

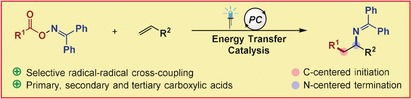
Abstract
An intermolecular, two‐component vicinal carboimination of alkenes has been accomplished by energy transfer catalysis. Oxime esters of alkyl carboxylic acids were used as bifunctional reagents to generate both alkyl and iminyl radicals. Subsequently, addition of the alkyl radical to an alkene generates a transient radical for selective radical–radical cross‐coupling with the persistent iminyl radical. Furthermore, this process provides direct access to aliphatic primary amines and α‐amino acids by simple hydrolysis.
Keywords: amination, carboxylic acids, cross-coupling, energy transfer, photocatalysis
Two birds, one stone: Oxime esters of aliphatic carboxylic acids were used as a bifunctional source of both C‐ and N‐radicals. The persistency of the N‐radicals enables highly selective intermolecular radical carboaminations of alkenes.
The 1,2‐difunctionalization of olefins, which allows the simultaneous construction of two new covalent bonds, is one of the most powerful and straightforward strategies for the rapid construction of molecular complexity.1 Given the ubiquity of C−C and C−N bonds in bioactive compounds, the carboamination of alkenes represents an important subset of such reactions, resulting in the concurrent installation of both carbon and nitrogen functionalities into the alkenyl framework.2, 3, 4 Ideally, cleavage of a C−N bond, followed by the addition of the two fragments into olefins, would be the most direct means of carboamination. However, the intrinsic inertness of C−N bonds, as well as the potential difficulties in C(sp3)−N reductive elimination over facile β‐hydride elimination, present a formidable challenge.5 Overcoming these issues, pioneering studies by Wolfe,6 Rovis,7 Liu,8 Engle,9 and our group10 have shown the prospect of transition metals in intermolecular carboamination reactions.
Alternatively, a radical approach by either N‐ or C‐centered initiation offers an attractive strategy to circumvent these problems.11, 12 Notwithstanding these significant breakthroughs, carboaminations with general alkyl groups for easily accessible primary amines remain an unsolved problem. This can be mainly attributed to the difficulty in generating a broad range of C‐centered radicals from alkylating reagents, such as alkyl halides.13 Additionally, the obligation to protect, and later deprotect, the amine functional group and the requirement for a radical–polar crossover step emphasize the retrosynthetic deficiencies of accessing primary amines through a C‐centered initiation.14
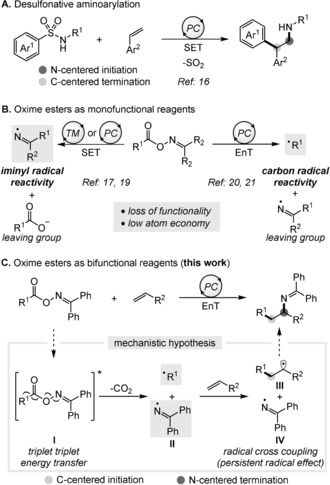
Similarly, N‐centered initiation often requires the use of excess alkene as N‐centered radicals exhibit higher reactivity and electrophilicity, which makes allylic hydrogen atom abstraction a kinetically competing process.15 To address this problem, Stephenson and co‐workers have targeted alkenes for single‐electron oxidation, followed by nucleophilic trapping with an arylsulfonylacetamide. In this approach, arylsulfonylacetamides were elegantly used as a single, bifunctional reagent for diastereoselective, intermolecular carboamination of alkenes by linear N‐addition (Figure 1 A).16
Figure 1.
Bifunctional reagents as both C‐ and N‐sources.
Unlike carboamination reactions, the carboimination of alkenes is still in its infancy and is mainly limited to intramolecular annulation reactions generating five‐ or six‐membered N‐heterocycles.17 Additionally, the high reactivity of iminyl radicals has been utilized in a plethora of 1,5‐hydrogen atom transfer (HAT) reactions.18, 19 In all of these cases, iminyl radicals are first generated from oxime esters through a single electron transfer (SET) event (Figure 1 B, left).19 Alternatively, oxime esters have also been employed successfully in the photosensitized generation of iminyl radicals through N−O bond homolysis.20 Recently, we have utilized this distinctive energy transfer (EnT) pathway to develop a general radical decarboxylation process applicable to both aromatic and aliphatic carboxylic acids (Figure 1 B, right).21 Nevertheless, all of these approaches are plagued by poor atom economy as either the carboxylate unit or the iminyl unit always become part of the waste stream.
The advantageous generation of two different radicals from N−O bond homolysis, coupled with our continuous interest in EnT catalysis,22 prompted us to investigate whether both C‐centered and N‐centered radicals could effectively leverage the vicinal carboimination of alkenes (Figure 1 C). During our previous investigation concerning the decarboxylation of oxime esters,21 we realized relatively long lifetimes of the benzophenone iminyl radicals through the observation of iminyl byproducts. Considering the triplet energies of the oxime esters of aliphatic carboxylic acids (E T=45.4 kcal mol−1, Section S2.1 in the Supporting Information), we envisioned that a triplet–triplet energy transfer (TTEnT) from the excited photosensitizer (with E T>46 kcal mol−1) should be thermodynamically favored. The excited oxime ester I would participate in a concerted decarboxylation/fragmentation process to generate a C‐centered alkyl radical and N‐centered diphenyliminyl radical pair II.21, 23 The long lifetime of the diphenyliminyl radical IV should allow the transient alkyl radical to escape the solvent cage and add to the terminal position of the alkene to generate a stabilized radical III. Lastly, a highly selective radical–radical cross‐coupling between III and IV would be kinetically feasible based on the persistent radical effect (PRE).24 However, such a radical–radical cross‐coupling approach possesses numerous challenges connected to potentially unproductive pathways outperforming the desired reaction.
Herein, we report an unprecedented intermolecular radical carboimination of alkenes involving benzophenone oxime carboxylates as a bifunctional source of both N‐ and C‐centered radicals with excellent regioselectivity for linear C‐radical addition. In the majority of reported protocols, N‐centered radicals are known to be highly reactive species favoring linear N‐radical additions.11 In our study, the use of benzophenone oxime esters is key to the generation of a long‐lived iminyl radical in order to achieve the inverted selectivity. Benzophenone imines are synthetic equivalents to ammonia and have been used in a variety of transition‐metal‐catalyzed coupling reactions.25 The diarylmethylene protecting group can be easily removed by hydrolysis or transamination to release primary amines, or it can be converted into a wide range of valuable amine building blocks.26, 27 Importantly, the use of benzophenone‐based oxime esters is also crucial to shut down unwanted SET reduction pathways, owing to their high reduction potential (E irr red=−2.05 V; Section S2.2), which is inaccessible by most of the commonly used photo(redox) catalysts. The use of an EnT pathway is also advantageous for two reasons: 1) Two different radicals are concurrently generated in equal rates by N−O bond homolysis of the oxime ester, and 2) the overall decarboxylation/fragmentation and carboamination steps should be independent of the intrinsic regeneration/turnover of the catalyst in the absence of any redox events. Expediently, this method uses highly abundant and stable aliphatic carboxylic acids (via their oxime ester derivative) in a highly atom‐economic fashion, generating carbon dioxide as the sole byproduct.28
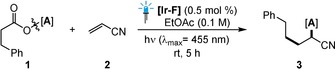
Upon exploring the various reaction parameters in line with our previous experience with oxime esters,21 we found that the oxime ester 1 a provided the desired carboimination product with acrylonitrile (2) when [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) ([Ir‐F], dF‐(CF3)ppy=2‐(2,4‐difluorophenyl)‐5‐trifluoromethylpyridine, dtbbpy=4,4′‐di‐tert‐butyl‐2,2′‐bipyridine) was used as the photosensitizer under irradiation with blue LEDs. The desired carboimination product 3 a was afforded in 57 % yield (1H NMR) with complete regioselectivity for the linear C‐radical addition (Table 1, entry 1). Encouraged by this, we evaluated a series of oxime esters with varying steric and electronic properties (entries 2–6). Pleasingly, simple benzophenone oxime ester 1 b was identified as the optimal choice in terms of reaction efficiency, affording the desired product 3 b in 71 % NMR yield. Control experiments with respect to the photosensitizer as well as light irradiation further demonstrated the necessity of each component. Additionally, to further identify the crucial parameters for our reaction, a systematic reaction‐parameter‐based sensitivity screen was performed (Table 1, radar diagram).29 Notably, the reaction was found to be sensitive towards low light intensity and high oxygen concentration, but robust in terms of the moisture sensitivity and temperature fluctuations (Section S3.2).
Table 1.
Effect of activators and sensitivity assessment.[a]
|
|
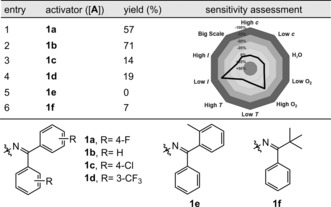
[a] Reaction conditions: 1 (0.15 mmol), 2 (0.10 mmol), [Ir‐F] (0.5 mol %) in ethyl acetate (1 mL), irradiation with 30 W blue LEDs (λ max=455 nm) under an argon atmosphere at room temperature for 5 h. Yields were determined by 1H NMR analysis using dibromomethane as an internal standard.
Notably, a critical distinction between an EnT and a hypothetical SET pathway is that the EnT‐induced reactivity should also be accessible by direct excitation with high‐energy light sources in the absence of any photocatalyst or electron source. Indeed, the desired carboiminated product 33 was observed upon direct photoirradiation with a 365 nm LED source, albeit in reduced yield (Section S2.3). Additionally, the use of different photocatalysts revealed a direct correlation of the product yield with the triplet energies of the photocatalysts (Section S2.4). All of these observations further substantiate the involvement of an EnT‐mediated carboimination process.
Next, with these optimized conditions in hand, the generality of the carboimination reaction was evaluated by exploring different substrates. First, we explored a range of alkenes with different electronic and steric properties (Table 2, compounds 4–18). Electron‐deficient alkenes such as acrylonitrile (4), vinyl ketone (5), and acrylate (15) reacted smoothly to provide the desired carboimination products. While styrenes bearing meta or para substituents worked efficiently, ortho‐substituted styrenes returned diminished yields (7–12). To our delight, electron‐poor nitrostyrene (11), pentafluorostyrene (13), as well as heterocyclic 2‐vinylpyridine (14) effectively delivered the desired products in good yields. Substituents at either the α‐ or the β‐position on the alkene could be accommodated (16–18) despite the greater steric demand and lower electrophilicity of these substrates, with a 1:1 diastereoselectivity for 17. Naturally occurring tulipalin A was successfully utilized to generate sterically congested imine 18 in moderate yield. However, cyclic alkenes and alkynes proved to be incompatible with our conditions.
Table 2.
Scope of the decarboxylative intermolecular carboimination of alkenes.[a]
|
|
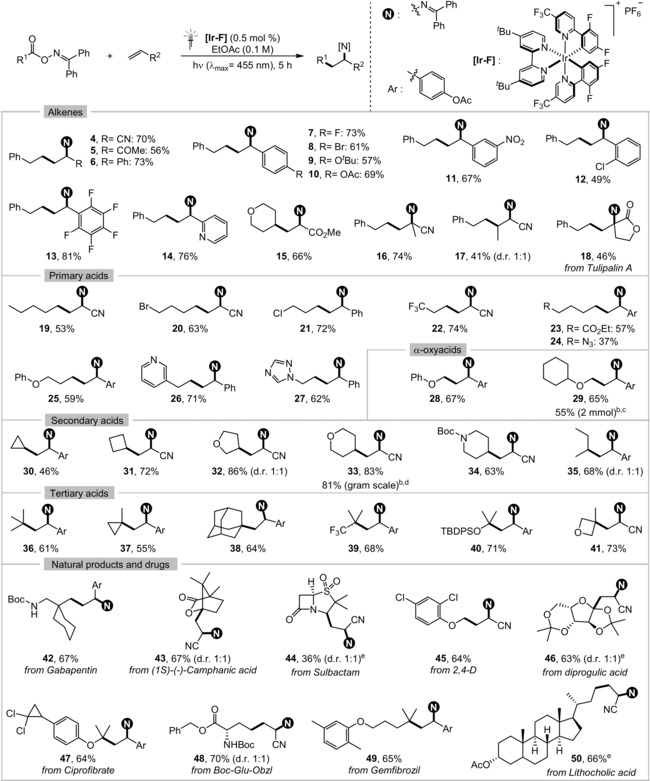
[a] Reaction conditions: alkene (0.30 mmol), oxime ester (0.45 mmol), [Ir‐F] (0.5 mol %) in ethyl acetate under an argon atmosphere, irradiation with 30 W blue LEDs (λmax=455 nm) at room temperature for 5 h. Yields of isolated products are given. [b] Reaction time: 16 h. [c] With 1.25 equiv of the oxime ester. [d] With 1.1 equiv of the oxime ester. [e] With 1.0 equiv of oxime ester.
Subsequently, we explored the carboxylic acid scope, including two large‐scale demonstrations. A plethora of carboxylic acids featuring primary, secondary, and tertiary carbon centers were found to be effective substrates for the carboimination reaction. Simple halides (20 and 21), trifluoromethyl (22), ester (23), azide (24), or ether (25) containing primary carboxylic acids were employed without any problem. Similarly, primary carboxylic acids comprising heterocycles such as pyridine (26) or 1,2,4‐triazole (27) were tolerated under the optimized conditions. Both aliphatic (28) and aromatic α‐oxy acids (29) could be applied successfully to form γ‐amino ethers. Different cyclic (30 and 31) and heterocyclic secondary carboxylic acids (32, 33, and 34) with varying ring sizes could be easily accommodated. Tertiary carboxylic acids starting from simple pivalic acid (36) to adamantane (38) or heterocyclic 3‐methyloxetane (41) derivatives reacted smoothly to generate quaternary carbon centers. Pleasingly, comparable reactivity was also observed in the case of sterically demanding acids (37–40). Unfortunately, carboiminations with oxime esters of aromatic carboxylic acids and phenylacetic acids were unsuccessful.
We next probed the range of densely functionalized substrates amenable to our reaction conditions. Pharmaceuticals and agrochemicals such as gabapentin, sulbactam, ciprofibrate, gemfibrozil, and 2,4‐D afforded the desired carboiminated products (42, 44, 45, 47, and 49) in synthetically useful yields. Naturally occurring carboxylic acids, such as diprogulic acid, lithocholic acid, chiral auxiliary (1S)‐(−)‐camphanic acid, and natural amino acid derived Boc‐Glu‐Obzl, also reacted smoothly (43, 46, 48, and 50).
Finally, after developing a convenient method for alkene carboimination, we sought to explore the reactivity and synthetic utility of the products of this reaction (Figure 2 A). Simple hydrolysis of the carboimination product 33 with 1.5 equiv of HCl in wet MeOH delivered the corresponding primary ammonium chloride salt in 89 % isolated yield (Figure 2 A, compound 51).25b Similarly, concomitant hydrolysis of both the nitrile and iminyl functional groupos produced the valuable α‐amino acid 52 in racemic fashion. Furthermore, the biologically relevant diarylmethylamine class27 of compounds could be easily generated from simple reduction of the iminyl moiety (53). Most interestingly, direct radical–radical cross‐coupled iminyl products were isolated in the absence of an alkene acceptor (Figure 2 B, compounds 54 and 55). Remarkably, the expensive iridium‐based photosensitizers could be replaced by simple organic sensitizers, albeit with diminished efficiency (Figure 2 C). As a result, a metal‐free, intermolecular carboimination process can be developed based on this study.
Figure 2.
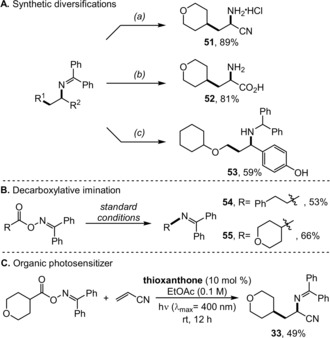
Synthetic utility and product diversification. Reagents and conditions: a) 1 n HCl (1.5 equiv) in MeOH; b) 6 n HCl; c) NaBH4 (3 equiv) in MeOH/CH2Cl2.
In summary, by using oxime esters of readily available alkyl carboxylic acids as bifunctional reagents, an intermolecular two‐component carboimination of olefins has been developed. A photosensitization/energy transfer strategy was used for effective generation of both C‐ and N‐centered radicals in a highly atom‐economic fashion. The longer lifetime of iminyl radicals underpins their exquisite regioselectivity. In addition, the mild reaction conditions, operational simplicity, and a broad scope provide an easy gateway towards unprotected alkyl amines.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Generous financial support from the Alexander von Humboldt Foundation (T.P.) and the Deutsche Forschungsgemeinschaft (Leibniz Award; SFB 858 for P.B. and SPP2102 for F.S.‐K.) are gratefully acknowledged. We thank Julian Heuer and Max Lübbesmeyer for experimental support and Frederik Sandfort, Toryn Dalton, Dr. Zackaria Nairoukh, and Dr. Anup Bhunia (all WWU Münster) for helpful discussions.
T. Patra, P. Bellotti, F. Strieth-Kalthoff, F. Glorius, Angew. Chem. Int. Ed. 2020, 59, 3172.
Dedicated to Professor Goutam K. Lahiri on the occasion of his 60th birthday
References
- 1.
- 1a. Chemler S. R., Fuller P. H., Chem. Soc. Rev. 2007, 36, 1153–1160; [DOI] [PubMed] [Google Scholar]
- 1b. Müller T. E., Hultzsch K. C., Yus M., Foubelo F., Tada M., Chem. Rev. 2008, 108, 3795–3892; [DOI] [PubMed] [Google Scholar]
- 1c. Lan X.-W., Wang N.-X., Xing Y., Eur. J. Org. Chem. 2017, 5821–5851; [Google Scholar]
- 1d. Giri R., Kc S., J. Org. Chem. 2018, 83, 3013–3022. [DOI] [PubMed] [Google Scholar]
- 2.For selected examples of alkene carboamination with intramolecular carbon sources, see:
- 2a. Li Y.-M., Wei X.-H., Li X.-A., Yang S.-D., Chem. Commun. 2013, 49, 11701–11703; [DOI] [PubMed] [Google Scholar]
- 2b. Matcha K., Narayan R., Antonchick A. P., Angew. Chem. Int. Ed. 2013, 52, 7985–7989; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8143–8147; [Google Scholar]
- 2c. Wu Z., Ren R., Zhu C., Angew. Chem. Int. Ed. 2016, 55, 10821–10824; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10979–10982. [Google Scholar]
- 3.For selected examples of alkene carboamination with intramolecular nitrogen sources, see:
- 3a. Rosewall C. F., Sibbald P. A., Liskin D. V., Michael F. E., J. Am. Chem. Soc. 2009, 131, 9488–9489; [DOI] [PubMed] [Google Scholar]
- 3b. Sunsdahl B., Smith A. R., Livinghouse T., Angew. Chem. Int. Ed. 2014, 53, 14352–14356; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14580–14584; [Google Scholar]
- 3c. Choi G. J., Knowles R. R., J. Am. Chem. Soc. 2015, 137, 9226–9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected examples of alkene carboamination with both intramolecular carbon and nitrogen sources, see:
- 4a. Yip K.-T., Yang M., Law K.-L., Zhu N.-Y., Yang D., J. Am. Chem. Soc. 2006, 128, 3130–3131; [DOI] [PubMed] [Google Scholar]
- 4b. Zeng W., Chemler S. R., J. Am. Chem. Soc. 2007, 129, 12948–12949; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Du W., Gu Q., Li Y., Lin Z., Yang D., Org. Lett. 2017, 19, 316–319. [DOI] [PubMed] [Google Scholar]
- 5. McDonald R. I., Liu G. S., Stahl S. S., Chem. Rev. 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Nakhla J. S., Kampf J. W., Wolfe J. P., J. Am. Chem. Soc. 2006, 128, 2893–2901; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Mai D. N., Wolfe J. P., J. Am. Chem. Soc. 2010, 132, 12157–12159; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Schultz D. M., Wolfe J. P., Synthesis 2012, 44, 351–361; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. White D. R., Hutt J. T., Wolfe J. P., J. Am. Chem. Soc. 2015, 137, 11246–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piou T., Rovis T., Nature 2015, 527, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Wang F., Qi X., Liang Z., Chen P., Liu G., Angew. Chem. Int. Ed. 2014, 53, 1881–1886; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1912–1917; [Google Scholar]
- 8b. Cheng J., Qi X., Li M., Chen P., Liu G., J. Am. Chem. Soc. 2015, 137, 2480–2483; [DOI] [PubMed] [Google Scholar]
- 8c. Wang D., Wu L., Wang F., Wan X., Chen P., Lin Z., Liu G., J. Am. Chem. Soc. 2017, 139, 6811–6814; [DOI] [PubMed] [Google Scholar]
- 8d. Zhang W., Chen P., Liu G., Angew. Chem. Int. Ed. 2017, 56, 5336–5340; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5420–5424. [Google Scholar]
- 9. Liu Z., Wang Y., Wang Z., Zeng T., Liu P., Engle K. M., J. Am. Chem. Soc. 2017, 139, 11261–11270. [DOI] [PubMed] [Google Scholar]
- 10. Lerchen A., Knecht T., Daniliuc C. G., Glorius F., Angew. Chem. Int. Ed. 2016, 55, 15166–15170; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15391–15395. [Google Scholar]
- 11.For examples of intermolecular carboaminations with N-radical initiation, see:
- 11a. Zhang H., Pu W., Xiong T., Li Y., Zhou X., Sun K., Liu Q., Zhang Q., Angew. Chem. Int. Ed. 2013, 52, 2529–2533; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2589–2593; [Google Scholar]
- 11b. Zhang Y., Liu H., Tang L., Tang H.-J., Wang L., Zhu C., Feng C., J. Am. Chem. Soc. 2018, 140, 10695–10699; [DOI] [PubMed] [Google Scholar]
- 11c. Jiang H., Seidler G., Studer A., Angew. Chem. Int. Ed. 2019, 58, 16528–16532; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16680–16684. [Google Scholar]
- 12.For examples of intermolecular carboamination with C-radical initiation, see:
- 12a. Fumagalli G., Boyd S., Greaney M. F., Org. Lett. 2013, 15, 4398–4401; [DOI] [PubMed] [Google Scholar]
- 12b. Yasu Y., Koike T., Akita M., Org. Lett. 2013, 15, 2136–2139; [DOI] [PubMed] [Google Scholar]
- 12c. Prasad Hari D., Hering T., König B., Angew. Chem. Int. Ed. 2014, 53, 725–728; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 743–747; [Google Scholar]
- 12d. Liu Y.-Y., Yang X.-H., Song R.-J., Luo S., Li J.-H., Nat. Commun. 2017, 8, 14720; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12e. Qian B., Chen S., Wang T., Zhang X., Bao H., J. Am. Chem. Soc. 2017, 139, 13076–13082. [DOI] [PubMed] [Google Scholar]
- 13. Gockel S. N., Buchanan T. L., Hull K. L., J. Am. Chem. Soc. 2018, 140, 58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Legnani L., Prina-Cerai G., Delcaillau T., Willems S., Morandi B., Science 2018, 362, 434. [DOI] [PubMed] [Google Scholar]
- 15. Zard S. Z., Chem. Soc. Rev. 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- 16. Monos T. M., McAtee R. C., Stephenson C. R. J., Science 2018, 361, 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Cai S.-H., Xie J.-H., Song S., Ye L., Feng C., Loh T.-P., ACS Catal. 2016, 6, 5571–5574; [Google Scholar]
- 17b. Davies J., Sheikh N. S., Leonori D., Angew. Chem. Int. Ed. 2017, 56, 13361–13365; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13546–13550; [Google Scholar]
- 17c. Jiang H., Studer A., Angew. Chem. Int. Ed. 2017, 56, 12273–12276; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12441–12444. [Google Scholar]
- 18. Mirjafary Z., Abdoli M., Saeidian H., Boroon S., Kakanejadifard A., RSC Adv. 2015, 5, 79361–79384. [Google Scholar]
- 19.
- 19a. Davies J., Morcillo S. P., Douglas J. J., Leonori D., Chem. Eur. J. 2018, 24, 12154–12163. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Hasebe M., Kogawa K., Tsuchiya T., Tetrahedron Lett. 1984, 25, 3887–3890; [Google Scholar]
- 20b. McBurney R. T., Walton J. C., J. Am. Chem. Soc. 2013, 135, 7349–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patra T., Mukherjee S., Ma J., Strieth-Kalthoff F., Glorius F., Angew. Chem. Int. Ed. 2019, 58, 10514–10520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10624–10630. [Google Scholar]
- 22.
- 22a. Strieth-Kalthoff F., James M. J., Teders M., Pitzer L., Glorius F., Chem. Soc. Rev. 2018, 47, 7190–7202; [DOI] [PubMed] [Google Scholar]
- 22b. Teders M., Henkel C., Anhäuser L., Strieth-Kalthoff F., Gómez-Suárez A., Kleinmans R., Kahnt A., Rentmeister A., Guldi D., Glorius F., Nat. Chem. 2018, 10, 981–988. [DOI] [PubMed] [Google Scholar]
- 23. Soni V. K., Lee S., Kang J., Moon Y. K., Hwang H. S., You Y., Cho E. J., ACS Catal. 2019, 9, 10454–10463. [Google Scholar]
- 24.
- 24a. Fischer H., Chem. Rev. 2001, 101, 3581–3610; [DOI] [PubMed] [Google Scholar]
- 24b. Leifert D., Studer A., Angew. Chem. Int. Ed. 2020, 59, 74–108; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 74–110. [Google Scholar]
- 25.
- 25a. Wolfe J. P., Åhman J., Sadighi J. P., Singer R. A., Buchwald S. L., Tetrahedron Lett. 1997, 38, 6367–6370; [Google Scholar]
- 25b. Peacock D. M., Roos C. B., Hartwig J. F., ACS Cent. Sci. 2016, 2, 647–652; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Mao R., Balon J., Hu X., Angew. Chem. Int. Ed. 2018, 57, 9501–9504; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9645–9648. [Google Scholar]
- 26.
- 26a. Kong D., Li M., Zi G., Hou G., He Y., J. Org. Chem. 2016, 81, 6640–6648; [DOI] [PubMed] [Google Scholar]
- 26b. Sakamoto R., Inada T., Sakurai S., Maruoka K., Org. Lett. 2016, 18, 6252–6255; [DOI] [PubMed] [Google Scholar]
- 26c. Kumarasamy E., Kandappa S. K., Raghunathan R., Jockusch S., Sivaguru J., Angew. Chem. Int. Ed. 2017, 56, 7056–7061; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7162–7167; [Google Scholar]
- 26d. Wang R., Ma M., Gong X., Fan X., Walsh P. J., Org. Lett. 2019, 21, 27–31. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Giardina G. A. M., Raveglia L. F., Grugni M., Sarau H. M., Farina C., Medhurst A. D., Graziani D., Schmidt D. B., Rigolio R., Luttmann M., Cavagnera S., Foley J. J., Vecchietti V., Hay D. W. P., J. Med. Chem. 1999, 42, 1053–1065; [DOI] [PubMed] [Google Scholar]
- 27b. Cai Q., Sun H., Peng Y., Lu J., Nikolovska-Coleska Z., McEachern D., Liu L., Qiu S., Yang C.-Y., Miller R., Yi H., Zhang T., Sun D., Kang S., Guo M., Leopold L., Yang D., Wang S., J. Med. Chem. 2011, 54, 2714–2726; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. He S., Xiao J., Dulcey A. E., Lin B., Rolt A., Hu Z., Hu X., Wang A. Q., Xu X., Southall N., Ferrer M., Zheng W., Liang T. J., Marugan J. J., J. Med. Chem. 2016, 59, 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Rodríguez N., Goossen L. J., Chem. Soc. Rev. 2011, 40, 5030–5048; [DOI] [PubMed] [Google Scholar]
- 28b. Johnston C. P., Smith R. T., Allmendinger S., MacMillan D. W. C., Nature 2016, 536, 322–325; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28c. Qin T., Cornella J., Li C., Malins L. R., Edwards J. T., Kawamura S., Maxwell B. D., Eastgate M. D., Baran P. S., Science 2016, 352, 801–805; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28d. Patra T., Maiti D., Chem. Eur. J. 2017, 23, 7382–7401; [DOI] [PubMed] [Google Scholar]
- 28e. Zhao W., Wurz R. P., Peters J. C., Fu G. C., J. Am. Chem. Soc. 2017, 139, 12153–12156; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28f. Murarka S., Adv. Synth. Catal. 2018, 360, 1735–1753; [Google Scholar]
- 28g. Schwarz J., König B., Green Chem. 2018, 20, 323–361; [Google Scholar]
- 28h. Yatham V. R., Bellotti P., König B., Chem. Commun. 2019, 55, 3489–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pitzer L., Schäfers F., Glorius F., Angew. Chem. Int. Ed. 2019, 58, 8572–8576; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8660–8664. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary