5 EDs to blood components, for example cryoprecipitate, fresh frozen plasma (FFP), was accepted.
2.3. Treatment
For prophylaxis, each patient received a starting dose of turoctocog alfa of 15‐50 IU/kg of body weight (BW) administered once weekly, which was increased gradually by the investigator and based on the patient's clinical profile and local treatment practices. Patients < 2 years old were required to start prophylaxis treatment no later than after ≤ 2 bleeding episodes requiring treatment, or once they had reached 2 years of age, whichever came first. Patients ≥ 2 years old at enrolment were required to start prophylaxis immediately. Despite this, some patients continued to receive on‐demand treatment for up to 23 EDs before initiating prophylaxis. All bleeds could be treated at home, regardless of severity. Individual doses for treatment of bleeds were determined by the investigators.
ITI treatment, if required, was initiated within 6 months (from inhibitor detection) and could continue for ≤24 months. Before any decision to initiate ITI treatment, patients could receive either on‐demand treatment (with bypassing agents or turoctocog alfa) or modified prophylaxis with turoctocog alfa. The ITI treatment regimen was at the investigator's discretion, and patients were allowed to continue in the extension phase after completion of ITI treatment and a negative inhibitor titre (<0.6 BU).
2.4. Study endpoints and assessments
The primary endpoint was the incidence rate of FVIII inhibitors (≥0.6 BU) during the main phase (≥50 EDs). Secondary safety endpoints for the main, extension and combined (main and extension) phases included frequency of adverse events (AEs) and serious AEs (SAEs), and incidence rate of FVIII inhibitors (≥0.6 BU) and high‐titre inhibitors (≥5 BU). Secondary efficacy endpoints included haemostatic response for treatment of bleeds, ABR during prophylaxis, number of infusions required per bleeding episode and consumption of turoctocog alfa. Haemostatic response was assessed by parents 8 hours after the first treatment for a bleed using a predefined four‐point scale (excellent, good, moderate and none). Further details are provided in the Appendix S1. FVIII inhibitors (Nijmegen‐modified Bethesda assay) and genotyping were assessed in a central laboratory. FVIII activity and recovery were both optional tests and not used to calculate endpoints and, consequently, have not been included here.
2.5. Statistical methods
No formal testing of statistical hypotheses was carried out, nor were any formal sample size calculations performed. It was expected that a target sample size of 50 patients completing the trial would adequately measure inhibitor development in the trial population. Consequently, 60 patients were enrolled in the trial which allowed for a 15% drop‐out rate. The full and safety analysis sets included all dosed patients with data after dosing.
Incidence rates for inhibitor development during each study phase were calculated and an exact one‐sided 97.5% upper confidence limit estimated using the binomial distribution. In cases of inhibitor recurrence, only the first occurrence was included in the analyses. Additional analyses evaluated relationships between the incidence rate of inhibitors and different patient characteristics (eg race, family history of inhibitors and FVIII gene mutation type) and first‐treatment characteristics (eg age, reason for FVIII treatment and intensity of treatment once initiated). Further details are provided in the Appendix S1. The protocol‐defined FVIII gene mutation classification was later deemed incomplete, and a revised classification was used in a post hoc analysis presented here. Patients who developed inhibitors were considered separately until their ITI treatment was completed, and their data were evaluated and reported independently from the trial population overall.
The incidence rates of high‐titre inhibitors were analysed in a similar way to the primary endpoint.
AEs and SAEs were summarized by frequency of events and frequency of patients with any event.
Secondary efficacy endpoints were analysed for the main and combined phases using descriptive statistics. ABR and 95% confidence intervals were estimated using a Poisson regression model allowing for over‐dispersion and using (log) trial duration as offset.
3. RESULTS
3.1. Patient exposure and treatment
In total, 60 patients were enrolled and exposed to treatment (Figure 2), receiving ≥1 dose of turoctocog alfa; 58 patients received turoctocog alfa prophylactically and 49 patients (81.7%) completed the trial. Baseline characteristics are shown in Table 1.
Figure 2.
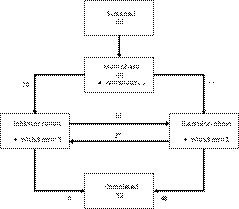
Participant flow. *One patient restarted ITI treatment and one developed inhibitors during the extension phase: both patients rejoined the extension phase. ITI, immune tolerance induction
Table 1.
Patient demographics and baseline characteristics
| Patients with inhibitors | Patients without inhibitors | Total | |
|---|---|---|---|
| Number of patients | 26 | 34 | 60 |
| Age, N (%) | |||
| 0‐<1 mo | 3 (11.5) | ‐ | 3 (5.0) |
| 1‐<6 mo | 5 (19.2) | 7 (20.6) | 12 (20.0) |
| 6‐<12 mo | 8 (30.8) | 19 (55.9) | 27 (45.0) |
| ≥12 mo | 10 (38.5) | 8 (23.5) | 18 (30.0) |
| Mean age at baseline (SD), months | 10.5 (8.86) | 10.0 (7.16) | 10.2 (7.88) |
| Ethnicity | |||
| Hispanic or Latino, N (%) | 1 (3.8) | 4 (11.8) | 5 (8.3) |
| Race (%) | |||
| White | 17 (65.4) | 27 (79.4) | 44 (73.3) |
| Asian | 7 (26.9) | 5 (14.7) | 12 (20.0) |
| Other | 2 (7.7) | 2 (5.9) | 4 (6.7) |
| Black/African descent | ‐ | ‐ | ‐ |
| Genotype—type of mutation,a N | |||
| High risk | 24 | 17 | 41 |
| Low risk | 1 | 14 | 15 |
| Genotype information missing | 1 | 1 | 2 |
| Family history of haemophilia, N (%) | 26 (100.0) | 34 (100.0) | 60 (100.0) |
| Yes | 13 (50.0) | 14 (41.2) | 27 (45.0) |
| Family history of inhibitor, N (%) | 13 (100.0) | 14 (100.0) | 27 (100.0) |
| Yes | 1 (7.7) | 4 (28.6) | 5 (18.5) |
| Unknown | 7 (53.8) | 4 (28.6) | 11 (40.7) |
| Previous exposure to blood componentsb (≤5 d), N (%) | |||
| Yes | 4 (15.4) | 7 (20.6) | 11 (18.3) |
Abbreviations: CI, confidence interval; d, days; mo, months; N, number of patients; SD, standard deviation.
FVIII gene mutation analysis was conducted post hoc.
Patients who had prior exposure (≤5 exposure days) to blood components, such as cryoprecipitate or fresh frozen plasma, were included in the trial.
3.2. Development of FVIII inhibitors
Overall, 25 of 58 (43.1%) patients who completed the main phase, and therefore were included in the primary analysis, developed FVIII inhibitors (Table 2). Inhibitors were detected in the main phase within 6‐24 EDs (from first dose), with a mean of 14.2 EDs before their first positive inhibitor test (Figure 3). One patient developed an inhibitor before starting prophylaxis treatment. One patient developed a low‐titre inhibitor during the extension phase (after 101 EDs); therefore, 26 patients developed inhibitors overall throughout the entire trial. High‐titre inhibitors accounted for 16 (61.5%) of the 26 inhibitor cases, with peak titres ranging from 5.5 to 334.6 BU. A post hoc analysis identified high‐risk genetic mutations (ie inversions, nonsense mutations, large deletions and certain small deletions that result in a lack of endogenous FVIII production;14, 15 Table 3) in 24 of the 26 inhibitor patients in the trial, including 15 of the 16 patients with high‐titre inhibitors. Genetic mutations classified as ‘high risk’ carried a statistically significant risk for inhibitor development (P = .003); other risk factors (eg race and family history) were not statistically significant. Inhibitor development was higher in patients of Asian descent than in other racial groups and the incidence of inhibitor development generally increased with the age of the patient when treatment was first initiated (statistically non‐significant in both cases).
Table 2.
Incidence rate of inhibitors
| Main phase | Extension phase | Total | ||||
|---|---|---|---|---|---|---|
| n/N (%) | 95% CI | n/N (%) | 95% CI | n/N (%) | 95% CI | |
| FVIII inhibitors (>0.6 BU) | 25/58 (43.1) | 30.2‐56.8 | 1/33 (3.0) | 0.1‐15.8 | 26/58 (44.8) | 31.7‐58.5 |
| High‐titre inhibitors (≥5 BU) | 16/58 (27.6) | 16.7‐40.9 | 0/33 (0.0) | 0.0‐10.6 | 16/58 (27.6) | 16.7‐40.9 |
| By patient characteristics | n/N (%) | OR | 95% CI | n/N (%) | OR | 95% CI | n/N (%) | OR | 95% CI |
|---|---|---|---|---|---|---|---|---|---|
| Age at first infusion of FVIII concentrate | |||||||||
| ≤5 mo | 3/10 (30) | 1.0 | NA | 1/7 (14.3) | ‐ | ‐ | 4/10 (40) | 1.0 | NA |
| 6‐11 mo | 8/22 (36.4) | 1.3 | 0.3‐6.7 | 0/14 (0.0) | ‐ | ‐ | 8/22 (36.4) | 0.9 | 0.2‐4.0 |
| 12‐18 mo | 10/20 (50.0) | 2.3 | 0.5‐11.7 | 0/10 (0.0) | ‐ | ‐ | 10/20 (50.0) | 1.5 | 0.3‐7.0 |
| >18 mo | 4/6 (66.7) | 4.7 | 0.5‐40.9 | 0/2 (0.0) | ‐ | ‐ | 4/6 (66.7) | 3.0 | 0.4‐24.9 |
| Mutation type | |||||||||
| High risk | 23/41 (56.1) | 1.0 | NA | 1/18 (5.6) | ‐ | ‐ | 24/41 (58.5) | 1.0 | NA |
| Low risk | 1/15 (6.7) | 0.1 | 0.0‐0.5 | 0/14 (0.0) | ‐ | ‐ | 1/15 (6.7) | 0.1 | 0.0‐0.4 |
| Missing | 1/2 (50.0) | 0.8 | 0.0‐13.4 | 0/1 (0.0) | ‐ | ‐ | 1/2 (50.0) | 0.7 | 0.0‐12.1 |
Percentage values are the incidence rates of inhibitor. One patient entered the extension phase but did not receive a dose of turoctocog alfa and was not included as ‘at risk’. Genotype testing was optional and data for 2 patients without assessment are not included.
Abbreviations: CI, confidence interval; FVIII, factor VIII; mo, months; N, number of patients at risk; n, number of patients with new inhibitor in the period; OR, odds ratio.
Figure 3.
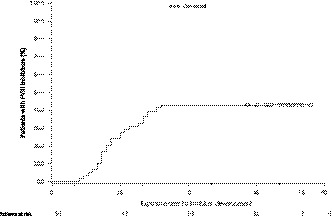
Time to development of confirmed inhibitor (during main phase). FVIII, factor VIII
Table 3.
Incidence of inhibitors by genetic mutation
| Risk level | Sub‐mutation | N | Developed inhibitor |
|---|---|---|---|
| n (%) | |||
| High‐risk | Intron 22a | 24 | 15 (62.5) |
| Small deletionb | 7 | 4 (57.1) | |
| Nonsense mutations | 5 | 1 (20.0) | |
| Intron 1 | 3 | 3 (100.0) | |
| Large deletion | 2 | 1 (50.0) | |
| Missense mutationsa | 1 | 0 (0.0) | |
| Low‐risk | Missense mutations | 6 | 0 (0.0) |
| Duplication | 2 | 0 (0.0) | |
| Splice site mutation | 2 | 0 (0.0) | |
| Large duplication | 1 | 1 (100.0) | |
| Large insertion | 1 | 0 (0.0) | |
| Small deletionb | 1 | 0 (0.0) | |
| Small duplication | 1 | 0 (0.0) | |
| Small insertionb | 1 | 0 (0.0) | |
| Missing | Missing | 2 | 1 (50.0) |
Abbreviations: N, number of mutations; n, number of patients with a given mutation who developed an inhibitor.
One patient had two mutations (intron 22 and missense mutation); as the patient had both a high‐ and low‐risk mutation, both mutations for this patient are listed under high risk.
Small deletion and insertion outside location 1213 or 1460 were considered high risk.
3.3. Frequency of AEs and SAEs
Overall, 721 AEs were reported in 60 patients exposed to turoctocog alfa with a total participation in the trial of 140.1 patient‐years. The most frequently reported SAEs are shown in Table 4. No cases of allergy or hypersensitivity related to turoctocog alfa were reported.
Table 4.
Most frequently reported SAEs (occurring in ≥ 5% of patients)
| AE | N % | E |
|---|---|---|
| FVIII inhibition | 25 (41.7) | 27 |
| Device‐related infection | 5 (8.3) | 7 |
| Traumatic haemorrhage | 4 (6.7) | 5 |
| Pyrexia | 4 (6.7) | 4 |
| Anaemia | 3 (5.0) | 3 |
| Head injury | 3 (5.0) | 3 |
Abbreviations: AE, adverse event; E, number of SAEs; N, number of patients with SAEs; SAE, serious adverse event.
3.4. Efficacy
A total of 402 bleeds in 52 patients on prophylaxis were reported during the combined phases; 98 bleeds occurred in 36 of 51 (70.6%) patients during the main phase, either before initiation of prophylaxis or development of inhibitors. Subcutaneous bleeds accounted for more than half (53.7%) of bleeds in patients on prophylaxis during the combined phases.
Haemostatic response was rated as ‘excellent’ or ‘good’ for 346 (86.1%) bleeds during prophylaxis overall (combined phases; including missing values as failure) (see Figure S1). Five severe bleeds were reported in patients on prophylaxis during the main phase; for some patients in this group with a poor haemostatic response, inhibitors were detected within a few days after the bleeding episode, suggesting that inhibitors may have already been present. After exclusion of bleeds with missing evaluations, the success rate for treatment of bleeds during prophylaxis was 88.5% overall and 89.6% in the main phase.
Patients on prophylaxis had an overall Poisson mean estimated ABR of 4.26 bleeds/patient/year (95% CI: 3.34‐5.44), with 5.63 bleeds/patient/year in the main phase and 3.81 bleeds/patient/year in the extension phase (see Figure S2). Further details of the prophylaxis treatment regimens at the start and end of the main and extension phases are provided in Table S1. In total, 34.5% and 18.3% of patients reported no bleeds during prophylaxis in the main phase and extension phase, respectively.
In total, 227 of 402 (56.5%) bleeds reported for patients on prophylaxis (combined phases) were controlled with one injection of turoctocog alfa; 22.6% were controlled with two injections and 7.5% with three injections.
3.5. Consumption of turoctocog alfa
Patients on prophylaxis had a mean of 167.7 EDs and received a mean of 172.4 injections/patient. Most patients received prophylaxis once or twice weekly during the main phase, and many continued on these regimens in the extension phase. Patients who were successfully tolerized and subsequently entered the extension phase typically received a higher‐frequency dosing regimen, for example three times weekly. Patients on prophylaxis consumed a mean of 5245.9 IU/kg of turoctocog alfa/patient/year in the combined main and extension phase. Further consumption details are provided in Table 5.
Table 5.
Consumption of turoctocog alfa
| Consumption (IU/kg) | Prophylaxis | |
|---|---|---|
| Main phase (n = 58) | Extension phase (n = 49) | |
| Preventive treatment per year/patient | ||
| Mean | 3522.0 | 5355.4 |
| Median (min.; max.) | 2724.8 (789; 20 390) | 4681.3 (1588; 13 208) |
| Preventive treatment per month/patient | ||
| Mean | 293.5 | 446.3 |
| Median (min.; max.) | 227.1 (66; 1699) | 390.1 (132; 1101) |
| Preventive treatment per week/patient | ||
| Mean | 68.3 | 103.8 |
| Median (min.; max.) | 52.8 (15; 395) | 90.7 (31; 256) |
| Total prophylaxis (n = 59) | On demand (n = 51) | Total (n = 60) | |
|---|---|---|---|
| Treatment of a bleed (start to stop of bleed) | |||
| Mean | 98.9 | 86.1 | 96.4 |
| Median (min.; max.) | 54.2 (16; 1471) | 48.7 (0; 803) | 53.4 (0; 1471) |
Abbreviations: Max., maximum; Min., minimum.
3.6. Treatment of inhibitors
Overall, 26 patients developed inhibitors in the trial. Of these, two patients withdrew from the trial without commencing ITI treatment and three patients achieved resolution of their low‐titre inhibitor and completed the trial without ITI treatment. Of the remaining 21 patients who started ITI treatment, 14 had a high‐titre inhibitor (≥5 BU). Eighteen of the twenty‐one patients (85.7%) completed the trial with a negative inhibitor titre, and three patients (14.3%) were withdrawn because either their inhibitor titre did not decline sufficiently during ITI treatment or they remained positive for inhibitors after 2 years of ITI.
During ITI treatment, patients were allowed to receive up to 200 IU/kg of turoctocog alfa daily. A variety of ITI dosing regimens were used, reflecting the diversity in ITI treatment practices in the participating centres. Of the 14 patients with high‐titre inhibitors, 11 received ITI once daily and three received ITI every second day or three times weekly. Six (of seven) patients with low‐titre inhibitors received ITI every second day or three times weekly, and one patient received once‐daily ITI treatment. ITI treatment administered three times weekly was generally at a low dose (approximately 50 IU/kg/dose) compared with ITI administered daily or every second day (100‐150 IU/kg/dose). The mean duration of ITI treatment was 312.9 and 425.7 days for patients with low‐ and high‐titre inhibitors, respectively. The median (interquartile range) time to first negative inhibitor test (<0.6 BU) was 140.0 (158.0) days (for the combined population of patients with low‐ and high‐titre inhibitors).
In patients who developed inhibitors, 179 bleeds were reported in 19 patients and most were classified as mild/moderate (95.5%) and were caused by trauma (86%). Mean ABR was slightly higher in patients with inhibitors compared with those patients who did not develop inhibitors (6.34 bleeds/patient/year [95% CI: 3.98‐10.13] vs 4.15 bleeds/patient/year [95% CI: 3.20‐5.39], respectively).
Three patients who completed ITI had recurrence of inhibitors during the extension phase. The first patient had a low‐titre inhibitor initially and again on recurrence (1.7 BU) and continued to receive prophylaxis once weekly in the extension phase. The second patient had a high‐titre inhibitor initially and had recurrence with a low‐titre inhibitor (0.7 BU); they received a second round of ITI treatment. The third patient had a high‐titre inhibitor originally and again on recurrence (5.8 BU) and continued to receive prophylaxis in the extension phase twice weekly. Further details are provided in Table S2.
4. DISCUSSION
This is the first trial within the guardian clinical trial programme to evaluate turoctocog alfa in PUPs, a patient population that is generally at an increased risk of developing antibodies to foreign protein such as exogenous FVIII. Treatment with turoctocog alfa was shown to be effective at preventing and controlling bleeds. ABR was well within the range reported in other recently published PUP studies with rFVIII.9, 10, 16, 17 In a prospective, open‐label study of moroctocog alfa (N = 37 PUPs) overall mean ABR was 5.9.10 A recent postmarketing surveillance study of rurioctocog alfa in PUPs (N = 114) in Japan reported mean ABR of 7.4 for patients with severe haemophilia A receiving prophylaxis.16, 17 In another study, interim ABR data for human‐cl hFVIII was 3.94 (standard deviation: 5.53).9 The dosing regimens used in guardian 4 reflect the standard clinical practices typically seen in clinics: most patients will usually receive prophylaxis on a lower‐frequency dosing regimen in their early years.
In guardian 4, 43.1% of patients who received turoctocog alfa developed an inhibitor during the main phase of the trial (within the first 50 EDs). An inherent limitation of the study is the small sample size and resultant large confidence intervals. The range of inhibitor incidence rates in the study population recruited for guardian 4 (ie PUPs) is within the range reported for rFVIII products in other key PUP studies, such as the UKHCDO,18 CANAL,19 RODIN,20 EUHASS21 and SIPPET22 (24.4%‐44.5%).
The risk of inhibitor development varies considerably between minimally treated patients (MTPs) (defined as ≤ 5 EDs)23 or PTPs and PUPs.24 Some PUP trials have included patients (up to 56%) with a certain amount of prior exposure (eg 4 EDs) to FVIII concentrate;25 by definition, these patients are MTPs, not PUPs. By contrast, in guardian 4, no patients had received FVIII concentrates and no more than 18.3% of patients had prior exposure (≤5 EDs) to blood components such as cryoprecipitate or FFP. Previous exposure to FVIII (endogenous or exogenous) will confer some degree of immunological tolerance.26 A recent sub‐analysis from the Survey of Inhibitors in Plasma‐Product Exposed Toddlers (SIPPET) trial showed that the inhibitor incidence rate during the first 5 EDs for PUPs treated with recombinant FVIII was 9.5%.8 Based on this analysis, MTPs with previous exposure of 5 EDs would have an approximately 30% risk of developing inhibitors, in contrast to a risk of 40% in true PUPs. Therefore, previous exposure to FVIII, however minimal, may affect the overall incidence of inhibitor development in a study population because patients who may develop inhibitors early would be excluded.
Older single‐product PUP studies have also included patients with mild or moderate haemophilia25, 27, 28 and several of them categorized patients with a FVIII activity of <2% as ‘severe’.27 In guardian 4, only patients with a FVIII level of ≤1% were included. These differences have considerable implications for how the results from PUP studies should be interpreted. The inhibitor risk for patients with mild/moderate haemophilia is considerably lower compared with those with severe haemophilia (from <5% to >30%, respectively6). It is also well known that even in severe patients, null vs non‐null mutations are associated with varying risks of inhibitor development; this underlies the direct inverse correlation between endogenous FVIII levels and inhibitor risk.29
The development of inhibitors in PUPs is a natural immune response to foreign protein, and several factors are known to be associated with inhibitor development.30 Large cohort studies in PUPs have identified the importance of modifiable risk factors for inhibitor development such as age at first infusion of FVIII concentrate, treatment intensity, treatment regimen (on‐demand treatment vs prophylaxis), and type of FVIII concentrate.20, 22, 31 Non‐modifiable risk factors include causative gene mutation, severity of disease, ethnicity, family history, and polymorphisms of immune response genes.6, 31 FVIII gene mutations considered to be a high risk for inhibitor development include intron 22 inversion, intron 1 inversion, nonsense mutations, and large deletions, as well as other mutations resulting in a complete absence of endogenous FVIII.32, 33 Genetic mutations are one of the strongest risk factors for inhibitor development, and this is supported by the findings in guardian 4. Most patients who developed inhibitors had an underlying high‐risk genetic mutation which was found to be the strongest statistically significant risk factor for inhibitor development (P = .003). Of the 25 patients who developed inhibitors in the main phase, 23 had a high‐risk genetic mutation. This finding is consistent with other PUP studies that used similar mutation categorization.19, 20
Inversions were found to be the most frequent type of mutation in this trial; of the 56 patients with a known mutation type, 24 had an intron 22 inversion and three patients had an intron 1 inversion. Patients with an intron 22 inversion express endogenous FVIII protein intracellularly providing central tolerance which in most cases prevents the immune system from reacting to exogenous FVIII.34 Consequently, intron 1 and intron 22 inversions are known to carry only approximately 20% risk for inhibitor development.15, 33 Conversely, almost two‐thirds of patients with either of these mutation types in guardian 4 developed inhibitors (63%). No other inhibitor risk factors were identified in this population.
No other patient or treatment characteristics (modifiable or non‐modifiable) were shown to be associated with an increased risk of inhibitor development in this trial; this may be due to the small sample size. Patients of African descent were not represented in the trial. This patient population is known to have an increased risk for inhibitor development and a poorer response to ITI treatment compared with other patient groups.35
Generally, there is a lack of consistency across PUP studies in the methods used for defining assay positivity, which may occasionally cause false‐positive results. Variation in methodology together with differences in patient population and statistical approach may account for the wide range of inhibitor rates (24.4%‐44.5%) reported in different PUP studies including this study.18, 22 Therefore, it is important to consider differences in study design, sample size and inhibitor‐testing methodologies when comparing PUP studies. Multiple laboratory tests are available for detecting inhibitors in haemophilia, and new methods for defining assay positivity have been suggested; however, there are still ongoing challenges in the areas of assay standardization and testing in the presence of modified and novel treatment products.36
In guardian 4, treatment with turoctocog alfa was effective at both preventing and stopping bleeds. The majority of patients who developed inhibitors in this study had a high‐risk genetic mutation, and many of these had high‐titre inhibitors. In total, 18 patients (85.7%) who developed inhibitors and commenced ITI therapy subsequently had a negative inhibitor titre. Treatment with turoctocog alfa was well tolerated and inhibitor development was within the expected range for PUPs with severe haemophilia A.
DISCLOSURES
HY has provided consultancy participating in advisory boards and received honoraria from Shire, Bayer, Novo Nordisk and Octapharma, and is on the speaker bureaus of Shire and Bayer; TM has participated in speaker bureaus and advisory boards for: Bayer, Shire, CSL Behring, Novo Nordisk, Chugai, Pfizer, Bioverativ, KM Biologistics and Nichiyaku; MFB has no competing interests; VJY has received reimbursement for attending symposia/congresses and/or honoraria for speaking and/or honoraria for consulting, and/or funds for research from Shire, Bayer, CSL Behring, Grifols, Novo Nordisk, Sobi, Roche, Octapharma and Pfizer; KK has received speaking fees, reimbursement for scientific congresses, funding as a study investigator and has advisory board membership with/from the sponsor (Novo Nordisk); LK is a former employee of Novo Nordisk A/S and held stocks in Novo Nordisk A/S at the time of writing; IM is an employee of Novo Nordisk A/S; CP has received research funding from Novo Nordisk, Bayer and Baxalta, has served as a consultant for Bayer, Novo Nordisk and Genentech, and serves on a Data Safety Monitoring Board for Spark; KR is an employee of Novo Nordisk A/S; RW has no competing interests.
AUTHOR CONTRIBUTIONS
HY performed research and reviewed the paper; TM performed research and reviewed the paper; MFB performed research and reviewed the paper; VJY performed research and reviewed the paper; KK performed research and reviewed the paper; LK performed research, analysed the data and reviewed the paper; IM performed the research, analysed the data and reviewed the paper; CP performed research and reviewed the paper; KR performed research, analysed the data and reviewed the paper; RW performed research and reviewed the paper.
Supporting information
ACKNOWLEDGEMENTS
The trial was sponsored by Novo Nordisk A/S (Bagsværd, Denmark). The authors thank all the participants, their families, investigators and trial staff who were involved in the trial. We also thank Niccolo Bassani, Kasper Lambeth from Novo Nordisk for their review and input to the manuscript, and Lisa Langley, MSc, and James McCary, BSc, (AXON Communications, London, UK) for medical writing and editorial support in preparation of the manuscript.
Yaish H, Matsushita T, Belhani M, et al. Safety and efficacy of turoctocog alfa in the prevention and treatment of bleeds in previously untreated paediatric patients with severe haemophilia A: Results from the guardian 4 multinational clinical trial. Haemophilia. 2020;26:64–72. 10.1111/hae.13883
Funding information
Novo Nordisk A/S.
Footnotes
Data on the last 3 patients completing ITI are based on information collated from sites prior to the final database lock.
Data on the last 3 patients completing ITI are based on information collated from sites prior to the final database lock.
REFERENCES
- 1. Thim L, Vandahl B, Karlsson J, et al. Purification and characterization of a new recombinant factor VIII (N8). Haemophilia. 2010;16:349‐359. [DOI] [PubMed] [Google Scholar]
- 2. Ahmadian H, Hansen EB, Faber JH, et al. Molecular design and downstream processing of turoctocog alfa (NovoEight), a B‐domain truncated factor VIII molecule. Blood Coagul Fibrinolysis. 2016;27:568‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lentz SR, Misgav M, Ozelo M, et al. Results from a large multinational clinical trial (guardian1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia. 2013;19:691‐697. [DOI] [PubMed] [Google Scholar]
- 4. Lentz SR, Janic D, Kavakli K, et al. Long‐term safety and efficacy of turoctocog alfa in prophylaxis and treatment of bleeding episodes in severe haemophilia A: Final results from the guardian 2 extension trial. Haemophilia. 2018;24:e391‐e394. [DOI] [PubMed] [Google Scholar]
- 5. Kulkarni R, Karim FA, Glamocanin S, et al. Results from a large multinational clinical trial (guardian3) using prophylactic treatment with turoctocog alfa in paediatric patients with severe haemophilia A: safety, efficacy and pharmacokinetics. Haemophilia. 2013;19:698‐705. [DOI] [PubMed] [Google Scholar]
- 6. Witmer C, Young G. Factor VIII inhibitors in hemophilia A: rationale and latest evidence. Ther Adv Hematol. 2013;4:59‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gouw SC, van den Berg HM, Oldenburg J, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta‐analysis. Blood. 2012;119:2922‐2934. [DOI] [PubMed] [Google Scholar]
- 8. Peyvandi F, Cannavo A, Garagiola I, et al. Timing and severity of inhibitor development in recombinant versus plasma‐derived factor VIII concentrates: a SIPPET analysis. J Thromb Haemost. 2018;16:39‐43. [DOI] [PubMed] [Google Scholar]
- 9. Liesner RJ, Abashidze M, Aleinikova O, et al. Immunogenicity, efficacy and safety of Nuwiq((R)) (human‐cl rhFVIII) in previously untreated patients with severe haemophilia A‐Interim results from the NuProtect Study. Haemophilia. 2018;24:211‐220. [DOI] [PubMed] [Google Scholar]
- 10. Rusen L, Kavakli K, Korth‐Bradley J, et al. Clinical experience with moroctocog alfa (AF‐CC) in younger paediatric patients with severe haemophilia A: Two open‐label studies. Haemophilia. 2018;24:604‐610. [DOI] [PubMed] [Google Scholar]
- 11. Kreuz W, Ettingshausen CE. Inhibitors in patients with haemophilia A. Thromb Res. 2014;134(Suppl 1):S22‐S26. [DOI] [PubMed] [Google Scholar]
- 12. World Medical Association . Declaration of Helsinki ‐ Ethical Principles for Medical Research Involving Human Subjects. Last amended by the 59th WMA General Assembly, Seoul, Republic of Korea. October 2008. 2008.
- 13. ICH Harmonised Tripartite Guideline . Good Clinical Practice. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed December 05, 2018.
- 14. Astermark J. FVIII Inhibitors: pathogenesis and avoidance. Blood. 2015;125:2045‐2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oldenburg J, Schroder J, Brackmann HH, Muller‐Reible C, Schwaab R, Tuddenham E. Environmental and genetic factors influencing inhibitor development. Semin Hematol. 2004;41:82‐88. [DOI] [PubMed] [Google Scholar]
- 16. Taki M, Fukutake K, Matsushita T, et al. Inhibitor development, safety, and efficacy of Advate((R)) in previously untreated patients with hemophilia A in a postmarketing surveillance in Japan. Int J Hematol. 2019;109:70‐78. [DOI] [PubMed] [Google Scholar]
- 17. Taki M, Fukutake K, Matsushita T, et al. Correction to: Inhibitor development, safety, and efficacy of Advate(R) in previously untreated patients with hemophilia A in a postmarketing surveillance in Japan. Int J Hematol. 2019;109:241. [DOI] [PubMed] [Google Scholar]
- 18. Collins PW, Palmer BP, Chalmers EA, et al. Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000–2011. Blood. 2014;124:3389‐3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gouw SC, van der Bom JG, Marijke van den Berg H. Treatment‐related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109:4648‐4654. [DOI] [PubMed] [Google Scholar]
- 20. Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368:231‐239. [DOI] [PubMed] [Google Scholar]
- 21. Fischer K, Lassila R, Peyvandi F, et al. Inhibitor development in haemophilia according to concentrate. Four‐year results from the European HAemophilia Safety Surveillance (EUHASS) project. Thromb Haemost. 2015;113:968‐975. [DOI] [PubMed] [Google Scholar]
- 22. Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374:2054‐2064. [DOI] [PubMed] [Google Scholar]
- 23. Gringeri A, Monzini M, Tagariello G, Scaraggi FA, Mannucci PM. Emoclot15 Study M. Occurrence of inhibitors in previously untreated or minimally treated patients with haemophilia A after exposure to a plasma‐derived solvent‐detergent factor VIII concentrate. Haemophilia. 2006;12:128‐132. [DOI] [PubMed] [Google Scholar]
- 24. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐47. [DOI] [PubMed] [Google Scholar]
- 25. Auerswald G, Thompson AA, Recht M, et al. Experience of Advate rAHF‐PFM in previously untreated patients and minimally treated patients with haemophilia A. Thromb Haemost. 2012;107:1072‐1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schep SJ, Schutgens REG, Fischer K, Boes ML. Review of immune tolerance induction in hemophilia A. Blood Rev. 2018;32:326‐338. [DOI] [PubMed] [Google Scholar]
- 27. Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N Engl J Med. 1993;328:453‐459. [DOI] [PubMed] [Google Scholar]
- 28. Yoshioka A, Fukutake K, Takamatsu J, Shirahata A. Kogenate Post‐Marketing Surveillance Study Group. Clinical evaluation of a recombinant factor VIII preparation (Kogenate) in previously untreated patients with hemophilia A. Int J Hematol. 2003;78:467‐474. [DOI] [PubMed] [Google Scholar]
- 29. Spena S, Garagiola I, Cannavo A, et al. Prediction of factor VIII inhibitor development in the SIPPET cohort by mutational analysis and factor VIII antigen measurement. J Thromb Haemost. 2018;16:778‐790. [DOI] [PubMed] [Google Scholar]
- 30. White GC 2nd, Kempton CL, Grimsley A, Nielsen B, Roberts HR. Cellular immune responses in hemophilia: why do inhibitors develop in some, but not all hemophiliacs? J Thromb Haemost. 2005;3:1676‐1681. [DOI] [PubMed] [Google Scholar]
- 31. Gouw SC, van der Bom JG, Auerswald G, Ettinghausen CE, Tedgard U, van den Berg HM. Recombinant versus plasma‐derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood. 2007;109:4693‐4697. [DOI] [PubMed] [Google Scholar]
- 32. Bardi E, Astermark J. Genetic risk factors for inhibitors in haemophilia A. Eur J Haematol. 2015;94(Suppl 77):7‐10. [DOI] [PubMed] [Google Scholar]
- 33. Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006;12(Suppl 6):15‐22. [DOI] [PubMed] [Google Scholar]
- 34. Sauna ZE, Lozier JN, Kasper CK, Yanover C, Nichols T, Howard TE. The intron‐22‐inverted F8 locus permits factor VIII synthesis: explanation for low inhibitor risk and a role for pharmacogenomics. Blood. 2015;125:223‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Callaghan MU, Rajpurkar M, Chitlur M, Warrier I, Lusher J. Immune tolerance induction in 31 children with haemophilia A: is ITI less successful in African Americans? Haemophilia. 2011;17:483‐489. [DOI] [PubMed] [Google Scholar]
- 36. Miller CH. Laboratory testing for factor VIII and IX inhibitors in haemophilia: A review. Haemophilia. 2018;24:186‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials